

Abstract
In this work, a series of benzylsulfone coumarin derivatives 5a–5o were synthesized and characterized. Kinase inhibitory activity assay indicated that most of the compounds showed considerable activity against PI3K. Anti-tumor activity studies of the active compounds were also carried out in vitro on the Hela, HepG2, H1299, HCT-116, and MCF-7 tumor cell lines by MTS assay. The structure–activity relationships (SARs) of these compounds were analyzed in detail. Compound 5h exhibited the most potent activities against the mentioned cell lines with IC50 values ranging from 18.12 to 32.60 μM, followed by 5m with IC50 values of 29.30–42.14 μM. Furthermore, 5h and 5m clearly retarded the migration of Hela cells in vitro. Next, an in silico molecular docking study was conducted to evaluate the binding models of 5h and 5m towards PI3Kα and PI3Kβ. Collectively, the above findings suggested that compounds 5h and 5m might be promising PI3K inhibitors deserving further investigation for cancer treatment.
Keywords: benzylsulfone, coumarins, phosphoinositide 3-kinase (PI3K), anticancer, cell migration, molecular docking
1. Introduction
The phosphoinositide 3-kinase (PI3K) pathway, one of the most frequently altered pathways in cancer, plays a momentous role in tumorigenesis and many other cellular processes [1,2,3,4]. Considering its contributions to the regulation of cell growth, survival and metastasis, targeting major molecules within this pathway represents a pivotal opportunity for cancer therapy [5,6,7]. As one key effector node, the intracellular lipid kinases PI3Ks have been studied thoroughly and validated as promising anticancer targets over the last 30 years [8,9]. Based on their structure and function, PI3K enzymes can be divided into three classes, of which class I has been the most studied, with the subdivisions of PI3Kα, PI3Kβ, PI3Kγ and PI3Kδ [10,11]. PI3Kα and PI3Kβ are universally expressed in all cells and tissues, whereas gamma and delta isoforms are exclusively enriched in immune cells [12,13].
In recent years, small molecule inhibitors of PI3K, including pan- and isoform-specific, are currently being evaluated and have exhibited significant anti-tumor activities in laboratory and clinic, either monotherapy or in combination with cytotoxic agents [14,15,16]. To our knowledge, pan-PI3K inhibitors include but are not limited to buparlisib [17], pictilisib [18] and pilaralisib [19], targeting all isoforms. Conversely, taselisib [20], alpelisib [21], and idelalisib [22] belong to isoform-specific inhibitors.
Rigosertib (Figure 1), also known as Estybon and ON01910.Na, is a synthetic non-ATP competitive, multi-kinase inhibitor and an anticancer agent developed by Onconova Therapeutics Inc. [23]. Studies demonstrated that rigosertib evidently inhibited PI3Kα and PI3Kβ isoforms between concentrations of 1 and 10 μM in vitro, yet showed moderate inhibition of γ and δ isoforms at high concentrations above 100 μM [24]. Furthermore, rigosertib induced cytotoxicity against varieties of tumor cell lines with IC50 (half maximal inhibitory concentration) values in the nanomolar ranges, while it rarely affects normal cells [25]. Rigosertib is one of the (E)-styrylbenzylsulfones (I in Figure 1), the biological activity of which is mainly dependent on the nature, position and number of the substituents on the two parent aromatic rings [26]. In phase I/II/III trials, rigosertib has revealed significant therapeutic results patients with solid tumors and hematological malignancies [27,28]. A phase III, open-label, randomized, controlled study of “Rigosertib versus physician’s choice of treatment in MDS (myelodysplastic syndrome) patients after failure of an HMA (hypomethylation agents)” is currently in the stage of recruiting volunteers (registered as NCT02562443).
Figure 1.

The design of the new benzylsulfone coumarins (II).
The coumarin (benzo-α-pyrone) skeleton, a common but important pharmacophore, can interact noncovalently with various active sites, which brings about the various biological activities of coumarins and their derivatives. Therefore, benzopyrone has been widely used as a structural subunit for the discovery of anti-cancer agents [29,30,31,32].
In an attempt to pursue new antitumor agents with better anticancer activities and higher selectivity, the target compounds were developed by molecular modification of the styrylbenzyl sulfones structure (I in Figure 1). A non-classic bioisosteric strategy [33] was used to design new sulfone structures by replacing styryl with a coumarin unit (Figure 1). Then, a series of new coumarin substituted benzylsulfone derivatives (II in Figure 1) were synthesized and assayed with the enzymatic activities against PI3Ks by ELISA, using rigosertib as a comparison. Furthermore, their inhibitory activities against five cancer cell lines were evaluated by MTS ([3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt]) assay. The most active candidates 5h and 5m were also screened by wound-healing assay and found to delay Hela cell migration clearly. In addition, the structure–activity relationships of the synthesized compounds and possible enzyme binding modes of 5h and 5m were also illustrated.
2. Results and Discussion
2.1. Chemistry
The target compounds 5a–5o were synthesized via a three-step synthetic route from substituted benzylchloride/bromide (1a–1j) as outlined in Scheme 1. The starting materials 1a–1j were treated with mercaptoacetic acid in the presence of sodium hydroxide to give benzylmercaptoacetic acids 2a–2j [34], which were oxidized by 30% hydrogen peroxide to give benzylsulfonylacetic acids 3a–3j [35]. Finally, the target compounds (5a–5o) were synthesized from knoevenagel reaction [36,37] of 3a–3j with substituted salicylaldehydes (4a–4c) in the presence of DCC (1,3-di-cyclohexylcarbodiimide) and DMAP (4-dimethylaminopyrid). All the target compounds were purified by recrystallization and their structures were confirmed by 1H-NMR, 13C-NMR and HRMS spectra analysis.
Scheme 1.

Reagents and conditions: (i) HSCH2COOH, NaOH, methanol, r.t., 1 h; (ii) H2O2, acetic acid, 40–45 °C, 4 h; (iii) DCC, DMAP, THF, r.t., 1 h.
2.2. Biological Evaluation
2.2.1. In vitro PI3K Inhibitory Assay
The 15 newly synthesized compounds 5a‒5o were first evaluated for PI3K inhibitory activity in cellular level by ELISA assay [38], using rigosertib as a comparison. Based on the results displayed in Table 1, compounds 5h and 5m exhibited the most potent activities with inhibition rates of 50.3% and 50.8% at concentration of 20 μM, respectively, which are comparable to that of rigosertib (53.2%). At 10 μM, 5h and 5m also showed moderate inhibitory effects, comparable to rigosertib. These valuable results suggested that 5h and 5m could be potent PI3K inhibitors and worthy of further investigation.
Table 1.
The PI3K inhibitory activity of compounds 5a‒5o.
| Compound | Inhibition Rates (%) | |
|---|---|---|
| 20 μM | 10 μM | |
| Rigosertib | 53.2 ± 3.7 | 40.9 ± 4.4 |
| 5a | 39.0 ± 5.1 | 28.8 ± 3.4 |
| 5b | 45.9 ± 6.7 | 26.4 ± 4.1 |
| 5c | 37.9 ± 7.5 | 25.1 ± 3.1 |
| 5d | 36.4 ± 0.4 | 18.7 ± 3.8 |
| 5e | 36.8 ± 4.1 | 23.4 ± 4.5 |
| 5f | 47.0 ± 1.4 | 31.7 ± 5.7 |
| 5g | 43.9 ± 3.4 | 23.4 ± 4.7 |
| 5h | 50.3 ± 4.8 | 38.0 ± 6.0 |
| 5i | 46.8 ± 4.4 | 20.0 ± 4.0 |
| 5j | 37.7 ± 4.5 | 25.5 ± 1.9 |
| 5k | 42.8 ± 2.4 | 22.8 ± 6.1 |
| 5l | 47.0 ± 5.7 | 24.1 ± 5.0 |
| 5m | 50.8 ± 7.0 | 40.3 ± 4.4 |
| 5n | 46.3 ± 6.3 | 23.7 ± 4.5 |
| 5o | 38.8 ± 7.7 | 27.2 ± 3.5 |
2.2.2. Cytotoxicity against Tumor Cells
To investigate the relationship between anticancer activity and PI3K inhibitory activity, the cytotoxicity of these compounds was primarily screened and evaluated by MTS assay in vitro according to the method of Munikrishnappa [39], working with five cancer cell lines (human cervical carcinoma cell line Hela, human liver carcinoma cell line HepG2, human non-small cell lung cancer cell line H1299, human colorectal cancer cell line HCT-116, and human breast carcinoma cell line MCF-7), and rigosertib was used as a reference compound. The in vitro antiproliferative activities are summarized and presented in Table 2. Each IC50 (mean ± SD) has been derived from at least three experiments in duplicate. From the recorded IC50 values, it was clear that all of the tested compounds showed favorable antitumor activities in low micromolar ranges. In addition, each active compound revealed better activities against Hela and HepG2 when compared to those against other tumor cells. Notably, compounds 5h and 5m exhibited remarkable antitumor activities with IC50 values of 18.1–32.6 μM and 29.3–42.1 μM, respectively. Furthermore, 5h was the most effective compound against HepG2 and MCF-7, scoring IC50 values of 18.1 μM and 20.5 μM. Nevertheless, other derivatives exhibited only moderate activity against H1299 and HCT-116.The analysis results of antiproliferation activities against tumor cell lines of 5h and 5m were in accordance with those of their PI3K inhibitory activities, which suggested that the potent anticancer activities of 5h and 5m were likely related to their PI3K inhibitory activities.
Table 2.
Cytotoxicity of compounds 5a‒5o against five cancer cell lines.
| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| Hela | HepG2 | H1299 | HCT-116 | MCF-7 | |
| 5a | 41.0 ± 4.5 | 43.0 ± 3.3 | 71.8 ± 5.7 | 83.9 ± 1.3 | 56.1 ± 0.7 |
| 5b | 39.5 ± 2.2 | 36.8 ± 1.1 | 54.3 ± 1.0 | 65.8 ± 2.0 | 32.9 ± 1.9 |
| 5c | 39.9 ± 0.9 | 36.8 ± 3.1 | 81.7 ± 5.5 | 33.5 ± 0.3 | >100 |
| 5d | 48.3 ± 4.3 | 33.2 ± 4.3 | 56.9 ± 1.0 | 56.6 ± 1.4 | 41.0 ± 0.4 |
| 5e | 51.8 ± 1.9 | 23.6 ± 2.4 | 38.0 ± 1.8 | 44.7 ± 0.9 | 39.9 ± 1.7 |
| 5f | 32.2 ± 0.7 | 29.6 ± 1.9 | 40.5 ± 2.1 | 30.6 ± 1.4 | 35.5 ± 0.9 |
| 5g | 41.4 ± 2.9 | 37.6 ± 3.7 | 46.3 ± 2.2 | 38.8 ± 1.8 | 41.1 ± 1.2 |
| 5h | 24.3 ± 2.8 | 18.1 ± 0.6 | 29.6 ± 0.7 | 32.6 ± 0.9 | 20.5 ± 0.8 |
| 5i | 37.5 ± 1.5 | 32.9 ± 4.1 | 32.9 ± 0.8 | 53.7 ± 1.4 | 36.6 ± 0.8 |
| 5j | 24.6 ± 1.0 | 31.1 ± 3.3 | 50.2 ± 2.8 | 41.8 ± 0.2 | 24.6 ± 1.7 |
| 5k | 35.9 ± 2.1 | 54.4 ± 1.4 | 63.9 ± 2.2 | >100 | 54.5 ± 4.3 |
| 5l | 22.6 ± 1.8 | 49.9 ± 1.5 | 62.6 ± 2.2 | 46.3 ± 1.0 | 50.7 ± 4.1 |
| 5m | 29.7 ± 2.1 | 30.6 ± 4.3 | 31.5 ± 1.1 | 42.1 ± 1.3 | 29.3 ± 1.8 |
| 5n | 33.8 ± 5.6 | 58.9 ± 2.1 | 81.9 ± 4.6 | 50.6 ± 1.4 | 69.5 ± 2.5 |
| 5o | 44.3 ± 4.3 | 41.9 ± 2.7 | 70.8 ± 7.2 | 54.7 ± 0.6 | 82.3 ± 5.4 |
| Regosertib | 0.01 ± 0 | 0.05 ± 0.01 | 0.14 ± 0.02 | 2.36 ± 0.21 | 0.83 ± 0.08 |
From the above data, some interesting structure–activity relationships can be observed: (i) the nature of substituent on the R2 position seemed to play an important role for the cytotoxic activity: nitro group > bromine atom > hydrogen atom. For example, in the case of the HepG2 cell line, compounds 5g and 5h, which linked with the nitro group, displayed better anticancer activities compared to 5k and 5m, which linked with the bromine atom; (ii) the different mono-substituents R1 of the benzyl group showed different effects on the anti-tumor activity: fluorine atom (5f) > bromine atom (5e) > chlorine atom (5d); (iii) moreover, the positions of R1 also had an evident effect on the activity: para-substitution (5h) > ortho-substitution (5f) > meta-substitution (5g). In short, when R1 was para-fluorine and R2 was the nitro group, compound (5h) showed the best antitumor activity.
2.2.3. Wound-Healing Assays of 5h and 5m
Wound-healing assays are some of the earliest and most common techniques to study collective cell migration in vitro due to their simplicity and ability to mimic cell migration during wound healing in vivo [40,41]. The conventional scratch assay is commonly used to measure cell repair rate and provides a simple method to study the directional cell migration in vitro [42,43].
Compounds 5h and 5m, with the most potent antitumor activity, were further assessed to determine their inhibitory effects on cell migration. Migration of Hela cells was observed at 12 and 24 h after wounding (Figure 2). Hela cells treated with different concentrations of 5h and 5m (10 μM and 5 μM) showed different migration rates (Table 2). In the presence of 5h and 5m, the wound closure rate of Hela cells was much lower compared to the control group, which suggested that 5h and 5m could obviously retard and inhibit Hela cell migration in vitro.
Figure 2.

Images from a scratch assay experiment at different time points (0 h, 12 h, 24 h) and the cell migration process of Hela cells in response to different concentration of 5h and 5m.
2.3. Molecular Docking Studies of 5h and 5m
Docking studies for the synthesized compounds 5h and 5m, which showed the most potent inhibitory activity against PI3K, were carried out using the AutoDock 4.2.6 protocol in order to show their binding modes and suggest the possible mechanism of their antiproliferative activity. As PI3K proteins were extracted from Hela cells, PI3Kα and PI3Kβ, which are universally expressed in all cells [12,13], became the focus of this study.
First of all, the validation of the docking protocol was done by re-docking the native ligand into the active sites of PI3Kα (PDB ID: 3HHM [44]) and PI3Kβ (PDB ID: 2Y3A [45]) with the root-mean-square deviations (RMSD) of 0.78 Å and 0.80 Å, respectively (Figure 3). These lower values of RMSD represented the reliability and accuracy of the docking procedure in reproducing the experimentally observed results. Results of the Gibbs free energies (ΔG values) of 5h and 5m with PI3Kα and PI3Kβ are summarized in Table 3, which reveal a rough correlation between their binding free energy and their activity.
Figure 3.

Native ligand (yellow) overlap with its best docked pose (black) in a re-docking protocol. (A) Native ligand of PI3Kα overlap with its best docked pose; (B) Native ligand of PI3Kβ overlap with its best docked pose.
Table 3.
The inhibitory effect of compounds 5h and 5m on cell migration.
| Compound | Wound Closure (%) a | |
|---|---|---|
| 12 h | 24 h | |
| Control | 10.3 ± 2.8 | 19.5 ± 4.5 |
| 5h (10 μM) | 4.2 ± 1.1 ** | 9.9 ± 1.5 ** |
| 5h (5 μM) | 5.1 ± 1.1 ** | 14.4 ± 3.9 ** |
| 5m (10 μM) | 3.1 ± 1.0 ** | 8.2 ± 1.6 ** |
| 5m (5 μM) | 3.9 ± 6.6 ** | 9.1 ± 1.1 ** |
a Data were expressed as the mean ± SD (n = 30) and analyzed using t-test with significance defined as ** p < 0.01.
2.3.1. Binding Modes of Different Compounds with PI3Kα (3HHM)
Figure 4, Figure 5 and Figure 6 demonstrate compounds rigosertib, 5h, and 5m docking into the binding site of PI3Kα. Docking results showed that the tested compounds formed interactions with the key amino acid residues such as LYS802, VAL851, ILE932 and ILE848 at its active site. In particular, compound 5h showed the lowest binding free energy of −8.47 kcal/mole, as compared to rigosertib and 5m (Table 4). The binding model of compound 5h into PI3Kα revealed several molecular interactions thought to be responsible for the observed affinity: (i) two hydrogen bond interactions between the two oxygen atoms of the sulfonyl group and VAL851 and SER854; (ii) pi–alkyl and pi–sigma interactions between the benzopyrone ring and ILE932 and ILE848; and (iii) other weak interactions, including C–H bonds, and Van der Waals.
Figure 4.

A 2D model of the interaction between rigosertib with the active site of PI3Kα.
Figure 5.

A 2D model of the interaction between compound 5h with the active site of PI3Kα.
Figure 6.

A 2D model of the interaction between compound 5m with the active site of PI3Kα.
Table 4.
Binding energies of compounds 5h and 5m with the PI3Kα and PI3Kβ enzymes.
| Compound | Chemical Structure | △G (PI3Kα) Kcal/mole | △G(PI3Kβ) Kcal/mole |
|---|---|---|---|
| Rigosertib |
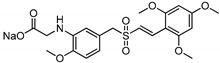
|
−7.19 | −7.26 |
| 5h |
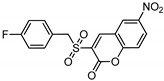
|
−8.47 | −7.74 |
| 5m |
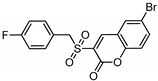
|
−7.01 | −7.28 |
Rigosertib and 5m formed more hydrogen bonds with PI3Kα than 5h, five and four respectively. Nevertheless, both rigosertib and 5m showed higher binding free energies of −7.19 and −7.01. This might be due to the formation of more efficacious hydrogen bonds with the active site. Furthermore, compared with the binding model of rigosertib and 5m (Figure 4), compound 5h interacted additionally with SER854. Thus, it seemed that SER854 played a vital role in the binding affinity of the ligand.
2.3.2. Binding Modes of Different Compounds with PI3Kβ (2Y3A)
Figure 7, Figure 8 and Figure 9 demonstrate compounds rigosertib, 5h, and 5m docking into the binding site of PI3Kβ. Similarly, compound 5h showed the lowest binding energy of −7.74. Some molecular interactions in Figure 8 were considered to be responsible for the observed affinity: (i) five hydrogen bond interactions between the ligand and LYS799, TYR833, TRP781 and SER851; (ii) pi–alkyl and pi–sigma interactions between aromatic rings and ILE797, ILE930, ILE845, MET920 and VAL847; and (iii) other weak interactions, including pi–anion, pi–pi T shaped bonds and Van der Waals.
Figure 7.

A 2D model of the interaction between rigosertib with the active site of PI3Kβ.
Figure 8.

A 2D model of the interaction between compound 5h with the active site of PI3Kβ.
Figure 9.

A 2D model of the interaction between compound 5m with the active site of PI3Kβ.
With five hydrogen bond interactions between the ligand and PI3Kβ, rigosertib displayed higher binding energy of −7.26, which is an interesting phenomenon worthy of serious consideration. Finally, it was observed that compound 5h showed additional bindings to TYR833 and SER851 through hydrogen bond interactions. Another strange example was compound 5m. There were no hydrogen bond interactions between 5m and PI3Kβ, but 5m exhibited binding energy value comparable to that of rigosertib. The halogen interactions, and pi–alkyl and pi–sigma interactions might contribute to this observed affinity.
The nice binding models of compound 5h and 5m with PI3K were consistent with the kinase assay data, which indicated that 5h and 5m might be potent PI3K inhibitors, especially targeting PI3Kα and PI3Kβ.
3. Materials and Methods
3.1. Chemistry
All the reagents and solvents were obtained from commercial suppliers (Beijing Innochem Technology Co., Ltd, Beijing, China) and used without further purification, unless otherwise stated. Rigosertib was synthesized and characterized by our research group using the reported method [26]. Reactions were monitored by thin layer chromatography (TLC) on pre-coated silica gel F254 plates with a UV indicator. Melting points were determined on a Uniscience Melting Point apparatus and were uncorrected. 1H-NMR and 13C-NMR spectra were obtained with a Bruker AM 400 and AM 500 MHz spectrometer (Palo Alto, CA, USA). Mass spectral data were obtained using electron spray ionization on a Micromass ZabSpec high-resolution mass spectrometer (Karlsruhe, Germany). The chemical shifts are reported in parts per million (δ) downfield using tetramethysilane (Me4Si) as the internal standard. Spin multiplicities are given as s (singlet), d (doublet), m (multiplet) and q (quartet). Coupling constants (J values) were measured in hertz (Hz).
Note: Only the synthesis and characterization of target compounds are presented in this article. The intermediates mentioned in Scheme 1 are described in the Supplementary Materials.
Synthesis of The Target Compounds (5a–5o)
3-[(4-Methylbenzyl)sulfonyl]-2H-chromen-2-one (5a). To an ice-cold solution of 2-[(4-methylbenzyl)sulfonyl]acetic acid (3i) (0.98 g, 4.3 mmol, 1.0 eq.) in THF was added salicylaldehyde (4a) (0.55 g, 4.5 mmol, 1.05 eq.), DCC (0.97 g, 4.7 mmol, 1.1 eq.) and DMAP (0.05 g, 0.4 mmol, 0.1 eq.). Stirring was continued at room temperature for 1h, after which time TLC showed the completion of reaction. The precipitate was filtered and the filtrate was concentrated in vacuo almost to dryness, then 50 mL ethyl acetate was added. The transparent solution was washed with dilute hydrochloric acid (20 mL × 4) and saturated brine (20 mL × 2) and dried with anhydrous sodium sulfate. The dried solution was concentrated to get the crude product, which was recrystallized in ethyl acetate to give a white product. Yield 37%, white solid; m.p. 168–170 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 2.25 (s, 3H, -CH3), 4.78 (s, 2H), 7.18 (dd, 4H, J = 8.0 Hz, Ar-H), 7.46 (t, 1H, J = 7.2 Hz, Ar-H), 7.56 (d, 1H, J = 8.4 Hz, Ar-H), 7.83 (t, 1H, J = 8.4 Hz, Ar-H), 8.01 (d, 1H, J = 7.8 Hz, Ar-H), 8.75 (s, 1H, =CH-). 13C-NMR (125 MHz, DMSO-d6) δ: 21.2, 58.7, 117.1, 117.7, 125.0, 125.8, 125.9, 129.7, 131.3, 131.6, 136.1, 138.7, 149.8, 155.2, 156.3. HRMS-ESI (m/z): calcd. for C17H14O4S [M + H]+ 315.0691, found: 315.0688.
3-[(4-Bromobenzyl)sulfonyl]-2H-chromen-2-one (5b). Yield 39%, white solid; m.p. 213–214 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 4.84 (s, 2H, -CH2-), 7.29 (d, 4H, J = 8.4 Hz, Ar-H), 7.47 (t, 1H, J = 7.2 Hz, Ar-H), 7.55–7.59 (m, 2H, Ar-H), 7.83 (t, 1H, J = 7.6 Hz, Ar-H), 8.02 (d, 1H, J = 7.8 Hz, Ar-H), 8.78 (s, 1H, =CH-). 13C-NMR (125 MHz, DMSO-d6) δ: 58.4, 117.0, 117.7, 122.8, 125.6, 126.0, 127.6, 131.7, 132.1, 133.7, 136.2, 150.1, 155.3, 156.2. HRMS-ESI (m/z): calcd. for C16H11BrO4S [M + H]+ 378.9640, found: 378.9634.
3-{[2-(Trifluoromethyl)benzyl]sulfonyl}-2H-chromen-2-one (5c). Yield 37%, pale yellow solid; m.p. 172–174 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 5.02 (s, 2H, -CH2-), 7.48 (t, 1H, J = 7.6 Hz, Ar-H), 7.57 (d, 1H, Ar-H), 7.63 (t, 1H, J = 7.2 Hz, Ar-H), 7.70-7.75 (m, 2H, Ar-H), 7.80-7.87 (m, 2H, Ar-H), 8.06 (d, 1H, J = 7.8 Hz, Ar-H), 8. 87 (s, 1H, =CH-). 13C-NMR (125 MHz, DMSO-d6) δ: 55.9, 117.0, 117.9, 125.5, 125.9, 126.6, 127.1 (q, J = 5 Hz), 129.3, 129.6, 130.0, 131.8, 133.0, 134.9, 136.2, 149.8, 155.3, 156.3. HRMS-ESI (m/z): calcd. for C17H11F3O4S [M + H]+ 369.0408, found: 369.0403.
3-[(2-Chlorobenzyl)sulfonyl]-6-nitro-2H-chromen-2-one (5d). Yield 7%, pale yellow solid; m.p. 258.5–260 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 5.01 (s, 2H, -CH2-), 7.40–7.43 (m, 2H, Ar-H), 7.50 (d, 1H, J = 2.0 Hz, Ar-H), 7.57 (d, 1H, J = 2.4 Hz, Ar-H), 7.79 (d, 1H, J = 8.8 Hz, Ar-H), 8.59 (d, 1H, J1 = 2.8 Hz, J2 = 9.2 Hz, Ar-H), 8.99 (s, 1H, =CH-), 9.04 (d, 1H, J = 2.8 Hz, Ar-H). 13C-NMR (125 MHz, DMSO-d6) δ: 56.5, 118.2, 118.7, 126.2, 127.4, 128.0, 128.3, 130.2, 131.3, 134.5, 135.0, 144.5, 149.1, 155.5, 158.7. HRMS-ESI (m/z): calcd. for C16H10ClNO6S [M + Na]+ 401.9815, found: 401.9813.
3-[(2-Bromobenzyl)sulfonyl]-6-nitro-2H-chromen-2-one (5e). Yield 13%, yellow solid; m.p. 252–254 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 5.01 (s, 2H, -CH2-), 7.34 (t, 1H, J = 8.0 Hz), 7.44 (t, 1H, J = 7.6 Hz, Ar-H), 7.56 (d, 1H, J = 7.6 Hz, Ar-H), 7.67 (d, 1H, J = 7.6 Hz, Ar-H), 7.79 (d, 1H, J = 9.2 Hz, Ar-H), 8.60 (d, 1H, J1 = 2.8 Hz, J2 = 9.0 Hz, Ar-H), 8.99 (s, 1H, =CH-), 9.05 (d, 1H, J = 2.8 Hz). 13C-NMR (125 MHz, DMSO-d6) δ: 58.9, 118.3, 118.7, 125.8, 127.4, 128.0, 128.3, 128.5, 130.2, 131.5, 133.5, 134.5, 144.5, 149.1, 155.5, 158.8. HRMS-ESI (m/z): calcd. for C16H10BrNO6S [M + Na]+ 445.9310, found: 445.9309.
3-[(2-Fluorobenzyl)sulfonyl]-6-nitro-2H-chromen-2-one (5f). Yield 17%, yellow solid; m.p. 257–259 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 4.90 (s, 2H, -CH2-), 7.24 (d, 2H, J = 7.2 Hz, Ar-H), 7.43 (t, 1H, Ar-H), 7.49 (t, 1H, Ar-H), 7.79 (d, 1H, J = 9.2 Hz, Ar-H), 8.60 (d, 1H, J1 = 2.8 Hz, J2 = 9.2 Hz, Ar-H), 8.98 (s, 1H, =CH-), 9.03 (d, 1H, J = 2.8 Hz, Ar-H). 13C-NMR (125 MHz, DMSO-d6) δ: 52.9, 115.2 (d, J = 15 Hz), 116.1 (d, J = 21 Hz), 118.2, 118.7, 125.3 (d, J = 3 Hz), 127.4, 127.9, 130.2, 131.9 (d, J = 8 Hz), 134.1 (d, J = 3 Hz), 144.5, 149.1, 155.4, 158.7, 161.5 (d, J = 246 Hz). HRMS-ESI (m/z): calcd. for C16H10FNO6S [M + H]+ 364.0291, found: 364.0290.
3-[(3-Fluorobenzyl)sulfonyl]-6-nitro-2H-chromen-2-one (5g). Yield 23%, yellow solid; m.p. 232–233 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 4.89 (s, 2H, -CH2-), 7.19–7.26 (q, 3H, Ar-H), 7.39–7.45 (q, 1H, Ar-H), 7.78 (d, 1H, J = 9.2 Hz, Ar-H), 8.59 (d, 1H, J1 = 2.8 Hz, J2 = 9.2 Hz, Ar-H), 9.00 (s, 1H, =CH-), 9.03 (d, 1H, J = 2.8 Hz, Ar-H). 13C-NMR (125 MHz, DMSO-d6) δ: 58.6, 116.2, 116.4, 118.3 (d, J = 16 Hz), 118.6 (d, J = 18 Hz), 127.4, 127.8, 127.9 (d, J = 3 Hz), 130.1, 130.3 (d, J = 9 Hz), 131.1 (d, J = 9 Hz), 144.5, 149.2, 155.5, 158.7, 162.4 (d, J = 243 Hz). HRMS-ESI (m/z): calcd. for C16H10FNO6S [M + Na]+ 386.0111, found: 386.0116.
3-[(4-Fluorobenzyl)sulfonyl]-6-nitro-2H-chromen-2-one (5h). Yield 28%, yellow solid; m.p. 258.5–260 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 4.85 (s, 2H, -CH2-), 7.22 (t, 2H, J = 8.8 Hz, Ar-H), 7.42 (t, 2H, J = 7.0 Hz, Ar-H), 7.80 (d, 1H, J = 9.2 Hz, Ar-H), 8.59 (d, 1H, J1 = 2.4 Hz, J2 = 9.2 Hz, Ar-H), 8.97 (s, 1H, =CH-), 9.03 (d, 1H, J = 2.8 Hz, Ar-H). 13C-NMR (125 MHz, DMSO-d6) δ: 58.3, 116.1 (d, J = 21 Hz), 118.2, 118.6, 124.0 (d, J = 3 Hz), 127.4, 127.8, 130.1, 133.8 (d, J = 9 Hz), 144.5, 149.1, 155.5, 158.5, 163.0 (d, J = 244 Hz). HRMS-ESI (m/z): calcd. for C16H10FNO6S [M + Na]+ 386.0111, found: 386.0109.
6-Nitro-3-{[2-(trifluoromethyl)benzyl]sulfonyl}-2H-chromen-2-one (5i). Yield 21%, pale yellow solid; m.p. 255–256 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 4.98 (s, 2H, -CH2-), 7.59–7.65 (t, 1H, Ar-H), 7.69–7.72 (t, 2H, Ar-H), 7.74–7.81 (q, 2H, Ar-H), 8.57 (d, 1H, J1 = 2.8 Hz, J2 = 9.2 Hz, Ar-H), 9.04 (s, 1H, Ar-H), 9.03 (s, 1H, =CH-). 13C-NMR (125 MHz, DMSO-d6) δ: 56.1, 118.4, 118.6, 125.6, 125.5, 127.1 (q, J = 6 Hz), 127.4, 128.7, 129.5 (q, J = 30 Hz), 130.0, 130.1, 133.1, 134.9, 144.5, 148.9, 155.5, 158.8. HRMS-ESI (m/z): calcd. for C17H10F3NO6S [M + Na]+ 436.0079, found: 436.0076.
6-Bromo-3-[(2-fluorobenzyl)sulfonyl]-2H-chromen-2-one (5j). Yield 37%, white solid; m.p. 223–224 °C.1H-NMR (400 MHz, DMSO-d6) δ: 4.89 (s, 2H, -CH2-), 7.19–7.26 (q, 2H, Ar-H), 7.40–7.50 (m, 2H, Ar-H), 7.55 (d, 1H, J = 8.9 Hz, Ar-H), 7.99 (d, 1H, J1 = 2.2 Hz, J2 = 8.6 Hz, Ar-H), 8.28 (d, 1H, J = 2.2 Hz, Ar-H), 8.73 (s, 1H, =CH-). 13C-NMR (125 MHz, DMSO-d6) δ: 52.8, 115.4 (d, J = 15 Hz), 116.0, 116.1, 117.4, 119.4 (d, J = 26 Hz), 125.2 (d, J = 3 Hz), 126.9, 131.8 (d, J = 8 Hz), 133.4, 134.1 (d, J = 3 Hz), 138.4, 148.8, 154.4, 155.7, 161.5 (d, J = 246 Hz). HRMS-ESI (m/z): calcd. for C16H10BrFO4S [M + H]+ 398.9525, found: 398.9523.
6-Bromo-3-[(3-fluorobenzyl)sulfonyl]-2H-chromen-2-one (5k). Yield 20%, white solid; m.p. 233–235 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 4.87 (s, 2H, -CH2-), 7.16–7.24 (m, 3H, Ar-H), 7.38–7.43 (m, 1H, Ar-H), 7.54 (d, 1H, J = 8.8 Hz, Ar-H), 7.98 (d, 1H, J1 = 2.4 Hz, J2 = 8.9 Hz, Ar-H), 8.28 (d, 1H, J = 2.4 Hz, Ar-H), 8.75 (s, 1H, =CH-). 13C-NMR (125 MHz, DMSO-d6) δ: 58.6, 116.2 (d, J = 20 Hz), 117.4, 118.3 (d, J = 21 Hz), 119.32, 119.58, 126.9, 127.8 (d, J = 3 Hz), 130.5 (d, J = 8 Hz), 131.1 (d, J = 9 Hz), 133.4, 138.3, 148.8, 154.4, 155.8, 162.3 (d, J = 243 Hz). HRMS-ESI (m/z): calcd. for C16H10BrFO4S [M + H]+ 398.9525, found: 398.9520.
6-Bromo-3-[(3-methylbenzyl)sulfonyl]-2H-chromen-2-one (5l). Yield 46%, white solid; m.p. 206–207 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 2.75 (s, 3H, -CH3), 4.77 (s, 2H, -CH2-), 7.11 (d, 1H, J = 7.6 Hz, Ar-H), 7.16 (s, 1H, Ar-H), 7.18 (s, 1H, Ar-H), 7.25 (t, 1H, J = 7.8 Hz, Ar-H), 7.54 (d, 1H, J = 8.8 Hz, Ar-H), 7.98 (d, 1H, J1 = 2.4 Hz, J2 = 8.8 Hz, Ar-H), 8.28 (d, 1H, J = 2.4 Hz, Ar-H), 8.74 (s, 1H, =CH-). 13C-NMR (125 MHz, DMSO-d6) δ: 21.3, 59.0, 117.4, 119.3, 119.6, 127.1, 127.6, 128.6, 129.0, 130.0, 132.2, 133.3, 138.2, 138.3, 148.6, 154.3, 155.9. HRMS-ESI (m/z): calcd. for C17H13BrO4S [M + H]+ 394.9776, found: 394.9772.
6-Bromo-3-[(4-fluorobenzyl)sulfonyl]-2H-chromen-2-one (5m). Yield 49%, white solid; m.p. 224–225 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 4.83 (s, 2H), 7.18–7.23 (t, 2H, Ar-H), 7.37–7.41 (t, 2H, Ar-H), 7.54 (t, 1H, J = 8.8 Hz, Ar-H), 7.98 (d, 1H, J1 = 2.4 Hz, J2 = 8.8 Hz, Ar-H), 8.28 (d, 1H, J = 2.4 Hz, Ar-H), 8.73 (d, 1H, =CH-). 13C-NMR (125 MHz, DMSO-d6) δ: 58.2, 116.1 (d, J = 21 Hz), 117.4, 119.3, 119.6, 124.2 (d, J = 3 Hz), 126.9, 133.4, 133.7 (d, J = 8 Hz), 138.3, 148.8, 154.3, 155.9, 162.9 (d, J = 244 Hz). HRMS-ESI (m/z): calcd. for C16H10BrFO4S [M + H]+ 396.9545, found: 396.9538.
6-Bromo-3-[(2-bromobenzyl)sulfonyl]-2H-chromen-2-one (5n). Yield 27%, white solid; m.p. 233–234 °C. 1H -NMR (400 MHz, DMSO-d6) δ: 5.0 (s, 2H, -CH2-), 7.33 (t, 1H, J = 7.6 Hz, Ar-H), 7.43 (t, 1H, J = 7.6 Hz, Ar-H), 7.52–7.56 (t, 2H, Ar-H), 7.66 (d, 1H, J = 8.0 Hz, Ar-H), 7.99 (d, 1H, J1 = 2.4 Hz, J2 = 8.8 Hz, Ar-H), 8.29 (d, 1H, J = 2.4 Hz, Ar-H), 8.75 (s, 1H, =CH-). 13C-NMR (125 MHz, DMSO-d6) δ: 58.8, 117.3, 119.3, 119.7, 125.8, 127.4, 128.1, 128.5, 131.4, 133.4, 133.4, 134.5, 138.3, 148.8, 154.4, 155.8. HRMS-ESI (m/z): calcd. for C16H10Br2O4S [M + H]+ 458.8724, found: 458.8718.
6-Bromo-3-[(3-bromobenzyl)sulfonyl]-2H-chromen-2-one (5o). Yield 19%, white solid; m.p. 235–236 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 4.86 (s, 2H, -CH2-), 7.33 (s, 1H, Ar-H), 7.34 (s, 1H, Ar-H), 7.53–7.59 (m, 3H, Ar-H), 7.99 (d, 1H, J1 = 2.4 Hz, J2 = 8.8 Hz, Ar-H), 8.29 (d, 1H, J = 2.4 Hz, Ar-H), 8.77 (s, 1H, =CH-). 13C-NMR (125 MHz, DMSO-d6) δ: 58.4, 117.4, 119.3, 119.6, 122.1, 126.9, 130.5, 130.7, 131.2, 132.1, 133.4, 134.3, 138.3, 148.9, 154.4, 155.8. HRMS-ESI (m/z): calcd. for C16H10Br2O4S [M + H]+ 458.8724, found: 458.8718.
3.2. Biological Evaluation
3.2.1. PI3K Inhibitory Activity Assay
The PI3K inhibitory activity assay was performed as described in the literature [36]. The ELISA kit for PI3K (purchased from Shanghai Biological Technology Inc., Shanghai, China) was used according to the manufacturer’s instructions. Hela cells were incubated with DMSO (vehicle control) and different concentrations (10 μM and 20 μM) of rigosertib, and compounds 5h and 5m, for 12 h separately. Total cell lysates were immunoprecipitated with anti-PI3K antibody, and PI3K activity was assessed by ELISA. Finally, the kinase assay was detected at 450 nm with microplate reader and the inhibition rate was calculated using the following equation [46].
| Inhibition rate (%) = (ODnegative control − ODcompounds)/(ODnegative control − ODblank) × 100%. |
3.2.2. Cytotoxic Activity Assays
Four cell lines (Hela, HepG2, H1299 and MCF-7) were cultured using Dulbecco’s Modified Eagle’s medium (DMEM), and HCT-116 was cultured in RPMI1640 medium. Both media were supplemented with 10% FBS (fetal bovine serum) and 1% penicillin and streptomycin (v/v). Tumor cells were seeded in 96-well tissue culture plates at a density of 4000–6000 cells/mL at 37 °C in a humidified, 5% CO2 atmosphere for 24 h. Then pre-set concentrations of tested compounds were added into 3 wells. After 48 h of incubation, the supernatant was replaced by fresh medium (100 μL/well), and 10 μL MTS reagent (some tetrazolium inner salt) was added to each well. After another 2 h of incubation at 37 °C, the optical density was measured at a wavelength of 492 nm on an ELISA microplate reader. The inhibition rates were calculated by the formula in Section 3.2.1. Next, IC50 values for the tested compounds on each cell line were calculated by nonlinear regression analysis using IBM SPSS Statistics v20.0 (IBM-SPSS, Chicago, IL, USA).
3.2.3. Wound Healing Assays
First, 6-well culture plates with markers on the outside bottom of the plates were used as reference points during image acquisition. Then, Hela cells were seeded into plates at a density of 1–1.3 × 106 per well in DMEM medium with 10% FBS. After the cells formed a 100% confluent monolayer, a 20-μL pipette tip was used to generate a wound by manually scraping, which was vertical to the parallel line marked on the plates. The cells were washed thrice with 1 mL of PBS to remove the cell debris from scratches. Next, 2 mL of DMEM with 1% FBS or 2 mL DMEM with 1% FBS and certain concentrations of the tested compounds (10 μM and 5 μM) were added to each well.
Images were obtained using an Olympus IX71 microscope (Olympus, Richmond Hill, ON, Canada) with 10× objective lenses based on the markers, which was in favor of comparing images of each sample at different time points. The wound area was quantified by Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD, USA). The migration of cells toward the wounds was displayed as a percentage of wound closure: wound closure (%) = [(At = 0 h − At = Δ h)/At = 0 h] × 100%, where, At = 0 h is the area of the wound measured immediately after scratching, and At = Δ h is the area measured 12 or 24 h later [47].
3.2.4. Molecular Docking Studies
Molecular docking of compounds 5h and 5m into the three-dimensional X-ray structures of PI3Kα (PDB code: 3HHM) and PI3Kβ (PDB code: (2Y3A) was carried out using AutoDock (version 4.2.6). The three-dimensional structures of compounds 5h and 5m were built using ChemBio 3D Ultra 12.0 software (Cambridge Soft Corporation, USA (2009)), which were then energetically minimized by using MMFF94. The protein crystal structures of PI3Kα and PI3Kβ were retrieved from the RCSB Protein Data Bank (http://www.rcsb.org). All ligands and bound waters were eliminated from the protein and the polar hydrogen was added. Docking parameters were set to default values, but only the generated conformations for each ligand were increased to 100. All docked poses of 5h and 5m were clustered using a tolerance of 2 Å for RMSD and were ranked in light of the binding docking energies. The desired conformation with lowest energy in the most populated cluster was selected for the following study. Discovery Studio 2017 R2 software was used to perform the visualization of the docking results [48].
4. Conclusions
In summary, to obtain effective lead compounds with new structures that can serve as anti-tumor agents, 15 coumarin substituted benzylsulfone derivatives 5a–5o were designed, synthesized, fully characterized and evaluated. It was concluded that most of these derivatives exhibited potent activities against enzymatic potencies and cell proliferation in vitro. In particular, compounds 5h and 5m displayed the most potent inhibitory activity against PI3K with inhibition rates of 50.3% and 50.8% at 20 μM, respectively. In addition, 5h and 5m, with broad spectrum antitumor activities, showed the greatest inhibitory activities against five tumor cell lines with IC50 values of 18.1–32.6 μM and 29.3–42.1 μM, respectively. Moreover, 5h and 5m could significantly inhibit the migration of Hela cells. The following in silico molecular docking investigations of compounds 5h and 5m into PI3Kα and PI3Kβ indicated that they could fit appropriately into active sites of PI3Kα and PI3Kβ, and interact well with important residues. The above results reveal that compounds 5h and 5m could be possible lead compounds against PI3K in cancer therapies and are worthy of further study and optimization.
Supplementary Materials
Supplementary materials are available online: the synthesis and identification of the intermediates mentioned in Scheme 1; 1H-NMR and 13C-NMR spectra of all the target compounds.
Author Contributions
Conceptualization, L.W. and T.W.; methodology, L.W., S.Z. and T.P.; software, T.W.; performing the chemistry and biological experiments, T.W.; writing—original draft preparation, T.W.; writing—review and editing, L.W., S.Z. and T.P.; data collection and analysis, X.W. and G.W. and Y.S.; funding acquisition, S.L. and L.W.
Funding
This research was performed under financial support from National Natural Science Foundation of China (Grant No. 81273431).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 5a–5o are available from the authors.
References
- 1.Dienstmann R., Rodon J., Serra V., Tabernero J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014;13:1021–1031. doi: 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- 2.Martini M., De Santis M.C., Braccini L., Gulluni F., Hirsch E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014;46:372–383. doi: 10.3109/07853890.2014.912836. [DOI] [PubMed] [Google Scholar]
- 3.LoRusso P.M. Inhibition of the PI3K/AKT/mTOR Pathway in Solid Tumors. J. Clin. Oncol. 2016;34:3803–3815. doi: 10.1200/JCO.2014.59.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayer I.A., Arteaga C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016;67:11–28. doi: 10.1146/annurev-med-062913-051343. [DOI] [PubMed] [Google Scholar]
- 5.Courtney K.D., Corcoran R.B., Engelman J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010;28:1075–1083. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartholomeusz C., Gonzalez-Angulo A.M. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin. Ther. Targets. 2012;16:121–130. doi: 10.1517/14728222.2011.644788. [DOI] [PubMed] [Google Scholar]
- 7.O’Donnell J.S., Massi D., Teng M., Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2018;48:91–103. doi: 10.1016/j.semcancer.2017.04.015. [DOI] [PubMed] [Google Scholar]
- 8.Engelman J.A., Luo J., Cantley L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 9.Rodon J., Dienstmann R., Serra V., Tabernero J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013;10:143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 10.Vanhaesebroeck B., Guillermet-Guibert J., Graupera M., Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 11.Liu X., Xu Y., Zhou Q., Chen M., Zhang Y., Liang H., Zhao J., Zhong W., Wang M. PI3K in cancer: Its structure, activation modes and role in shaping tumor microenvironment. Future Oncol. 2018;14:665–674. doi: 10.2217/fon-2017-0588. [DOI] [PubMed] [Google Scholar]
- 12.Katso R., Okkenhaug K., Ahmadi K., White S., Timms J., Waterfield M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 13.Janku F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat. Rev. 2017;59:93–101. doi: 10.1016/j.ctrv.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 14.Fruman D.A., Rommel C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X., Ding J., Meng L.H. PI3K isoform-selective inhibitors: Next-generation targeted cancer therapies. Acta Pharmacol. Sin. 2015;36:1170–1176. doi: 10.1038/aps.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mabuchi S., Kuroda H., Takahashi R., Sasano T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 2015;137:173–179. doi: 10.1016/j.ygyno.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Rodon J., Brana I., Siu L.L., De Jonge M.J., Homji N., Mills D., Di Tomaso E., Sarr C., Trandafir L., Massacesi C., et al. Phase I dose-escalation and-expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig. New Drugs. 2014;32:670–681. doi: 10.1007/s10637-014-0082-9. [DOI] [PubMed] [Google Scholar]
- 18.Sarker D., Ang J.E., Baird R., Kristeleit R., Shah K., Moreno V., Clarke P.A., Raynaud F.I., Levy G., Ware J.A., et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015;21:77–86. doi: 10.1158/1078-0432.CCR-14-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tolaney S., Burris H., Gartner E., Mayer I.A., Saura C., Maurer M., Ciruelos E., Garcia A.A., Campana F., Wu B., et al. Phase I/II study of pilaralisib (SAR245408) in combination with trastuzumab or trastuzumab plus paclitaxel in trastuzumab-refractory HER2-positive metastatic breast cancer. Breast Cancer Res. Treat. 2015;149:151–161. doi: 10.1007/s10549-014-3248-4. [DOI] [PubMed] [Google Scholar]
- 20.Juric D., Krop I., Ramanathan R.K., Wilson T.R., Ware J.A., Sanabria B.S., Savage H.M., Sampath D., Salphati L., Lin R.S., et al. Phase I Dose-Escalation Study of Taselisib, an Oral PI3K Inhibitor, in Patients with Advanced Solid Tumors. Cancer Discov. 2017;7:704–715. doi: 10.1158/2159-8290.CD-16-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenwell I.B., Flowers C.R., Blum K.A., Cohen J.B. Clinical use of PI3K inhibitors in B-cell lymphoid malignancies: Today and tomorrow. Expert Rev. Anticancer Ther. 2017;17:271–279. doi: 10.1080/14737140.2017.1285702. [DOI] [PubMed] [Google Scholar]
- 22.Westin J.R. Status of PI3K/Akt/mTOR pathway inhibitors in lymphoma. Clin. Lymphoma Myeloma Leuk. 2014;14:335–342. doi: 10.1016/j.clml.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gumireddy K., Reddy M.V., Cosenza S.C., Boominathan R., Baker S.J., Papathi N., Jiang J., Holland J., Reddy E.P. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell. 2005;7:275–286. doi: 10.1016/j.ccr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Prasad A., Park I.W., Allen H., Zhang X., Reddy M.V., Boominathan R., Reddy E.P., Groopman J.E. Styryl sulfonyl compounds inhibit translation of cyclin D1 in mantle cell lymphoma cells. Oncogene. 2009;28:1518–1528. doi: 10.1038/onc.2008.502. [DOI] [PubMed] [Google Scholar]
- 25.Chapman C.M., Sun X., Roschewski M., Aue G., Farooqui M., Stennett L., Gibellini F., Arthur D., Perez-Galan P., Wiestner A. ON 01910.Na is selectively cytotoxic for chronic lymphocytic leukemia cells through a dual mechanism of action involving PI3K/AKT inhibition and induction of oxidative stress. Clin. Cancer Res. 2012;18:1979–1991. doi: 10.1158/1078-0432.CCR-11-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reddy M.V., Venkatapuram P., Mallireddigari M.R., Pallela V.R., Cosenza S.C., Robell K.A., Akula B., Hoffman B.S., Reddy E.P. Discovery of a clinical stage multi-kinase inhibitor sodium (E)-2-{2-methoxy-5-[(2′,4′,6′-trimethoxystyrylsulfonyl)methyl]phenylamino}acetate (ON 01910.Na): Synthesis, structure-activity relationship, and biological activity. J. Med. Chem. 2011;54:6254–6276. doi: 10.1021/jm200570p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antonio J., Jing L., Messersmith W.A., Daniel L., Rudek M.A., Manoj M., Manuel H., Baker S.D., Donehower R.C. Phase I study of ON 01910.Na, a novel modulator of the Polo-like kinase 1 pathway, in adult patients with solid tumors. J. Clin. Oncol. 2008;26:5504–5510. doi: 10.1200/JCO.2008.17.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jimeno A., Chan A., Cusatis G., Zhang X., Wheelhouse J., Solomon A., Chan F., Zhao M., Cosenza S.C., Ramana R.M., et al. Evaluation of the novel mitotic modulator ON 01910.Na in pancreatic cancer and preclinical development of an ex vivo predictive assay. Oncogene. 2009;28:610–618. doi: 10.1038/onc.2008.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tian Y., Liang Z., Xu H., Mou Y., Guo C. Design, Synthesis and Cytotoxicity of Novel Dihydroartemisinin-Coumarin Hybrids via Click Chemistry. Molecules. 2016;21:758. doi: 10.3390/molecules21060758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morsy S.A., Farahat A.A., Nasr M., Tantawy A.S. Synthesis, molecular modeling and anticancer activity of new coumarin containing compounds. Saudi Pharm. J. 2017;25:873–883. doi: 10.1016/j.jsps.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z., Gu L., Wang B., Huang W., Zhang Y., Ma Z., Zeng S., Shen Z. Discovery of novel coumarin derivatives as potent and orally bioavailable BRD4 inhibitors based on scaffold hopping. J. Enzyme Inhib. Med. Chem. 2019;34:808–817. doi: 10.1080/14756366.2019.1587417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dimic D.S., Markovic Z.S., Saso L., Avdovic E.H., Dorovic J.R., Petrovic I.P., Stanisavljevic D.D., Stevanovic M.J., Potocnak I., Samolova E., et al. Synthesis and Characterization of 3-(1-((3,4-Dihydroxyphenethyl)amino)ethylidene)-chroman-2,4-dione as a Potential Antitumor Agent. Oxid. Med. Cell. Longev. 2019;2019:2069250. doi: 10.1155/2019/2069250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lima L.M., Barreiro E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005;12:23–49. doi: 10.2174/0929867053363540. [DOI] [PubMed] [Google Scholar]
- 34.Ning X., Guo Y., Wang X., Ma X., Tian C., Shi X., Zhu R., Cheng C., Du Y., Ma Z. Design, synthesis, and biological evaluation of (e)-3,4-dihydroxystyryl aralkyl sulfones and sulfoxides as novel multifunctional neuroprotective agents. J. Med. Chem. 2014;57:4302–4312. doi: 10.1021/jm500258v. [DOI] [PubMed] [Google Scholar]
- 35.Zhou N., Feng T., Shen X., Cui J., Wu R., Wang L., Wang S., Zhang S., Chen H. Synthesis, characterization, and radioprotective activity of alpha, beta-unsaturated aryl sulfone analogs and their Tempol conjugates. Medchemcomm. 2017;8:1063–1068. doi: 10.1039/C7MD00017K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freeman F. Properties and reactions of ylidenemalononitriles. Chem. Rev. 1979;80:329–350. doi: 10.1021/cr60326a004. [DOI] [Google Scholar]
- 37.Pedro V., Francisco S., Emilia T. Knoevenagel reaction in [MMIm] [MSO4]: Synthesis of coumarins. Molecules. 2011;16:4379–4388. doi: 10.3390/molecules16064379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prasad A., Khudaynazar N., Tantravahi R.V., Gillum A.M., Hoffman B.S. ON 01910.Na (rigosertib) inhibits PI3K/Akt pathway and activates oxidative stress signals in head and neck cancer cell lines. Oncotarget. 2016;7:79388–79400. doi: 10.18632/oncotarget.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Munikrishnappa C.S., Puranik S.B., Kumar G.V., Prasad Y.R. Part-1: Design, synthesis and biological evaluation of novel bromo-pyrimidine analogs as tyrosine kinase inhibitors. Eur. J. Med. Chem. 2016;119:70–82. doi: 10.1016/j.ejmech.2016.04.056. [DOI] [PubMed] [Google Scholar]
- 40.Riahi R., Yang Y., Zhang D.D., Wong P.K. Advances in wound-healing assays for probing collective cell migration. J. Lab. Autom. 2012;17:59–65. doi: 10.1177/2211068211426550. [DOI] [PubMed] [Google Scholar]
- 41.Trepat X., Chen Z., Jacobson K. Cell Migration. Compr. Physiol. 2012;2:2369–2392. doi: 10.1002/cphy.c110012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang M., Li H., Ma H., Qin J. A simple microfluidic strategy for cell migration assay in an in vitro wound-healing model. Wound Repair Regen. 2013;21:897–903. doi: 10.1111/wrr.12106. [DOI] [PubMed] [Google Scholar]
- 43.Yarrow J.C., Perlman Z.E., Westwood N.J., Mitchison T.J. A high-throughput cell migration assay using scratch wound healing, a comparison of image-based readout methods. BMC Biotechnol. 2004;4:21. doi: 10.1186/1472-6750-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mandelker D., Gabelli S.B., Schmidt-Kittler O., Zhu J., Cheong I., Huang C.H., Kinzler K.W., Vogelstein B., Amzel L.M. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc. Natl. Acad. Sci. USA. 2009;106:16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X., Vadas O., Perisic O., Anderson K., Clark J., Hawkins P., Stephens L., Williams R. Structure of Lipid Kinase p110β/p85β Elucidates an Unusual SH2-Domain-Mediated Inhibitory Mechanism. Mol. Cell. 2011;41:567–578. doi: 10.1016/j.molcel.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang L., Peng T., Wang G., Wen X., Sun Y., Zhang S., Liu S., Wang L. Design, Synthesis and Preliminary Biological Evaluation of Novel Benzyl Sulfoxide 2-Indolinone Derivatives as Anticancer Agents. Molecules. 2017;22:1979. doi: 10.3390/molecules22111979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yue P.Y., Leung E.P., Mak N.K., Wong R.N. A simplified method for quantifying cell migration/wound healing in 96-well plates. J. Biomol. Screen. 2010;15:427–433. doi: 10.1177/1087057110361772. [DOI] [PubMed] [Google Scholar]
- 48.Mu C., Wu M., Li Z. Anti-Inflammatory Effect of Novel 7-Substituted Coumarin Derivatives through Inhibition of NF-kappaB Signaling Pathway. Chem. Biodivers. 2019;16:e1800559. doi: 10.1002/cbdv.201800559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.