

Abstract
Many immune diseases occur at different rates among people with schizophrenia compared to the general population. Here, we evaluated whether this phenomenon might be explained by shared genetic risk factors. We used data from large genome-wide association studies to compare the genetic architecture of schizophrenia to 19 immune diseases. First, we evaluated the association with schizophrenia of 581 variants previously reported to be associated with immune diseases at genome-wide significance. We identified five variants with potentially pleiotropic effects. While colocalization analyses were inconclusive, functional characterization of these variants provided the strongest evidence for a model in which genetic variation at rs1734907 modulates risk of schizophrenia and Crohn’s disease via altered methylation and expression of EPHB4—a gene whose protein product guides the migration of neuronal axons in the brain and the migration of lymphocytes towards infected cells in the immune system. Next, we investigated genome-wide sharing of common variants between schizophrenia and immune diseases using cross-trait LD score regression. Of the 11 immune diseases with available genome-wide summary statistics, we observed genetic correlation between six immune diseases and schizophrenia: inflammatory bowel disease (rg = 0.12 ± 0.03, P = 2.49 × 10−4), Crohn’s disease (rg = 0.097 ± 0.06, P = 3.27 × 10−3), ulcerative colitis (rg = 0.11 ± 0.04, P = 4.05 × 10–3), primary biliary cirrhosis (rg = 0.13 ± 0.05, P = 3.98 × 10−3), psoriasis (rg = 0.18 ± 0.07, P = 7.78 × 10–3) and systemic lupus erythematosus (rg = 0.13 ± 0.05, P = 3.76 × 10–3). With the exception of ulcerative colitis, the degree and direction of these genetic correlations were consistent with the expected phenotypic correlation based on epidemiological data. Our findings suggest shared genetic risk factors contribute to the epidemiological association of certain immune diseases and schizophrenia.
Introduction
Despitex recent advances in identifying key biomarkers and genetic loci for schizophrenia, its pathophysiology remains poorly understood (1,2). One interesting epidemiological observation is that the risk of developing many immune-mediated diseases is increased among patients with schizophrenia (3–5) and vice versa (6,7). Here, we use the term immune disease to broadly encompass both autoimmune and inflammatory disorders. While there are discrepancies among studies regarding which immune diseases are most strongly correlated with schizophrenia, there is converging evidence that these diseases co-occur at a greater rate than is expected by chance (3–7). A notable exception is rheumatoid arthritis (RA), where a consistent inverse association with schizophrenia has been observed (5,8).
Genetic factors have long been proposed as an explanation for the differing prevalence of immune diseases among patients with schizophrenia compared to the general population (5,6). The recently reported role of complement component 4 (C4) variation in schizophrenia (9) illustrates a potential shared genetic mechanism in the development of immune and psychiatric disorders. Genetic variants conferring increased C4 expression protect against developing systemic lupus erythematosus (SLE), possibly by increased tagging of apoptotic cells—which are the trigger for autoantibody development in SLE—leading to more effective clearance by macrophages (10). The same genetic mechanism may increase the risk of developing schizophrenia, by increased tagging of neuronal synapses for elimination by microglia leading to excessive synaptic pruning (9). We hypothesize that similar shared genetic mechanisms may occur throughout the genome, with cellular manifestations in immune cells and neurons influencing the development of immune and psychiatric disorders, respectively. Previously, we found that susceptibility to schizophrenia does not appear to be driven by the broad set of loci harboring immune genes (11). However, not all genetic variants conferring risk of immune disease fall within immune loci. Here, we evaluated whether common genetic variants influencing the risk of 19 different immune diseases may also be involved in schizophrenia.
Our cross-disorder genetic approach is supported by recent successes in identifying shared genetic risk variants (pleiotropy) across a variety of human diseases (12–18). The biological interpretation of pleiotropy is challenging, given the various molecular mechanisms that can drive shared genetic risk variants across complex traits. For instance, pleiotropy can result from a single-nucleotide polymorphism (SNP) independently influencing two unrelated traits (horizontal pleiotropy), a SNP influencing one trait that is related to or a risk factor for additional traits (vertical pleiotropy), or a SNP influencing one trait that has a high rate of misclassification with or represents a subgroup of a second trait (clinical heterogeneity) (19,20). Here, we use the term pleiotropy to refer broadly to a single genetic variant affecting multiple traits, regardless of the underlying molecular basis and biological implications. Pleiotropy in this broad sense is emerging as a pervasive phenomenon in the human genome (21–23), and cross-disorder studies characterizing the nature of genotype–phenotype relationships have the potential to yield significant insights into disease etiology. For instance, cross-trait genetic analyses have shed new light on cardiovascular disease and lipid biology, shifting attention away from high-density lipoprotein cholesterol (HDL) as a potential treatment target by demonstrating that genetically increased HDL levels do not reduce the risk of myocardial infarction (14). In psychiatry, cross-disorder analyses have identified pleiotropic variants between schizophrenia, bipolar disorder and major depressive disorder, indicating that these diseases are not as distinct at a pathophysiological level as current diagnostic criteria suggest (12,13,24).
While previous studies have investigated genome-wide pleiotropy between schizophrenia and immune disorders, results have been inconsistent (Supplementary Data, Table S1). Genetic correlation has been reported between schizophrenia and Crohn’s disease [CRO, (25–29)], multiple sclerosis [MS, (30)], primary biliary cirrhosis [PBC, (27)], psoriasis [PSO, (27,31)], RA (25,26), SLE (26,27), type 1 diabetes [T1D, (25)] and ulcerative colitis [UC, (26–29)] in some studies, but not in others (8,13,16,26,32). Interestingly, negative genetic correlation (whereby genetic risk protects against developing schizophrenia) has also been reported for RA (33), in keeping with the inverse epidemiological association (5,8). Potential explanations for these inconsistent results across studies include differences in statistical power due to diverse methodologies and sample sizes (Supplementary Data, Table S1) and varying degrees of influence by confounding variables such as population stratification and linkage disequilibrium (LD). Furthermore, although immune diseases have a significant sex bias with women at greater risk overall (34), potential sex-specific effects have not been explored in cross-trait analyses to date. If sex-specific effects are present, differences in the proportion of male:female samples across genome-wide association studies (GWAS) may also contribute to some of the differences across studies.
Additional studies are needed to reconcile the inconsistencies in existing cross-trait analyses of schizophrenia and immune disorders, with careful attention towards potential confounding variables (e.g. population stratification, LD, non-independence of GWAS samples and sex-specific effects). To this end, we have performed a comprehensive cross-disorder analysis of schizophrenia and 19 immune diseases, using data from large genetic studies in European samples. Our findings add to a growing body of literature supporting pervasive pleiotropy between schizophrenia and immune diseases. We extend existing literature by including 10 immune diseases that have not previously been compared with schizophrenia, prioritizing pleiotropic genes through integrative analyses of multi-omics data and estimating how much of the phenotypic correlation between schizophrenia and immune diseases was explained by the genetic correlations we observed.
Results
Defining immune risk variants
We identified immune-mediated diseases with robust GWAS findings using ImmunoBase (http://www.immunobase.org; accessed 7 June 2015), an online resource providing curated GWAS data for immune-related human diseases. These included the following 19 diseases: alopecia areata (AA), ankylosing spondylitis (AS), autoimmune thyroid disease (ATD), celiac disease (CEL), CRO, inflammatory bowel disease (IBD), juvenile idiopathic arthritis (JIA), MS, narcolepsy (NAR), PBC, primary sclerosing cholangitis (PSC), PSO, RA, Sjögren’s syndrome (SJO), SLE, systemic sclerosis (SSC), T1D, ulcerative colitis (UC) and vitiligo (VIT). Notably, the majority of IBD risk variants were also risk variants for CRO and/or UC. For 11 of these immune diseases (see Table 1), we also obtained full GWAS summary statistics allowing us to conduct additional cross-trait linkage disequilibrium score regression (LDSC) analyses (16).
Table 1.
Description of data sets analyzed
| Abr | Genome-wide significant SNPs a | Available GWAS summary statistics b | Cases | Controls | Number of SNPs with available summary statistics | |
|---|---|---|---|---|---|---|
| Schizophrenia | SCZ | − | (1) | 35 476 | 46 839 | 9 444 230 |
| Bipolar disorder (+) | BP | − | (12) | 6990 | 4820 | 1 233 534 |
| Height (−) | HGT | − | (36) | 253 288 | − | 2 085 602 |
| Alopecia areata | AA | 11 | − | − | − | − |
| Ankylosing spondylitis | AS | 23 | − | − | − | − |
| Autoimmune thyroid disease | ATD | 7 | − | − | − | − |
| Celiac disease | CEL | 38 | (37) | 4533 | 10 750 | 523 398 |
| Crohn’s disease | CRO | 119 | (38) | 5956 | 14 927 | 12 276 506 |
| Inflammatory bowel disease | IBD | 145 | (38) | 12 882 | 21 770 | 12 716 150 |
| Juvenile idiopathic arthritis | JIA | 22 | − | − | − | − |
| Multiple sclerosis | MS | 103 | − | − | − | − |
| Narcolepsy | NAR | 3 | − | − | − | − |
| Primary biliary cirrhosis | PBC | 19 | (39) | 2764 | 10 475 | 1 038 537 |
| Primary sclerosing cholangitis | PSC | 12 | − | − | − | − |
| Psoriasis | PSO | 34 | (40) | 2178 | 5175 | 7 586 779 |
| Rheumatoid arthritis | RA | 77 | (41) | 5539 | 20 169 | 2 090 825 |
| Sjögren’s syndrome | SJO | 6 | − | − | − | − |
| Systemic lupus erythematosus | SLE | 19 | (42) | 4036 | 6959 | 7 915 251 |
| Systemic sclerosis | SSC | 4 | (43) | 1486c | 3 477c | 253 179 |
| Type 1 diabetes | T1D | 56 | (44) | 9934 | 16 956 | 1 943 760 |
| Ulcerative colitis | UC | 96 | (38) | 6968 | 20 464 | 12 255 263 |
| Vitiligo | VIT | 16 | (45) | 1381 | 14 518 | 8 790 155 |
aWe obtained lists of genome-wide significant SNPs for each immune disease from ImmunoBase and processed them as described in Supplementary Data.
bBecause genome-wide summary statistics were required for the LDSC analysis, we were unable to estimate genetic correlation with schizophrenia for eight immune diseases for which these data were not available (AA, AS, ATD, JIA, MS, NAR, PSC, SJO); −, negative control; +, positive control.
cOnly the US cohort from this study was available for analysis; Abr, abbreviation; −, not analyzed.
Given that human leukocyte antigen (HLA) alleles within the major histocompatibility complex (MHC) region (chromosome 6: 25–34 Mb) account for a significant proportion of heritability of immune and inflammatory disorders (35), we considered HLA and non-HLA risk variants separately in our analyses. Within the MHC region, we considered only the most strongly associated HLA variant (including SNPs, imputed HLA amino acid sites and classical alleles) for each disease based on univariate analysis in previously published studies (see Table 2), because multivariate conditional analyses reporting adjusted effect sizes of independent HLA variants were not available for all immune diseases. Outside of the MHC region, we considered all non-HLA variants curated in ImmunoBase for each of the 19 immune diseases.
Table 2.
Association of top HLA variants for immune diseases in schizophrenia
| Immune Disease | Schizophrenia | |||||
|---|---|---|---|---|---|---|
| Disease | HLA variant | P | OR | P | OR | r 2 with top SCZ SNP a |
| AA (46) | HLA-DRB1#37Asn | 4.99×10 −73 | 0.42 | 4.85×10 −9 | 0.91 | 0.04 |
| AS (47) | HLA-B*27 | <1×10−100 | 46 | 0.13 | 1.05 | 0 |
| ATD (48) | rs2281388 (tags HLA-DPB1*05:01) | 1.50×10−65 | 1.64 | 0.39 | 1.04b | 0 |
| CEL (49) | HLA-DQB1#74Ala | n.r. | 2.14 | 2.16×10 −12 | 0.89 | 0.11 |
| CRO (50) | HLA-DRB1*01:03 | 3.00×10−62 | 2.51 | 0.61 | 0.96 | 0 |
| IBD (50) | HLA-DRB1*01:03 | 1.93×10−112 | 3.01 | 0.61 | 0.96 | 0 |
| JIA (51) | rs7775055 | 3.14×10−174 | 6.01 | 0.12 | 0.94 | 0 |
| MS (52) | HLA-DRB1*15:01 | 1.40×10−234 | 2.92 | 5.10×10−3 | 1.06 | 0 |
| NAR (53) | HLA-DQB1*06:02 | 1.04×10−120 | 251 | 7.30×10−3 | 1.06 | 0 |
| PBC (54) | HLA-DQA1*04:01 | 5.90×10−45 | 3.06 | 0.20 | 0.95 | 0 |
| PSC (55) | HLA-B*08:01 | 3.70×10 −246 | 2.82 | 5.65×10 −16 | 0.84 | 0.2 |
| PSO (56) | HLA-C*06:02 | 2.10×10−201 | 3.26 | 0.55 | 0.99 | 0 |
| RA (57) | HLA-DRB1#11Val | <1×10−581 | 3.80 | 2.68×10−4 | 1.07 | 0 |
| SJO (58) | HLA-DQB1*02:01 | 1.38×10 −95 | 3.36 | 3.84×10 −15 | 0.85 | 0.11 |
| SLE (59) | HLA-DRB1#13Arg | 7.99×10−10 | 1.55c | 5.81×10−4 | 1.07 | 0 |
| SSC (60) | rs17500468 (TAP2) | 5.87×10−62 | 2.87 | 6.76×10−4 | 1.07 | 0 |
| T1D (61) | HLA-DQB1#57Ala | <1×10−1000 | 5.17 | 7.80×10−4 | 0.95 | 0.06 |
| UC (50) | rs6927022 | 8.00×10−154 | 1.49 | 3.37×10−4 | 1.06 | 0.03 |
| VIT (45) | rs9271597 (4.7kb upstream of HLA-DQA1) | 3.15×10−89 | 1.77 | 0.01 | 1.04 | 0 |
ar2 with rs1233578, the top HLA variant in schizophrenia, was obtained from the GAIN schizophrenia cohort (mgs2).
bEffect size estimate is for HLA-DPB1*05:01.
cEffect size estimate obtained from Asian sample. n.r., not reported; disease abbreviations as defined in Table 1. Bold font indicates statistically significant association with schizophrenia.
The number of genome-wide significant non-HLA risk loci for each of the 19 immune diseases varied from three (NAR) to 144 (IBD). Several variants were associated with more than one immune disease. In total, we identified 581 unique variants (563 non-HLA variants and 18 HLA variants) associated with any immune disease at genome-wide significance. We refer to these variants as immune risk variants. For the complete list of non-HLA immune risk variants, see Supplementary Data, Table S2.
Identifying pleiotropic variants implicated in both immune disease and schizophrenia
First, we evaluated whether there was any evidence of overall risk allele sharing between each of the 19 immune diseases and schizophrenia using a binomial sign test. To do this, we used previously published findings from a GWAS conducted by the Schizophrenia Working Group of the Psychiatric Genomics Consortium (1,11). This GWAS represented a meta-analysis of 52 cohorts, comprising a total of 35 476 cases and 46 839 controls and the full data set is referred to here as the PGC2 study. Overall, the direction of effect for the sets of non-HLA SNPs associated with each of the 19 immune diseases at genome-wide significance was not shared with schizophrenia more than expected by chance (all binomial sign test P > 0.05; Supplementary Data, Fig. S1). Thus, we did not observe evidence of risk allele sharing between any immune disease and schizophrenia when using a stringent genome-wide significance threshold to define immune risk variants. We also evaluated the collective association of 261 LD-independent, non-HLA immune risk variants associated with at least one of the 19 immune-mediated diseases, for which LD score and minor allele frequency (MAF) information were available in the European LD score database (16). We found significant deviation from the theoretical null in schizophrenia for immune risk SNPs (λ = 1.46). However, when we compared the association of immune risk SNPs to that of similar randomly selected SNP sets (Supplementary Material), we observed no evidence of enrichment (P = 0.66; Supplementary Data, Fig. S2), indicating that immune risk SNPs were not associated with schizophrenia more than expected by chance given the polygenic nature of schizophrenia.
Next, we identified potential pleiotropic variants by evaluating the association of individual immune risk variants with schizophrenia. We considered SNPs associated with schizophrenia at P < 8.6×10−5 (Bonferroni correction for 581 tests, 563 non-HLA and 18 HLA variants) to have pleiotropic effects. Given the size of the schizophrenia GWAS, we had over 80% power to detect pleiotropic SNPs assuming an OR≥1.12 in schizophrenia.
Within the MHC region, we observed four HLA risk alleles associated with both immune disease and schizophrenia, particularly in the class II HLA region (Table 2; Supplementary Data, Fig. S3). These HLA risk alleles were the strongest MHC region associations for AA (HLA-DRB1 #37 Asn), CEL (HLA-DQB1 #74 Ala), PSC (HLA-B*08:01) and SJO (HLA-DQB1*02:01). The presence of HLA-DRB1 #37 Asn conferred a protective association in both AA and schizophrenia, but the remaining HLA variants showed the opposite direction of effect in schizophrenia compared to immune disease (Table 2; Supplementary Data, Fig. S3). Notably, none of these four HLA variants were significantly associated with schizophrenia in previous conditional analyses (9,11), suggesting that their association with schizophrenia may be driven by LD with other causal variants in the region rather than true pleiotropy. Thus, we did not focus additional analyses on these variants.
Outside of the MHC region, five immune risk variants showed potential pleiotropic effects, with the risk allele for immune disease also conferring risk for schizophrenia (for regional association plots in schizophrenia and the immune diseases of interest, see Supplementary Data, Fig. S4). These variants have been previously implicated in CRO (rs6738825, rs13126505, rs1734907), MS (rs7132277) and CEL (rs296547). To evaluate the pleiotropic potential of these non-HLA variants, we performed Bayesian colocalization analysis (62) to estimate the posterior probability (PP) that the corresponding regional associations were driven by the same SNP in both schizophrenia and the immune disease of interest. We used available summary statistics from the PGC2 GWAS and the GWAS reporting these immune risk variants as genome-wide significant in CRO (63,64), MS (65) and CEL (37). Notably, the immune disease GWAS summary statistics were not imputed [total number of SNPs: 953 241 (63), 25 075 (64), 155 756 (65) and 523 398 (37)]. Low density SNP coverage across the genome results in a loss of statistical power to detect colocalization, as the true causal variants are unlikely to be genotyped (62). We observed statistically significant colocalization (PP > 0.50) in the regions of association on chromosomes 2, 4 and 12. Only the region on chromosome 2 colocalized to the immune SNP of interest, rs6738825 (PP = 0.23, Table 3); this region also colocalized to rs7587251 (PP = 0.25), a SNP in high LD with rs6738825 [r2 = 0.98 in 1000 Genomes Phase 3 CEU Population (66)]. We did not observe statistically significant colocalization in the region of association on chromosome 1 (PP = 0.06) or chromosome 7 (PP = 0.05, Table 3). Given that our colocalization analyses were susceptible to type II error for the reasons discussed above, we cannot exclude the possibility of a shared variant underlying the associations seen in schizophrenia and immune diseases for these regions.
Table 3.
Immune disease risk SNPs showing potentially pleiotropic effects in schizophrenia
| SNP (chr:bp) | Immune disease | Risk/non-risk allele | Immune OR (95% CI); P a | Schizophrenia OR (95% CI); P | Nearby genes | Colocalization b | Conditional analysis c | eQTL d | mQTL e | Genomic associations colocalizing to this gene f |
|---|---|---|---|---|---|---|---|---|---|---|
| rs296547g (chr1:200892137) | CEL (37) | G/A | 1.12 (1.09–1.16); 4.11×10−9 |
1.04 (1.02–1.07); 6.17×10−5 |
CAMSAP2
C1orf106 KIF21B CACNA1S ASCL5 |
PPregional=0.06 PPrs296547<10−20 |
rs111530734, P = 1.2×10−3 | n.s. | C1orf106, decreased methylation | SCZ-mQTL |
| rs6738825 (chr2: 198896895) | CRO (63) | A/G | 1.06 (1.02–1.11); 3.50×10−9 | 1.05 (1.03–1.07); 3.02×10−6 | SF3B1 COQ10B HSPD1 MOB4 HSPE1 RFTN2 MARS2 BOLL PLCL1 | PPregional=0.57 PPrs6738825=0.23 |
rs111744017, P=8.0×10−6 |
PLCL1, increased expression |
PLCL1, decreased methylation RFTN2, decreased methylation |
SCZ-mQTL, SCZ-eQTL SCZ-mQTL |
| rs13126505 (chr4:102865304) | CROh (64) | A/G | 1.17 (1.10–1.25); 2.33×10−10 |
1.14 (1.10-1.19); 1.19×10−8 |
BANK1
SLC39A8 NFKB1 |
PPregional=0.99 PPrs13126505<10−100 |
rs35225200, P=1.8×10−3 | SLC39A8, decreased expression | SLC39A8, increased methylation | SCZ-eQTL, SCZ-mQTL |
| rs1734907 (chr7:100315517) | CROh (64) | A/G | 1.16 (1.11–1.21); 1.67×10−13 |
1.07 (1.04–1.10); 7.55×10−6 |
TFR2
ACTL6B GNB2 GIGYF1 POP7 EPO ZAN EPHB4 SLC12A9 |
PPregional=0.05 PPrs1734907l<10−30 |
rs11539289, P=1.8×10−3 |
EPHB4, decreased expression TRIP6, decreased expression |
EPHB4, increased methylation TRIP6, inconsistent effect across probes |
SCZ-eQTL, SCZ-mQTL, eQTL-mQTL SCZ-eQTL, eQTL-mQTL |
| rs7132277 (chr12:123593382) |
MS (65) | A/G | 1.12 (n.r.); 1.90×10−13 |
1.07 (1.04–1.09); 2.52×10−6 |
ABCB9
ARL6IP4 MIR4304 OGFOD2 PITPNM2 MPHOSPH9 CDK2AP1 SBNO1 |
PPregional=0.94 PPrs7132277<10−30 |
rs74240770, P1.0×10−8 |
ABCB9, increased expression ARL6IP4, decreased expression MPHOSPH9, increased expression |
ABCB9, increased methylation PITPNM2, decreased methylation |
SCZ-eQTL, SCZ-mQTL, eQTL-mQTL |
aEffect sizes and P-values reported based on Immunobase curation, which reports statistics from meta-analysis of discovery and replication data sets where available;
bResults of Bayesian colocalization analyses using the coloc2 method (62), reported as posterior probabilities for each region of association (PPregional) and for each immune risk variant (PPrsid);
cResults of conditional analyses using COJO (67), reported as the top SNP association remaining in the region after conditioning on the immune risk variant of interest. P>8.6×10−5 indicates no significant associations remain after conditioning on the immune risk variant.
deQTL data was obtained from the CAGE study (72) that measured gene expression in peripheral blood. Effect on expression (increased/decreased) corresponds to the risk allele.
emQTL data was obtained from a meta-analysis of the Brisbane Systems Genetics Study (70) and Lothian Birth Cohorts of 1921 and 1936 (71), which measured DNA methylation in peripheral blood. Effect on expression (increased/decreased) corresponds to the risk allele.
fSignificant SMR and HEIDI (68,69) results indicating colocalization of genomic associations with the gene of interest in schizophrenia-eQTL (SCZ-eQTL), schizophrenia-mQTL (SCZ-mQTL) and eQTL-mQTL (eQTL-mQTL) data sets.
geQTL data were unavailable for rs296547, and rs404339 was used as a proxy SNP (r2=0.85 in 1000 Genomes Phase 3 CEU Population) (66).
hAlso associated with inflammatory bowel disease; n.s., no statistically significant findings; disease abbreviations as defined in Table 1.
Next, we used conditional and joint analysis (COJO) (67) to perform association analyses in the PGC2 schizophrenia GWAS conditioning on each of the five immune risk variants (Supplementary Data, Fig. S5). If the immune risk variants (or SNPs in high LD with them) accounted for the regional associations seen in schizophrenia, no significant associations should remain after conditioning on these variants (statistically, all P > 8.6×10−5). We observed no remaining associations with schizophrenia after conditioning on rs296547 (top SNP after conditioning: rs111530734, P = 1.19×10−3), rs1734907 (top SNP after conditioning: rs11768688, P = 9.79×10−4) and rs13126505 (top SNP after conditioning: rs112786981, P = 4.58×10−4). Significant associations with schizophrenia remained after conditioning on rs6738825 (top SNP after conditioning: rs111744017, P = 8.03×10−6) and rs7132277 (top SNP after conditioning: rs74240770, P = 1.37×10−8), suggesting there may be additional independent causal variants contributing to the associations in these regions for schizophrenia.
In order to prioritize genes underlying the five potentially pleiotropic SNPs, we performed an integrative analysis of GWAS summary statistics with methylation quantitative trait loci (mQTL) and expression quantitative trait loci (eQTL) studies using Summary-data-based Mendelian Randomization (SMR) and heterogeneity in dependent instruments (HEIDI) (68,69) (Materials and Methods). Briefly, we obtained summary-level mQTL and eQTL SNP data described in Wu et al. (69). The mQTL data were from 1 980 individuals with DNA methylation measured in peripheral blood (70,71), and eQTL data were from 2 765 individuals with gene expression levels measured in peripheral blood (72). Notably, rs296547 was not genotyped in the eQTL data set, and we used rs404339 as a proxy SNP [r2 = 0.85 in 1000 Genomes Phase 3 CEU Population (66)] in SMR analyses of gene expression for rs296547.
We observed that rs1734907, a SNP associated with both CRO and schizophrenia, was an mQTL (β = 0.47, P = 2.13×10−26) and eQTL (β = −0.24, P = 3.54×10−10) for EPHB4 in peripheral blood (Supplementary Data, Table S3; Fig. 1). rs1734907 showed consistent pleiotropic associations with schizophrenia and EPHB4 DNAm (βSMR = −0.14, pSMR = 3.58×10−5, pHEIDI = 0.12), schizophrenia and EPHB4 expression (βSMR = −0.28, pSMR = 2.63×10−4, pHEIDI = 0.17) and EPHB4 DNAm and EPHB4 expression (βSMR = 1.98, pSMR = 6.56×10−8, pHEIDI = 0.011). These consistent associations across molecular phenotypes and schizophrenia at the EPHB4 locus suggest EPHB4 may be driving the association of rs1734907 in schizophrenia (Fig. 1). Notably, TRIP6 is also a candidate functional gene underlying the association of rs1734907 with schizophrenia. We observed pleiotropic association for rs1734907 with schizophrenia and TRIP6 DNAm with inconsistent direction of effect (βSMR = 0.15, pSMR = 5.00×10−5, pHEIDI = 0.17 for probe cg18683606; βSMR = −0.12, pSMR = 2.32×10−5, pHEIDI = 0.18 for probe cg27396824), a trend for association with schizophrenia and TRIP6 expression (βSMR = −0.33, pSMR = 6.38×10−4, pHEIDI = 0.14), but no significant association with TRIP6 DNAm and TRIP6 expression. Colocalization analyses were inconclusive with respect to rs1734907 showing pleiotropic association with schizophrenia and CRO as described above, while conditional analyses suggested this variant explained the regional association seen on chromosome 7 in schizophrenia (Table 3).
Figure 1.

Prioritizing genes driving the pleiotropic association of rs1734907 in CRO and schizophrenia. Associations for SNP and SMR analyses across GWAS, eQTL and mQTL data sets for rs1734907. Top plot gray circles illustrate SNP association (−log10P-value) with schizophrenia in the PGC2 GWAS, while pink diamonds and blue circles indicate results of SMR tests (−log10P-value) for association of gene expression and DNAm with schizophrenia, respectively, with solid shading indicating probes passing the HEIDI test. Middle plot illustrates SNP association (−log10P-value) with gene expression from peripheral blood eQTL data set. Lower plots illustrate SNP association (−log10P-value) with gene methylation from peripheral blood mQTL data set.
We also observed that rs7132277, a SNP associated with both MS and schizophrenia (Table 3), was an mQTL (β = 0.27, P = 2.87×10−11) and eQTL (β = 0.32, P = 5.23×10−19) for ABCB9 in peripheral blood (Supplementary Data, Table S3). Furthermore, we observed consistent pleiotropic associations for rs7132277 with schizophrenia and ABCB9 DNAm (βSMR = −0.24, pSMR = 1.20×10−4, pHEIDI = 0.55), schizophrenia and ABCB9 expression (βSMR = 0.20, pSMR = 3.10×10−5, pHEIDI = 0.17) and ABCB9 DNAm and ABCB9 expression (βSMR = −0.83, pSMR = 9.81×10−8, pHEIDI = 0.48). Thus, there was consistent association across molecular phenotypes and schizophrenia at the ABCB9 locus, suggesting this gene may be driving the association of rs7132277 in schizophrenia. Although the region of association on chromosome 12 showed statistically significant evidence of colocalization in schizophrenia and MS, rs7132277 was not the pleiotropic variant driving this association (Table 3). Thus, our analyses highlight ABCB9 as a candidate gene underlying the association of rs7132277 in schizophrenia, but do not implicate this SNP as a pleiotropic immune variant.
The other potentially pleiotropic SNPs did not demonstrate consistent localization to a particular gene across traits and molecular phenotypes (Table 3; Supplementary Data, Table S3).
Detecting genetic correlations between immune disease and schizophrenia
Our immune risk variant set captured only those variants associated with immune diseases at genome-wide significance in current GWAS. Given the polygenicity of immune-related diseases, there are 100s to 1000s of additional variants associated with each disease that have not yet been identified (73). To evaluate sharing of risk alleles between immune diseases and schizophrenia using a broader set of variants, we used LDSC (16).
LDSC provides an interpretable and comparable estimation of genetic sharing between two traits in the form of genetic correlation (rg) values; the method uses genome-wide summary statistics, effectively accounts for linkage-disequilibrium and is robust to non-independence of GWAS samples (16). We therefore used LDSC to estimate pairwise genome-wide genetic correlations between schizophrenia and immune diseases. In addition to the 11 immune diseases with available genome-wide summary statistics, we included bipolar disorder as a positive control and height as a negative control. We used summary statistics from the 49 European-ancestry cohorts in the PGC2 study (33 640 cases and 43 456 controls) to ensure comparable LD structure across samples (1). Notably, LDSC is less sensitive than other methods of estimating genetic correlation (e.g. polygenic risk scoring (PRS), genome-based restricted maximum likelihood (GREML)) and is not robust when applied to genetic data obtained from specialty chips (e.g. Immunochip) (16). We considered immune diseases with rgP < 0.05 to show genetic overlap with schizophrenia.
As previously reported (16), our positive control (bipolar disorder) showed significant genetic overlap with schizophrenia (rg = 0.75±0.05, P = 8.5×10−60, Fig. 2, Table 4) and our negative control (height) showed no such overlap (rg = −0.004±0.02, P = 0.84, Fig. 2, Table 4). With respect to immune diseases, we observed genetic overlap with schizophrenia for CRO, IBD, PBC, PSO, SLE and UC (rg = 0.10–0.18, Fig. 2, Table 4). Notably, genetic correlations for PSO and UC did not survive correction for the 11 tests performed (Table 4). Unsurprisingly, the genetic correlations between schizophrenia and immune diseases were smaller in magnitude than those of commonly overlapping immune diseases (e.g. RA and SLE: rg = 0.55±0.08, P = 3.60×10−11).
Figure 2.

Genetic correlation between schizophrenia and other traits. Genetic correlation between schizophrenia, bipolar disorder, height and 14 immune diseases was estimated using cross-trait LDSC (16). Colour intensity and size of square are proportional to strength of genetic correlation (red, negative correlation; blue, positive correlation). Asterisks indicate genetic correlations that are statistically significant at P < 0.05 (*), P < 0.004 (**) and P < 0.0002 (***) thresholds. BP, bipolar disorder; CEL, celiac disease; CRO, Crohn’s disease; HGT, height; IBD, inflammatory bowel disease; PBC, primary biliary cirrhosis; PSO, psoriasis, RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SSC, systemic sclerosis; T1D, type 1 diabetes; UC, ulcerative colitis; VIT, vitiligo.
Table 4.
Estimated phenotypic and genome-wide genetic correlations between schizophrenia and other traits, r2 and h2 are reported on the observed scale for all diseases.
| Total sample | Male-specific | Female-specific | ||||||
|---|---|---|---|---|---|---|---|---|
| Trait | h2 ± SEa | rp | rg ± SE | P | rg ± SE | P | rg ± SE | P |
| BP (+) | 0.46 ± 0.02 | 0.75 ± 0.05 | 4.02×10 −57 | 0.657 ± 0.04 | 3.23×10 −52 | 0.820 ± 0.06 | 1.69×10 −49 | |
| HGT (−) | 0.34 ± 0.02 | 7.47×10−5 ± 0.02 | 0.99 | −0.012 ± 0.02 | 0.53 | 0.012 ± 0.02 | 0.60 | |
| CEL | 0.23 ± 0.05 | 0.04 | 0.107 ± 0.06 | 0.05 | 0.055 ± 0.06 | 0.34 | 0.075 ± 0.07 | 0.29 |
| CRO | 0.37 ± 0.04 | 0.04 | 0.097 ± 0.03 | 3.27×10−3 | 0.103 ± 0.03 | 9.17×10−4 | 0.110 ± 0.04 | 8.85×10−3 |
| IBD | 0.32 ± 0.04 | n.a. | 0.117 ± 0.03 | 2.49×10−4 | 0.114 ± 0.03 | 1.14×10−4 | 0.139 ± 0.04 | 6.09×10−4 |
| PBC | 0.46 ± 0.08 | 0.11 | 0.131 ± 0.05 | 4.00×10−3 | 0.141 ± 0.04 | 1.50×10−3 | 0.073 ± 0.05 | 0.15 |
| PSO | 0.27 ± 0.09 | 0.13 | 0.182 ± 0.07 | 7.80×10−3 | 0.205 ± 0.07 | 4.10×10−3 | 0.212 ± 0.09 | 0.01 |
| RA | 0.18 ± 0.03 | −0.04 | −0.032 ± 0.04 | 0.78 | 0.017 ± 0.04 | 0.70 | −0.067 ± 0.05 | 0.19 |
| SLE | 0.13 ± 0.05 | 0.05 | 0.130 ± 0.05 | 3.76×10−3 | 0.153 ± 0.05 | 1.51×10−3 | 0.065 ± 0.05 | 0.22 |
| SSC | 0.26 ± 0.08 | n.a. | 0.086 ± 0.07 | 0.16 | 0.190 ± 0.09 | 0.04 | 0.011 ± 0.09 | 0.91 |
| T1D | 0.20 ± 0.04 | 0.15 | −0.008 ± 0.05 | 0.86 | 0.041 ± 0.05 | 0.38 | −0.013 ± 0.05 | 0.81 |
| UC | 0.23 ± 0.03 | − 0.001 | 0.106 ± 0.04 | 4.00×10−3 | 0.121 ± 0.04 | 9.87×10−4 | 0.153 ± 0.05 | 8.98×10−4 |
| VIT | 0.86 ± 0.15 | n.a. | 0.011 ± 0.05 | 0.84 | 0.060 ± 0.05 | 0.23 | 0.045 ± 0.07 | 0.51 |
ah2 was estimated using LDSC.
(+), positive control; (−), negative control; n.a., not available due to lack of data regarding incidence rate ratio of this immune disease in schizophrenia; SE, standard error; rg, genetic correlation; rp, expected phenotypic correlation based on epidemiological data (see Materials and Methods for details of rp estimation). Bold font indicates significant genetic correlation with schizophrenia (P < 0.05) r2 and h2 are reported on the observed scale for all diseases.
Given the significant sex bias of immune diseases, with women at greater risk overall (34), we hypothesized that there may be sex-dependent genetic overlap between schizophrenia and some immune-mediated diseases. We therefore performed exploratory sex-stratified LDSC, estimating genetic correlation between immune diseases and schizophrenia separately in males (33 097 schizophrenia cases and 35 190 controls) and females (17 760 schizophrenia cases and 36 903 controls) of European ancestry from the PGC2 study and additional PGC samples. We found consistent genetic correlation estimates across male and female subsamples compared to the total sample for 9 of the 11 immune diseases investigated (Table 4). For SLE, we observed significant genetic correlation with schizophrenia in the total sample and in the male sample, but not in the female schizophrenia sample. For SSC, we observed genetic correlation with schizophrenia only in the male schizophrenia sample. For both SLE and SSC, there was a trend for the same direction of effect (positive genetic correlation) among females that did not reach statistical significance.
Benchmarking genetic correlations between immune disease and schizophrenia with epidemiological data
To determine how much of the phenotypic correlation between schizophrenia and immune diseases was explained by the genetic correlations we observed, we benchmarked significant genetic correlations between schizophrenia and immune-mediated disorders relative to the expected phenotypic correlations from epidemiological data (Materials and Methods). Using incidence of immune diseases in schizophrenia reported in a large population-based study (3), we estimated phenotypic correlations between schizophrenia and immune diseases with available incidence rate ratios in schizophrenia. For CRO, PBC, PSO and SLE, we observed small positive genetic correlations with schizophrenia that were consistent with the epidemiological data (Table 4). For UC, we observed a small positive estimate of genetic correlation (rg = 0.106 ± 0.04) while there was no strong evidence for any correlation between UC and schizophrenia in the epidemiological data (rp = −0.001). Importantly, while the MHC region contains risk factors for both schizophrenia and immune diseases, our genetic correlation estimates were obtained considering only SNPs outside of the MHC due to unusual LD in this region (74).
Discussion
Using a variety of statistical approaches, we provide evidence of shared genetic risk for schizophrenia and immune diseases. Within the MHC region, we identified four HLA variants showing statistically significant association with schizophrenia. An important caveat is that these four variants were not the top variants in their respective regions of association with schizophrenia and were not primary drivers of the MHC association in schizophrenia in stepwise conditional analyses (9,11). Therefore, the biological significance of these particular HLA variants in schizophrenia is likely limited.
Outside of the MHC region, we identified five SNPs with potential pleiotropic effects—influencing risk for both schizophrenia and CEL (rs296547), CRO (rs1734907, rs13126505, rs6738825) or MS (rs7132277). These variants do not appear to be broadly pleiotropic across human traits, as there were no phenome-wide significant results reported for any of these SNPs in the PheWAS catalog (all P > 1.2×10−8; all false discovery rate (FDR) > 0.1 (75)). Integration of GWAS, mQTL and eQTL data implicated ABCB9 and EPHB4/TRIP6 as functional candidates underlying the association of rs7132277 and rs1734907, respectively. Although ABCB9 emerged as a functional candidate driving the association of rs7132277 in schizophrenia, the significant colocalization of this region between schizophrenia and MS (PP = 0.94) was not driven by rs7132277 (PP = 1.38×10−39). Overall, our findings provide the strongest evidence for rs1734907 as a functional, pleiotropic immune variant associated with both CRO and schizophrenia. In particular, our results suggest a model in which genetic variation at rs1734907 (~85 kb upstream of EPHB4) increases DNA methylation, upregulates EPHB4 expression and decreases the risk of schizophrenia. While DNA methylation is classically associated with gene silencing, the effect of methylation on transcription depends on the genomic context (76); for instance, methylation of silencers or insulators eliminates transcription-blocking activity thereby promoting gene expression (77,78). EPHB4 is a transmembrane tyrosine kinase receptor that coordinates cell movement via bidirectional intercellular signaling at sites of direct cell-to-cell contact (79). In the brain, ephrin signaling mediates synaptic plasticity by initiating and stabilizing neuronal synapse formation (reviewed in 80). An analogous role has not yet been discovered in the immune system, possibly due to the much shorter lifespan of immunological synapses between lymphocytes and antigen presenting cells (minutes) as compared to neuronal synapses (years) (81,82). Interestingly, ephrin signaling attenuates the migration responses of both neurons and immune cells toward chemoattractants in vitro (83,84). Thus, pathfinding may be a shared risk mechanism by which EPHB4 contributes to immune disease and schizophrenia. The hypotheses raised by our findings require further validation. If the association of rs1734907 with CRO and schizophrenia is robustly replicated in future GWAS, functional studies will be needed to investigate both the genetic mechanism by which rs1734907 (or a causal variant in LD with this SNP) influences EPHB4 transcription and the biological mechanism by which increased EPHB4 expression influences susceptibility to CRO and schizophrenia. With the multi-kinase inhibitor dasatinib already on the market for treatment of chronic myeloid leukemia (85) and other EphB4 inhibitors currently in phase II trials (86–89), the potential for future drug repurposing makes EPHB4 an attractive candidate for further investigation.
We observed genome-wide sharing of risk variants for schizophrenia and six immune diseases (IBD including both CRO and UC, PBC, PSO, SLE) using LDSC, all of which have been previously reported to co-occur with schizophrenia in epidemiological studies (3,5). With the exception of UC, the small positive genetic correlations observed between these immune diseases and schizophrenia (rg~0.1) were consistent with phenotypic correlations observed in epidemiological data. Thus, currently available genetic data suggest that shared genetic risk contributes to the co-occurrence of CRO, PBC, PSO and SLE in schizophrenia. We note that most of the phenotypic correlation between these immune diseases and schizophrenia appears to be captured by common genetic variation. Interestingly, phenotypic correlations for the remaining five immune diseases were similar in magnitude (rp = 0.04–0.15), but no genetic correlation was detected. Possible explanations for this include inadequate statistical power to detect genetic correlations for these diseases in our study or a stronger environmental component contributing to the epidemiological relationship of these disorders with schizophrenia. Some of the immune diseases that did show significant genetic correlation with schizophrenia (CRO, UC) are considered autoinflammatory diseases (90), and the others (PBC, PSO, SLE) have a strong inflammatory component (90–92). This raises the possibility that the genetic risk we observed between these particular immune diseases and schizophrenia reflects a subgroup of inflammation-driven schizophrenia cases and/or sharing of specific innate immunity pathways between schizophrenia and these particular immune diseases.
To our knowledge, this is the first time that sex-dependent genetic correlation with immune diseases has been investigated in schizophrenia. Interestingly, SLE and SSC showed significant genetic correlation with schizophrenia only among males in sex-stratified analyses. Our findings raise the possibility of male-specific pleiotropy between schizophrenia and certain immune diseases. Interestingly, animal studies indicate that sex hormones have opposing effects on predisposition to schizophrenia and autoimmunity; estrogen has been reported to protect against the development of schizophrenia (93), while androgens appear to protect against the development autoimmune diseases (94,95). We emphasize that our sex-dependent findings require validation in independent samples.
Our work was subject to several important limitations. Firstly, we did not have access to imputed data sets for the original immune disease GWAS reporting the potentially pleiotropic SNPs (Table 3). Lack of dense SNP coverage in the regions of interest resulted in low power to detect colocalizing association in schizophrenia, and thus our analyses are inconclusive with respect to pleiotropic potential for the two regions that did not show significant colocalization. Secondly, genome-wide summary statistics were not available for all of the immune diseases, resulting in a more limited analysis of 11 diseases for estimating genetic correlations. Thirdly, we used LDSC to estimate genetic correlations because of its robustness to non-independence of GWAS samples as many of the samples analyzed included Wellcome Trust Case Control Consortium samples. Compared to alternative methods for estimating genetic correlation that use individual-level genotype data, LDSC has lower statistical power. Thus, we cannot exclude the possibility of additional immune-schizophrenia genetic relationships not identified in our analyses. Finally, we had greater statistical power to detect genetic correlation with immune diseases in the male-specific schizophrenia GWAS compared to the female-specific GWAS. This difference in power may account for the male-specific genetic correlations we observed for SLE and SSC, as opposed to sex-dependent pleiotropic effects.
Despite these limitations, our work adds to a growing body of evidence suggesting that schizophrenia and immune diseases share genetic risk factors. There are conflicting reports in the literature with respect to the specific immune diseases demonstrating genetic overlap with schizophrenia and the direction of effect (positive or negative genetic correlation). Genetic overlap with schizophrenia has been previously investigated for 9 of the 19 immune diseases studied here. Genome-wide genetic correlation with schizophrenia has been previously reported for CRO (25–29), MS (30), PBC (27), PSO (27,31), RA [both positive (25,26) and negative (33) genetic correlations], SLE (26,27), T1D (25) and UC (26–29) (see Supplementary Data, Table S1 for a summary of previous studies). Our results are consistent with previously reported genetic overlap between schizophrenia and CRO (25–29), PBC (27), PSO (27,31), SLE (26,27) and UC (26–29). We did not find any significant genetic correlation between schizophrenia and CEL, T1D, RA, SSC or VIT. For RA in particular, there is a robust inverse epidemiological association with schizophrenia (8). However, the genetic association is less consistent. Using methods based on summary statistics (including PRS and LDSC), six previous studies reported no evidence of pleiotropy between schizophrenia and RA (8,16,27–29,32), while two studies reported positive genetic correlation (25,26). Notably, Lee et al. (33) reported an inverse genetic correlation—in keeping with the observed epidemiological effect—using GREML, a method utilizing full genotype data that has greater statistical power to detect small pleiotropic effects than PRS or LDSC. Given the modest genetic correlations observed in the present study, subtle differences in statistical power across studies using different statistical methods and GWAS data sets may explain these discrepant findings. As genetic samples continue to grow, and our understanding of the degree of genetic overlap expected among complex traits evolves, it will be worthwhile to revisit these analyses.
Overall, our analyses provide statistical evidence supporting extensive pleiotropy between immune diseases and schizophrenia. Our results highlight EPHB4, a transmembrane receptor that coordinates cell migration and has dual roles in immune cell and neuronal pathfinding, as a promising candidate for future functional studies. More broadly, our findings indicate that common genetic variants influencing the risk of immune diseases—in particular CRO, PBC, PSO, SLE and UC—are also involved in schizophrenia. Studies identifying the cell types and biological pathways that may be driving this genetic overlap are needed and will hopefully provide further insights into the pathophysiology of schizophrenia. In the meantime, our work supports the emerging hypothesis that pathogenic mechanisms are shared across immune and central nervous system disorders.
Materials and Methods
Samples and quality control
We used either imputed genotype data or summary statistics generated as described in the original GWAS. For sample details, see Table 1.
Schizophrenia data set
We used data from the PGC2 study (1). For analyses of non-HLA genome-wide significant risk variants for immune diseases, we used publicly available summary statistics from the total data set (52 cohorts, 35 476 cases and 46 839 controls) (1). For LDSC analyses, we used all 49 European ancestry case-control cohorts in PGC2 (33 640 cases and 43 456 controls). For analyses including HLA variants, we used a further refined 31 European ancestry case-control cohorts (20 253 cases and 25 011 controls) with high-quality coverage of the MHC region, as previously described (11).
Immune disease data sets
To estimate the extent of genetic overlap between schizophrenia and immune diseases, we obtained full GWAS summary statistics for 11 of the 19 immune diseases (eight immune diseases were not included in LDSC analyses due to lack of available summary statistics). We obtained publicly available summary statistics for five immune diseases (see URLs): CRO (38), IBD (38), RA (41), SLE (42) and UC (38). For the following six immune diseases, we obtained summary statistics with permission from the authors: CEL (65), PBC (39), PSO (40), SSC (43), T1D (44) and VIT (45).
Testing the association of genome-wide significant risk alleles for 19 immune diseases in schizophrenia
For each of the 19 immune diseases, we defined risk loci outside of the MHC region (chromosome 6: 25–34 Mb) using curated GWAS results from ImmunoBase (http://www.immunobase.org; accessed 7 June 2015. For details, see Supplementary Data). Notably, the majority of IBD risk variants were also risk variants for CRO and/or UC. Within the MHC region, we considered only the most strongly associated HLA variant (including SNPs, imputed HLA amino acid sites and classical alleles) for each disease based on univariate analysis in previously published studies (see Table 2), because multivariate conditional analyses reporting adjusted effect sizes of independent HLA variants were not available for all immune diseases. In total, there were 581 unique variants (563 non-HLA variants and 18 HLA variants) associated with any immune disease at genome-wide significance. A complete list of non-HLA and HLA immune risk variants is provided in Table 2 and Supplementary Data, Table S2, respectively.
First, we tested for shared direction of effect with schizophrenia among SNPs associated with each of the 19 immune diseases using the binomial sign test. Because some immune risk SNPs were associated with multiple diseases with inconsistent direction of effect, we could not evaluate shared direction of effect among the collective set of immune risk SNPs in schizophrenia.
Next, we evaluated the collective association of SNPs associated with any immune disease. First, we extracted the P-values from the PGC2 GWAS for a pruned set of 261 LD-independent, non-HLA immune risk SNPs with LD score and MAF information available in the European LD score database (16). We then quantified enrichment of these immune risk SNP associations in schizophrenia using the genomic inflation value λ. We obtained an empirical enrichment P-value by comparing this to λ values from 1000 equal-sized sets of SNPs drawn from the schizophrenia GWAS summary data and matched to the immune SNP set for MAF and LD score, as these parameters are correlated with GWAS test statistics (see Supplementary Data for details).
Finally, we evaluated the association of each of the 581 variants with schizophrenia using previously published association results for non-HLA (1) and HLA variants (11). We considered SNPs associated with schizophrenia at P < 8.6×10−5 (Bonferroni correction for 581 tests, 563 non-HLA and 18 HLA variants) to have pleiotropic effects.
To evaluate the pleiotropic potential of immune risk variants significantly associated with schizophrenia, we tested for colocalization of association signals in immune diseases and schizophrenia using the Bayesian coloc2 method implemented in the R package coloc (63). We used window sizes that captured all SNPs showing r2 > 0.2 with each immune risk variant (chr1:200.8-201.2Mb, chr2:198.0-199.0Mb, chr4:102.6-103.4Mb, chr7:100.2-100.5Mb, chr12:123.4-123.9Mb). We used default prior probabilities for immune risk variants being associated with immune disease (P = 1×10−4), schizophrenia (P = 1×10−4) and both immune disease and schizophrenia (P = 1×10−5) as recommended. In addition to colocalization testing, we performed COJO using GCTA (96). Specifically, we used COJO to perform association analyses in the PGC2 schizophrenia GWAS conditioning on the immune risk variants of interest (i.e. SNPs that were significantly associated with both an immune disease and schizophrenia). If the immune risk variants explained the regional associations in schizophrenia, no significant associations with schizophrenia should remain after conditioning on these variants (statistically, all P > 8.6×10−5). We used the 1000 Genomes Phase 3 European data set as a reference panel to calculate LD between SNPs.
To prioritize genes and regulatory elements driving the pleiotropic GWAS loci we identified (associated with both immune disease and schizophrenia, see Table 3), we followed the analytic approach described by Wu et al. (69). This approach integrates summary statistics from independent mQTL studies, eQTL studies and GWAS to identify SNPs associated with gene expression, DNA methylation and disease through shared genetic effects.
We obtained mQTL and eQTL data used in Wu et al. (69) for genetic regions within a 2 Mb window of each pleiotropic SNP. These data and the quality control measures applied have been described in detail elsewhere (70). Briefly, mQTL summary-level SNP data were from a meta-analysis of the Brisbane Systems Genetics Study (70) and Lothian Birth Cohorts of 1921 and 1936 (71), which comprised 1 980 individuals with DNA methylation measured in peripheral blood. eQTL summary-level SNP data were from the Consortium for the Architecture of Gene Expression (CAGE) study (72), which comprised 2 765 individuals with gene expression levels measured in peripheral blood. GWAS summary-level SNP data for schizophrenia was from the PGC2 study (1).
We applied SMR using GCTA (96) to test for shared associations between the pleiotropic SNPs with DNAm probes and gene expression probes, DNAm probes and schizophrenia and gene expression probes and schizophrenia. We considered significant associations as those with pSMR < 1.30×10−4 (0.05/385 tagged genes) for mQTLs and pSMR < 4.31×10−4 for eQTLs (0.05/116 tagged genes). Next, we applied the HEIDI test (68) using GCTA (96) to evaluate whether significant shared associations between DNAm, gene expression and schizophrenia were driven by linkage (i.e. separate causal variants in LD exerting genetic effects on DNAm, gene expression and schizophrenia) or a shared pleiotropic causal variant. We considered genetic effects that passed the HEIDI test (pHEIDI > 0.01) to be driven by a single causal variant. We looked for consistent SMR and HEIDI results across GWAS, mQTL and eQTL studies to prioritize genes for future functional studies.
Estimating the degree of genetic correlation between schizophrenia and 14 immune diseases
To evaluate whether common variants influencing risk of immune diseases collectively contribute to schizophrenia, we used cross-trait LDSC (16). Cross-trait LDSC estimates the genetic correlation (rg) between two traits using GWAS summary statistics. Our main analysis included 11 immune diseases with available genome-wide summary statistics. To benchmark the amount of genetic overlap between schizophrenia and immune disease, we included bipolar disorder as a positive control (12) and human height as a negative control (36) based on their previously reported genetic correlations with schizophrenia using cross-trait LDSC (16).
The statistical framework for cross-trait LDSC has been described in detail previously (16). Briefly, LDSC leverages the relationship between LD and association test statistics to estimate heritability as the slope of the regression of z-scores against LD scores (97). Cross-trait LDSC is a bivariate extension of this method that estimates genetic covariance as the slope of the regression of the products of z-scores against LD scores using the following equation (16):
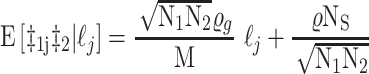 |
where  denotes the z score for study i and SNPj,
denotes the z score for study i and SNPj, 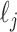 is the LD score (96),
is the LD score (96),  is the sample size for study i,
is the sample size for study i,  is the genetic covariance,
is the genetic covariance, 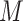 is the number of SNPs in the reference panel with MAF between 5% and 50%,
is the number of SNPs in the reference panel with MAF between 5% and 50%, 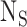 is the number of individuals included in both studies and
is the number of individuals included in both studies and  is the phenotypic correlation among the
is the phenotypic correlation among the  overlapping samples. Genetic covariance
overlapping samples. Genetic covariance  is estimated by regressing
is estimated by regressing  against
against  and multiplying the resulting slope by
and multiplying the resulting slope by  . Statistical significance is assessed using block jackknifing over 200 equally sized blocks of SNPs (16). Importantly, the MHC region is excluded from LDSC analyses due to its unusual LD structure and genetic architecture (74).
. Statistical significance is assessed using block jackknifing over 200 equally sized blocks of SNPs (16). Importantly, the MHC region is excluded from LDSC analyses due to its unusual LD structure and genetic architecture (74).
Because LDSC is robust to sample sharing across GWAS (16), we used summary statistics from the 49 European-ancestry cohorts in the PGC2 study (33 640 cases and 43 456 controls) (1). We used LD scores from the ‘eur_w_ld_chr/’ files available from https://data.broadinstitute.org/alkesgroup/LDSCORE, computed using 1000 Genomes Project (66) Europeans as a reference panel as previously described (74). To ensure we were using well-imputed SNPS, we filtered all GWAS as previously described (16), including limiting the analysis to HapMap 3 (98) SNPs as implemented in the LDSC script munge_sumstats.py (https://github.com/bulik/ldsc). We estimated h2 for each trait on the observed scale (Supplementary Data, Table S4). We considered traits with rgP < 0.05 to have significant genetic correlation with schizophrenia.
Benchmarking with epidemiological data
To determine how much of the phenotypic correlation between schizophrenia and immune-mediated diseases was explained by the genetic correlations we observed, we used the approach previously described by Lee et al. (33). Briefly, we benchmarked our genetic correlation estimates between schizophrenia and immune diseases relative to the expected phenotypic correlations from epidemiological data. We obtained estimates of the population risk of schizophrenia ( ), the population risk of each immune disease (
), the population risk of each immune disease ( ) and the probability of each immune disease among patients with schizophrenia (
) and the probability of each immune disease among patients with schizophrenia ( ) from the literature as referenced in Supplementary Data, Table S4. We estimated the phenotypic correlation between schizophrenia and the immune disease of interest (RSCZ-IMMUNE) using the following formula, as derived by Lee et al. (33) assuming that the phenotypic liabilities of schizophrenia (
) from the literature as referenced in Supplementary Data, Table S4. We estimated the phenotypic correlation between schizophrenia and the immune disease of interest (RSCZ-IMMUNE) using the following formula, as derived by Lee et al. (33) assuming that the phenotypic liabilities of schizophrenia ( ) and immune disease (
) and immune disease (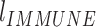 ) follow a bivariate normal distribution with mean = 0 and standard deviation = 1:
) follow a bivariate normal distribution with mean = 0 and standard deviation = 1:
 |
where:
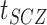 is the liability threshold for schizophrenia: Z-score of the (
is the liability threshold for schizophrenia: Z-score of the (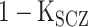 )th percentile
)th percentile
 is the liability threshold for immune disease: Z-score of the (
is the liability threshold for immune disease: Z-score of the ( )th percentile
)th percentile
 is the liability threshold for immune disease in those with schizophrenia: Z-score of the (
is the liability threshold for immune disease in those with schizophrenia: Z-score of the ( )th percentile
)th percentile
 is the ‘height’ of the normal distribution at the schizophrenia liability threshold: probability density function of
is the ‘height’ of the normal distribution at the schizophrenia liability threshold: probability density function of 
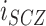 is the mean phenotypic liability of those with schizophrenia:
is the mean phenotypic liability of those with schizophrenia:
 |
Statistical power
Power to detect association of individual non-HLA and HLA immune risk variants in schizophrenia was calculated using the Genetic Power Calculator (99) assuming a risk allele frequency (RAF) of 0.05, disease prevalence of 1% and significance threshold (α) of 8.6×10−5. We used a RAF of 0.05 as this was the point at which statistical power remained >80% given the prevalence of schizophrenia and size of our sample. The vast majority of immune variants had MAF >0.05 (551/563 variants).
URLs
LD score database: ftp://atguftp.mgh.harvard.edu/brendan/1k_eur_r2_hm3snps_se_weights.RDS.
Publicly available GWAS summary statistics:
Funding
This research was supported in part by a number of funding sources. This research uses resources provided by the Genetic Association Information Network (GAIN), obtained from the database of Genotypes and Phenotypes (dbGaP) found at http://www.ncbi.nlm.nih.gov/gap through dbGaP accession number phs000021.v3.p2; samples and associated phenotype data for this study were provided by the Molecular Genetics of Schizophrenia Collaboration (PI: Pablo V. Gejman, Evanston Northwestern Healthcare and Northwestern University, Evanston, IL, USA). Fulbright Canada, the Weston Foundation, and Brain Canada through the Canada Brain Research Fund—a public-private partnership established by the Government of Canada (to J.G.P.); the National Research Foundation of Korea (NRF) [grant 2016R1C1B2013126 to B.H.] and the Bio & Medical Technology Development Program of the NRF [grant 2017M3A9B6061852 to B.H.] funded by the Korean government, Ministry of Science and ICT; the Finnish Cultural Foundation and Academy of Finland [grant 309643 to H.M.O.]; the Spanish Ministry of Economy and Competitiveness and P12-BIO-1395 from Consejería de Innovación, Ciencia y Tecnología, Junta de Andalucía (Spain) [grant SAF2015-66761-P to J.M.]; the US National Institutes of Health (NIH) [grants R01AR045584, R01AR056292, X01HG007484 and P30AR057212 to Y.J., S.A.S. and R.S.]; the US NIH [grants N01AR02251 and R01AR05528 to M.D.M.]; the US NIH [grants 1R01AR063759, 1R01AR062886, 1UH2AR067677-01 and U19AI111224-01 to S.R.] and Doris Duke Charitable Foundation [grant 2013097 to S.R.]. Funding for the GAIN schizophrenia sample was provided by the US NIH [grants R01 MH67257, R01 MH59588, R01 MH59571, R01 MH59565, R01 MH59587, R01 MH60870, R01 MH59566, R01 MH59586, R01 MH61675, R01 MH60879, R01 MH81800, U01 MH46276, U01 MH46289, U01 MH46318, U01 MH79469 and U01 MH79470] and the genotyping of samples was provided through GAIN. The funding sources did not influence the study design, data analysis or writing of this manuscript.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Supplementary Material
Acknowledgements
We thank all study participants, and all research staff at the numerous study sites that made this research possible. Computations were performed on the Genetic Cluster Computer hosted by SURFsara (www.surfsara.nl). We thank ImmunoBase for curating genetic risk factors for the immune diseases investigated. We thank the Canadian-US PBC Consortium, the Italian PBC Genetics Study Group and the UK-PBC Consortium for sharing their summary data for the PBC GWAS. We thank the International Inflammatory Bowel Disease Genetics Consortium for providing publicly available genome-wide summary statistics for CRO, UC and IBD. We also thank the investigators leading the GWAS for RA and SLE for providing genome-wide summary statistics from their studies on ImmunoBase.
References
- 1. Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chan M.K., Krebs M.O., Cox D., Guest P.C., Yolken R.H., Rahmoune H., Rothermundt M., Steiner J., Leweke F.M., van Beveren N.J. et al. (2015) Development of a blood-based molecular biomarker test for identification of schizophrenia before disease onset. Transl. Psychiatry, 5, e601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Benros M.E., Nielsen P.R., Nordentoft M., Eaton W.W., Dalton S.O. and Mortensen P.B. (2011) Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am. J. Psychiatry, 168, 1303–1310. [DOI] [PubMed] [Google Scholar]
- 4. Chen S.J., Chao Y.L., Chen C.Y., Chang C.M., Wu E.C., Wu C.S., Yeh H.H., Chen C.H. and Tsai H.J. (2012) Prevalence of autoimmune diseases in in-patients with schizophrenia: nationwide population-based study. Br. J. Psychiatry, 200, 374–380. [DOI] [PubMed] [Google Scholar]
- 5. Benros M.E., Pedersen M.G., Rasmussen H., Eaton W.W., Nordentoft M. and Mortensen P.B. (2014) A nationwide study on the risk of autoimmune diseases in individuals with a personal or a family history of schizophrenia and related psychosis. Am. J. Psychiatry, 171, 218–226. [DOI] [PubMed] [Google Scholar]
- 6. Eaton W.W., Byrne M., Ewald H., Mors O., Chen C.Y., Agerbo E. and Mortensen P.B. (2006) Association of schizophrenia and autoimmune diseases: linkage of Danish national registers. Am. J. Psychiatry, 163, 521–528. [DOI] [PubMed] [Google Scholar]
- 7. Eaton W.W., Pedersen M.G., Nielsen P.R. and Mortensen P.B. (2010) Autoimmune diseases, bipolar disorder, and non-affective psychosis. Bipolar Disord., 12, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Euesden J., Breen G., Farmer A., McGuffin P. and Lewis C.M. (2015) The relationship between schizophrenia and rheumatoid arthritis revisited: genetic and epidemiological analyses. Am. J. Med. Genet. B. Neuropsychiatr. Genet., 168B, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sekar A., Bialas A.R., de Rivera H., Davis A., Hammond T.R., Kamitaki N., Tooley K., Presumey J., Baum M., Van Doren V. et al. (2016) Schizophrenia risk from complex variation of complement component 4. Nature, 530, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yih Chen J., Ling Wu Y., Yin Mok M., Jan Wu Y.J., Lintner K.E., Wang C.M., Chung E.K., Yang Y., Zhou B., Wang H. et al. (2016) Effects of complement C4 gene copy number variations, size dichotomy, and C4A deficiency on genetic risk and clinical presentation of systemic lupus erythematosus in East Asian populations. Arthritis Rheumatol., 68, 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pouget J.G., Goncalves V.F., Spain S.L., Finucane H.K., Raychaudhuri S., Kennedy J.L. and Knight J. (2016) Genome-wide association studies suggest limited immune gene enrichment in schizophrenia compared to 5 autoimmune diseases. Schizophr. Bull., 42, 1176–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cross-Disorder Group of the Psychiatric Genomics Consortium (2013) Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet, 381, 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cross-Disorder Group of the Psychiatric Genomics Consortium (2013) Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet., 45, 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Voight B.F., Peloso G.M., Orho-Melander M., Frikke-Schmidt R., Barbalic M., Jensen M.K., Hindy G., Holm H., Ding E.L., Johnson T. et al. (2012) Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet, 380, 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Do R., Willer C.J., Schmidt E.M., Sengupta S., Gao C., Peloso G.M., Gustafsson S., Kanoni S., Ganna A., Chen J. et al. (2013) Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat. Genet., 45, 1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bulik-Sullivan B., Finucane H.K., Anttila V., Gusev A., Day F.R., Loh P.R., Duncan L., Perry J.R., Patterson N., Robinson E.B. et al. (2015) An atlas of genetic correlations across human diseases and traits. Nat. Genet., 47, 1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ellinghaus D., Jostins L., Spain S.L., Cortes A., Bethune J., Han B., Park Y.R., Raychaudhuri S., Pouget J.G., Hübenthal M. et al. (2016) Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet., 48, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brainstorm Consortium, Anttila V., Bulik-Sullivan B., Finucane H.K., Walters R.K., Bras J., Duncan L., Escott-Price V., Falcone G.J., Gormley P. et al. (2018) Analysis of shared heritability in common disorders of the brain. Science, 360, eaap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paaby A.B. and Rockman M.V. (2013) The many faces of pleiotropy. Trends Genet., 29, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han B., Pouget J.G., Slowikowski K., Stahl E., Lee C.H., Diogo D., Hu X., Park Y.R., Kim E., Gregersen P.K. et al. (2016) A method to decipher pleiotropy by detecting underlying heterogeneity driven by hidden subgroups applied to autoimmune and neuropsychiatric diseases. Nat. Genet., 48, 803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chesmore K., Bartlett J. and Williams S.M. (2018) The ubiquity of pleiotropy in human disease. Hum. Genet., 137, 39–44. [DOI] [PubMed] [Google Scholar]
- 22. Verbanck M., Chen C.Y., Neale B. and Do R. (2018) Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet., 50, 693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jordan D.M., Verbanck M. and Do R. (2018) The landscape of pervasive horizontal pleiotropy in human genetic variation is driven by extreme polygenicity of human traits and diseases. bio Rxiv, https://doi.org/10.1101/416545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gandal M.J., Haney J.R., Parikshak N.N., Leppa V., Ramaswami G., Hartl C., Schork A.J., Appadurai V., Buil A., Werge T.M. et al. (2018) Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science, 359, 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stringer S., Kahn R.S., de Witte L.D., Ophoff R.A. and Derks E.M. (2014) Genetic liability for schizophrenia predicts risk of immune disorders. Schizophr. Res., 159, 347–352. [DOI] [PubMed] [Google Scholar]
- 26. Wang Q., Yang C., Gelernter J. and Zhao H. (2015) Pervasive pleiotropy between psychiatric disorders and immune disorders revealed by integrative analysis of multiple GWAS. Hum. Genet., 134, 1195–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tylee D.S., Sun J., Hess J.L., Tahir M.A., Sharma E., Malik R., Worrall B.B., Levine A.J., Martinson J.J., Nejenstev S. et al. (2018) Genetic correlations among psychiatric and immune-related phenotypes based on genome-wide association data. Am. J. Med. Genet. B. Neuropsychiatr. Genet., 177, 641–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duncan L.E., Shen H., Ballon J.S., Hardy K.V., Noordsy D.L. and Levinson D.F. (2018) Genetic correlation profile of schizophrenia mirrors epidemiological results and suggests link between polygenic and rare variant (22q11.2) cases of schizophrenia. Schizophr. Bull., 44, 1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pickrell J.K., Berisa T., Liu J.Z., Segurel L., Tung J.Y. and Hinds D.A. (2016) Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet., 48, 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Andreassen O.A., Harbo H.F., Wang Y., Thompson W.K., Schork A.J., Mattingsdal M., Zuber V., Bettella F., Ripke S., Kelsoe J.R. et al. (2015) Genetic pleiotropy between multiple sclerosis and schizophrenia but not bipolar disorder: differential involvement of immune-related gene loci. Mol. Psychiatry, 20, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yin X., Wineinger N.E., Wang K., Yue W., Norgren N., Wang L., Yao W., Jiang X., Wu B., Cui Y. et al. (2016) Common susceptibility variants are shared between schizophrenia and psoriasis in the Han Chinese population. J. Psychiatry Neurosci., 41, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O’Donovan M.C., Sullivan P.F. and Sklar P. (2009) Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature, 460, 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee S.H., Byrne E.M., Hultman C.M., Kahler A., Vinkhuyzen A.A., Ripke S., Andreassen O.A., Frisell T., Gusev A., Hu X. et al. (2015) New data and an old puzzle: the negative association between schizophrenia and rheumatoid arthritis. Int. J. Epidemiol., 44, 1706–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fairweather D., Frisancho-Kiss S. and Rose NR. (2008) Sex differences in autoimmune disease from a pathological perspective. Am J Pathol, 173, 600–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fernando M.M., Stevens C.R., Walsh E.C., De Jager P.L., Goyette P., Plenge R.M., Vyse T.J. and Rioux J.D. (2008) Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet., 4, e1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wood A.R., Esko T., Yang J., Vedantam S., Pers T.H., Gustafsson S., Chu A.Y., Estrada K., Luan J., Kutalik Z. et al. (2014) Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet., 46, 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dubois P.C., Trynka G., Franke L., Hunt K.A., Romanos J., Curtotti A., Zhernakova A., Heap G.A., Adany R., Aromaa A. et al. (2010) Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet., 42, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu J.Z., van Sommeren S., Huang H., Ng S.C., Alberts R., Takahashi A., Ripke S., Lee J.C., Jostins L., Shah T. et al. (2015) Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet., 47,979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cordell H.J., Han Y., Mells G.F., Li Y., Hirschfield G.M., Greene C.S., Xie G., Juran B.D., Zhu D., Qian D.C. et al. (2015) International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat. Commun., 6, 8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Strange A., Capon F., Spencer C.C., Knight J., Weale M.E., Allen M.H., Barton A., Band G., Bellenguez C., Bergboer J.G. et al. (2010) A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet., 42, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stahl E.A., Raychaudhuri S., Remmers E.F., Xie G., Eyre S., Thomson B.P., Li Y., Kurreeman F.A., Zhernakova A., Hinks A. et al. (2010) Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet., 42, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bentham J., Morris D.L., Graham D.S.C., Pinder C.L., Tombleson P., Behrens T.W., Martin J., Fairfax B.P., Knight J.C., Chen L. et al. (2015) Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet., 47, 1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Radstake T.R., Gorlova O., Rueda B., Martin J.E., Alizadeh B.Z., Palomino-Morales R., Coenen M.J., Vonk M.C., Voskuyl A.E., Schuerwegh A.J. et al. (2010) Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat. Genet., 42, 426–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bradfield J.P., Qu H.Q., Wang K., Zhang H., Sleiman P.M., Kim C.E., Mentch F.D., Qiu H., Glessner J.T., Thomas K.A. et al. (2011) A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet., 7, e1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jin Y., Andersen G., Yorgov D., Ferrara T.M., Ben S., Brownson K.M., Holland P.J., Birlea S.A., Siebert J., Hartmann A. et al. (2016) Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat. Genet., 48, 1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Betz R.C., Petukhova L., Ripke S., Huang H., Menelaou A., Redler S., Becker T., Heilmann S., Yamany T., Duvic M. et al. (2015) Genome-wide meta-analysis in alopecia areata resolves HLA associations and reveals two new susceptibility loci. Nat. Commun., 6, 5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cortes A., Hadler J., Pointon J.P., Robinson P.C., Karaderi T., Leo P., Cremin K., Pryce K., Harris J., Lee S. et al. (2013) Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet., 45, 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chu X., Pan C.M., Zhao S.X., Liang J., Gao G.Q., Zhang X.M., Yuan G.Y., Li C.G., Xue L.Q., Shen M. et al. (2011) A genome-wide association study identifies two new risk loci for Graves’ disease. Nat. Genet., 43, 897–901. [DOI] [PubMed] [Google Scholar]
- 49. Gutierrez-Achury J., Zhernakova A., Pulit S.L., Trynka G., Hunt K.A., Romanos J., Raychaudhuri S., van Heel D.A., Wijmenga C. and de Bakker P.I. (2015) Fine mapping in the MHC region accounts for 18% additional genetic risk for celiac disease. Nat. Genet., 47, 577–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goyette P., Boucher G., Mallon D., Ellinghaus E., Jostins L., Huang H., Ripke S., Gusareva E.S., Annese V., Hauser S.L. et al. (2015) High-density mapping of the MHC identifies a shared role for HLA-DRB1*01:03 in inflammatory bowel diseases and heterozygous advantage in ulcerative colitis. Nat. Genet., 47, 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hinks A., Cobb J., Marion M.C., Prahalad S., Sudman M., Bowes J., Martin P., Comeau M.E., Sajuthi S., Andrews R. et al. (2013) Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat. Genet., 45, 664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Patsopoulos N.A., Barcellos L.F., Hintzen R.Q., Schaefer C., van Duijn C.M., Noble J.A., Raj T., Gourraud P.A., Stranger B.E., Oksenberg J. et al. (2013) Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet., 9, e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tafti M., Hor H., Dauvilliers Y., Lammers G.J., Overeem S., Mayer G., Javidi S., Iranzo A., Santamaria J., Peraita-Adrados R. et al. (2014) DQB1 locus alone explains most of the risk and protection in narcolepsy with cataplexy in Europe. Sleep, 37, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu J.Z., Almarri M.A., Gaffney D.J., Mells G.F., Jostins L., Cordell H.J., Ducker S.J., Day D.B., Heneghan M.A., Neuberger J.M. et al. (2012) Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat. Genet., 44, 1137–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu J.Z., Hov J.R., Folseraas T., Ellinghaus E., Rushbrook S.M., Doncheva N.T., Andreassen O.A., Weersma R.K., Weismuller T.J., Eksteen B. et al. (2013) Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat. Genet., 45, 670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Okada Y., Han B., Tsoi L.C., Stuart P.E., Ellinghaus E., Tejasvi T., Chandran V., Pellett F., Pollock R., Bowcock A.M. et al. (2014) Fine mapping major histocompatibility complex associations in psoriasis and its clinical subtypes. Am. J. Hum. Genet., 95, 162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Raychaudhuri S., Sandor C., Stahl E.A., Freudenberg J., Lee H.S., Jia X., Alfredsson L., Padyukov L., Klareskog L., Worthington J. et al. (2012) Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet., 44, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lessard C.J., Li H., Adrianto I., Ice J.A., Rasmussen A., Grundahl K.M., Kelly J.A., Dozmorov M.G., Miceli-Richard C., Bowman S. et al. (2013) Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjogren’s syndrome. Nat. Genet., 45, 1284–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim K., Bang S.Y., Lee H.S., Okada Y., Han B., Saw W.Y., Teo Y.Y. and Bae S.C. (2014) The HLA-DRbeta1 amino acid positions 11-13-26 explain the majority of SLE-MHC associations. Nat. Commun., 5, 5902. [DOI] [PubMed] [Google Scholar]
- 60. Mayes M.D., Bossini-Castillo L., Gorlova O., Martin J.E., Zhou X., Chen W.V., Assassi S., Ying J., Tan F.K., Arnett F.C. et al. (2014) Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am. J. Hum. Genet., 94, 47–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hu X., Deutsch A.J., Lenz T.L., Onengut-Gumuscu S., Han B., Chen W.M., Howson J.M., Todd J.A., de Bakker P.I., Rich S.S. et al. (2015) Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat. Genet., 47, 898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C. and Plagnol V. (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet., 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Franke A., McGovern D.P., Barrett J.C., Wang K., Radford-Smith G.L., Ahmad T., Lees C.W., Balschun T., Lee J., Roberts R. et al. (2010) Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet., 42, 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., Lee J.C., Schumm L.P., Sharma Y., Anderson C.A. et al. (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature, 491, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Beecham A.H., Patsopoulos N.A., Xifara D.K., Davis M.F., Kemppinen A., Cotsapas C., Shah T.S., Spencer C., Booth D., Goris A. et al. (2013) Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet., 45, 1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. 1000 Genomes Project Consortium, Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang J., Ferreira T., Morris A.P., Medland S.E., Genetic Investigation of ANthropometric Traits (GIANT) Consortium, DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Madden P.A., Heath A.C., Martin N.G., Montgomery G.W. et al. (2012) Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet., 44, 369–375S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., Montgomery G.W., Goddard M.E., Wray N.R., Visscher P.M. et al. (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet., 48, 481–487. [DOI] [PubMed] [Google Scholar]
- 69. Wu Y., Zeng J., Zhang F., Zhu Z., Qi T., Zheng Z., Lloyd-Jones L.R., Marioni R.E., Martin N.G., Montgomery G.W. et al. (2018) Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun., 9, 918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Powell J.E., Henders A.K., McRae A.F., Caracella A., Smith S., Wright M.J., Whitfield J.B., Dermitzakis E.T., Martin N.G., Visscher P.M. et al. (2012) The Brisbane Systems Genetics Study: genetical genomics meets complex trait genetics. PLoS One, 7, e35430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen B.H., Marioni R.E., Colicino E., Peters M.J., Ward-Caviness C.K., Tsai P.C., Roetker N.S., Just A.C., Demerath E.W., Guan W. et al. (2016) DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY), 8, 1844–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lloyd-Jones L.R., Holloway A., McRae A., Yang J., Small K., Zhao J., Zeng B., Bakshi A., Metspalu A., Dermitzakis M. et al. (2017) The genetic architecture of gene expression in peripheral blood. Am. J. Hum. Genet., 100, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stahl E.A., Wegmann D., Trynka G., Gutierrez-Achury J., Do R., Voight B.F., Kraft P., Chen R., Kallberg H.J., Kurreeman F.A. et al. (2012) Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat. Genet., 44, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.R., Anttila V., Xu H., Zang C., Farh K. et al. (2015) Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet., 47, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Denny J.C., Bastarache L., Ritchie M.D., Carroll R.J., Zink R., Mosley J.D., Field J.R., Pulley J.M., Ramirez A.H., Bowton E. et al. (2013) Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol., 31, 1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jones P.A. (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet., 13, 484–492. [DOI] [PubMed] [Google Scholar]
- 77. Bell A.C. and Felsenfeld G. (2000) Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf 2 gene. Nature, 405, 482–485. [DOI] [PubMed] [Google Scholar]
- 78. Eden S., Constancia M., Hashimshony T., Dean W., Goldstein B., Johnson A.C., Keshet I., Reik W. and Cedar H. (2001) An upstream repressor element plays a role in Igf 2 imprinting. EMBO J., 20, 3518–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Davis S., Gale N.W., Aldrich T.H., Maisonpierre P.C., Lhotak V., Pawson T., Goldfarb M. and Yancopoulos G.D. (1994) Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science, 266, 816–819. [DOI] [PubMed] [Google Scholar]
- 80. Hruska M. and Dalva M.B. (2012) Ephrin regulation of synapse formation, function and plasticity. Mol. Cell. Neurosci., 50, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dustin M.L. (2012) Signaling at neuro/immune synapses. J. Clin. Invest., 122, 1149–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Basu R. and Huse M. (2017) Mechanical communication at the immunological synapse. Trends Cell Biol., 27, 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sharfe N., Freywald A., Toro A., Dadi H. and Roifman C. (2002) Ephrin stimulation modulates T cell chemotaxis. Eur. J. Immunol., 32, 3745–3755. [DOI] [PubMed] [Google Scholar]
- 84. Lu Q., Sun E.E., Klein R.S. and Flanagan J.G. (2001) Ephrin-B reverse signaling is mediated by a novel PDZ-RGS protein and selectively inhibits G protein-coupled chemoattraction. Cell, 105, 69–79. [DOI] [PubMed] [Google Scholar]
- 85. U.S. Food and Drug Administration 2006 June 28Centre for Drug Evaluation and Research Sprycel (dasatinib) NDA 21986/22072 approval letter. Retrieved November 4, 2018 fromhttps://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021986s000_Sprycel__APPROV.pdf.
- 86. Clinical Trials.gov [Internet] Bethesda (MD): National Library of Medicine (US) 2016 March 23. Identifier: NCT02717156, Combination therapy with Pembrolizumab and sEphB4-HSA in previously treated urothelial carcinoma Available from: https://clinicaltrials.gov/ct 2/show/NCT02717156.
- 87. Clinical Trials.gov [Internet] Bethesda (MD): National Library of Medicine (US) 2016 June 15. Identifier NCT02799485, EphB4-HSA in treating patients with Kaposi sarcoma. Available from: https://clinicaltrials.gov/ct2/show/NCT02799485.
- 88. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US) 2017 May 10. Identifier NCT03146871, Recombinant EphB4-HSA fusion protein and Azacitidine or Decitabine in treating patients with relapsed or refractory myelodysplastic syndrome, chronic myelomonocytic leukemia, or acute myeloid leukemia previously treated with a hypomethylating agent. Available from: https://clinicaltrials.gov/ct2/show/NCT03146871.
- 89. ClinicalTrials.gov [Internet], C. Bethesda (MD): National Library of Medicine (US) 2017 February 10. Identifier: NCT03049618, Recombinant EphB4-HSA fusion protein and Pembrolizumab in treating patients with locally advanced or metastatic non-small cell lung cancer or locally recurrent or metastatic head and neck squamous cell cancer. Available from: https://clinicaltrials.gov/ct2/show/NCT03049618.
- 90. Kontaki E. and Boumpas D.T. (2010) Innate immunity in systemic lupus erythematosus: sensing endogenous nucleic acids. J. Autoimmun., 35, 206–211. [DOI] [PubMed] [Google Scholar]
- 91. McGonagle D. and McDermott M.F. (2006) A proposed classification of the immunological diseases. PLoS Med., 3, e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lleo A. and Invernizzi P. (2013) Apotopes and innate immune system: novel players in the primary biliary cirrhosis scenario. Dig. Liver Dis., 45, 630–636. [DOI] [PubMed] [Google Scholar]
- 93. Arad M. and Weiner I. (2009) Disruption of latent inhibition induced by ovariectomy can be reversed by estradiol and clozapine as well as by co-administration of haloperidol with estradiol but not by haloperidol alone. Psychopharmacology (Berl)., 206, 731–40. [DOI] [PubMed] [Google Scholar]
- 94. Zhu ML., Bakhru P., Nelson JS., Free M., Martin A. et al. (2016) Sex bias in CNS autoimmune disease mediated by androgen control of autoimmune regulator. Nat Commun, 7, 11350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Markle JG., Frank DN., Mortin-Toth S., Robertson CE., Feazel LM., Rolle-Kampczyk U. et al. (2013) Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science, 339, 1084–8. [DOI] [PubMed] [Google Scholar]
- 96. Yang J., Lee S.H., Goddard M.E. and Visscher P.M. (2011) GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet., 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bulik-Sullivan B.K., Loh P.R., Finucane H.K., Ripke S., Yang J., Patterson N., Daly M.J., Price A.L. and Neale B.M. (2015) LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet., 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. International Hap Map 3 Consortium, Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F. et al. (2010) Integrating common and rare genetic variation in diverse human populations. Nature, 467, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Purcell S., Cherny S.S. and Sham P.C. (2003) Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics, 19, 149–150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.