

Abstract
Purpose:
Preclinical studies have demonstrated that post-irradiation tumor revascularization is dependent on a stromal cell-derived factor-1 (SDF-1)/C-X-C chemokine receptor type 4 (CXCR4)-driven process in which myeloid cells are recruited from bone marrow. Blocking this axis results in survival improvement in preclinical models of solid tumors, including glioblastoma (GBM). We conducted a phase I/II study to determine the safety and efficacy of Macrophage Exclusion after Radiation Therapy (MERT) using the reversible CXCR4 inhibitor plerixafor in newly diagnosed glioblastoma patients.
Patients and Methods:
We enrolled 9 patients to the phase I study and an additional 20 patients to phase II using a modified toxicity probability interval (mTPI) design. Plerixafor was continuously infused intravenously via PICC line for four consecutive weeks beginning at day 35 of conventional treatment with concurrent chemo-radiation. Blood serum samples were obtained for pharmacokinetic analysis. Additional studies included relative cerebral blood volume (rCBV) analysis using MRI and histopathology analysis of recurrent tumors.
Results:
Plerixafor was well tolerated with no drug-attributable grade 3 toxicities observed. At the maximum dose of 400 μg/kg/day, biomarker analysis found suprathreshold plerixafor serum levels and an increase in plasma SDF-1 levels. Median overall survival was 21.3 months (95% Confidence Interval (CI) 15.9, NA) with a progression-free survival of 14.5 months (95% CI 11.9, NA). MRI and histopathology support the mechanism of action to inhibit post-irradiation tumor revascularization.
Conclusions:
Infusion of the CXCR4 inhibitor plerixafor was well tolerated as an adjunct to standard chemo-irradiation in newly diagnosed GBM patients and improves local control of tumor recurrences.
Keywords: Glioblastoma, SDF-1/CXCR4, Phase I/II, Bone marrow derived cells (BMDC), Radiation enhancement, Plerixafor
INTRODUCTION
Glioblastoma (GBM) is a highly aggressive brain cancer for which irradiation and chemotherapy with temozolomide remain the foundation of treatment and local recurrence remains the primary reason for treatment failure. A number of strategies have attempted to address the problem of local control, including escalating radiation doses and brachytherapy [1, 2], though these have not been found to impact in-field recurrence rates. At present, focused radiation fields combined with temozolomide chemotherapy is standard of care, though the percentage of tumors recurring locally remains approximately 80%. [3]
One potential mechanism underlying recurrence within the irradiation field (defined as <2cm from the primary tumor site) [3] is thought to be the result of the presence of radioresistant hypoxic cells in the residual tumor after surgery as well as radiation-induced HIF-1 levels that lead to enhanced cytokine production and endothelial cell radioresistance [4]. We have proposed an additional mechanism: namely that the increased local hypoxia induced by radiation prompts an increase in stromal cell-derived factor-1 (SDF-1/CXCL12) secretion, which subsequently recruits bone marrow-derived monocytes into the tumors by binding to their chemokine receptors CXCR4 and CXCR7, resulting in new tumor vessel production (vasculogenesis) and tumor recurrence [5-7]. Preclinical studies with intracranial gliomas in both mice and rats clearly demonstrate that the marked increase in monocyte/macrophage entry into irradiated tumors is mediated by the SDF-1/CXCR4-CXCR7 pathway and that inhibition of this pathway blocks macrophage entry thereby improving local control and survival after radiation therapy [8-10], a strategy we have termed Macrophage Exclusion after Radiation Therapy (MERT).
To explore whether MERT may improve survival outcomes in patients, we conducted an open label Phase I/II study using an inhibitor of the SDF-1/CXCR4 pathway, plerixafor (Mozobil; AMD3100). A small molecule CXCR4 reversible antagonist, plerixafor is currently FDA approved for use as a subcutaneous injection to mobilize stem cells for collection during bone marrow transplantation. Signaling via the CXCR4 receptor is also an important mediator of inflammatory cell trafficking and retention in tissues as well as a driver for cancer cell invasion [11-13]. Using a novel continuous infusion schema, we sought to improve local control by inhibiting post-irradiation vasculogenesis by blocking the SDF-1/CXCR4 axis. Regional cerebral blood volume measured via dynamic susceptibility contrast perfusion MRI (DSC-MRI) served as a biomarker for plerixafor activity. Here we report on the safety and efficacy of a 4-week course of plerixafor treatment for patients with newly diagnosed GBM. We observed a prolonged decrease in cerebral blood volume within the irradiated field that was associated with a decreased in-field recurrence rate.
PATIENTS & METHODS
Patient eligibility
Patients with histologically confirmed newly diagnosed WHO grade IV GBM, between ages 18 and 75 years were eligible. Inclusion criteria included Karnofsky performance score (KPS) of 60 or greater, and adequate bone marrow, renal, and hepatic function. All patients of childbearing potential were required to use contraception. Exclusion criteria included pregnancy, history of clinically significant cardiac disease, or concurrent malignancy. The protocol was approved by the Stanford Institutional Review Board and Stanford Cancer Institute’s Scientific Review Committee. The study was in accordance with institutional and federal guidelines for human investigations. All patients provided written informed consent prior to study enrollment.
Treatment plan
The objective of the phase I clinical trial was to determine the toxicity profile and the recommended phase II dose in patients with newly diagnosed WHO grade IV glioblastoma (GBM). The objective of the phase II component was to assess efficacy of plerixafor, with a primary study endpoint of six-month progression free survival among these patients. This Phase I/II non-randomized study () was designed to treat patients within the standard treatment framework [14].
The fractionated radiotherapy field was per EORTC/Stupp volumes, with gross tumor volume (GTV) including the resection cavity plus residual enhancing and non-enhancing tumor. Edema was not specifically targeted. The clinical treatment volume (CTV) was a 1.7 cm margin, shaved at natural anatomic boundaries of tumor spread such as the falx, calvarium and tentorium. The planning treatment volume (PTV) was an isotropic margin of 0.3 cm. There was a single field to 60 Gy in 30 fractions. All treatments were delivered with intensity modulated radiotherapy with daily image-guidance. VMAT IMRT with daily kv/kv IGRT. There was no cone down. All patients also received concurrent temozolomide 75 mg/m2 for 42 days, starting within one day of radiation initiation (Figure 1A).
Figure 1. Trial Design & Phase I Biomarker Results.
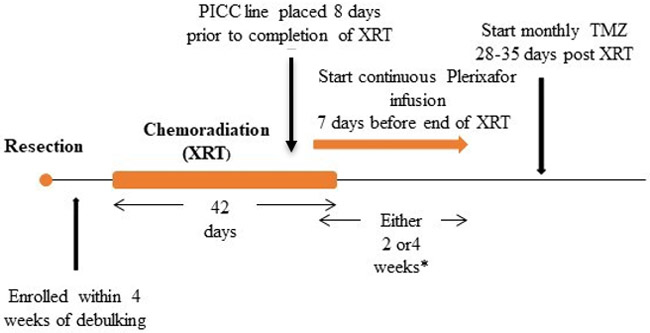
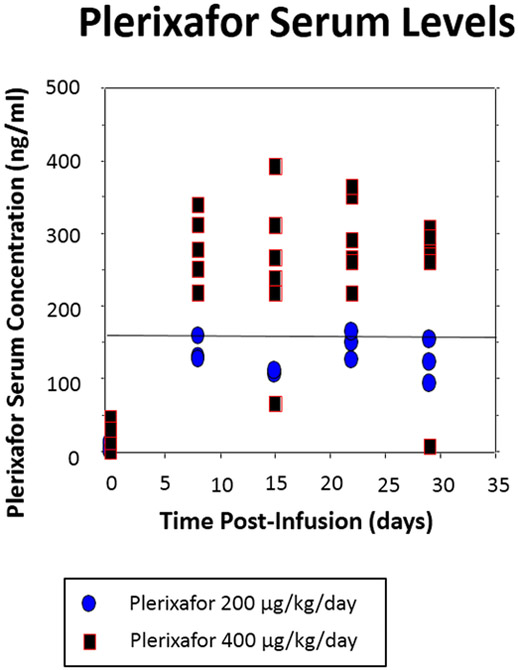
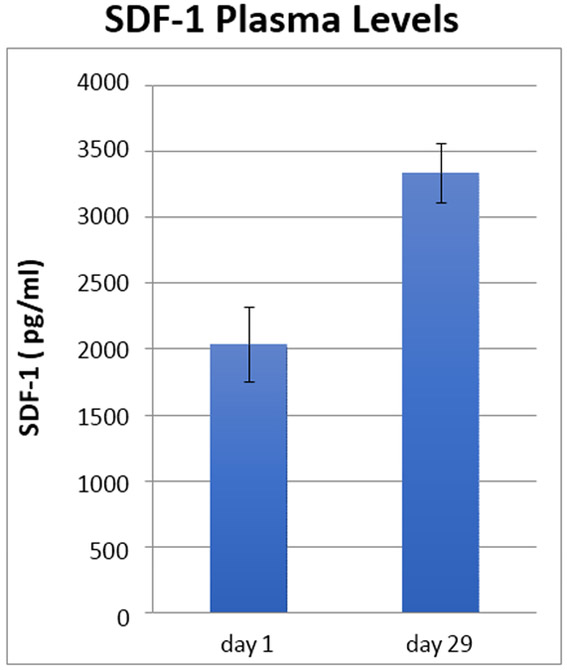
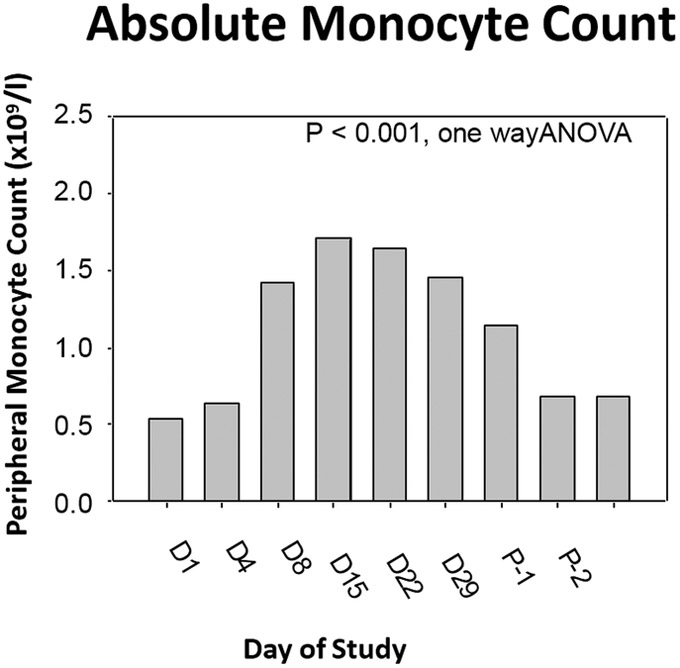
(A) Trial design. (B) Plerixafor levels performed at various time points. A significant difference between dose levels (400 and 200 μg/kg/day) measuring above 150 ng/ml was evident (P < 0.01, t-test) (C) Changes in SDF-1 levels 1 and 29 days after initiation of plerixafor infusion (P < 0.01, paired t-test). (D) Evolution of peripheral monocyte counts during plerixafor infusion (P < 0.001, one way ANOVA). P-1 and P-2 timepoints represent the first two complete blood counts performed after infusion cessation (approximately 1 and 4 weeks).
*Infusion for either two or four weeks in Phase I
Phase II dose and duration determined in the Phase I
Patients underwent PICC line placement 7–10 days prior to the completion of irradiation. Plerixafor intravenous infusion was initiated at the requisite dosage of 200 μg/kg/day in the first three patients and escalated to 400 μg/kg/day for all subsequent patients. All patients received a continuous infusion of plerixafor via PICC line for a total of 28 days in an outpatient setting. While receiving plerixafor treatment, patients were evaluated weekly per the study schedule. Patients underwent MR imaging one week after plerixafor completion, after which they were treated with monthly adjuvant temozolomide (150 mg/m2/day x 5 days) for up to six months [15].
Dose levels were determined based on a prior open-label dose escalation study evaluating continuous infusion of plerixafor in patients with HIV for ten days [16]. Using the mean serum levels from this study, we extrapolated our doses using data of efficacy and radiation enhancement in preclinical animal models [8] (the effective dose of 5mg/kg/day in the mouse predicts an equivalent dose of 400 μg/kg/day based on the mouse to human conversion faction of 0.08 [17]). The drug plerixafor (AMD3100) was supplied by Sanofi as infusion ready vials containing 10 mL of 20 mg/mL plerixafor solution. Plerixafor dose escalation was based on the mTPI table [18]. There were four potential dose levels planned within the mTPI dose escalation framework: (1) 200 μg/kg/day x 2 weeks, (2) 100 μg/kg/day x 4 weeks, (3) 200 μg/kg/day x 4 weeks and (4) 400 μg/kg/day x 4 weeks. Since all three of the first patients tolerated the starting dose of 200 μg/kg/day x 4 weeks without any dose limiting toxicities, all subsequent patients were treated at the goal dose of 400 μg/kg/day x 4 weeks.
Pharmacokinetic and pharmacodynamic biomarker assessments
Phase I patients underwent blood draws at 1 hour and 8, 15, 22 and 29 days post-initiation of infusion to monitor SDF-1 and plerixafor levels. Serum was collected by venipuncture and a Ficoll centrifugation technique was performed under sterile conditions for removal of the plasma layer. Plasma was frozen at −80°C for future assessment of SDF-1 protein and plerixafor levels. For the SDF-1 ELISA measurements, samples were thawed and centrifuged for 10 minutes at 10,000xg to ensure the removal of platelets and processed with the Quantikine ELISA kit DSA00 (R&D systems). Each sample was measured in triplicate and the experiment was performed at least twice.
To assess Plerixafor levels, 50 μl of sodium heparin human plasma was added to 20 μL of an internal standard solution (AMD16617 at 250 ng/mL in methanol), and 30 μl EDTA. Samples were then vortexed, and 50 μl of 1% TFA in acetonitrile was added. After 10 mins at room temperature, 50 μl of aliquot was added to 0.300 mL of 2:1 (v/v) 1% TFA in water and then injected onto a UPLC system equipped with a triple quadrupole tandem mass spectrometer (AB/MDS Sciex API-5000) detector operated in positive TurboIonSpray® mode. Separation of plerixafor from extracted matrix materials was accomplished using a Varian Polaris C18-A column (50 × 2.0 mm, 3.0 μm particle size) operating at 40°C. The gradient mobile phase system consisted of 0.09% TFA in water (MPA) and 0.09% TFA in acetonitrile (MPB) at a total flow rate of 0.300 mL/min.
Perfusion imaging acquisition and analysis
MRIs were performed on either a 1.5 (Signa; GE Healthcare, Milwaukee, WI, USA) or 3 Tesla (MR750; GE Healthcare) scanner. Images were acquired as part of our institution’s brain tumor imaging protocol (BTIP) [19], using gadobenate dimeglumine (MultiHance, Bracco, Milan, Italy) as the gadolinium-based contrast agent. All of the patients included for perfusion analysis had dynamic susceptibility contrast (DSC) perfusion-weighted imaging, which was acquired with non-preload single-echo gradient echo-planar imaging (TR=1800 ms, TE=35–40 ms, slice thickness=5 mm, slice gap=0 mm, echo train length=1, flip angle=60 degrees, number of averages=1, matrix=128×128 mm, FOV=240 mm). Dynamic bolus was acquired by using single-dose contrast (0.1 mmol/kg), which was intravenously administered via a power injector with an injection rate of 4 mL/sec. Between 14 and 18 axial slices of 5 mm slice thickness with skip 1.5 mm covered the brain.
Data processing for DSC-MRIs was performed with a widely available commercial software package using gamma-variate fitting (BrainStatGVF, GE Healthcare, Milwaukee, WI). Gamma-variate fitting is a post-processing method that can help to correct for contrast agent recirculation effects and minimize the effects of contrast leakage into the extravascular space [20]. Relative cerebral blood volume (rCBV) values were determined in the treatment field by region-of-interest (ROI) analysis as established in prior studies [21-23]. Based on fusion images of post-gadolinium axial 3D T1-weighted spoiled gradient recalled echo (SPGR) and radiation treatment planning maps, which were acquired using MIM MaestroTM software (MIM Software Inc., Cleveland, OH), four regions of interest were chosen in the white matter (WM) of the affected hemisphere: two within the irradiation field represented by the 95% isodose radiation line (one point adjacent to the resection cavity or tumor if there was no surgery and the other point remote from the tumor bed), one within the 50% isodose line, and one outside of the 50% isodose line. A reference point outside of the radiation field in the contralateral white matter (avoiding areas of T2/FLAIR signal abnormality) was also chosen to normalize the CBV for each patient since CBV is a relative and not absolute measurement. After selection of the points on the fused radiation planning MRI, ROIs were visually chosen in the respective and corresponding target regions on post-gadolinium axial 3D SPGR images, which were acquired as part of the DSC-MRI, using BrainStatGV perfusion software. These target points were then co-registered to the corresponding areas on the processed rCBV map. 50 mm2-sized ROIs were manually drawn at each selected point, and the mean rCBV for each ROI was recorded. Normalized rCBV ratios were obtained by dividing the target ROI by the reference ROI in the contralateral white matter.
Measurements of rCBV were quantified by DSC-MRI ROI analysis in 25 (4 did not have perfusion imaging) plerixafor-treated and 11 contemporaneously treated GBM patients at baseline, one, and six months in four brain regions: WM area adjacent to resection cavity or tumor within the 95% dose curve, WM area distal from the resection cavity or tumor within the 95% dose curve, WM area within the 50% dose curve, and WM area outside of the radiation field..
Statistical analysis
The primary objective for the Phase I study was to assess the safety of using continuous infusion plerixafor beginning one week prior to the end of concurrent chemotherapy with temozolomide and radiation therapy in patients with newly diagnosed GBM. The maximum sample size considered was 9, with a target toxicity level of 30% and precision of 5%. The primary outcome was dose-limiting toxicity, defined as cardiac arrhythmia or grade III or IV adverse events. The modified toxicity probability interval (mTPI) design was used [18]
The primary objective for the Phase II study was to assess the efficacy of plerixafor as measured by progression free survival at 6 months (PFS6) from time of diagnosis, assessed using Response Assessment in Neuro-Oncology (RANO) criteria. The analysis population included all 20 patients enrolled in the Phase II study at the final dose determined by the Phase I study. In addition, toxicity was monitored at each increment of 5 patients. If a dose limiting toxicity (DLT) was observed in one out of the first 5 patients, or 2 of the first 10, or 2 of the first 15, toxicity was reviewed to assess whether the trial should continue. This stopping rule had a 5% chance of being triggered if the true DLT toxicity rate was 5%, a 62% chance of being triggered if the true DLT rate was 20% and an 88% chance of being triggered if the true DLT rate was 30%.
The primary outcome for the Phase II study was 6-month progression free survival (PFS). The secondary outcomes were overall survival (OS) from initial diagnosis as well as safety and tolerability of the study drug. Time to progression and time to death were assessed using the Kaplan-Meier curve. If neither progression nor death occurred by the end of the study, the patient’s outcome was considered censored. Median progression free survival and median overall survival with 95% confidence intervals were presented. An age-adjusted Cox regression model was used to determine if MGMT status or extent of resection was associated with OS. All statistical analyses and figures were generated in the R (version 3.3.3) statistical computing framework.
RESULTS
Between August 2014 and April 2017, 29 patients with pathologically verified GBM were enrolled in the Phase I (n = 9) and II (n = 20) components of this study. Nine (31%) of these patients were women and the median age for all patients was 60 years [95% CI 54, 67]. A visually complete resection was accomplished in 48%, while 24% underwent biopsy only. MGMT promoter hyper-methylation was present in 45% (13 of 28 patients tested), and 92% (19 of 21) of those tested were not IDH1/2 mutated (Table 1).
Table 1.
Patient Demographics.
| Demographics | |
|---|---|
| n | 29 |
| Dose=400 μg/kg/day (%) | 26 (89.7) |
| Dose=200 μg/kg/day (%) | 3 (10.3) |
| Age (median [IQR]) | 60 [54, 67] |
| IDH1 status (%) | |
| Wild Type | 19 (65.5) |
| Mutant | 2 (6.9) |
| Unknown | 8 (27.6) |
| MGMT Methylation Positive (%) | 13 (44.8) |
| Extent of Resection (%) | |
| Biopsy | 7 (24.1) |
| Subtotal Resection | 8 (27.6) |
| Gross Total Resection | 14 (48.3) |
| Karnofsky Performance Score | |
| 60 | 2 (6.9) |
| 70 | 5 (17.2) |
| 80 | 5 (17.2) |
| 90 | 11 (37.9) |
| 100 | 6 (20.7) |
Nine patients participated in the Phase I component. The initial three patients were infused at 200 μg/kg/day and the remainder at the maximum targeted level of 400 μg/kg/day. Weekly plerixafor measurements revealed that at the lower dosage, only 27% (3 of 11) of measurements performed after week 1 exceeded the targeted minimum concentration of 150 ng/ml compared to 87% (20 of 23) at the higher dose (P < 0.01, Fishers exact test) (Figure 1B). Furthermore, mean SDF-1 levels increased almost two-fold (p < 0.01) by the end of the plerixafor infusion compared to the levels at infusion onset, providing evidence of CXCR4 blockade (P < 0.01, paired t-test) (Figure 1C). In association with this elevation we also noted a marked increase in circulating myeloid (i.e., neutrophils and monocytes) cells, consistent with a significant release of these cells from the bone marrow (Figure 1D).
Plerixafor infusion was well tolerated, with no grade 3–5 toxicities noted; according to the mTPI plan, only two (of potentially four) dose levels needed to be assessed. Therefore, in the Phase II component, twenty additional patients were treated at the 400 μg/kg/day infusion rate, without a grade 3 toxicity being encountered. For the purposes of outcome analysis, both Phase I and II patients were pooled. Twenty-seven (93%) patients achieved the primary endpoint of 6 month PFS (Figure 2A). As of February 28, 2019, median progression-free survival was 14.5 months (11.9, NA) and median overall survival was 21.3 months (15.9, NA) (Figure 2B). On multivariate analysis, survival did not significantly correlate with extent of resection, age, or performance status (Figure 2C).
Figure 2. Progression Free- and Survival Analysis.
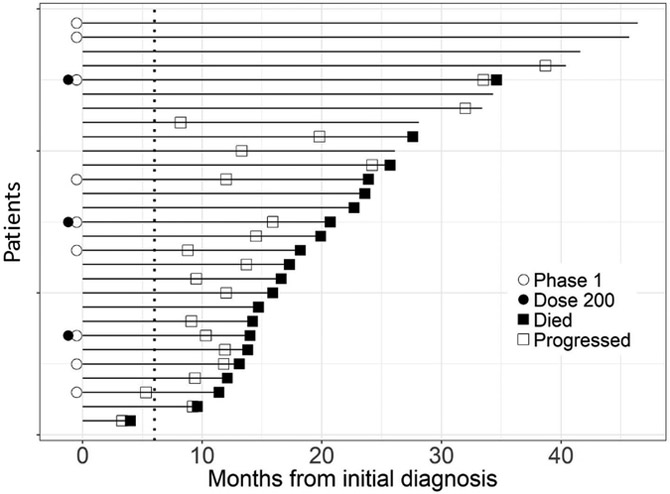
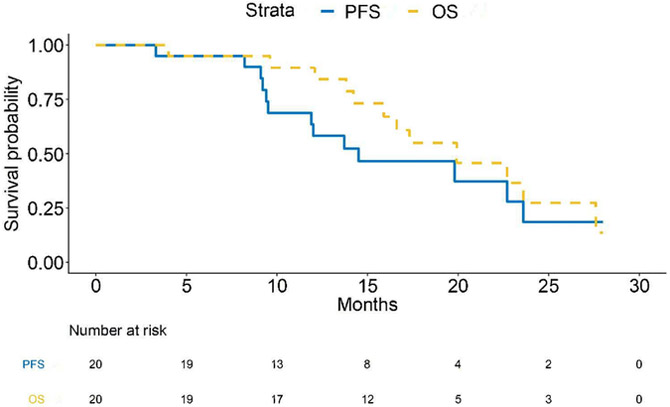
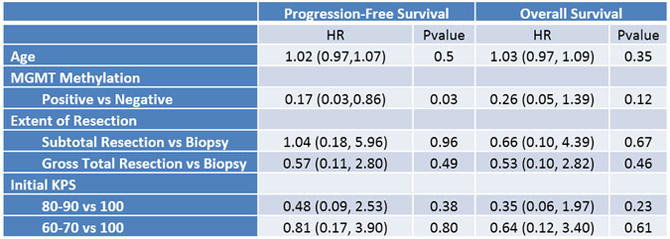
(A) Swimmer’s plot. Open squares indicate time of progression. 93% of patients attained the primary endpoint of 6-month progression free survival. (B) PFS and overall survival for the cohort. PFS for the 29 patients was 14.5 months (95% CI:11.9,NA) and overall survival 21.3 months (95% CI:15.9,NA). (C) Multivariate analysis of survival.
A distinct pattern of first recurrence was noted outside of the irradiation field including within the meninges (Figure 3). For all patients (Phase I and II), first progression occurred within the irradiation field in 47% (9 out of 19 patients). The high rate of out-of-field recurrence was also correlated with a radiographic finding of decreased relative cerebral blood volume (rCBV) on DSC-MRI. Of note, compared with a contemporaneously treated group of patients (the demographics of which are tabulated in Supplementary Table 1), rCBV decreased an average of 18% and 31% within the 95% isodose field in plerixafor-treated patients at 1- and 6-months post-completion of radiation, respectively (P < 0.03 and <0.01, 2 tailed t-test, respectively). Outside of the irradiation field, mean rCBV did not decrease (mean −9% and −2%, respectively, P not significant, 2 tailed t-test) (Figure 4A,B). Interestingly, if one assessed this effect as the percent local control (such as is done in other solid neoplasms such as head and neck [24]), a significant difference was noted between the Plerixafor treated and the contemporaneous control group (P < 0.001, log rank statistic) (Figure 4C).
Figure 3. Out-of-field recurrence.
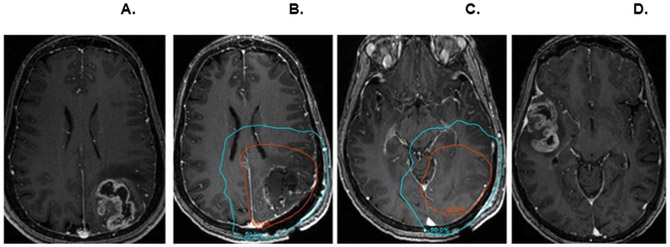
(A) Original pre-operative contrast-enhanced axial 3D T1-weighted SPGR image demonstrating a left parieto-occipital mass. (B, C). Radiation planning MRI following resection with 95% (red) and 50% (blue) isodose lines. (D) MRI performed 12 months later demonstrating a new contralateral mass in the right temporal lobe outside of the radiation field; resection of this mass confirmed GBM.
Figure 4. Impact of plerixafor infusion on CBV in the irradiated field.
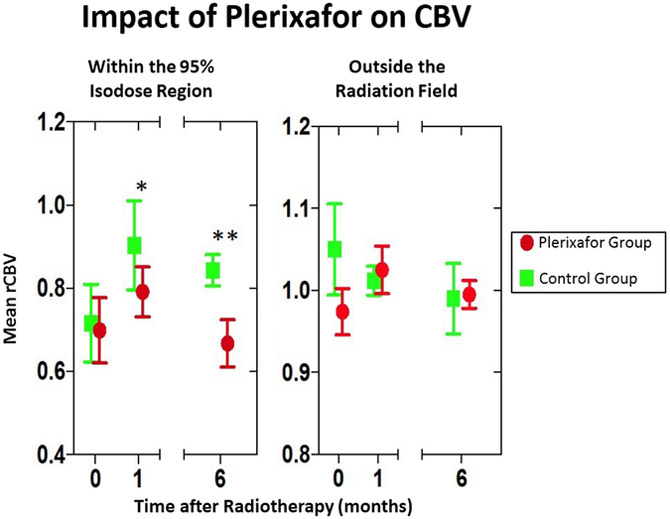
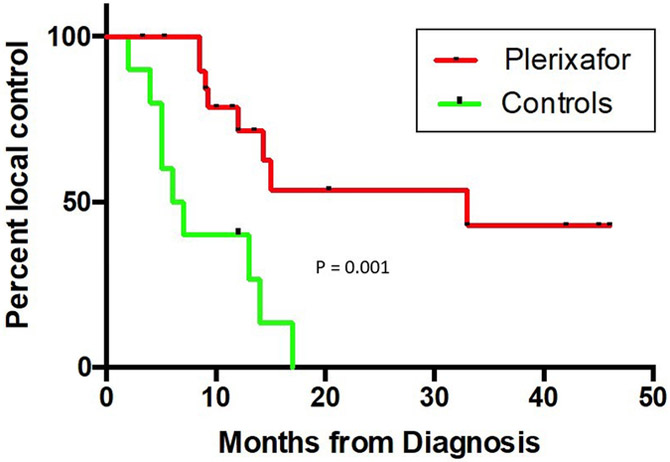
(A) Within the 95% isodose field, there is a decrease in rCBV in the plerixafor treated group compared to a contemporaneously group treated with standard RT and temozolomide. The zero time point represents data obtained prior to radiotherapy. (B) Increased time to local recurrence (defined as the development of tumor within the 95% isodose field at the time of first recurrence) between Plerixafor-treated (n = 29) and the contemporaneously assembled control group (see Supplementary Table 1 for demographic information). The differences between the groups is significant (P = 0.001, log rank statistic).
In nine (31%) patients, a pathologic assessment of changes after plerixafor treatment was available, either from a second surgery (n = 5) or autopsy (n = 4) (Figure 5A). In three of four surgical cases and one of four autopsies, only treatment related changes were noted. Interestingly, in three of the 4 autopsy cases where residual neoplasm was noted, three showed an absence of microvascular proliferation (Figure 5B,C). One surgery specimen (Pt. #4) sampled from an out of field recurrence showed a similar histologic appearance to the original tumor, with abundant microvascular proliferations noted (Figure 5D). In comparison, in another patient in whom a re-resection was performed with the irradiated field (Case 2), there was a notable decrease in microvascular proliferation, with only one specimen showing minimal microvascular changes (Figure 5E). While these results are not definitive, they do suggest that plerixafor administration resulted in aberrant revascularization of the irradiated tumor.
Figure 5. Impact of plerixafor/RT treatment on tumor vasculature.


(A) Demographic and pathological data for the nine patients in this series who underwent a second pathologic examination after Plerixafor infusion. (B,C) Representative tissue sections obtained at time of surgery and autopsy within the isodose field demonstrating original microvascular proliferation (B; arrows) and the absence of same within areas of viable tumor at autopsy performed 38 months (C). (D) Representative section from an out-of-field first recurrence in the patient whose images are illustrated in Figure 5. Microvascular proliferations (arrows) are noted within the recurrent tumor. (E) Representative section from a surgical resection from a site within the 95% isodose field, demonstrating the only region where there were marginal examples of microvascular proliferation.
DISCUSSION
Although early therapeutic studies utilized whole brain irradiation for malignant gliomas [25-27], this strategy was abandoned around 1990 when it was noted that first recurrences occurred within the pre-operative tumor site in 80% of patients [28] and that partial brain was as effective as whole brain irradiation in a randomized study [29]. Partial brain irradiation remains the standard of care and recent studies have noted that even stereotactic radiosurgery techniques continue to demonstrate that local recurrence remains the primary site of treatment failure [30]
Progression outside of the irradiation field after GBM treatment historically occurs in less than 20% of patients, with the addition of temozolomide and other therapies not significantly changing this rate [31-40]. Nevertheless, there have been case series suggesting that out-of-field recurrence rates may be higher when MGMT promoter methylation is positive [41, 42], complete resection of enhancing tumor is performed [35, 43], local RT is intensified [44], and tumor involvement of the subventricular zone (SVZ) is noted radiographically [45].
The current study was based on strong preclinical data that noted MERT provided marked improvements in tumor control and survival using both orthotopically implanted and neurocarcinogen-induced gliomas with continuous SDF-1/CXCR4-CXCR7 blockade for three to eight weeks following cranial irradiation [9, 10, 46]. We utilized plerixafor, the only currently FDA approved CXCR4 antagonist, to perform the first clinical test of MERT in primary GBM treatment. Since plerixafor is primarily administered as an intermittent daily injection at a dose that produces serum levels below that used in our preclinical studies, there were some empirical decisions that needed to be made concerning trial design. First, since this agent had never been administered for more than ten days continuously, we limited the infusion length to 28 days. To ensure maximal blockade of SDF-1 recruited myeloid cells into tumors, we therefore began treatment at the tail end of radiation and used infusion doses designed to produce continuous drug levels above our estimated effective dose of 150 ng/ml to produce a much higher AUC over a four week period than utilized earlier [16].
Our Phase I experience with plerixafor infusion indicated that it was very well tolerated at the highest dose of 400 μg/kg/day with no Grade 3 or higher toxicities being attributable to the infusion. Additionally, we noted a rapid sustained elevation of plerixafor levels at this dosage with levels being consistently higher than the targeted 150 ng/ml and confirmed CXCR4 blockade with the observation of elevated serum SDF-1 levels and increased intravascular monocyte and neutrophil counts.
As with any treatment assessment that addresses a small number of patients, one must interpret results cautiously. Nevertheless, we found the preliminary results encouraging, wherein we noted a median survival of over 21 months in a population of GBM patients of which a quarter underwent biopsy only and had KPS scores of ≤70. Importantly and consistent with the hypothesized mechanism of action was the high rate of local control with a concomitant increase of first recurrences out of the radiation field, which was defined stringently as occurring outside the 95% isodose field. This finding further correlates with a persistently diminished rCBV within the 95% isodose field that persisted for at least 6 months post-infusion in plerixafor treated patients compared with contemporaneous controls, in which first recurrence occurred in-field 90% of the time (9 of 10) (Figure 4B). This diminished blood volume mirrors a pronounced failure of blood flow to return to normal levels in irradiated tumors in mice treated with plerixafor [8]. Additionally, in the current study pathologic review also suggested that a marked perturbation in tumor vascularity occurred within irradiated fields. These findings all support the hypothesis that post-radiation CXCR4 blockade increases local control via inhibition of revascularization within the irradiated field.
Blockade of local tumor revascularization does not fully explain the high out-of-field recurrence rate observed in this study. One possibility is that CXCR4 blockade alone has no effect on tumor growth without concomitant irradiation. In a mouse xenograft model of GBM, inhibition of vasculogenesis only occurred when animals received both irradiation and SDF-1 blockade, but not with SDF-1 blockade alone [8]. A recent clinical trial assessing plerixafor with bevacizumab, without irradiation, noted minimal impact of CXCR4 blockade [47]. At the time of diagnosis, tumor cells may already be dispersed throughout the neuraxis, and therefore, are not covered by current, conventional GBM radiation fields.
This mechanism could also explain the relatively modest increase in overall survival observed in this human study when compared to preclinical studies in autochthonous rat brain tumors [9, 10]. In animal models there is a marked effect on local control and subsequent dramatic increase in median survival. Critically, the rats receive whole brain irradiation, preventing the growth of small undetectable tumors away from the primary tumor, which is a common feature of this tumor model [48]. In humans, a notable sanctuary site for tumor is the subventricular zone (SVZ) [49, 50], the radiographic involvement of which has been linked with both poorer outcome and an increased incidence of out-of-field recurrence. This suggests a possible next step that would capitalize on our findings; namely to widen the irradiated area to include at least the SVZ. While this approach might cause a higher incidence of cognitive sequelae, experimental studies using anti-SDF-1/CXCR4–7 treatment results in significant radioprotection of the skin and GI tract [51, 52]. Another recent experimental study has demonstrated that neurocognitive impairment produced by whole brain irradiation in mice could be prevented by depletion of resident macrophages (microglia) using a CSF-1 inhibitor [53].
In summary, our data suggest that the addition of plerixafor to standard chemoradiotherapy has altered the pattern of failure in GBM, a finding that was correlated with a marked decrease of rCBV within the irradiated field, and an increase in overall survival, thus providing supportive evidence for the clinical efficacy of MERT. Further studies to assess ways of exploiting this finding are indicated, not only for the malignant glioma but also for other solid cancers in which improving radiotherapeutic local control are needed.
Supplementary Material
Statement of translational relevance:
Based on strong preclinical evidence, we propose that an important factor in post-irradiation tumor recurrence in glioma and other tumor models is the entry of bone marrow derived cells, especially macrophages. Preventing entry of these cells, which can be accomplished via blockade of the CXCL12 (SDF-1)/CXCR4/7 chemokine axis, markedly improves subject survival in these preclinical studies. We have termed this strategy Macrophage Exclusion after Radiation Therapy (MERT). The current study represents the first clinical assessment of this strategy using continuous infusion plerixafor, a CXCR4 antagonist, and demonstrates that MERT is safe and well tolerated. Furthermore, the demonstration of a decreased blood flow within the local radiation field relative to a contemporaneously treated set of patients as well as a high local control rate, provides evidence that this strategy is effective and merits further study, not only in glioblastoma but in any tumor type where local radiation control is challenging.
ACKNOWLEDGEMENTS
We acknowledge the help provided by clinical research coordinators Priya Yeraballa, Stephanie Lewis, and Cathy Recht and Neuro Oncology fellows Drs. Zachary Corbin, Abdulrazag Ajlan, Tresa McGranahan, and Linda Xu. We also acknowledge the financial support of Sanofi-Aventis, the Stanford Cancer Institute, and the Stanford SPARK group including Dr. Kevin Grimes.
Footnotes
Research Support for this study was provided by Sanofi/Aventis and Stanford SPARK program.
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Gutin PH, et al. , Interstitial brachytherapy and hyperthermia for malignant gliomas. J Neurooncol, 1993. 17(2): p. 161–6. [DOI] [PubMed] [Google Scholar]
- 2.Souhami L, et al. , Randomized comparison of stereotactic radiosurgery followed by conventional radiotherapy with carmustine to conventional radiotherapy with carmustine for patients with glioblastoma multiforme: report of Radiation Therapy Oncology Group 93–05 protocol. Int J Radiat Oncol Biol Phys, 2004. 60(3): p. 853–60. [DOI] [PubMed] [Google Scholar]
- 3.Liang BC, et al. , Malignant astrocytomas: focal tumor recurrence after focal external beam radiation therapy. J Neurosurg, 1991. 75(4): p. 559–63. [DOI] [PubMed] [Google Scholar]
- 4.Barker HE, et al. , The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer, 2015. 15(7): p. 409–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown JM, Vasculogenesis: a crucial player in the resistance of solid tumours to radiotherapy. Br J Radiol, 2014. 87(1035): p. 20130686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russell JS and Brown JM, The irradiated tumor microenvironment: role of tumor-associated macrophages in vascular recovery. Front Physiol, 2013. 4: p. 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tseng D, Vasquez-Medrano DA, and Brown JM, Targeting SDF-1/CXCR4 to inhibit tumour vasculature for treatment of glioblastomas. Br J Cancer, 2011. 104(12): p. 1805–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kioi M, et al. , Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest, 2010. 120(3): p. 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu SC, et al. , Blockade of SDF-1 after irradiation inhibits tumor recurrences of autochthonous brain tumors in rats. Neuro Oncol, 2014. 16(1): p. 21–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walters MJ, et al. , Inhibition of CXCR7 extends survival following irradiation of brain tumours in mice and rats. Br J Cancer, 2014. 110(5): p. 1179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDermott DH, et al. , Plerixafor for the Treatment of WHIM Syndrome. N Engl J Med, 2019. 380(2): p. 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salazar N and Zabel BA, Support of Tumor Endothelial Cells by Chemokine Receptors. Front Immunol, 2019. 10: p. 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gravina GL, et al. , The novel CXCR4 antagonist, PRX177561, reduces tumor cell proliferation and accelerates cancer stem cell differentiation in glioblastoma preclinical models. Tumour Biol, 2017. 39(6): p. 1010428317695528. [DOI] [PubMed] [Google Scholar]
- 14.Stupp R, et al. , Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med, 2005. 352: p. 987–996. [DOI] [PubMed] [Google Scholar]
- 15.National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: central nervous system cancers V.I.2016. 2016. [cited 2017 July 12]; Available from: https://www.nccn.org/professionals/physician_gls/PDF/cns.pdf.
- 16.Hendrix CW, et al. , Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr, 2004. 37(2): p. 1253–62. [DOI] [PubMed] [Google Scholar]
- 17.Nair AB and Jacob S, A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm, 2016. 7(2): p. 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji Y and Wang SJ, Modified toxicity probability interval design: a safer and more reliable method than the 3 + 3 design for practical phase I trials. J Clin Oncol, 2013. 31(14): p. 1785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellingson BM, et al. , Consensus recommendations for a standardized Brain Tumor Imaging Protocol in clinical trials. Neuro Oncol, 2015. 17(9): p. 1188–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orsingher L, Piccinini S, and Crisi G, Differences in dynamic susceptibility contrast MR perfusion maps generated by different methods implemented in commercial software. J Comput Assist Tomogr, 2014. 38(5): p. 647–54. [DOI] [PubMed] [Google Scholar]
- 21.Caseiras GB, et al. , Relative cerebral blood volume measurements of low-grade gliomas predict patient outcome in a multi-institution setting. Eur J Radiol, 2010. 73(2): p. 215–20. [DOI] [PubMed] [Google Scholar]
- 22.Law M, et al. , Glioma grading: sensitivity, specificity, and predictive values of perfusion MR imaging and proton MR spectroscopic imaging compared with conventional MR imaging. AJNR Am J Neuroradiol, 2003. 24(10): p. 1989–98. [PMC free article] [PubMed] [Google Scholar]
- 23.Law M, et al. , Gliomas: predicting time to progression or survival with cerebral blood volume measurements at dynamic susceptibility-weighted contrast-enhanced perfusion MR imaging. Radiology, 2008. 247(2): p. 490–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nordsmark M and Overgaard J, A confirmatory prognostic study on oxygenation status and loco-regional control in advanced head and neck squamous cell carcinoma treated by radiation therapy. Radiother Oncol, 2000. 57(1): p. 39–43. [DOI] [PubMed] [Google Scholar]
- 25.Andersen AP, Postoperative irradiation of glioblastomas. Results in a randomized series. Acta Radiol Oncol Radiat Phys Biol, 1978. 17(6): p. 475–84. [DOI] [PubMed] [Google Scholar]
- 26.Walker MD, et al. , Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg, 1978. 49(3): p. 333–43. [DOI] [PubMed] [Google Scholar]
- 27.Shapiro WR and Young DF, Treatment of malignant glioma. A controlled study of chemotherapy and irradiation. Arch Neurol, 1976. 33(7): p. 494–50. [DOI] [PubMed] [Google Scholar]
- 28.Wallner KE, et al. , Patterns of failure following treatment for glioblastoma multiforme and anaplastic astrocytoma. Int J Radiat Oncol Biol Phys, 1989. 16(6): p. 1405–9. [DOI] [PubMed] [Google Scholar]
- 29.Shapiro WR, et al. , Randomized trial of three chemotherapy regimens and two radiotherapy regimens and two radiotherapy regimens in postoperative treatment of malignant glioma. Brain Tumor Cooperative Group Trial 8001. J Neurosurg, 1989. 71(1): p. 1–9. [DOI] [PubMed] [Google Scholar]
- 30.Grossman SA, et al. , Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res, 2010. 16(8): p. 2443–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gebhardt BJ, et al. , Patterns of failure for glioblastoma multiforme following limited-margin radiation and concurrent temozolomide. Radiat Oncol, 2014. 9: p. 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paulsson AK, et al. , Limited margins using modern radiotherapy techniques does not increase marginal failure rate of glioblastoma. Am J Clin Oncol, 2014. 37(2): p. 177–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petrecca K, et al. , Failure pattern following complete resection plus radiotherapy and temozolomide is at the resection margin in patients with glioblastoma. J Neurooncol, 2013. 111(1): p. 19–23. [DOI] [PubMed] [Google Scholar]
- 34.McDonald MW, et al. , Pattern of failure after limited margin radiotherapy and temozolomide for glioblastoma. Int J Radiat Oncol Biol Phys, 2011. 79(1): p. 130–6. [DOI] [PubMed] [Google Scholar]
- 35.Konishi Y, et al. , Patterns of intracranial glioblastoma recurrence after aggressive surgical resection and adjuvant management: retrospective analysis of 43 cases. Neurol Med Chir (Tokyo), 2012. 52(8): p. 577–86. [DOI] [PubMed] [Google Scholar]
- 36.Chang EL, et al. , Evaluation of peritumoral edema in the delineation of radiotherapy clinical target volumes for glioblastoma. Int J Radiat Oncol Biol Phys, 2007. 68(1): p. 144–50. [DOI] [PubMed] [Google Scholar]
- 37.Milano MT, et al. , Patterns and timing of recurrence after temozolomide-based chemoradiation for glioblastoma. Int J Radiat Oncol Biol Phys, 2010. 78(4): p. 1147–55. [DOI] [PubMed] [Google Scholar]
- 38.Pope WB, et al. , Patterns of progression in patients with recurrent glioblastoma treated with bevacizumab. Neurology, 2011. 76(5): p. 432–7. [DOI] [PubMed] [Google Scholar]
- 39.Pan H, et al. , Patterns of imaging failures in glioblastoma patients treated with chemoradiation: a retrospective study. Med Oncol, 2012. 29(3): p. 2040–5. [DOI] [PubMed] [Google Scholar]
- 40.Chamberlain MC, Radiographic patterns of relapse in glioblastoma. J Neurooncol, 2011. 101(2): p. 319–23. [DOI] [PubMed] [Google Scholar]
- 41.Brandes AA, et al. , Recurrence pattern after temozolomide concomitant with and adjuvant to radiotherapy in newly diagnosed patients with glioblastoma: correlation With MGMT promoter methylation status. J Clin Oncol, 2009. 27(8): p. 1275–9. [DOI] [PubMed] [Google Scholar]
- 42.Niyazi M, et al. , FET-PET assessed recurrence pattern after radio-chemotherapy in newly diagnosed patients with glioblastoma is influenced by MGMT methylation status. Radiother Oncol, 2012. 104(1): p. 78–82. [DOI] [PubMed] [Google Scholar]
- 43.De Bonis P, et al. , The influence of surgery on recurrence pattern of glioblastoma. Clin Neurol Neurosurg, 2013. 115(1): p. 37–43. [DOI] [PubMed] [Google Scholar]
- 44.Nakagawa K, et al. , High-dose conformal radiotherapy influenced the pattern of failure but did not improve survival in glioblastoma multiforme. Int J Radiat Oncol Biol Phys, 1998. 40(5): p. 1141–9. [DOI] [PubMed] [Google Scholar]
- 45.Lim DA, et al. , Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro Oncol, 2007. 9(4): p. 424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kioi M, et al. , Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clinical Investigation, 2010. 120: p. 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee EQ, et al. , Phase I and biomarker study of plerixafor and bevacizumab in recurrent high-grade glioma. Clin Cancer Res, 2018. [DOI] [PubMed] [Google Scholar]
- 48.Jang T, et al. , A distinct phenotypic change in gliomas at the time of magnetic resonance imaging detection. J Neurosurg, 2008. 108(4): p. 782–90. [DOI] [PubMed] [Google Scholar]
- 49.Weinberg BD, et al. , Location of subventricular zone recurrence and its radiation dose predicts survival in patients with glioblastoma. J Neurooncol, 2018. 138(3): p. 549–556. [DOI] [PubMed] [Google Scholar]
- 50.Lee JH, et al. , Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature, 2018. 560(7717): p. 243–247. [DOI] [PubMed] [Google Scholar]
- 51.Kim JH, et al. , Plerixafor, a CXCR4 antagonist, mitigates skin radiation-induced injury in mice. Radiat Res, 2012. 178(3): p. 202–6. [DOI] [PubMed] [Google Scholar]
- 52.Chaudary N, et al. , Plerixafor Improves Primary Tumor Response and Reduces Metastases in Cervical Cancer Treated with Radio-Chemotherapy. Clin Cancer Res, 2017. 23(5): p. 1242–1249. [DOI] [PubMed] [Google Scholar]
- 53.Acharya MM, et al. , Elimination of microglia improves cognitive function following cranial irradiation. Sci Rep, 2016. 6: p. 31545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.