

Abstract
Natural 5,8-quinolinedione antibiotics exhibit a broad spectrum of activities including anticancer, antibacterial, antifungal, and antimalarial activities. The structure–activity research showed that the 5,8-quinolinedione scaffold is responsible for its biological effect. The subject of this review report is a presentation of the pharmacological activity of synthetic 5,8-quinolinedione compounds containing different groups at C-6 and/or C-7 positions. The relationship between the activity and the mechanism of action is included if these data have been included in the original literature. The review mostly covers the period between 2000 and 2019. Previously published literature data were used to present historical points.
Keywords: 5,8-quinolinedione; streptonigrin; biological activity; NAD[P]H-quinone oxidoreductase
1. Introduction
Natural products are an important source of medicinal substances used in the therapy of various ailments including infectious, cardiovascular, neurological, and oncological diseases. Such substances are produced by plants, microorganisms, and invertebrates. About half of the currently used antibiotics come from the strain of Streptomyces bacteria, a microorganism which is widespread in nature [1].
In 1959, the bioactive 7-aminoquinone called Streptonigrin 1 (Figure 1) was isolated from Streptomyces flocculus [2]. This alkaloid was also found in Actinomyces albus var bruneomycini, Streptomyces rufochromogenes, and Streptomyces echinatus [3,4].
Figure 1.

Structure of natural 5,8-quinolinedione antibiotics 1–5.
Based on chemical degradative methods and spectral analysis, Rao et al. developed the chemical structure of Streptonigrin 1 [5]. Finally, the structure of compound 1 was confirmed by x-ray crystallography in 1975 [6]. The total synthesis of the alkaloid 1 has been carried out by two research groups (Weinreb et al. and Kende et al.) from 1980 to 1982 [7,8].
In the following years, Lavendamycin 2 [9,10] and Streptonigron 3 were isolated from the Streptomyces species (Figure 1) [9,11]. Another two compounds of 5,8-quinolinedione named Ascidiathiazones A 4 and B 5 were obtained from a New Zealand ascidian Aplidium species in 2005 (Figure 1) [12,13]. Naturally occurring 5,8-quinolinediones 1–5 exhibit a wide spectrum of biological properties including anticancer, antimicrobial, antiviral, antimalarial, and anti-inflammatory activities [9,12,14,15,16,17,18,19,20,21,22]. The most attractive anticancer activity is shown by compound 1, which has been tested as an anti-leukemia drug. However, a high degree of toxicity caused the termination of this research at phase II of clinical trials [9].
The multidirectional biological activity of natural compounds 1–5 results from their structural diversity and the possibility of interaction with various molecular targets. The mechanism of biological action of the compounds containing the 5,8-quinolinedione moiety largely depends on their ability to form radicals in vivo. Numerous studies have confirmed that nicotinamide adenosine diphosphate (NADP) or NADPH-dependent quinone oxidoreductase (NQO1) are important molecular targets for new derivatives based on the structure of 5,8-quinolinedione [18,20,23].
Research into the structure–activity relationship for natural antibiotics 1–5 has shown that the 5,8-quinolinedione scaffold is essential for ensuring biological activity [9,18,24,25]. Introduction of various groups at the C-6 and C-7 positions significantly affects the biological properties of the compounds. In many cases, modification of the 5,8-quinolinedione moiety at the C-2 position reduces activity when compared to compounds not substituted at this position [26,27,28].
Most synthetic compounds containing the 5,8-quinolinedione scaffold exhibit better activity and lower toxicity than natural antibiotics 1–3 [17,29,30]. This review presents selected examples of synthetic derivatives of 5,8-quinolinedione and their biological activity.
2. Synthesis of 5,8-Quinolinedione Compounds
Synthetic derivatives of 5,8-quinolinedione are commonly obtained from 5,8-quinolinedione 7 or 6,7-dihalogen-5,8-quinolinediones 8–9. The synthetic pathways of compounds 8–9 from 8-hydroxyquinoline 6 are depicted in Scheme 1 and reaction conditions are presented in Table 1.
Scheme 1.

Synthesis of compounds 7–9.
Table 1.
Reaction conditions for the preparation of compounds 7–9.
| Compound. | Reaction Conditions | Yield [%] | Ref. |
|---|---|---|---|
| 7 | Bis-[(trifluoroacetoxy)iodo]benzene, water-acetonitrile, 0 ℃ | 82–89 | [31,32] |
| oxygen, methanol-dichloromethane, 15 ℃ | 64 | [33] | |
| Potassium dihydrogenphosphate, potassium nitrososulfonate, methanol, room temperature | 92 | [34] | |
| phenyliodine(III) diacetate, water-acetonitrile, 0 ℃ | 86 | [35] | |
| 5,10,15,20-Tetrakisphenylporphyrin, oxygen, dichloromethane, 20 ℃ | 82 | [36] | |
| [Bis(acetoxy)iodo]benzene, water-acetonitrile, 0 ℃ | 82 | [37] | |
| 8 | Hydrogenchloride, sodium chlorate, 30–60 ℃ | 17–30 | [26,30,38,39,40] |
| Hydrogenchloride, nitric acid, 90 ℃ | 39 | [41] | |
| Multi-step reaction with 3 steps: 1: hydrogenchloride, sodium nitrite, water, 0–4 °C 2: sodium dithionite, sulfuric acid, sodium hydroxide 3: hydrogenchloride, sodium chlorate |
No date | [42,43] | |
| 9 | Multi-step reaction with 3 steps: 1: hydrogenchloride, sodium nitrite, water, 0–4 °C 2: sodium dithionite, sulfuric acid, sodium hydroxide 3: sodium bromate, hydrogen bromide |
No date | [42,44] |
Treatment of compounds 7–9 with different nucleophilic agents like amines, alcohols, or thiols, led to the formation of mono- or disubstituted derivatives (Scheme 2, Scheme 3 and Scheme 4).
Scheme 2.

Synthesis of amine derivatives of 5,8-quinolinedione.
Scheme 3.

Synthesis of alkoxy derivatives of 5,8-quinolinedione.
Scheme 4.

Synthesis of thiol derivatives of 5,8-quinolinedione.
Reaction of 5,8-quinolinediones 7–9 with an amine under basic conditions led to a mixture of 6- or 7-mono-substituted compounds, but the ratio of the products depended on the type of solvent. Use of an aprotic solvent such as tetrahydrofuran (THF) or dimethylformamide (DMF) led to the 7-amino compound as a major product. The 6-substituted compounds were formed in a protic solvent like water or ethanol (Scheme 2) [40,45,46,47,48]. Moreover, 6-amino-5,8-quinolinedione could be obtained selectively in reaction with the addition of a catalyst like CeCl3 or NiCl2 in the presence of ethanol (Scheme 2) [24,26,49,50]. Modification of 6-amino-5,8-quinolinedione derivatives resulted in 6-amino-7-thiol-5,8-quinolinedione and multicyclic compounds [24,26,38].
The alkoxy derivatives were obtained in the reaction of 6,7-dichloro-5,8-quinolinedione 8 with alcohol in the presence of potassium carbonate. However, if the reaction was conducted in THF or dimethyl sulfoxide (DMSO) as a solvent, then 7-mono or 6,7-disubstituted products were obtained, respectively (Scheme 3) [28,51,52].
Treatment of compound 8 with arylthiols in the presence of ethanol resulted in 6,7-diarylthiol derivatives (Scheme 4) [53].
3. Biological Activity
3.1. Anticancer Activity
The World Health Organization (WHO) reports that cancer is the first or second main cause of death in 71 of 127 countries. The International Agency for Research on Cancer (IARC) estimated that only in 2018, 18.1 million new cases were diagnosed and 9.6 million people died of cancer. According to the GLOBOCAN 2018 research, the most common types of cancers are lung and breast cancer, which accounted for over 23% of new oncological cases [54].
Several compounds containing the 5,8-quinolinedione moiety that exhibit anticancer activity have been described in the literature. Ling et al. obtained a series of 6- and 7-arylamino-5,8-quinolinediones that were tested against drug sensitive (HeLeS3) and multidrug resistant (KB-vin) cell lines [37].
Compounds 10–11 (Figure 2) were characterized by high anticancer activity against both cell lines and their IC50 values were within the range of 0.59–1.52 µM. The 7-substituted derivative 10 exhibited 1.4-time and 1.6-time higher activity than compound 11 against HeLeS3 and KB-vin, respectively. Further research has shown that both compounds cause an NQO1-dependent antiproliferative effect and induce a dose-dependent lethal mitochondrial dysfunction in the tested cell lines. Derivative 10 exhibited anticancer activity, but did not affect a normal cell line. Moreover, it induced apoptosis by upregulating Bcl-2 protein and reducing Bax protein and cleaved caspase-3 [37].
Figure 2.

Structure of [4-(4-methylpiperazin-1-yl)phenyl]aminoquinoline-5,8-diones 10–11.
A small library of arylamine derivatives of 5,8-quinolinedione were prepared by Ryu et al. [50,55,56].
Derivatives 12–23 (Figure 3) were tested against lung cancer (A549), ovary adenocarcinoma (SK-OV-3), and malignant melanoma (SK-MEL-2) cell lines. In the series of 12–15, compound 13 exhibited high activity against all tested cell lines with the IC50 range of 0.36–0.85 µg/mL. Moreover, this derivative had a higher activity than cisplatin. Compound 12 demonstrated a selective activity against the A549 cell line, which was 2- and 3-times higher than Streptonigrin 1 and cisplatin, respectively. Additionally, for compounds 12–15, the effect on NQO1 activity was evaluated using the cytosolic fractions of the A549 cell line. This research showed that the type of substituent affects the NQO1 activity as follows: propyl (15) > chloride (12) > ethyl (14) > trifluorometoxy (13) [50]. In order to check the influence of the substituent at the C-7 position, 6-arylamino-7-halogen-5,8-quinolinediones 16–20 were obtained (Figure 3) [55]. Compounds 12, 16, and 19 differed only in the C-7 position substituent. Comparing their anticancer activity against the A549, SK-OV-3, and SK-MEL-2 cell lines, it showed that the introduction of a halogen atom at the C-7 position causes a decrease in activity [50,55]. However, the derivatives containing a chlorine atom (16–17) at this position exhibited higher activity than those containing a bromine atom (19–20). The structure–activity relationship showed that the R group affects the anticancer activity, but there was no correlation between the properties of the R group and the activity against the tested cell lines [55].
Figure 3.

Structure of arylamino-5,8-quinolinediones 12–23.
Natural 5,8-quinolinedione antibiotics contain an amine group at the C-7 position; for this reason, Ryu et al. obtained 7-arylamine-6-chloro-5,8-quinolinedione 21–23 (Figure 3) [56]. The study showed that compounds 16–18 and 21–23 had a similar activity against the SK-OV-3 and SK-MEL-2 cell lines. The 7-arylamine-6-chloro-5,8-quinolinedines 21–23 was characterized by a lower activity against lung cancer (A549) than derivatives 16–18 (Figure 3) [55,56]. In the series of 7-arylamine compounds 21–23, the lowest IC50 value against the A549 cell line was exhibited by 6-chloro-7-(4-fluorphenylamine)-5,8-quinolinedione 22, having activity comparable with that of cisplatin (IC50 = 1.80 µg/mL) [56].
The 6-arylamine derivatives 24–26 were used as substrates for the synthesis of tetracyclic compounds 27–29 (Figure 4). Both series of compounds were tested against ovary adenocarcinoma (SK-OV-3), malignant melanoma (SK-MEL-2), lung (A549), brain cancer (XF 498), and colon (HCT 15) cancer cell lines [24].
Figure 4.

Structure of 6-arylamine 24–26 and multicyclic compounds of 5,8-quinoliniedione 27–34.
A comparison of the activity of derivatives 24–25 and 27–28 showed that tetracyclic compounds 27–28 exhibited a higher anticancer activity in relation to all tested cell lines. The IC50 value for 29 was in the range of 0.12–0.21 µM, and was comparable with the activity of doxorubicin against the SK-OV-3 and XF 498 cell lines [24].
Suh et al. described tri- and tetracyclic (30–32 and 33–34, respectively) (Figure 4) imidazo-5,8-quinolinedione analogues, which exhibited potential anticancer activity against the A549, SK-OV-3, SK-MEL-2, XF 498, and HCT 15 cell lines. A comparison of the IC50 values of 30–32 derivatives against the tested cell lines suggested that side chain elongation of the substituent led to a decrease in activity. Compounds 33 and 34 showed a similar anticancer effect against the tested cell lines. In the case of the SK-OV-3 line, compound 33 exhibited an activity 3.5-times higher than 34 [21].
The indoleamine 2,3-dioxygenase 1 (IDO1) enzyme catalyzes degradation of tryptophan to N-formyl-kynurenine. This reaction initiates the kynurenine pathway leading to the synthesis of nicotinamide adenine dinucleotide (NAD), which is used to transfer electrons in many enzymatic reactions. Many studies have confirmed that IDO1 has an influence on the development of many types of human tumors [57,58,59]. One of the group that exhibited promising IDO1 inhibitory activity was imidazo-5,8-quinolindione compounds 35–36 and thiazolo-5,8-quinolinediones 37–38 (Figure 5) [38].
Figure 5.

Structure of imidazo-5,8-quinolindiones 35–36 and thiazolo-5,8-quinolinediones 37–38.
The inhibitory rates of compounds 35–36 and 37–38 at 1 µM were 65.5–76.0%. However, derivatives containing 4-thiazolyl substituent as the R group (36 and 38) showed a higher activity against IDO1 than epacadostat (INCB24360) used as the positive control. The IC50 values determined for 36 and 38 were equal to 61 nM and 18 nM, respectively. The cytotoxic activity was evaluated against human breast (MCF-7) and cervical (HeLa) cancer cell lines. The in vivo test showed that 38 reduced kynurenine levels in plasma by 30%. The CC50 values were significantly higher than the IC50 values, meaning that the tested compounds were not cytotoxic in their effective concentration against IDO1 [38].
Enzymes controlling the cell cycle process include phosphatase Cdc25, which exists as three homologues denoted as Cdc24A, Cdc25B, and Cdc25c. Overexpression of Cdc25B and Cdc25c has been observed in many human tumors and is considered to be oncogenic [60]. Lazo et al. examined 5,8-quinolinedione compounds as potential inhibitors of recombinant human Cdc25B2 [47].
In the series of tested compounds, the highest inhibitory activity was shown by compounds containing the 2-morpholin-4-ylethylamine substituent, 39 and 40 (Figure 6). A comparison of the activity of these two compounds showed that the position of amine substituent affects the activity. The IC50 value for 39 was 3-times lower than that calculated for 40, and they were equal to 0.21 µM and 0.82 µM, respectively [47]. The in vitro tests against breast cancer cells showed that derivative 39 selectively and irreversibly inhibits Cdc25 phosphatases, which results in a decrease in the Cdc2 kinase level and causes the arrest of the cells in the G1 and G2/M phases of the cell cycle [47,61].
Figure 6.

Structure of alkylamino-5,8-quinolinediones 39–42.
Compounds 41–42 (Figure 6) were designed as inhibitors of Cdc2 and ERK proteins [62]. Extracellular signal-regulated kinase (ERK) plays an important role in the signal pathway, which controls cancer cell proliferation and transformation [63]. The preliminary test showed that only compound 42 induces cytotoxicity in the lung cancer cell line (A549) significantly, and the IC50 value was estimated at 5 µM. Hsu et al. found that 42 inhibited the phosphorylated sites of Cdc2 and increased the G2/M arrest in lung cancer cells. The blockade of ERK by 42 may result in the inhibition of proliferation and apoptosis of A549 cells [62].
The alkyne moiety, which is one of the most important groups in medical chemistry, affects the biological and chemical properties of compounds that contain this fragment [64,65,66]. For this reason, acetylenic amine derivatives of 5,8-quinolinedione 43–46 (Figure 7) were prepared. The antiproliferative activity was tested against melanoma (C-32), glioblastoma (SNB-19), and breast cancer (T47D) cell lines [48].
Figure 7.

Structure of alkynylamino-5,8-quinolinediones 43–46.
Derivatives 43–44 and 45–46 exhibited a higher efficiency against the tested cell lines than cisplatin. In both groups of compounds, it was observed that an introduction of the methyl group led to a decrease in the activity. A comparison of the activity of 6- and 7-amino substituted derivatives showed that 43–44 had a lower anticancer efficiency than 45–46 [48].
Streptonigrin 1 has been tested in vitro and in vivo studies as an anti-leukemia drug, however due to its toxicity, these studies were terminated [15,67]. For this reason, alkylamine derivatives 47–48 and 49–50 (Figure 8) were evaluated against promyelocytic leukemia (HL60) and T-cells [26].
Figure 8.

Structure of alkylamino-5,8-quinolinediones 47–50 and their modifications 51–54.
The regioisomers slightly differed in their activity against HL60 and T-cells, and the IC50 values of compounds 47–48 and 49–50 (Figure 8) were in the range of 1.62–4.30 µM and 2.27–5.21 µM, respectively. The derivatives containing a 2-chloroethylamine group (48 and 50) exhibited higher activity against both cancer cell lines than those with a methylamine group (47 and 49). Compounds 47 and 49 were used to synthesize derivatives 51 and 52. The introduction of a thiomethyl group at the C-6 or C-7 position of the 5,8-quinolinedione moiety (51 and 52) caused a decrease in the activity against the tested cell lines. Cyclization of 48 and 50 led to the formation of 53 and 54. A comparison of the activity of compounds 48, 50, 53, and 54 showed that insertion of a 1,4-thiazynyl ring had an adverse impact on the anticancer activity [26].
Mitomycin C, porfiromycin, carzinophilin, and imexon are natural anticancer agents containing the aziridin moiety [68,69,70,71]. Connection of 5,8-quinolinedione with a 2-methylaziridinyl fragment led to compounds 55–57 and 58–60 (Figure 9), which exhibited a moderate activity depending on the R group. The research showed that the substituent type affects cytotoxic activity as follows: amine (57 and 60) > methoxy (56 and 59) > chloride (55 and 58) [72].
Figure 9.

Structure of 2-methylaziridinyl-5,8-quinolinediones 55–60.
A comparison of activities in both series of compounds showed that the derivatives containing the aziridinyl moiety at the C-6 position (58–60) were characterized by a higher cytotoxicity against the SK-OV-3, SK-MEL-2, and XF498 cell lines than the C-7 analogues 55–57 [72].
In the literature data, compounds containing an alkoxy, alkynyloxy, or enediyne substituent at the C-7 or/and C-6 position were described. Derivatives of this type seem to be potential anticancer agents [28,51,52].
The anticancer activity of compounds 61–64 and 65–68 (Figure 10) was tested by using melanoma (C-32), glioblastoma (SNB-19), and breast cancer (MDA-MB-231) cell lines. The obtained derivatives exhibited a higher activity against all tested cell lines than 6,7-dichloro-5,8-quinolinedione 8. A comparison of the activity of 61–63 and 65–67 showed that the introduction of an alkoxy group at the C-6 position led to an increase in the activity against MDA-MB-231 cells. For the SNB-19 line, the activity of mono- 61–63 and dialkoxy 65–67 derivatives increased together with the elongation of the alkoxy chain. Normal fibroblast (HFF-1) cells were used to evaluate the toxicity of the target derivatives. Compounds containing a propoxy (63 and 67) and propenoxy (64 and 68) group did not show toxic effects in the tested concentration range [52].
Figure 10.

Structure of alkoxy-5,8-quinolinediones 61–74.
Compounds 69–74 (Figure 10) containing substituents with a triple carbon-carbon bond were tested in relation to their anticancer activity against C-32, SNB-19, MDA-MB231, and MCF-7 cell lines. Alkynyloxy derivatives 69–71 and 72–74 exhibited a higher activity against C-32, SNB-19, and MCF-7 cells than cisplatin and 6,7-dichloro-5,8-quinolinedione 8. The activity of compounds 69–71 and 72–74 against C-32 cells depended on the substituent type and the order was as follows: 2-propynoxy (69 and 72) > 3-phenyl-2-propynyloxy (70 and 73) > 4-chloro-2-butynyloxy (71 and 74). The alkynyloxy group at the C-6 position (72–74) was an important structural element and caused an increase in the activity against the melanoma (C-32) cell line. Mono- and dialkynyloxy compounds (69–71 and 72–74) possessed different activity against breast cancer cell lines and were more cytotoxic for MCF-7 than the MDA-MB-231 cell line. The highest activity against the MCF-7 line was shown by derivative 71, for which the IC50 was equal to 0.04 µM. Additionally, the disubstituted compounds 72–74 had a higher selectivity index than monosubstituted derivatives 69–71. Molecular docking study of an NQO1 protein showed that the 5,8-quinolinedione moiety interacted with the hydrophobic active site of the enzyme by binding with Trp105, Tyr128, and Phe178. However, the disubstituted derivatives 72–74 created more stable complexes than monosubstituted compounds 69–71 [51].
The activity of compounds 63, 64, and 69 (Figure 10) against the C-32 cell line increased in the following order: propoxy (63) > 2-propenoxy (64) > 2-propynoxy (69) derivative (Figure 10). For disubstituted compounds (65, 68, and 72), no correlation between the type of bond in the substituent and activity was observed [28,51,52].
Natural enediyne antibiotics contain a nine or ten-membered unsaturated ring composed of two triple bonds connected through a double bond. Their mechanism of action is based on the transformation of the unsaturated moiety into a 1,4-benzenoid diradical, which can interact with DNA [73,74,75]. Compounds 73 (acyclic) and 74 (cyclic) (Figure 11), obtained as a result of condensation of enediyne-1,8-diol with 6,7-dichloro-5,8-quinolinedione 8, have been tested as cytotoxic agents on C-32, SNB-19, and MDA-MB-231 cell lines [28].
Figure 11.

Structure of enediyne derivatives of 5,8-quinolinedione 73–74.
Compound 74 exhibited a cytotoxic activity against MDA-MB-231 4-times higher than 73. The acyclic derivative 73 had a higher cytotoxicity than 74 against C-32 and SNB-19, for which the IC50 was equal to 3.1 µM and 2.4 µM, respectively. Additionally, acyclic compound 73 did not show any cytotoxic activity against normal fibroblast (HFF-1) cells [28].
Betulin was one of the first chemically pure compounds isolated from plant material. Betulin and its semi-synthetic derivatives are characterized by high biological activity including anticancer, antimicrobial, and antiviral activities [76,77,78]. The 5,8-quinolinedione moiety was connected with betulin derivatives to obtain hybrids 75–77 (Figure 12). The compounds were tested for their anticancer activity against glioblastoma (SNB-19), melanoma (C-32 and Colo-829), breast (MCF-7, T47D and MDA-MB-231), and lung (A549) cancer cell lines [79].
Figure 12.

Structure of hybrids betulin-5,8-quinolinedione 75–77.
Hybrids 75–77 had different activities against the SNB-19, C-32, MDA-MB-231, and A549 cells lines. The cytotoxic effect was dependent on the location of the 6-chloro-5,8-quinolinedione moiety and the order was as follows: 28-propynoyl-3-betulinyloxy (76) > 3,28-diacetyl-30-betulinyloxy (77) > 28-betulinyloxy (75). However, compounds 75–77 exhibited the highest activity against the A549 cell line; the IC50 values were in the range 0.45–8.58 µM. An apoptosis assay showed that compound 76 caused a significant increase in TP53 gene expression in A549 and MCF-7 cells. The TP53 gene encodes the p53 protein, which can affect expression of proapoptotic (Bax) and antiapoptotic (Bcl-2) protein [80,81,82]. Hybrid 76 induced a significant expression of Bax and did not affect the expression of Bcl-2 and the ratio of Bax and Bcl-2. The obtained results suggested that the cytotoxic effect of derivative 76 is associated with the mitochondrial apoptosis pathway in the A549 and MCF-7 cell lines. Molecular docking study showed that hybrid 76 was bonded to the NQO1 active site by hydrophobic interaction between the enzyme and the 5,8-quinolinedione moiety [79].
Condensation of 6,7-dichloro-5,8-quinolinedione 8 with phenolic derivatives affords benzofuro-5,8-quinolinedione compounds 78–79 (Figure 13). These compounds were tested for cytotoxic activity against lung (A549), stomach (SNU-638), colon (HCT116), ovarian (SK-OV-3), and melanoma (SK-MEL-2) cell lines [83].
Figure 13.

Structure of benzofuro-5,8-quinolinediones 78–79.
The kind of substituent at the nitrogen atom in derivatives 78–79 affected their anticancer activity. Compound 78, which contained an ethyl group, exhibited a higher activity against all tested cell lines. In addition, this derivative had activity that was 3-times and 10-times higher against A549 and HCT116 cells than that of doxorubicin, respectively [84]. Most chemotherapeutic agents interact or inhibit topoisomerase I and II, which are involved in the DNA replication process [84]. Compounds 78–79 at a 5 µM concentration strongly inhibited the activity of Topo II, and did not interact with Topo I [83].
3.2. Antimicrobial Activity
Infections caused by antibiotic-resistant bacteria are one of the most serious problems of modern medicine. The EU and the European Economic Area estimates that in 2015, the median number of infections was 671,689 and more than 33,000 people died of infection [85]. Only one antibiotic, daptomycin, belonging to lipopeptide antibiotics, has been discovered and registered as a drug in the past 50 years [86]. The search for new antibiotics is therefore a particularly important research problem.
Many compounds containing the 1,4-quinone moiety are being investigated for their antibacterial and antifungal properties [87,88,89]. One of the groups that has exhibited promising antibacterial and antifungal activity was constituted by the 5,8-quinolinedione compounds containing the arylamine group [90].
The tested compounds 80–82 (Figure 14) did not exhibit any activity against Gram-negative bacteria like E. coli and P. aeruginosa. Activity of derivatives 80–82 against Gram-positive bacteria were higher or comparable with that of ampicillin, and depended on the R substituent. The research showed that the R group affects the activity against MRS aureus, and this effect decreases in the following order: bromine (81) > chloride (82) > fluorine (80). The arylamine compounds 80–82 had a potent antifungal activity against C. albicans, C. albicans L., and A. niger. Their activities were higher than those of fluconazole and griseofulvin, respectively [90].
Figure 14.

Structure of arylamine (80–82) and thiol (83–85 and 86–89) derivatives of 5,8-qinolinedione.
Continuing research on antifungal substances, Ryu et al. synthesized two groups of compounds containing a sulfur atom: 83–85 with a thiomethyl moiety at the C-7 position and 86–89 with 6,7-bisarylthiol substituents (Figure 14) [53,91]. Derivatives 83–85 exhibited a potent activity against Candidia species and minimum inhibitory concentration (MIC) was within the range of 3.2–25 µg/mL. Conversion of the hydroxy group (83) to the ethoxy group (85) led to a decrease in the activity against Candidia krusei [91]. Bisarylthiol compounds 86–89 exhibited a lower activity against Candida albicans than flucytosine. The antifungal potency against Candida krusei depended on the R group and was as follows: ethyl (87) > methyl (86) > chloride (88) > fluorine (89) [53]. A comparison of the activity of derivatives 80–89 showed that bisarylthiol 86–89 exhibited higher antifungal activity [53,90,91].
Hybrids of the 5,8-quinolinedione moiety with dihydropyrrolyl fragment 90–93 (Figure 15) were tested for antifungal activity against the Candida species [92].
Figure 15.
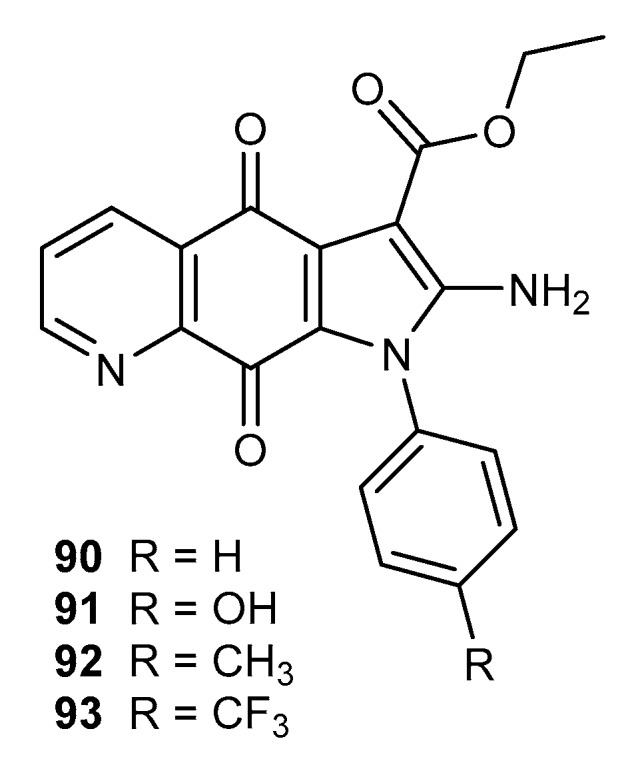
Structure of dihydropyrrole derivatives of 5,8-quinolinedione 90–93.
Compounds 90–93 exhibited the highest antifungal activity against Candida tropicalis and the MIC was within the range of 0.6–6.3 µg/mL. For these species, the order of activities was as follows: 91 > 93 > 92 > 90 [92].
Natural enediyne antibiotics are characterized by a high antibacterial activity against Gram-positive and Gram-negative strains [73,74]. The 5,8-quinolinedione moiety contains a double bond between the C-6 and C-7 carbon atom, which enables the enediyne fragment to be obtained by a reaction with terminal alkynes. Ezeokonkwa et al. described the synthesis of mono- (94–96) (Figure 16) and disubstituent (97–99) (Figure 16) hybrids of 5,8-quinolinedione with terminal alkynes and their antibacterial activity against E. coli 12, K. pneumonia, S. aureus, and P. aeruiginosa [93,94].
Figure 16.

Structure of hybrids alkyne-5,8-quinolinedione 94–99.
Compounds 94–96 and 97–99 exhibited higher activities than the reference substances, ampicillin and gentamicin. In the series of monoalkynyl derivatives 94–96, potent antibacterial activity against E. coli, K. Pneumonia, and P. aeruiginosa was shown by 94 with a MIC within the range of 0.16–0.32 mg/mL. Replacement of the bromine atom with an alkyne group influenced the antibacterial activity. Unfortunately, the correlation between structure and antibacterial activity of 94–96 and 97–99 was not observed. In the enediyne compounds 97–99, the strongest activity against E. coli and K. pneumonia was shown by 6,7-bis(phenylethynyl)-5,8-quinolinedione 98.
Gram-negative bacteria possess a specific outer membrane, lipid A, as a major component. The LpxC enzyme is responsible for the synthesis of lipid A and is a promising target for the preparation of selective anti-Gram-negative antibiotics [95,96]. The molecular docking study showed that compounds 94–99 could bind to LpxC and the obtained ΔG was higher than that of the co-crystalized inhibitor. However, dialkyne derivatives 97–99 interacted slightly less with the active site of enzyme [95,96].
The most deadly bacterial disease is tuberculosis, which caused the death of 1.3 million people in 2017. People infected by the HIV virus suffer from the active form of tuberculosis 20–30 times more often than people without HIV [97]. Tuberculosis in HIV-negative people is caused by Mycobacterium tuberculosis, but in HIV-positive patients, the etiological factor of the disease may also be Mycobacterium bovis, Mycobacterium africanum, Mycobacterium canetti, and Mycobacterium microti [98].
One of the first compounds containing the 5,8-quinolinedione moiety that exhibited activity against M. tuberculosis was compound 100 (Figure 17) with a MIC equal to 3.5 µM. Mulchin et al. synthesized a series of amine and thiol derivatives and tested these compounds regarding their anticancer and tuberculostatic activities [26,40].
Figure 17.

Structure of aliphatic amine derivatives of 5,8-quinolinedione 100–105.
In the series of compounds 47–50 (Figure 8) and 101–105 (Figure 17), the highest activity against M. bovis was shown by 6-amino-7-chloro-5,8-quinolinedione 101. Introduction of a thiomethyl or thiophenyl group at the C-7 position decreases the activity. Derivatives containing an amine group at the C-6 position (50, 105) were characterized by higher activity against M. bovis than 7-aminosubstituted compounds (48, 104). A comparison of the activities of 2-chloroethylamine (48 and 50) (Figure 8) and 2-bromoethylamine (104–105) (Figure 17) compounds showed that replacing the chloride atom with a bromine atom increased the tuberculostatic activity [26].
Tuberculostatic activities of 6- 106–109 and 7-aminosubstituted 110–113 5,8-quinolinediones (Figure 18) were tested against M. tuberculosis [40].
Figure 18.

Structure of alkylamino-5,8-quinolinediones 106–113.
Analyzing the obtained results, a general trend was observed. The research proved that the type of substituent and its position had an influence on the activity. A comparison of activities of compounds 106–107 and 110–111 showed that the derivatives containing a phenyl moiety at the C-7 position (106 and 107) had higher activity than the 6-substituted 110–111. An inverse relationship was observed for alkylamine derivatives, and the MIC values for 112–113 were lower than those for 108–109. The highest activity against M. tuberculosis was exhibited by derivative 45 (Figure 7) with a MIC equal to 8 µM [40].
The most important problems in Sub-Saharan Africa include malaria caused by the Plasmodium parasite. Quinine isolated from cinchona trees was the first anti-malarial drug and is the scaffold for obtaining more effective substances [99,100]. Lanfranchi et al. synthetized a series of derivatives that were tested on two types of P. falciparum (Figure 19) [101].
Figure 19.

Structure of methyl derivatives of 5,8-quinolinedione 114–117.
Compounds 115 and 117 (Figure 19) containing a phenyl substituent at the C-6 position did not exhibit any activity against chloroquine-sensitive 3D7 and multi-resistance Dd2 strains of P. falciparum. Derivative 114 showed a higher activity against the 3D7 strain with an IC50 equal to 727 nM. Compound 116 exhibited a comparable activity against both strains of P. falciparum [101].
The 6-substituted derivatives of 7-bromo-5,8-quinolinedione (118–120) and 5,8-quinolinedione (121–123) (Figure 20) were tested on a chloroquine-sensitive strain of P. falciparum (NF54) [102].
Figure 20.

Structure of biphenyl-4-amino-5,8-quinolinediones 118–123.
In the series of tested compounds, only 118 showed antiplasmodial activity (IC50 equal to 3 µM). Comparing the activities of 5,8-quinoinediones 118 and 122, it was found that the bromine atom at the C-7 position was necessary to maintain the antimalarial activity [102].
3.3. Anti-HIV Activity
Human immunodeficiency virus (HIV), which causes acquired immune deficiency syndrome (AIDS), is one of the most serious health problems in the worldwide. It is estimated that in 2018, 37.9 million people lived with the HIV virus and 5000 people are infected by this virus every day [103]. Streptonigrin 1 and its derivatives inhibit HIV reverse transcriptase (RT) and the postulated mechanism of action assumes an interaction of the quinone moiety with the active site of this enzyme [104]. Derivatives 124–125 at a concentration 10 µg/mL caused over 80% inhibition of HIV reverse transcriptase activity (Figure 21). The activity of 7-methoxy-5,8-quinolinedione 124 was slightly higher. At the same concentration, derivatives 124–125 did not affect cellular DNA polymerases, which could suggest a non-toxic effect of these compounds [104].
Figure 21.

Structure of disubstituted derivatives of 5,8-quinolinedione 124–127.
Alfadhli et al. tested compounds 8 and 126–127 as inhibitors of RNA binding to the HIV-1 matrix (MA) protein (Figure 21) [105]. All tested derivatives inhibited MA-RNA binding and the percentage of RNA binding inhibition was equal to 99–73%. Moreover, 8 and 127 caused the crosslinking of cytosine-containing proteins, MA and PrGap proteins in the HIV-1 virus. Unfortunately, the compounds were characterized by a high toxicity against the human T-cell line [105].
The 5,8-quinolinedione compounds were also tested as anti-inflammatory agents [26], inhibitors of enzymes such as nitric synthase, cyclooxygenase-2 [106], acetylcholine induced vasorelaxation of rat aorta with the endothelium [107,108], tyrosine phosphatase [109], and picornavirus 3C protease [110].
4. Conclusions
The 5,8-quinolinedione scaffold occurs in many natural and synthetic compounds. In recent years, interest in its potential therapeutic properties against many diseases has increased. A comparison of activities of the 6- and 7-substituted compounds showed that it depended on the type of substituent and the substitution site in the 5,8-quinolinedione moiety. According to this review, in most cases, the 6-substituted derivatives had higher activities, but is not a rule.
Despite the large number of natural and synthetic active derivatives, only a few of them have qualified for clinical trials [7,8,9,10]. The most important problem in this class of compounds is due to their toxicity. For this reason, further research in all areas of the presented biological activity of 5,8-quinolinedione compounds is still needed to design novel, more effective, and less toxic structures.
The discovery of novel modifications of the 5,8-quinolinedione moiety at C-6 and C-7 positions may lead to interesting medical applications in the future.
Author Contributions
Conceptualization and writing, M.K.-T.; Writing, E.B. and E.C.; and Review, S.B.
Funding
This research was funded by the Medical University of Silesia in Katowice, Poland, grant no. KNW-2-004/N/9/K.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Nepal K.K., Wang G. Streptomycetes: Surrogate hosts for the genetic manipulation of biosynthetic gene clusters and production of natural products. Biotechnol. Adv. 2019;37:1–20. doi: 10.1016/j.biotechadv.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rao K.V., Cullen W.P. Streptonigrin, an antitumor substance. I. Isolation and characterization. Antibiot. Annu. 1959;7:950–953. [PubMed] [Google Scholar]
- 3.Kudrina E.S., Ol’khovatova O.L., Murav’eva L.I., Gauze G.F. Systematic position and variability of the producent of antitumor antibiotic bruneomycin. Antibiotiki (Mocsow) 1966;11:400–405. [PubMed] [Google Scholar]
- 4.Societe des Usines Chemiques Rhone-Poulenc. 8,722,61. British Patent. 1961 Jul 5;
- 5.Rao K.V., Biemann K., Woodward R.B. The structure of Streptonigrin. J. Am. Chem. Soc. 1963;85:2532–2533. doi: 10.1021/ja00899a051. [DOI] [Google Scholar]
- 6.Chiu Y.Y., Lipscomb W.N. Molecular and crystal structure of Streptonigrin. J. Am. Chem. Soc. 1975;97:2525–2530. doi: 10.1021/ja00842a033. [DOI] [PubMed] [Google Scholar]
- 7.Weinreb S.M., Basha F.Z., Hibino S., Khatri N.A., Kim D., Pye W.E., Wu T.T. Total synthesis of the antitumor antibiotic streptonigrin. J. Am. Chem. Soc. 1982;104:536–544. doi: 10.1021/ja00366a028. [DOI] [Google Scholar]
- 8.Kende A.S., Lorah D.P., Boatman R.J. A new and efficient total synthesis of Streptonigrin. J. Am. Chem. Soc. 1981;103:1271–1273. doi: 10.1021/ja00395a071. [DOI] [Google Scholar]
- 9.Bringmann G., Reichert Y., Kane V.V. The total synthesis of streptonigrin and related antitumor antibiotic natural products. Tetrahedron. 2004;60:3539–3574. doi: 10.1016/j.tet.2004.02.060. [DOI] [Google Scholar]
- 10.Balitz D.M., Bush J.A., Bradner W.T., Doyle T.W., O’Herron F.A., Nettleton D.E. Isolation of lavendamycin, a new antibiotic from Streptomyces lavendulae. J. Antibiot. (Tokyo) 1982;35:259–265. doi: 10.7164/antibiotics.35.259. [DOI] [PubMed] [Google Scholar]
- 11.Herlt A.J., Rickards R.W., Wu J.P. The structure of streptonigrone, and a comment on the biosynthesis of the streptonigrin antibiotics. J. Antibiot. (Tokyo) 1985;38:516–518. doi: 10.7164/antibiotics.38.516. [DOI] [PubMed] [Google Scholar]
- 12.Pearce A.N., Chia E.W., Berridge M.V., Clark G.R., Harper J.L., Larsen L., Maas E.W., Page M.J., Perry N.B., Webb V.L., et al. Anti-inflammatory thiazine alkaloids isolated from the New Zealand ascidian Aplidium sp.: Inhibitors of the neutrophil respiratory burst in a model of gouty arthritis. J. Nat. Prod. 2007;70:936–940. doi: 10.1021/np060626o. [DOI] [PubMed] [Google Scholar]
- 13.Lam C.F., Pearce A.N., Tan S.H., Kaiser M., Copp B.R. Discovery and evaluation of thiazinoquinones as anti-protozoal agents. Mar. Drugs. 2013;11:3472–3499. doi: 10.3390/md11093472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolzán A.D., Bianchi M.S. Genotoxicity of streptonigrin: A review. Mutat. Res. 2001;488:25–37. doi: 10.1016/S1383-5742(00)00062-4. [DOI] [PubMed] [Google Scholar]
- 15.Donohoe T.J., Jones C.R., Kornahrens A.F., Barbosa L.C.A., Walport L.J., Tatton M.R., O’Hagan M., Rathi A.H., Baker D.B. Total synthesis of the antitumor antibiotic (±)-streptonigrin: First- and second-generation routes for de novo pyridine formation using ring-closing metathesis. J. Org. Chem. 2013;78:12338–12350. doi: 10.1021/jo402388f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hassani M., Cai W., Holley D.C., Lineswala J.P., Maharjan B.R., Ebrahimian G.R., Seradj H., Stocksdale M.G., Mohammadi F., Marvin C.C., et al. Novel lavendamycin analogues as antitumor agents: Synthesis, in vitro cytotoxicity, structure-metabolism, and computational molecular modeling studies with NAD(P)H:quinone oxidoreductase 1. J. Med. Chem. 2005;48:7733–7749. doi: 10.1021/jm050758z. [DOI] [PubMed] [Google Scholar]
- 17.Cai W., Hassani M., Karki R., Walter E.D., Koelsch K.H., Seradj H., Lineswala J.P., Mirzaei H., York J.S., Olang F., et al. Synthesis, metabolism and in vitro cytotoxicity studies on novel lavendamycin antitumor agents. Bioorg. Med. Chem. 2010;18:1899–1909. doi: 10.1016/j.bmc.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boger D.L., Yasuda M., Mitscher L.A., Drake S.D., Kitos P.A., Thompson S.C. Streptonigrin and lavendamycin partial structures. Probes for the minimum, potent pharmacophore of streptonigrin, lavendamycin, and synthetic quinoline-5,8-diones. J. Med. Chem. 1978;30:1918–1928. doi: 10.1021/jm00393a040. [DOI] [PubMed] [Google Scholar]
- 19.Chan B.K., Ciufolini M.A. Total synthesis of streptonigrone. J. Org. Chem. 2007;72:8489–8495. doi: 10.1021/jo701435p. [DOI] [PubMed] [Google Scholar]
- 20.Chia E.W., Pearce A.N., Berridge M.V., Larsen L., Perry N.B., Sansom C.E., Godfrey C.A., Hanton L.R., Lu G.L., Walton M., et al. Synthesis and anti-inflammatory structure–activity relationships of thiazine-quinoline-quinones: Inhibitors of the neutrophil respiratory burst in a model of acute gouty arthritis. Bioorg. Med. Chem. 2008;16:9432–9442. doi: 10.1016/j.bmc.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 21.Suh M.E., Kang M.J., Yoo H.W., Park S.Y., Lee C.O. Synthesis and cytotoxicity of 2-methyl-1-substituted-imidazo [4,5-g]quinoline-4,9-dione and 7,8-dihydro-10H-[1,4]oxazino[3′,4′:2,3]imidazo[4,5-g]quinoline-5,12-dione derivatives. Bioorg. Med. Chem. 2000;8:2079–2083. doi: 10.1016/S0968-0896(00)00132-2. [DOI] [PubMed] [Google Scholar]
- 22.McBride T.J., Oleson J.J., Woolf D. The activity of streptonigrin against the Rauscher murine leukemia virus in vivo. Cancer Res. 1966;26:727–732. [PubMed] [Google Scholar]
- 23.Parkinson E., Bair J.S., Cismesia M., Hergenrother P.J. Efficient NQO1 substrates are potent and selective anticancer agents. ACS Chem. Biol. 2013;8:2173–2183. doi: 10.1021/cb4005832. [DOI] [PubMed] [Google Scholar]
- 24.Kim Y.S., Park S.Y., Lee H.J., Suh M.E., Schollmeyer D., Lee C.O. Synthesis and cytotoxicity of 6,11-dihydro-pyrido- and 6,11-dihydro-benzo[2,3-b]phenazine-6,11-dione derivatives. Bioorg. Med. Chem. 2003;11:1709–1714. doi: 10.1016/S0968-0896(03)00028-2. [DOI] [PubMed] [Google Scholar]
- 25.Kende A.S., Naegely P.C. Total synthesis of the streptonigrin quinone carbon framework. Tetrahedron Lett. 1978;19:4775–4778. doi: 10.1016/S0040-4039(01)85728-2. [DOI] [Google Scholar]
- 26.Mulchin B.J., Newton C.G., Baty J.W., Grasso C.H., Martin W.J., Walton M.C., Dangerfield E.M., Plunkett C.H., Berridge M.V., Harper J.L., et al. The anti-cancer, anti-inflammatory and tuberculostatic activities of a series of 6,7-substituted-5,8-quinolinequinones. Bioorg. Med. Chem. 2010;19:3238–3251. doi: 10.1016/j.bmc.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 27.Kadela-Tomanek M., Pawełczak B., Jastrzębska M., Bębenek E., Chrobak E., Latocha M., Kusz J., Książek M., Boryczka S. Structural, vibrational and quantum chemical investigations for 6,7-dichloro-2-methyl-5,8-quinolinedione. Cytotoxic and molecular docking studies. J. Mol. Struct. 2018;1168:73–83. doi: 10.1016/j.molstruc.2018.05.031. [DOI] [Google Scholar]
- 28.Kadela-Tomanek M., Bębenek E., Chrobak E., Latocha M., Boryczka S. Alkoxy and enediyne derivatives containing 1,4-benzoquinone subunits-synthesis and antitumor activity. Molecules. 2017;22:447. doi: 10.3390/molecules22030447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizuno N. Mechanism of Action of Antieukaryotic and Antiviral Compounds. Volume 5. Springer; Berlin/Heidelberg, Germany: 1979. Streptonigrin; pp. 372–384. [Google Scholar]
- 30.Shaikh I.A., Johnson F., Grollman A.P. Streptonigrin. 1. Structure–activity relationships among simple bicyclic analogs. Rate dependence of DNA degradation on quinone reduction potential. J. Med. Chem. 1986;298:1329–1340. doi: 10.1021/jm00158a002. [DOI] [PubMed] [Google Scholar]
- 31.Barret R., Daudon M. Oxidation of phenols to quinones by bis(trifluoroacetoxy)iodobenzene. Tetrahedron Lett. 1990;31:4871–4872. doi: 10.1016/S0040-4039(00)97755-4. [DOI] [Google Scholar]
- 32.Morin C., Besset T., Moutet J., Fayolle M., Brückner M., Limosin D., Becker K., Davioud-Charvet E. The aza-analogues of 1,4-naphthoquinones are potent substrates and inhibitors of plasmodial thioredoxin and glutathione reductases and of human erythrocyte glutathione reductase. Org. Biomol. Chem. 2008;6:2731–2742. doi: 10.1039/b802649c. [DOI] [PubMed] [Google Scholar]
- 33.Amarasekara A.S. A new synthesis of quinoline-5,8-quinone. Synth. Commun. 1999;29:3063–3066. doi: 10.1080/00397919908085930. [DOI] [Google Scholar]
- 34.Cameron D.W., Deutscher K.R., Feutrill G.I. Nucleophilic alkenes. IX. Addition of 1,1-dimethoxyethene to azanaphthoquinones: Synthesis of bostrycoidin and 8-O-methylbostrycoidin. Aust. J. Chem. 1982;35:1439–1450. doi: 10.1071/CH9821439. [DOI] [Google Scholar]
- 35.Wada Y., Harayama Y., Kamimura D., Yoshida M., Shibata T., Fujiwara K., Morimoto K., Fujioka H., Kita Y. The synthetic and biological studies of discorhabdins and related compounds. Org. Biomol. Chem. 2011;9:4959–4976. doi: 10.1039/c1ob05058c. [DOI] [PubMed] [Google Scholar]
- 36.Cossy J., Belotti D. Efficient synthesis of substituted quinoline-5,8-quinones from 8-hydroxyquinolines by photooxygenation. Tetrahedron Lett. 2001;42:4329–4331. doi: 10.1016/S0040-4039(01)00727-4. [DOI] [Google Scholar]
- 37.Ling Y., Yang Q.X., Teng Y.N., Chen S., Gao W.J., Guo J., Hsu P.L., Liu Y., Morris-Natschke S.L., Hung C.C., et al. Development of novel amino-quinoline-5,8-dione derivatives as NAD(P)H:quinone oxidoreductase 1 (NQO1) inhibitors with potent antiproliferative activities. Eur. J. Med. Chem. 2018;154:199–209. doi: 10.1016/j.ejmech.2018.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan L., Zheng Q., Chen Y., Yang R., Yang Y., Li Z., Meng X. Design, synthesis and biological evaluation of novel naphthoquinone derivatives as IDO1 inhibitors. Eur. J. Med. Chem. 2018;157:423–436. doi: 10.1016/j.ejmech.2018.08.013. [DOI] [PubMed] [Google Scholar]
- 39.Meng T., Qin Q.P., Chen Z.L., Zou H.H., Wang K., Liang F.P. Discovery of high in vitro and in vivo antitumor activities of organometallic ruthenium(II)-arene complexes with 5,7-dihalogenated-2-methyl-8-quinolinol. Dalton Trans. 2019;48:5352–5360. doi: 10.1039/C9DT00866G. [DOI] [PubMed] [Google Scholar]
- 40.Santoso K.T., Menorca A., Cheung C.Y., Cook G.M., Stocker B.L., Timmer M.S.M. The synthesis and evaluation of quinolinequinones as anti-mycobacterial agents. Bioorg. Med. Chem. 2019;27:3532–3545. doi: 10.1016/j.bmc.2019.06.002. [DOI] [PubMed] [Google Scholar]
- 41.Cheng Y., An L.K., Wu N., Wang X.D., Bu X.Z., Huang Z.S., Gu L.Q. Synthesis, cytotoxic activities and structure–activity relationships of topoisomerase I inhibitors: Indolizinoquinoline-5,12-dione derivatives. Bioorg. Med. Chem. 2008;16:4617–4625. doi: 10.1016/j.bmc.2008.02.036. [DOI] [PubMed] [Google Scholar]
- 42.Egu A., Okoro U.C., Efeturi O. New aryl derivatives of quinolinedione and related heterocyclic compounds. J. Heterocycl. Chem. 2017;54:1572–1577. doi: 10.1002/jhet.2745. [DOI] [Google Scholar]
- 43.Li Z., Zhou J., Gan Y., Yin Y., Zhang W., Yang J., Tang Y., Dai Y. Synthesis of a novel platinum(II) complex with 6,7-dichloro-5,8-quinolinedione and the study of its antitumor mechanism in testicular seminoma. J. Inorg. Biochem. 2019;197:110701–110709. doi: 10.1016/j.jinorgbio.2019.110701. [DOI] [PubMed] [Google Scholar]
- 44.Ezema B.E., Ezema C.G., Ayogu J.I., Ugwu D.I., Onoabedje E.A. Synthesis of aza derivatives of isomers of angular and complex phenothiazine rings. Orient J. Chem. 2015;31:379–385. doi: 10.13005/ojc/310145. [DOI] [Google Scholar]
- 45.Yoon E.Y., Choi H.Y., Shin K.J., Yoo K.H., Chi D.Y., Kim D.J. The regioselectivity in the reaction of 6,7-dihaloquinoline-5,8-diones with amine nucleophiles in various solvents. Tetrahedron Lett. 2000;41:7475–7480. doi: 10.1016/S0040-4039(00)01278-8. [DOI] [Google Scholar]
- 46.Jastrzębska M., Boryczka S., Kadela M., Wrzalik R., Kusz J., Nowak M. Synthesis, crystal structure and infrared spectra of new 6- and 7-propylamine-5,8-quinolinediones. J. Mol. Struct. 2014;1067:160–168. doi: 10.1016/j.molstruc.2014.03.031. [DOI] [Google Scholar]
- 47.Lazo J.S., Aslan D.C., Southwick E.C., Cooley K.A., Ducruet A.P., Joo B., Vogt A., Wipf P. Discovery and biological evaluation of a new family of potent inhibitors of the dual specificity protein phosphatase Cdc25. J. Med. Chem. 2001;44:4042–4049. doi: 10.1021/jm0102046. [DOI] [PubMed] [Google Scholar]
- 48.Kadela-Tomanek M., Jastrzębska M., Bębenek E., Chrobak E., Latocha M., Kusz J., Tarnawska D., Boryczka S. New acetylenic amine derivatives of 5,8-quinolinediones: Synthesis, crystal structure and antiproliferative activity. Crystal. 2017;7:15. doi: 10.3390/cryst7010015. [DOI] [Google Scholar]
- 49.Pratt Y.T. Quinolinequinones. VI. reactions with aromatic amines. J. Org. Chem. 1962;271:3905–3910. doi: 10.1021/jo01058a036. [DOI] [Google Scholar]
- 50.Ryu C.K., Jeong H.J., Lee S.K., You H.J., Choi K.U., Shim J.Y., Heo Y.H., Lee C.O. Effects of 6-arylamino-5,8-quinolinediones and 6-chloro-7-arylamino-5,8-isoquinolinediones on NAD(P)H: Quinone oxidoreductase (NQO1) activity and their cytotoxic potential. Arch. Pharm. Res. 2001;24:390–396. doi: 10.1007/BF02975181. [DOI] [PubMed] [Google Scholar]
- 51.Kadela-Tomanek M., Jastrzębska M., Pawełczak B., Bębenek E., Chrobak E., Latocha M., Książek M., Kusz J., Boryczka S. Alkynyloxy derivatives of 5,8-quinolinedione: Synthesis, in vitro cytotoxicity studies and computational molecular modeling with NAD(P)H:Quinone oxidoreductase 1. Eur. J. Med. Chem. 2017;126:969–982. doi: 10.1016/j.ejmech.2016.12.031. [DOI] [PubMed] [Google Scholar]
- 52.Kadela M., Jastrzębska M., Bębenek E., Chrobak E., Latocha M., Kusz J., Książek M., Boryczka S. Synthesis, structure and cytotoxic activity of mono- and dialkoxy derivatives of 5,8-quinolinedione. Molecules. 2016;21:156. doi: 10.3390/molecules21020156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ryu C.K., Sun Y.J., Shim J.Y., You H.J., Choi K.U., Lee H. Synthesis and antifungal activity of 6,7-bis-[S-(aryl)thio]-5,8-quinolinediones. Arch. Pharm. Res. 2002;25:795–800. doi: 10.1007/BF02976994. [DOI] [PubMed] [Google Scholar]
- 54.Bray F., Ferlay J., Soerjomataram I., Siegel R., Torre L., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 55.Ryu C.K., Kang H.Y., Yi Y.J., Lee C.O. Cytotoxic activities of 6-arylamino-7-halo-5,8-quinolinediones against human tumor cell lines. Arch. Pharm. Res. 2000;23:42–45. doi: 10.1007/BF02976464. [DOI] [PubMed] [Google Scholar]
- 56.Ryu C.K., Lee I.K., Jung S.H., Lee C.O. Synthesis and cytotoxic activities of 6-chloro-7-arylamino-5,8-isoquinolinediones. Bioorg. Med. Chem. Lett. 1999;9:1075–1080. doi: 10.1016/S0960-894X(99)00152-3. [DOI] [PubMed] [Google Scholar]
- 57.Timosenko E., Hadjinicolaou A.V., Cerundolo V. Modulation of cancer-specific immune responses by amino acid degrading enzymes. Immunotherapy. 2017;9:83–97. doi: 10.2217/imt-2016-0118. [DOI] [PubMed] [Google Scholar]
- 58.Munn D.H., Mellor A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34:137–143. doi: 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brochez L., Chevolet I., Kruse V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer. 2017;76:167–182. doi: 10.1016/j.ejca.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 60.Galaktionov K., Lee A.K., Eckstein J., Draetta G., Meckler J., Loda M., Beach D. CDC25 phosphatases as potential human oncogenes. Science. 1995;269:1575–1577. doi: 10.1126/science.7667636. [DOI] [PubMed] [Google Scholar]
- 61.Tamura K., Southwick E.C., Kerns J., Rosi K., Carr B.I., Wilcox C., Lazo J.S. Cdc25 inhibition and cell cycle arrest by a synthetic thioalkyl vitamin K analogue. Cancer Res. 2000;60:1317–1325. [PubMed] [Google Scholar]
- 62.Hsu T.H., Chen C., Lee P.T., Chiu S.J., Liu H.F., Tsai C.C., Chao J.I. 7-Chloro-6-piperidin-1-yl-quinoline-5,8-dione (PT-262), a novel synthetic compound induces lung carcinoma cell death associated with inhibiting ERK and Cdc25 phosphorylation via a p53-independent pathway. Cancer Chemother. Pharmacol. 2008;62:799–808. doi: 10.1007/s00280-007-0667-5. [DOI] [PubMed] [Google Scholar]
- 63.Sawhney R.S., Cookson M.M., Omar Y., Hauser J., Brattain M.G. Integrin alpha2-mediated ERK and calpain activation play a critical role in cell adhesion and motility via focal adhesion kinase signaling: Identification of a novel signaling pathway. J. Biol. Chem. 2006;281:8497–8510. doi: 10.1074/jbc.M600787200. [DOI] [PubMed] [Google Scholar]
- 64.Liu L.T., Yuan T.T., Liu H.H., Chen S.F., Wu Y.T. Synthesis and biological evaluation of substituted 6-alkynyl-4-anilinoquinazoline derivatives as potent EGFR inhibitors. Bioorg. Med. Chem. Lett. 2007;17:6373–6377. doi: 10.1016/j.bmcl.2007.08.061. [DOI] [PubMed] [Google Scholar]
- 65.Bouhfid R., Essassi E.M. Synthesis of 6-chloro-2-(propargyloxy)quinoline-4-carboxylic acid and propargyl 6-chloro-2-(propargyloxy)quinoline-4-carboxylate. Molbank. 2006;2006:M486. doi: 10.3390/M486. [DOI] [Google Scholar]
- 66.Yin H.Y., Kong F., Wang S., Yao Z.J. Assembly of pentacyclic pyrido[4,3,2-mn]acridin-8-ones via a domino reaction initiated by Au(I)-catalyzed 6-endo-dig cycloisomerization of N-propargylaminoquinones. Tetrahedron Lett. 2012;53:7078–7082. doi: 10.1016/j.tetlet.2012.10.065. [DOI] [Google Scholar]
- 67.Bringmann G., Reichert M., Hemberger Y. The absolute configuration of streptonigrin. Tetrahedron. 2008;64:515–528. doi: 10.1016/j.tet.2007.11.015. [DOI] [Google Scholar]
- 68.Wakaki S., Maruma H., Tomioka K., Shimizu G., Kato E., Kamada H., Kudo S., Fujimoto Y. Isolation of new fractions of antitumor mitomycins. Antibiot. Chemother. 1958;8:228–240. [PubMed] [Google Scholar]
- 69.Herr R.R., Bergy M.E., Eble T.E., Jahnke H.K. Porfiromycin, a new antibiotic. II. Isolation and characterization. Antimicrob. Agents Ann. 1960;39:14968–14975. [Google Scholar]
- 70.Zang H., Gates K.S. DNA binding and alkylation by the “left half” of azinomycin B. Biochemistry. 2000;39:14968–14975. doi: 10.1021/bi001998d. [DOI] [PubMed] [Google Scholar]
- 71.Dvorakova K., Payne C.M., Tome M.E., Briehl M.M., McClure T., Dorr R.T. Induction of oxidative stress and apoptosis in myeloma cells by the aziridine-containing agent imexon. Biochem. Pharmacol. 2000;60:749–758. doi: 10.1016/S0006-2952(00)00380-4. [DOI] [PubMed] [Google Scholar]
- 72.Yoo K.H., Yoon E.Y., Park Y.Y., Park S.W., Lee C.O., Lee W.K., Chi D.Y., Kim D.J. Synthesis and SAR of aziridinylquinoline-5,8-diones as agents against malignant tumor cells. Bull. Korean Chem. Soc. 2001;22:1067–1068. [Google Scholar]
- 73.Siddiq A., Dembitsky V. Acetylenic anticancer agents. Anticancer Agents Med. Chem. 2008;8:132–170. doi: 10.2174/187152008783497073. [DOI] [PubMed] [Google Scholar]
- 74.Kuklev D.V., Domb A.J., Dembitsky V.M. Bioactive acetylenic metabolites. Phytomedicine. 2013;20:1145–1159. doi: 10.1016/j.phymed.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 75.Gredicak M., Jerić I. Enediyne compounds—New promises in anticancer therapy. Acta Pharm. 2007;57:133–150. doi: 10.2478/v10007-007-0011-y. [DOI] [PubMed] [Google Scholar]
- 76.Kovač-Bešović E.E., Durić K., Kalođera Z., Sofić E. Identification and isolation of pharmacologically active triterpenes in Betuale Cortex, Betula Pendula Roth., Betulaceae. Bosnian J. Basic Med. Sci. 2009;9:31–38. doi: 10.17305/bjbms.2009.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Król S.K., Kiełbus M., Rivero-Muller A., Stepulak A. Comprehensive review on betulin as a potent anticancer agent. Biomed. Res. Int. 2015;2015:584189. doi: 10.1155/2015/584189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hordyjewska A., Ostapiuk A., Horecka A., Kurzepa J. Betulin and betulinic acid: Triterpenoids derivatives with a powerful biological potential. Phytochem. Rev. 2019;18:929–951. doi: 10.1007/s11101-019-09623-1. [DOI] [Google Scholar]
- 79.Kadela-Tomanek M., Bębenek E., Chrobak E., Marciniec K., Latocha M., Kuśmierz D., Jastrzębska M., Boryczka S. Betulin-1,4-quinone hybrids: Synthesis, anticancer activity and molecular docking study with NQO1 enzyme. Eur. J. Med. Chem. 2019;177:302–315. doi: 10.1016/j.ejmech.2019.05.063. [DOI] [PubMed] [Google Scholar]
- 80.Chipuk J.E., Kuwana T., Bouchier-Hayes L., Droin N.M., Newmeyer D.D., Schuler M., Green D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;13:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 81.Hemann M.T., Lowe S.W. The p53–Bcl-2 connection. Cell Death Differ. 2006;13:1256–1259. doi: 10.1038/sj.cdd.4401962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karagül M.I., Aktaş S., Yetkin D., Bayrak G., Çelikcan D. P53, Bcl2 and Bax expression and apoptosis in perifosine and vitamin D-treated endometrial cancer cell line (HEC1A) Proceedings. 2018;2:1564. doi: 10.3390/proceedings2251564. [DOI] [Google Scholar]
- 83.Rhee H.K., Park H.J., Lee S.K., Lee C.O., Choo H.Y. Synthesis, cytotoxicity, and DNA topoisomerase II inhibitory activity of benzofuroquinolinediones. Bioorg. Med. Chem. 2007;15:1651–1658. doi: 10.1016/j.bmc.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 84.Gustafson D., Page R. Withrow and MacEwen’s Small Animal Clinical Oncology. 5th ed. Elsevier Inc.; Amsterdam, The Netherlands: 2013. 11—Cancer Chemotherapy; pp. 157–179. [Google Scholar]
- 85.Cassini A., Högberg L.D., Plachouras D., Quattrocchi A., Hoxha A., Simonsen G.S., Colomb-Cotinat M., Kretzschmar M.E., Devleesschauwer B., Cecchini M., et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet. Infect. Dis. 2019;19:56–66. doi: 10.1016/S1473-3099(18)30605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lewis K. Antibiotics: Recover the lost art of drug discovery. Nature. 2012;485:439–440. doi: 10.1038/485439a. [DOI] [PubMed] [Google Scholar]
- 87.Tandon V.K., Maurya H.K., Yadav D.B., Tripathi A., Kumar M., Shukla P.K. Naphtho[2,3-b][1,4]-thiazine-5,10-diones and 3-substituted-1,4-dioxo-1,4-dihydronaphthalen-2-yl-thioalkanoate derivatives: Synthesis and biological evaluation as potential antibacterial and antifungal agents. Bioorg. Med. Chem. Lett. 2006;16:5883–5887. doi: 10.1016/j.bmcl.2006.08.060. [DOI] [PubMed] [Google Scholar]
- 88.Ibis C., Tuyun A.F., Ozsoy-Gunes Z., Bahar H., Stasevych M.V., Musyanovych R.Y., Komarovska-Porokhnyavets O., Novikov V. Design, synthesis and biological evaluation of novel nitrogen and sulfur containing hetero-1,4-naphthoquinones as potent antifungal and antibacterial agents. Eur. J. Med. Chem. 2011;46:5861–5867. doi: 10.1016/j.ejmech.2011.09.048. [DOI] [PubMed] [Google Scholar]
- 89.Riffel A., Medina L.F., Stefani V., Santos R.C., Bizani D., Brandelli A. In vitro antimicrobial activity of a new series of 1,4-naphthoquinones. Braz. J. Med. Biol. Res. 2002;35:811–818. doi: 10.1590/S0100-879X2002000700008. [DOI] [PubMed] [Google Scholar]
- 90.Ryu C.K., Kim H.J. The synthesis of 6-(N-arylamino)-7-chloro-5,8-quinolinedione derivatives for evaluation of antifungal activities. Arch. Pharm. Res. 1994;17:139–144. doi: 10.1007/BF02974248. [DOI] [PubMed] [Google Scholar]
- 91.Ryu C.K., Choi J.A., Kim S.H. Synthesis and antifungal evaluation of 6-(N-arylamino)-7-methylthio-5,8-quinolinediones. Arch. Pharm. Res. 1998;21:440–444. doi: 10.1007/BF02974640. [DOI] [PubMed] [Google Scholar]
- 92.Ryu C.K., Lee J.Y., Jeong S.H., Nho J.H. Synthesis and antifungal activity of 1H-pyrrolo[3,2-g]quinoline-4,9-diones and 4,9-dioxo-4,9-dihydro-1H-benzo[f]indoles. Bioorg. Med. Chem. Lett. 2009;19:146–148. doi: 10.1016/j.bmcl.2008.10.131. [DOI] [PubMed] [Google Scholar]
- 93.Ezugwu J.A., Ezeokonkwo M.A., Okafor S.N., Godwin-Nwakwasi E.U., Ibeanu F.N. Palladium-catalyzed sonogashira synthesis of alkynyl derivatives of quinoline-5,8-dione. Asian J. Appl. Sci. 2017;5:1137–1144. [Google Scholar]
- 94.Ezeokonkwo M.A., Ezugwu J.A., Okafor S.N., Onoabedje E.A., Godwin-Nwakwasi E.U., Ibeanu F.N. Double alkynylation of quinoline-5,8-diones and their in-silico and antimicrobial studies. J. Appl. Sci. Environ. Manag. 2018;22:1121–1127. doi: 10.4314/jasem.v22i7.20. [DOI] [Google Scholar]
- 95.Barb A.W., Zhou P. Mechanism and inhibition of LpxC: An essential zinc-dependent deacetylase of bacterial lipid A synthesis. Curr. Pharm. Biotechnol. 2008;9:9–15. doi: 10.2174/138920108783497668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Erwin A.L. Antibacterial drug discovery targeting the lipopolysaccharide biosynthetic enzyme LpxC. Cold Spring Harb. Perspect. Med. 2016;6:025304–025318. doi: 10.1101/cshperspect.a025304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.World Health Organization. [(accessed on 30 September 2019)]; Available online: https://www.who.int/gho/tb/epidemic/cases_deaths/en.
- 98.Jameson J., Fauci A., Kasper D., Hauser S., Longo D., Loscalzo J. Harrison’s Principles of Internal Medicine. 20th ed. McGraw-Hill Education; New York, NY, USA: 2018. [Google Scholar]
- 99.Kumar V., Mahajan A., Chibale K. Synthetic medicinal chemistry of selected antimalarial natural products. Bioorg. Med. Chem. 2009;17:2236–2275. doi: 10.1016/j.bmc.2008.10.072. [DOI] [PubMed] [Google Scholar]
- 100.Hsu E. Reflections on the discovery of the antimalarial qinghao. Br. J. Clin. Pharmacol. 2006;61:666–670. doi: 10.1111/j.1365-2125.2006.02673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lanfranchi D.A., Cesar-Rodo E., Bertrand B., Huang H.H., Day L., Johann L., Elhabiri M., Becker K., Williams D.L., Davioud-Charvet E. Synthesis and biological evaluation of 1,4-naphthoquinones and quinoline-5,8-diones as antimalarial and schistosomicidal agents. Org. Biomol. Chem. 2012;10:6375–6387. doi: 10.1039/c2ob25812a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hussain H., Specht S., Sarite S.R., Hoerauf A., Krohn K. New quinoline-5,8-dione and hydroxynaphthoquinone derivatives inhibit a chloroquine resistant Plasmodium falciparum strain. Eur. J. Med. Chem. 2012;54:936–942. doi: 10.1016/j.ejmech.2012.06.046. [DOI] [PubMed] [Google Scholar]
- 103.Global Statistic. [(accessed on 30 September 2019)]; Available online: https://www.hiv.gov/hiv-basics/overview/data-and-trends/global-statistics.
- 104.Take Y., Inouye Y., Nakamura S., Allaudeen H.S., Kubo A. Comparative studies of the inhibitory properties of antibiotics on human immunodeficiency virus and avian myeloblastosis virus reverse transcriptases and cellular DNA polymerases. J. Antibiot. (Tokyo) 1989;42:107–115. doi: 10.7164/antibiotics.42.107. [DOI] [PubMed] [Google Scholar]
- 105.Alfadhli A., Mack A., Harper L., Berk S., Ritchie C., Barklis E. Analysis of quinolinequinone reactivity, cytotoxicity, and anti-HIV-1 properties. Bioorg. Med. Chem. 2016;27:5618–5625. doi: 10.1016/j.bmc.2016.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lim K.M., Lee J.Y., Lee S.M., Bae O.N., Noh J.Y., Kim E.J., Chung S.M., Chung J.H. Potent anti-inflammatory effects of two quinolinedione compounds, OQ1 and OQ21, mediated by dual inhibition of inducible NO synthase and cyclooxygenase-2. Br. J. Pharmacol. 2009;156:328–337. doi: 10.1111/j.1476-5381.2008.00028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ryu C.K., Jung S.H., Lee J.A., Kim H.J., Lee S.H., Chung J.H. 6-arylamino-5,8-quinolinediones and 7-arylamino-5,8-isoquinolinediones as inhibitors of endothelium-dependent vasorelaxation. Bioorg. Med. Chem. Lett. 1999;9:2469–2472. doi: 10.1016/S0960-894X(99)00411-4. [DOI] [PubMed] [Google Scholar]
- 108.Choo H.Y., Choi S., Ryu C.K., Kim H.J., Lee I.Y., Paeb A.N., Koh H.Y. QSAR study of quinolinediones with inhibitory activity of endothelium-dependent vasorelaxation by CoMSIA. Bioorg. Med. Chem. 2003;11:2019–2023. doi: 10.1016/S0968-0896(03)00045-2. [DOI] [PubMed] [Google Scholar]
- 109.Dewang P.M., Hsu N.M., Peng S.Z., Li W.R. Protein tyrosine phosphatases and their inhibitors. Curr. Med. Chem. 2015;12:1–22. doi: 10.2174/0929867053363504. [DOI] [PubMed] [Google Scholar]
- 110.Jung E., Lee J.Y., Kim H.J., Ryu C.K., Lee K.I., Kim M., Lee C.K., Go Y.Y. Identification of quinone analogues as potential inhibitors of picornavirus 3C protease in vitro. Bioorg. Med. Chem. Lett. 2018;28:2533–2538. doi: 10.1016/j.bmcl.2018.05.046. [DOI] [PubMed] [Google Scholar]