

Abstract
(1) Background. Snake venom phosphodiesterases (SVPDEs) are among the least studied venom enzymes. In envenomation, they display various pathological effects, including induction of hypotension, inhibition of platelet aggregation, edema, and paralysis. Until now, there have been no 3D structural studies of these enzymes, thereby preventing structure–function analysis. To enable such investigations, the present work describes the model-based structural and functional characterization of a phosphodiesterase from Crotalus adamanteus venom, named PDE_Ca. (2) Methods. The PDE_Ca structure model was produced and validated using various software (model building: I-TESSER, MODELLER 9v19, Swiss-Model, and validation tools: PROCHECK, ERRAT, Molecular Dynamic Simulation, and Verif3D). (3) Results. The proposed model of the enzyme indicates that the 3D structure of PDE_Ca comprises four domains, a somatomedin B domain, a somatomedin B-like domain, an ectonucleotide pyrophosphatase domain, and a DNA/RNA non-specific domain. Sequence and structural analyses suggest that differences in length and composition among homologous snake venom sequences may account for their differences in substrate specificity. Other properties that may influence substrate specificity are the average volume and depth of the active site cavity. (4) Conclusion. Sequence comparisons indicate that SVPDEs exhibit high sequence identity but comparatively low identity with mammalian and bacterial PDEs.
Keywords: snake venom, phosphodiesterases, amino acid sequence and three-dimensional structural analysis, variable substrate specificity, PDE_Ca structure–function relationship
1. Introduction
Snake venom is a crude mixture that contains enzymatic and non-enzymatic proteins, peptides, organic compounds of low molecular weight, and inorganic compounds [1,2]. Proteins constitute the major portion (about 90%) of the total dry mass of crude snake venom, with or without catalytic activity, including neurotoxins, cardiotoxins, C-type lectins, proteinases, metalloproteinases, serine proteinases, phospholipases, hyaluronidases, acetylcholinesterases, L-amino acid oxidases, three-finger toxins, phospholipase A2s, and nucleases [3,4,5,6,7,8,9]. Metalloproteinases, serine proteinases, phospholipases, and neurotoxins are the most widely studied snake venom proteins, as they occur in high concentrations and are relatively easy to purify [1,2,3,4,5,10,11,12]. Other enzymes, such as nucleases, exist in small quantities and are the least studied. Nucleases are capable of cleaving phosphodiester bonds in nucleic acids, and, in snake venom, they have been classified as endonucleases and exonucleases [13,14]. Phosphodiesterases are generally considered exonucleases [14].
Phosphodiesterases (E.C. No. 3.1.4.1) belong to the Ectonucleotide pyrophosphatase/phosphodiesterase (E-NPP) family of metalloenzymes [13]. Generally, viperid venoms contain more phosphodiesterases (PDEs) than crotalid or elapid venoms [15,16]. Phosphodiesterases cleave phosphodiester bonds in polynucleotides in a sequential manner, starting at the 3′-end, and release 5′-mononucleotides [16]. PDEs have been shown to hydrolyze a wide variety of nucleotides, such as ATP, ADP, NAD+, NADP+, and GDP [4,17]. Because this enzyme degrades oligonucleotide fragments, there is increasing demand for purified PDE for use in the structural analysis of nucleic acids [17,18].
PDEs have been isolated from various snake venoms, including Deinagkistrodon acutus [19], Bothrops atrox [20], Bothrops alternatus [21], Cerastes vipera [22], Crotalus atrox [23], Crotalus adamanteus [24,25] Crotalus durissus terrificus [25], Protobothrops flavoviridis [26], and Vipera aspis [27]. In addition to snake venoms, they also occur in spider venoms [28]. The structures of human, mouse, and bacterial PDEs have been well studied compared to snake and spider venom PDEs.
Snake venom PDE was first reported by Uzawa [29]. This is one of the least studied enzymes in snake venom due to the fact that earlier reports showed it to be non-toxic and involved only in digestion [13]. However, recent reports indicate that PDE has a major role in envenomation by hydrolyzing DNA and RNA, releasing adenosine and other purine nucleosides [30,31]. Adenosine induces a variety of pathological and pharmacological effects, such as increased vascular permeability, hypotension, inhibition of platelet aggregation, edema, and paralysis [32,33]. Snake venom phosphodiesterases (SVPDEs) have also been used as therapeutic agents in various diseases and conditions, such as cerebrovascular and cardiovascular diseases, hypertension, and atherosclerosis [34].
SVPDEs are monomeric proteins of high molecular weight (98–140 kDa), with basic pIs (8.4–9.2), and metal cofactors, usually zinc, which are essential for catalytic activity [35,36,37,38]. Some studies report a dimeric structure [13,39,40]. Sometimes, a single venom may contain multiple PDE isoforms [13,37,40].
Although the amino acid sequences of PDEs from various snake species are available in the literature, there is no information regarding their three-dimensional (3D) structures. Therefore, it is difficult to correlate function with structure. In order to enable structure-function studies, here we present a model-based 3D structural characterization of the phosphodiesterase from Crotalus adamanteus venom. The PDE_Ca structure model was produced and validated using various software (I-TESSER, MODELLER 9v19, Swiss-Model, PROCHECK, ERRAT, and Verif3D). The sequence alignment, structure-based substrate specificity, maturation, and comparison with PDEs from other organisms are also discussed.
2. Results and Discussion
2.1. Sequence Alignment Analysis
The PDE_Ca precursor contains 851 amino acids with 830 amino acid residues in the mature form. Sequence alignment indicates high sequence identity among SVPDEs and comparatively low sequence identity (<65%) with their mammalian counterparts (Table 1). The average sequence identities among SVPDEs and mammalian phosphodiesterases are 90.6% and 58.3%, respectively. The metal ion-binding/active site residues (Zn+2 1 (D153, T191, D358, H359), Zn+2 2 (D311, H315, and H476), and Ca+2 (N751, D753, H755, D757) (PDE_Ca precursor numbering scheme) are fully conserved among all phosphodiesterases examined, except N751 and H755 in SVPDEs, where these have mutated to D751 and R755, respectively, in mammalian homologs (Figure 1). Amino acid residues around the metal ion binding and active sites are highly conserved among all phosphodiesterases examined. The amino acid sequence of PDE_Ca contains 33 cysteine residues, of which 32 form 16 disulfide bridges (Table 2). The generated model and DiANNA web server [38] also confirmed the presence of sixteen disulfide bridges in PDE_Ca.
Table 1.
Percent sequence identities among SVPDEs and their mammalian counterparts.
| Proteins | PDE_Ca | PDE_Pm | PDE_Ml | PDE_Pe | PDE_Oo | 5GZ4 | 6CO1 | 4B56 | 5IJQ |
|---|---|---|---|---|---|---|---|---|---|
| PDE_Ca | ---- | 85% | 93% | 92% | 96% | 85% | 64% | 50% | 47% |
| PDE_Pm | 85% | ----- | 92% | 94% | 95% | 84% | 63% | 50% | 48% |
| PDE_Ml | 93% | 92% | ----- | 87% | 90% | 86% | 63% | 50% | 46% |
| PDE_Pe | 92% | 94% | 87% | ------ | 95% | 85% | 64% | 50% | 49% |
| PDE_Oo | 96% | 95% | 90% | 95% | ------ | 85% | 64% | 50% | 48% |
| 5GZ4 | 85% | 84% | 86% | 85% | 85% | ----- | 64% | 50% | 47% |
| 6CO1 | 64% | 63% | 63% | 64% | 64% | 64% | ----- | 53% | 46% |
| 4B56 | 50% | 50% | 50% | 50% | 50% | 50% | 53% | ----- | 44% |
| 5IJQ | 47% | 48% | 46% | 49% | 48% | 47% | 46% | 44% | ----- |
PDE_Ca: Phosphodiesterase Crotalus adamanteus, PDE_Pm: Phosphodiesterase Protobothrops mucrosquamatus, PDE_Ml: Phosphodiesterase Macrovipera lebetina, PDE_Pe: Phosphodiesterase Protobothrops elegans, PDE_Oo: Phosphodiesterase Ovophis okinavensis, 5GZ4: Phosphodiesterase Naja atra atra, 6C01: human Ectonucleotide pyrophosphatase / phosphodiesterase 3 (ENPP3), 4B56: Ectonucleotide pyrophosphatase phosphodiesterase-1 (NPP1) (Mus musculus), 5IJQ: human Autotaxin (ENPP2).
Figure 1.



Sequence alignment of snake venom phosphodiesterases (SVPDEs) and mammalian homologs. 5IJQ; crystal of human Autotaxin (ENPP2); 4B56: Ectonucleotide pyrophosphatase phosphodiesterase-1 (NPP1) (Mus musculus); 6C01: human Ectonucleotide pyrophosphatase/phosphodiesterase 3 (ENPP3); 5GZ4, phosphodiesterase Naja atra atra (Gene Bank ID: A0A2D0TC04), PDE_Ml, phosphodiesterase Macrovipera lebetina (Vl) (Gene Bank ID: AHJ80885.1), PDE_Oo, phosphodiesterase Ovophis okinavensis (Gene Bank ID: BAN89426.1), PDE_Ca, phosphodiesterase Crotalus adamanteus (Gene Bank ID: JAS04699.1), PDE_Pm, phosphodiesterase Protobothrops mucrosquamatus (Gene Bank ID: XP_015675293.1), PDE_Pe, phosphodiesterase Protobothrops elegans (Gene Bank ID: BAP39928.1). Residues involved in catalysis and metal ion binding are underlined with blue and black, respectively. Cysteine residues that form disulfide bridges are linked (yellow lines). Putatively N-glycosylated amino acid residues are underlined in brown. Amino acid residues in the somatomedin B domain, the somatomedin B-like domain, the ectonucleotide pyrophosphatase/phosphodiesterase domain (also called autotaxin), and the DNA/RNA non-specific domain, are colored green, light green, blue, and red, respectively. Secondary structural elements (alpha helices and beta strands) are shown above the sequence. Sequence numbering corresponds to the PDE_Ca precursor protein.
Table 2.
Cysteine residues participating in disulfide bridges.
| 1st Cysteine | 2nd Cysteine |
|---|---|
| 34 | 51 |
| 38 | 75 |
| 49 | 62 |
| 55 | 61 |
| 84 | 101 |
| 89 | 119 |
| 99 | 112 |
| 105 | 111 |
| 130 | 176 |
| 138 | 350 |
| 366 | 466 |
| 415 | 819 |
| 555 | 618 |
| 569 | 675 |
| 571 | 660 |
| 767 | 777 |
2.2. Domain Analysis
The primary structure of mature PDE_Ca contains four domains—the somatomedin B domain (residues 33–79), somatomedin B-like domain (residues 81–124), ectonucleotide pyrophosphatase/phosphodiesterase domain (also called autotaxin) (residues 147–479), and the DNA/RNA non-specific domain (residues 534–867) (Figure 1). The regions of amino acid residues from 124–146, 480–533, and 868–877 are connecting segments.
The ThreaDom (Threading-based Protein Domain Prediction) online web server [41] for domain conservation indicated that the domain architecture of PDE_Ca is fully conserved, with other proteins in the Protein Data Bank (PDB) containing a similar structure fold (Figure 2). The molecular weights of PDE_Ca in zymogen and the mature forms were 96.37 and 93.10 kDa, with corresponding pIs of 8.13 and 8.05, respectively [42]. The theoretically calculated molecular weights and pIs accord with the experimentally measured values for other SVPDEs [43,44,45,46,47].
Figure 2.

The ThreaDom-based domain prediction in the domain conservation score profile. Four domains are separated by three connecting segments. Vertical dotted lines indicate the start and end locations of each putative connecting segment. Vertical solid lines denote predicted boundaries at the middle of the connecting segments.
2.3. Glycosylation Sites
The primary structure of PDE_Ca contains 60 asparagine (N) residues (Protparam, [48]), of which nine were identified as potential glycosylation sites using the NetNGlyc 1.0 Server (N39, N222, N265, N276, N412, N526, N613, N695, and N771) (Figure 1). These asparagine residues are fully conserved among SVPDEs (Figure 1). Glycosylation sites were also confirmed with the Scan Prosite tool [49]. The primary structure of Vipera lebetina has also been shown to contain nine putative N-glycosylation sites [43]. PDEs of B. jararaca and Walterinnesia aegyptia contain 33% and 24% carbohydrates, respectively [48,49].
2.4. Homology Modeling and Model Evaluation
The homology model for three-dimensional (3D) structure characterization was produced using various online modeling servers (I-TESSER [50], MODELLER 9v19 program [51], and SWISS Model [52], using atomic coordinates of human ectonucleotide pyrophosphatase/phosphodiesterase 3 (PDB ID: 6C01, amino acid sequence identity 63.39%) as a template [53]. The best model was chosen based on analyses using the PROCHECK, ERRAT, and Verif3D software [54,55,56,57].
The PROCHECK analysis of the best model shows that >95% of the amino acid residues are in the most favored region of the graph (Figure 3). The remaining 5% are in the allowed region with no residues in the forbidden/disallowed region. ERRAT analysis shows an overall quality factor of 87.88 for the PDE_Ca model, which lies in the average quality range for 3D protein structures [55].
Figure 3.

A Ramachandran plot of the modeled structure of PDE_Ca. In total, >95% of the amino acid residues are in the most favored region, while the remaining 5% are in the allowed region with no residues in the forbidden/disallowed region. Quadrant I displays a region where multiple conformations are allowed. Quadrant II shows the biggest region in the graph, with the most favorable conformations of atoms. Quadrant III shows the next biggest region in the graph, where the right-handed alpha helices lie. Quadrant IV has almost no outlined region. This conformation is disfavored due to steric clash.
2.5. Molecular Dynamics Simulation
GROMACS, AMBER16, MDWeb, and MDMobby [58,59,60] all produced the same results. MD simulation analysis indicated that all structural parameters, such as chirality, disulfide bonds, and the absence of steric clashes, were correct (Figure S1A). The two basic assessments (root mean square deviation (RMSD) and radius of gyration (RG)) used to validate the structures through MD simulation were analyzed. The RMSD deviation indicated that the PDE_Ca structure did not deviate more than 1 Å from the initial structure, and the RG was also maintained at around 31.7 Å (Figure S1B,C). Flexibility analysis (Bfactor and RMSD per residue), identified some flexible regions, located mostly in loop regions (Figure S1C,D). All these analyses indicate that the simulated structure does not exhibit critical structural deformations.
2.6. Overall Structure of PDE_Ca
The 3D structure of PDE_Ca is similar to that of the other members of the alkaline phosphatase-like superfamily (ALP-like superfamily) [25,61,62,63]. PDE_Ca has a complex structure. It is a multi-domain protein that consists of four domains—a somatomedin B domain, a somatomedin B-like domain, an ectonucleotide pyrophosphatase/phosphodiesterase domain (also called autotaxin), and a DNA/RNA non-specific domain (Figure 4). These domains are briefly described below.
Figure 4.

Overall structure of PDE_Ca: (A) cartoon representation. The active site and metal ion-binding residues are shown as green sticks. Zn+2 and Ca+2 ions are shown as gray and green spheres, respectively. Disulfide bridges are represented by yellow sticks. (B–D), residues involved in Zn+2 ion-binding, catalysis, and Ca+2 ion-binding are highlighted. Parts of the secondary structure belonging to the somatomedin B domain, somatomedin B-like domain, ectonucleotide pyrophosphatase/phosphodiesterase domain, DNA/RNA non-specific domain, and connecting are colored in green, light green, blue, red, and black, respectively.
2.6.1. Somatomedin B Domain
The somatomedin B domain (SMB) is located at the N-terminus of the protein and comprises amino acid residues 33–79 (Figure 1, Figure 4 and Figure 5). It has two alpha helices (Figure 4 and Figure 5). The SMB domain is stabilized by four intrachain disulfide bridges, one salt bridge (between Asp52-Arg58) (Table 3), and extensive hydrogen bonding (14 intrachain H-bonds).
Figure 5.

Topology diagram of PDE_Ca. The alpha helices (1–20) and beta strands (A–L) are represented as cylinders and arrows, respectively. Short alpha helices and beta strands are shown as primes (e.g., 17’). Secondary structures and amino acid residues in alpha helices and beta strands were assigned from the primary sequence using the program DSSP [66] and were confirmed with PyMOL from the tertiary structure. Parts of the secondary structure belonging to the somatomedin B, somatomedin B-like, ectonucleotide pyrophosphatase/phosphodiesterase, and DNA/RNA non-specific domains, as well as the connecting segments (UN), are colored in green, light green, blue, red, and black, respectively.
Table 3.
Salt bridges in the PDE_Ca 3D structure.
| Residue 1 | Residue 2 | Distance |
|---|---|---|
| NH1 ARG A 58 | OD1 ASP A 52 | 3.59 |
| NH1 ARG A 58 | OD2 ASP A 52 | 2.60 |
| NH2 ARG A 58 | OD1 ASP A 52 | 2.69 |
| NH2 ARG A 58 | OD2 ASP A 52 | 3.30 |
| NH2 ARG A 82 | OE1 GLU A 85 | 3.39 |
| NH1 ARG A 87 | OD1 ASP A 104 | 2.58 |
| NH1 ARG A 87 | OD2 ASP A 104 | 3.54 |
| NH2 ARG A 87 | OD1 ASP A 98 | 2.84 |
| NH2 ARG A 87 | OD2 ASP A 98 | 3.93 |
| NH2 ARG A 87 | OD1 ASP A 104 | 3.31 |
| NH2 ARG A 87 | OD2 ASP A 104 | 2.62 |
| NZ LYS A 102 | OD2 ASP A 98 | 2.69 |
| NZ LYS A 168 | OD2 ASP A 158 | 2.87 |
| ND1 HIS A 189 | OD2 ASP A 352 | 3.42 |
| NE2 HIS A 189 | OD2 ASP A 352 | 3.82 |
| NH1 ARG A 278 | OE1 GLU A 302 | 2.85 |
| NH2 ARG A 278 | OE1 GLU A 302 | 2.88 |
| NE2 HIS A 309 | OD1 ASP A 305 | 2.89 |
| NE2 HIS A 309 | OD2 ASP A 305 | 2.95 |
| NZ LYS A 337 | OD2 ASP A 122 | 3.84 |
| NH2 ARG A 339 | OD1 ASP A 287 | 3.88 |
| NH2 ARG A 339 | OD2 ASP A 287 | 2.58 |
| NE2 HIS A 353 | OD1 ASP A 147 | 3.04 |
| NE2 HIS A 353 | OD1 ASP A 352 | 3.46 |
| NE2 HIS A 353 | OD2 ASP A 352 | 2.96 |
| NH1 ARG A 384 | OD2 ASP A 205 | 2.62 |
| NH2 ARG A 384 | OD2 ASP A 205 | 3.69 |
| NH2 ARG A 384 | OD2 ASP A 436 | 3.73 |
| NZ LYS A 425 | OD2 ASP A 727 | 2.56 |
| NH1 ARG A 426 | OD1 ASP A 465 | 3.74 |
| NH1 ARG A 426 | OE1 GLU A 467 | 3.98 |
| NH2 ARG A 426 | OD1 ASP A 465 | 2.70 |
| NH2 ARG A 426 | OD2 ASP A 465 | 3.11 |
| NH2 ARG A 426 | OE1 GLU A 467 | 3.26 |
| NE2 HIS A 428 | OD1 ASP A 816 | 2.79 |
| NH2 ARG A 434 | OD2 ASP A 210 | 2.94 |
| NE2 HIS A 462 | OD1 ASP A 305 | 3.83 |
| NE2 HIS A 462 | OD2 ASP A 305 | 2.68 |
| NZ LYS A 469 | OE1 GLU A 155 | 3.97 |
| NE2 HIS A 515 | OD1 ASP A 502 | 2.80 |
| NE2 HIS A 515 | OD2 ASP A 502 | 2.97 |
| NH1 ARG A 588 | OE1 GLU A 534 | 3.19 |
| NH2 ARG A 588 | OE1 GLU A 534 | 2.60 |
| NH2 ARG A 645 | OD1 ASP A 643 | 3.26 |
| NH2 ARG A 645 | OD2 ASP A 643 | 2.59 |
| NZ LYS A 710 | OE1 GLU A 804 | 3.97 |
| NH1 ARG A 786 | OE1 GLU A 791 | 3.76 |
| NH2 ARG A 786 | OD1 ASP A 788 | 3.97 |
| NE2 HIS A 810 | OE1 GLU A 791 | 3.37 |
| NH1 ARG A 813 | OE1 GLU A 492 | 3.24 |
| NH2 ARG A 813 | OD1 ASP A 816 | 2.69 |
| NH1 ARG A 815 | OE1 GLU A 818 | 2.75 |
| NZ LYS A 840 | OE1 GLU A 818 | 3.80 |
NH1 and NH2: Nitrogen atoms (amino groups) of the arginine side chain, OD1, and OD2: Oxygen atoms of aspartic acid side chains, OE: Oxygen atoms of glutamic acid side chains, NZ: Nitrogen atoms (amino groups) of lysine side chains.
2.6.2. Somatomedin B-like Domain
The somatomedin B domain connects to another domain called the Somatomedin B-like domain (SMB-like). This domain consists of residues 81 to 124 (Figure 1). Like the somatomedin B domain, it also contains eight cysteine residues in four disulfide bridges (Figure 1). The secondary structure of this domain contains two alpha helices (Figure 4 and Figure 5). Beside its disulfides, the SMB-like domain is stabilized by four salt bridges (Arg82-Glu85, Arg87-Asp104, Arg87-Asp98, Lys102-Asp98), and thirteen interchain H-bonds (four with the SMB domain and nine with the PDE domain), as well as 24 intrachain H-bonds.
Both the SMB and SMB-like domains are highly compact. Disulfide bridges are arranged in the centers of both domains, forming covalently bonded cores (Figure 4).
2.6.3. Ectonucleotide Pyrophosphatase/Phosphodiesterase Domain
The SMB-like domain connects to the catalytic domain, called the ectonucleotide pyrophosphatase/phosphodiesterase (ENPP/PDE) domain, through a short connecting segment of 22 amino acid residues (125–146) (Figure 1, Figure 4 and Figure 5).
The ENPP/PDE domain consists of amino acid residues 147–479 (Figure 1). It contains five cysteine residues, two of which make an intrachain disulfide bridge and three of which make interchain disulfide bridges (two with the connecting segment and one with the DNA/RNA non-specific domain) (Figure 1 and Figure 4) (Table 2). This domain also contains the active site residues, together with the two zinc ions (Figure 4). One of the zinc ions (Zn+21) is coordinated by four amino acids (Asp153, Thr191, Asp358, and His59), and the other one (Zn+22) is coordinated by three residues (Asp311, His315, and His476). All these catalytic amino acid residues are fully conserved in mouse NPP1 [62], human NPP3 (61), human autotaxin ENPP2 [64], and bacterial PDE [29] (Figure 6).
Figure 6.

Active site comparison of (A) PDE_Ca with (B) human Ectonucleotide pyrophosphatase/phosphodiesterase 3 (ENPP3), (C) Ectonucleotide pyrophosphatase-phosphodiesterase-1 (NPP1) (Mus musculus), (D) ENPP2 (human Autotaxin), (E) Xac Nucleotide Pyrophosphatase/Phosphodiesterase (Xanthomonas axonopodis), and (F) Phosphodiesterase Naja atra. Metal ion-binding amino acid residues are displayed as sticks and metal ions as grey spheres.
The secondary structure of this domain contains 14 beta strands and 14 alpha helices (Figure 5). Of the fourteen alpha helices and beta strands, seven and five are short alpha helices and beta strands, respectively. This domain is stabilized by interchain disulfide bridges (one with the SMB-like domain and seven with the DNA/RNA non-specific domain), 13 salt bridges (Table 3), and numerous interchain H-bonds.
2.6.4. DNA/RNA Non-Specific Domain
This domain comprises residues 603 to 867 (Figure 1). It contains nine cysteines that form one intrachain and three interchain disulfides (one with the PDE domain and two with the connecting segment) (Figure 4). This domain also contains the Ca2+-binding loop (Figure 4). The secondary structure of this domain contains seven beta strands and ten alpha helices (Figure 4 and Figure 5). This domain is connected to the PDE domains through a long loop, called the lasso loop [65]. This domain is stabilized by four disulfides, eight salt bridges, and numerous H-bonds.
2.6.5. Metal Ion-Binding Sites
SVPDEs are metalloenzymes that contain zinc and calcium ions [6,26,35,36,67]. The zinc ion participates in the active site and is important for catalytic activity [26,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67]. In the modeled structure of PDE_Ca, two zinc ions and one calcium ion were found (Figure 4). Of the two zinc ions, one (Zn+21) is coordinated by amino acid residues Asp153, Thr191, His359, and Asp358 (Figure 4), and the other (Zn+22) is coordinated by amino acid residues Asp311, His315, and His476 (Figure 4). The Ca+2 is coordinated by amino acid residues Asn751, Asp753, His755, and Asp757 (Figure 4). The metal ion comparison with the human Ectonucleotide pyrophosphatase/phosphodiesterase 3, Ectonucleotide pyrophosphatase-phosphodiesterase-1 (Mus musculus), ENPP2 (Human Autotaxin), Xac Nucleotide Pyrophosphatase/Phosphodiesterase (Xanthomonas axonopodis), and Taiwan cobra (Naja atra atra) PDE (PDB ID: 5GZ4 and 5GZ5) shows that the amino acid residues coordinating these metal ions are fully conserved (Figure 1A–F and Figure 6A–F).
2.7. Structural Basis for Substrate Specificity of Snake Venom Phosphodiesterases
For substrates other than oligonucleotides, SVPDEs display variable substrate specificity [43,44,45,46,47]. Among the SVPDEs for which substrate specificity has been studied, the PDEs from Vipera lebetina and Daboia russelli russelli hydrolyze ADP [43], while PDEs from Crotalus adamanteus, Trimeresurus stejnegeri and Bothrops jararaca hydrolyze ATP [45,46,47].
To explain these broad specificities of SVPDEs, the structures of two other PDEs from Vipera lebetina (PDE_Vl) and Bothrops atrox (PDE_Ba) were modeled, using the same modeling and validation programs used for the PDE_Ca structural model. The atomic coordinate of human Ectonucleotide pyrophosphatase/phosphodiesterase 3 (PDB ID: 6C01, amino acid sequence identity 64.09% and 62.91% with PDE_Vl and PDE_Ba, respectively) was used as a template. The Ramachandran plot analysis indicates that in both the modeled structures, 98% of amino acid residues were in the favored region of the plot, and 2% were in the allowed region (Figures S2 and S3). The ERRAT analysis shows an overall quality factor of 90 for the modeled structure, which lies in the average quality range for the protein 3D structures (Figures S4 and S5) [55]. The modeled structures of the PDE_Ca, PDE of Vipera lebetina (PDE_Vl), and Bothrops atrox (PDE_Ba) were compared, taking into account the active site residue composition, active site cavity volume and average depth, and the surface charge distribution (Figure 7, Table 4). The active site’s amino acid residues are the same for these enzymes. However, the average active site’s cavity volume, the average depth of the active site, and the surface charge distribution vary considerably (Figure 7, Table 4).
Figure 7.

Surface charge distributions of (A) PDE_Ca, (B) PDE_Vl, (C) human Ectonucleotide pyrophosphatase/phosphodiesterase 3 (ENPP3), (D) Ectonucleotide pyrophosphatase- phosphodiesterase-1 (NPP1) (Mus musculus), (E) ENPP2 (human Autotaxin), and (F) Xac Nucleotide Pyrophosphatase/Phosphodiesterase (Xanthomonas axonopodis). Blue, red, and white represent the positive, negative, and neutral regions, respectively. Black circles indicate the location of the active-site pocket.
Table 4.
Average active site cavity volumes and average active site cavity depths of SVPDEs and their mammalian and bacterial counterparts.
| Protein | Average Volume (Å3) | Average Depth (Å) |
|---|---|---|
| PD_Ca model | 6608.25 | 21.39 |
| PD_Vl model | 3985.03 | 12.54 |
| 6C01 | 14,690.95 | 19.13 |
| 4B56 | 3651.33 | 16.30 |
| 2GSN | 10,107.28 | 18.58 |
| 4ZG7 | 11,367.42 | 17.94 |
The average active site cavity volumes of PDE_Ca, PDE_Vl, and PDE_Ba are 6608.25, 3985.03, and 2243.11 Å3, respectively, with corresponding average depths of 21.39, 12.54, and 9.86 Å, respectively (Table 4). These values indicate that the average active site cavity volume and depth of PDE_Ca is much larger than that of either PDE_Vl or PDE_Ba. These characteristics permit larger substrates (ATP) to access the active site of PDE_Ca, while preventing it for PDE_Vl.
Another factor that affects the substrate specificity of these enzymes is the surface charge distribution (Figure 7). The surface charge of PDE_Ca (overall and around the active site) is highly positive (Figure 7A), for PDE_Vl it is partially positive and negative (Figure 7B). Analysis of the active site cavity volume and its average depth indicate that SVPDEs with small active site cavity volumes and average depths (like Vipera lebetina, Daboia russelli russelli, and Cerastes cerastes ) (Table 4) show a high preference for ADP, while other SVPDEs with large active site volumes and average depths (like PDEs from Crotalus adamanteus, Trimeresurus stejnegeri, and Bothrops jararaca) show a high preference for ATP.
2.8. Structural Alignment between PDE_Ca, Human ENPP3, Mouse NPP1, Human Autotaxin, Xa NPP1, PDE_Vl, PDE_Ba and Naja atra atra PDE.
The structural alignment between PDE_Ca, human ENPP3 [53], mouse NPP1 [61], human autotaxin [63], Xa NPP1 [62], phosphodiesterase from Vipera lebetina (PDE_Vl), Bothrops atrox (PDE_Ba), and the PDE from Taiwan cobra (Naja atra atra; PDB ID: 5GZ4 and 5GZ5) shows that the three-dimensional structural folds of these enzymes are similar and that all of them align well, with an RMSD value range between 0.21 and 0.92 (average RMSD value of 0.61) (Table 5) (Figure 8). They have the same active site residues (Figure 1 and Figure 6) and disulfide bridges (Figure 1). However, the amino acid residues in the loop regions vary considerably, both in composition and length (Figure 8). For this reason, the surface charge distribution also varies (Figure 7A–G), which may impart variable substrate specificity to these enzymes [45,68,69]. The overall surface charge for the SVPDEs is positive, while it is negative for human ENPP3, mouse NPP1, human autotaxin, and Xa NPP1 (Figure 7C–F). The average active site cavity volume and average depth also vary among these enzymes (Table 4).
Table 5.
Root mean square deviation values of PDE_Ca, PDE_Vl, PDE_Ba, and their mammalian counterparts.
| Protein | RMSD Value |
|---|---|
| PDE_Ca aligned 6C01 | 0.21 |
| PDE_Ca aligned 4B56 | 0.72 |
| PDE_Ca aligned 4ZG7 | 0.73 |
| PDE_Ca aligned 2GSN | 0.92 |
| PDE_Ca aligned PDE_Vl | 0.57 |
| PDE_Ca aligned PDE_Ba | 0.56 |
| PDE_Ca aligned 5GZ4 | 0.60 |
Figure 8.
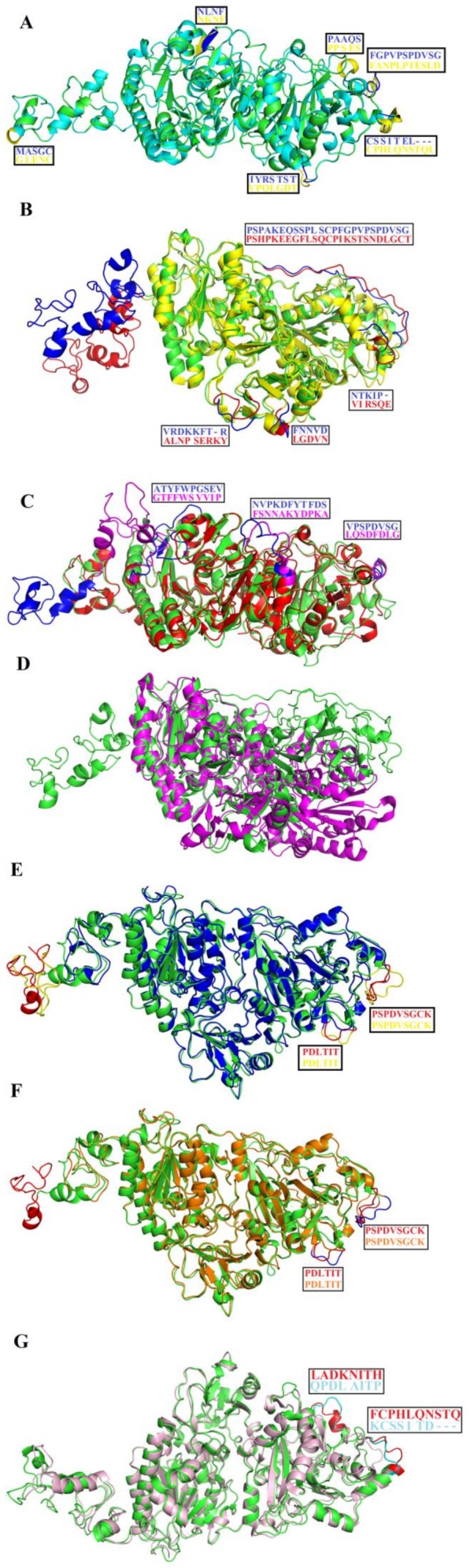
Structural alignment among SVPDEs (PDE_Ca, PDE_Vl, and PDE_Ba) and their mammalian and bacterial counterparts; (A) PDE_Ca (green) align human Ectonucleotide pyrophosphatase/phosphodiesterase 3 (ENPP3) (Cyan), (B) PDE_Ca (green) align Ectonucleotide pyrophosphatase-phosphodiesterase-1 (NPP1) (Mus musculus) (yellow), (C) PDE_Ca (green) align ENPP2 (human Autotaxin) (red), (D) PDE_Ca (green) align Xac Nucleotide Pyrophosphatase/Phosphodiesterase (Xanthomonas axonopodis) (magenta), (E) PDE_Ca (green) align Vipera lebetina phosphodiesterase (blue), (F) PDE_Ca (green) align Bothrops atrox phosphodiesterase (orange), (G) PDE_Ca (green) align 5ZG4 (light pink). Loops exhibiting differences and their corresponding amino acid residues are shown in boxes and colored in blue, red, magenta, orange, yellow, and cyan for PDE_Ca, ENPP3, NPP1, PDE_Vl, PDE_Ba, and 5GZ4, respectively.
The phylogenetic tree analysis indicates that PDE_Ca has a close evolutionary relationship with SVPDEs and PDEs from human beings and mice (Figure S6).
2.9. Maturation Mechanism for SVPDEs
PDE_Ca, like other SVPDEs, is synthesized as a precursor protein (zymogen) [43,45]. The immature PDE_Ca contains 851 amino acid residues [64] (Figure 1), in which the first 23 amino acid residues belong to a signal peptide (confirmed with SignalP 3.0 [70], Figure 9A), eight amino acid residues to the activation peptide, and the remaining 820 amino acid residues to the mature protein [48]. The signal peptide prevents the protein from proper folding and is removed cotranslationally or by signal peptidases [71,72]. The Kyte and Doolittle hydropathy plot [42] indicates that this region is located in the hydrophilic part (Figure 9B). The function of the activation peptide is unknown. However, this is considered important for proper folding of the protein, as described for spider venom and plant proteins [2,73]. This part is removed by endopeptidases [74] (Figure 9D). It is also located in the hydrophilic region of the Kyte and Doolittle hydropathy plot [42], and it is exposed on the surface of the protein (and thereby accessible to peptidases). The remaining peptide does not undergo further processing.
Figure 9.

Maturation/processing mechanism for PDE_Ca. (A) A signalP-HMM prediction plot for PDE_Ca. (B) A Kyte and Doolittle plot for signal and activation peptides. (C) Ribbon representation of PDE_Ca colored by B-factor (temperature). (D) The prepropeptide of PDE_Ca with the signal peptide (colored in yellow), activation peptide (colored in brown), and the mature protein with four domains colored in green (somatomedin B domain), light green (somatomedin B-like domain), blue (Ectonucleotide pyrophosphatase/Phosphodiesterase domain), and red (DNA/RNA non-specific domain). The connecting segments are colored in black.
3. Materials and Methods
3.1. Sequence Retrieval and Multiple Sequence Alignment
The amino acid sequence of PDE_Ca (851 amino acid residues) (gene bank accession no. JAS04699.1; UniProt ID: A0A0F7Z2Q3) [64] was obtained from the NCBI (National Centre for Biotechnology Information) protein database (http://www.ncbi.nlm.nih.gov/protein). The signal peptide was identified using the SignalP 3.0 server [70] with default parameters. The amino acid sequence of PD_Ca was used as a query for searching homologous proteins from the non-redundant database by searching with the NCBI Protein BLAST using default parameters. The multiple sequence alignment of the selected homologous protein sequences, including the amino acid sequence of PD_Ca, was generated using MUSCLE [75]. The aligned sequences were edited and colored in Box-shade V3.21 [76].
3.2. Sequence Logo Generated from Multiple Sequence Alignment
The Weblogo 3.2 [77,78] was used to illustrate the conservation patterns of amino acids in the protein sequence, and graphically represent the multiple sequence alignment, using default parameters, except for the composition, which was done without adjustment.
3.3. In Silico Analysis of the Domain and Biochemical Properties of the PDE_Ca
The PDE_Ca primary sequence was analyzed for the presence of domains/motifs using the conserved domain search tool [79], available at http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi. The protparam and Compute pI/MW tools from the ExPASy Proteomics server (http://web.expasy.org/compute_pi/) [42] were used to compute the isoelectric point (pI) and molecular weight of the protein.
3.4. Prediction of Ligand Binding and Glycosylation Sites
The 3DLigandSite-Ligand binding site prediction Server [80] was used for ligand binding amino acid residues in PDE_Ca, while putative glycosylation sites were predicted using the NetNGlyc 1.0 Server [48] and ScanProsite tool [81], with default parameters.
3.5. Disulfide Bond Prediction
The DiANNA web server [38] and Dinosolve [82,83,84,85,86] were used for prediction of disulfide bridges in PDE_Ca.
3.6. Homology Model Building of PDE_Ca
The 3D model of PDE_Ca was generated using various online proteins modeling programs, such as I-TESSER [87], the MODELLER 9v19 program [51], and SWISS Model [52], using the atomic coordinates of human Ectonucleotide pyrophosphatase/phosphodiesterase 3 (PDB ID: 6C01, amino acid sequence identity 63.39%) as a template [53]. The final model was selected based on the quality and validation reports generated by PROCHECK [50].
3.7. Molecular Dynamics Simulation
The modeled structure of PDE_Ca was validated through MD simulation using various programs, like AMBER16 [58], GROMACS [59], MDweb, and MDMoby [60]. The all-atom protein interaction was determined using the FF14SB force field [85]. The web-server H++ [84] was used to determine the protonation states of the amino acid side chain at pH 7.0. Chloride ions were used for system neutralization and were placed in a rectangular box of TIP3P water and extended to at least 15 Å from any protein atom. For the removal of bad contacts from the structure, the system was energy minimized for 500 conjugate gradients steps by applying a constant force constraint of 15 kcal/mol. Å2. The system was then heated gradually from 0 to 300 K for 250 ps with a constant atom number, volume, and temperature (NVT) ensemble, at the same time that the protein was restrained with a constant force of 10 kcal/mol. Å2. The equilibration step was carried out using the constant atom number, pressure, and temperature (NPT) ensemble for 500 ps, and the simulation was done for 100 ns with a 4 fs time step. The temperature and pressure were kept constant at 300 K and 1 atm, respectively, by Langevin coupling. The long-range electrostatic interactions were computed with the Particle–Mesh Ewald method (PME) [85], keeping the cut-off distance of 10 Å to Van der Waals interactions.
3.8. Model Validation
The build model of PDE_Ca was validated using the PROCHECK software [50,54], ERRAT version 2.0 [55], and Verify 3D [56,57].
3.9. Structure Superimposition
The build PDE_Ca protein model was aligned to homologous proteins using the PyMOL molecular graphics visualization program [86].
3.10. Surface Charge Analysis
Charge and radius calculations were carried out using the PDB2PQR server program [88]. The surface and charge were then visualized in ABPS Tools from the PyMOL molecular graphics visualization program [86].
4. Conclusions
In conclusion, a sequence and structural analysis of PDE_Ca was carried out using various computational biology programs. The amino acid sequence comparison analysis indicated that SVPDEs display high sequence identity (90.6%) with one another and comparatively low sequence identity (58.33%) with mammalian and bacterial PDEs. The three-dimensional model of PDE_Ca, produced using various modeling programs, was of good quality, as shown by the PROCHECK and ERRAT analysis. The modeled structure was further analyzed by molecular dynamic simulation, and the analysis indicated that all important structural parameters, such as chirality, disulfide bonds, and the absence of steric clashes, were correct. The root mean square deviation and radius of the gyration did not suffer significantly during model building and were maintained at 1 Å and 31.7 Å, respectively. The structural analysis indicated that the complex structure of PDE_Ca is folded into a multi-domain protein that comprises four domains—a somatomedin B domain, a somatomedin B-like domain, an Ectonucleotide pyrophosphatase/Phosphodiesterase domain (also called autotaxin), and a DNA/RNA non-specific domain. Structural comparisons with PDEs from other snake venoms, and mammalian and bacterial counterparts indicated that the surface charge distribution and the average active site cavity volume and depth vary considerably, which may contribute to their variable substrate specificity. Finally, during the maturation process, venom PDEs lose their signal and activation peptides to convert into the fully active mature forms. The structure of PDE_Cα presented in this paper is only a predicted structure. These conclusions need to be confirmed with experimental evidence [89].
Acknowledgments
The authors acknowledge financial support by the Cluster of Excellence “Advanced Imaging of Matter” of the Deutsche Forschungs-gemeinschaft (DFG) - EXC 2056 - project ID 390715994.
Supplementary Materials
The following are available online at https://www.mdpi.com/2072-6651/11/11/625/s1. Figure S1. Molecular dynamic simulation analysis of PDE_Ca; Figure S2. Ramachandran plot of the modeled structures of Vipera lebentia phosphodiesterase model; Figure S3. Ramachandran plot of the modeled structures of Bothrops atrox phosphodiesterase model; Figure S4. Errors plot for the modeled structure of Vipera lebentia. The plot was generated by ERRAT2. The amino acid residues showing errors were shown by black lines; Figure S5. Errors plot for the modeled structure of Bothrops atrox model. The plot was generated by ERRAT2. The amino acid residues showing errors were shown by black lines; Figure S6. Phylogenetic relationships of PDEs based on protein sequences according to the neighbor-joining method without distance corrections.
Author Contributions
A.U. design of the work, conceptualization, supervision, and methodology; C.B. Funding Acquisition, Validation, analysis, and interpretation of data; K.U., writing—review; H.A., manuscript drafting and S.u.R. substantial revision and editing of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Key Contribution
This is the first report on snake venom phosphodiesterase (SVPDE) that describes the overall structural properties, makes structural comparisons, and examines the structural basis of substrate specificity.
References
- 1.Kang T.S., Georgieva D., Genov N., Murakami M.T., Sinha M., Kumar R.P., Kaur P., Kumar S., Dey S., Sharma S., et al. Enzymatic toxins from snake venom: Structural characterization and mechanism of catalysis. FEBS J. 2011;278:4544–4576. doi: 10.1111/j.1742-4658.2011.08115.x. [DOI] [PubMed] [Google Scholar]
- 2.Ullah A., Masood R., Ali I., Ullah K., Ali H., Akbar H., Betzel C. Thrombin-like enzymes from snake venom: Structural characterization and mechanism of action. Int. J. Biol. Macromol. 2018;114:788–811. doi: 10.1016/j.ijbiomac.2018.03.164. [DOI] [PubMed] [Google Scholar]
- 3.Ogawa T., Chijiwa T., Oda-Ueda N., Ohno M. Molecular diversity and accelerated evolution of C-type lectin-like proteins from snake venom. Toxicon. 2005;45:1–14. doi: 10.1016/j.toxicon.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 4.Kini R.M., Doley R. Structure, function and evolution of three finger toxins: Mini proteins with multiple targets. Toxicon. 2010;56:855–867. doi: 10.1016/j.toxicon.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 5.Fox J.W. A brief review of the scientific history of several lesser-known snake venom proteins: L-amino acid oxidases, hyaluronidases and phosphodiesterases. Toxicon. 2013;62:75–82. doi: 10.1016/j.toxicon.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Ullah A., Masood R., Spencer P.J., Murakami M.T., Arni R.K. Crystallization and preliminary X-ray diffraction studies of an L-amino-acid oxidase from Lachesis muta venom. Acta Crystallogr. Sect. F Struct. Boil. Commun. 2014;70:1556–1559. doi: 10.1107/S2053230X14017877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ullah A., Magalhães G.S., Masood R., Mariutti R.B., Coronado M.A., Murakami M.T., Barbaro K.C., Arni R.K. Crystallization and preliminary X-ray diffraction analysis of a novel sphingomyelinase D from Loxosceles gaucho venom. Acta Crystallogr. Sect. F Struct. Boil. Commun. 2014;70:1418–1420. doi: 10.1107/S2053230X14019207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Oliveira L.M.F., Ullah A., Masood R., Zelanis A., Spencer P.J., Serrano S.M., Arni R.K. Rapid purification of serine proteinases from Bothrops alternatus and Bothrops moojeni venoms. Toxicon. 2013;76:282–290. doi: 10.1016/j.toxicon.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 9.Ullah A., Coronado M., Murakami M.T., Betzel C., Arni R.K. Crystallization and preliminary X-ray diffraction analysis of an L-amino-acid oxidase from Bothrops jararacussu venom. Acta Crystallogr. Sect. F Struct. Boil. Cryst. Commun. 2012;68:211–213. doi: 10.1107/S1744309111054923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ullah A., Souza T.D.A.C.B.D., Masood R., Murakami M.T., Arni R.K. Purification, crystallization and preliminary X-ray diffraction analysis of a class P-III metalloproteinase (BmMP-III) from the venom of Bothrops moojeni. Acta Crystallogr. Sect. F Struct. Boil. Cryst. Commun. 2012;68:1222–1225. doi: 10.1107/S1744309112036603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ullah A., Souza T.A., Betzel C., Murakami M.T., Arni R.K. Crystallographic portrayal of different conformational states of a Lys49 phospholipase A2 homologue: Insights into structural determinants for myotoxicity and dimeric configuration. Int. J. Biol. Macromol. 2012;51:209–214. doi: 10.1016/j.ijbiomac.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Ullah A., Souza T.A., Abrego J.R., Betzel C., Murakami M.T., Arni R.K. Structural insights into selectivity and cofactor binding in snake venom L-amino acid oxidases. Biochem. Biophys. Res. Commun. 2012;421:124–128. doi: 10.1016/j.bbrc.2012.03.129. [DOI] [PubMed] [Google Scholar]
- 13.Mori N., Nikai T., Sugihara H. Phosphodiesterase from the venom of crotalus ruber ruber. Int. J. Biochem. 1987;19:115–119. doi: 10.1016/0020-711X(87)90321-1. [DOI] [PubMed] [Google Scholar]
- 14.Stoynov S.S., Bakalova A.T., Dimov S.I., Mitkova A.V., Dolapchiev L.B. Single-strand-specific DNase activity is an inherent property of the 140-kDa protein of the snake venom exonuclease. FEBS Lett. 1997;409:151–154. doi: 10.1016/S0014-5793(97)00489-4. [DOI] [PubMed] [Google Scholar]
- 15.da Silva N.J., Jr., Aird S.D. Prey specificity, comparative lethality and compositional differences of coral snake venoms. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2001;128:425–456. doi: 10.1016/S1532-0456(00)00215-5. [DOI] [PubMed] [Google Scholar]
- 16.Sales R.B., Santoro M.L. Nucleotidase and DNase activities in Brazilian snake venoms. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2008;147:85–95. doi: 10.1016/j.cbpc.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Dhananjaya B.L., D’Souza C.J.M. An overview on nucleases (DNase, RNase, and phosphodiesterase) in snake venoms. Biochemistry. 2010;75:1–6. doi: 10.1134/s0006297910010013. [DOI] [PubMed] [Google Scholar]
- 18.Philipps G.R. Purification and Characterization of Phosphodiesterase from Crotalus Venom. Hoppe Seylers Z. Physiol. Chem. 1975;356:1085–1096. doi: 10.1515/bchm2.1975.356.2.1085. [DOI] [PubMed] [Google Scholar]
- 19.Sugihara H., Nikai T., Komori Y., Katada H., Mori N. Purification and characterization of the phosphodiesterase from the venom of Akistrodon acutus (China) Jpn. J. Trop. Med. Hyg. 1984;12:247–254. doi: 10.2149/tmh1973.12.247. [DOI] [Google Scholar]
- 20.Björk W. Purification of Phosphodiesterase from Bothrops atrox venoms, with special consideration of the elimination of monophosphatases. J. Biol. Chem. 1963;238:2487–2490. [PubMed] [Google Scholar]
- 21.Valério A.A., Corradini A.C., Panunto P.C., Mello S.M., Hyslop S. Purification and Characterization of a Phosphodiesterase from Bothrops alternatus Snake Venom. J. Protein Chem. 2002;21:495–503. doi: 10.1023/A:1022414503995. [DOI] [PubMed] [Google Scholar]
- 22.Saad S.M., Khan S., Ashraf M. Purification of phosphodiesterase I from Cerastes Vipera venom, biochemical and biological properties of the purified enzyme. Proc. Pak. Acad. Sci. 2009;46:1–12. [Google Scholar]
- 23.Oka J., Ueda K., Hayaishi O. Snake venom phosphodiesterase. Simple purification with blue sepharose and its application to poly (ADP-ribose) study. Biochem. Biophys. Res. Commun. 1978;80:841–848. doi: 10.1016/0006-291X(78)91321-9. [DOI] [PubMed] [Google Scholar]
- 24.Tatsuki T., Iwanaga S., Suzuki T. A simple method for preparation of snake venom phosphodiesterase almost free from 5’-nucleotidase. J. Biochem. 1975;77:831–836. doi: 10.1093/oxfordjournals.jbchem.a130790. [DOI] [PubMed] [Google Scholar]
- 25.Laskowski M., Sr. Purification and properties of venom phosphodiesterase. Methods Enzymol. 1980;65:276–284. doi: 10.1016/s0076-6879(80)65037-x. [DOI] [PubMed] [Google Scholar]
- 26.Kini R.M., Gowda T.V. Rapid method for separation and purification of four isoenzymes of Phosphodiesrerase from Trimeresurus flavorviridis (Habu snake) venom. J. Chromatogr. 1984;291:299–305. doi: 10.1016/S0021-9673(00)95032-5. [DOI] [PubMed] [Google Scholar]
- 27.Ballario P., Bergami M., Pedone F. A simple method for the purification of phosphodiesterase from Vipera aspis venom. Anal. Biochem. 1977;80:646–651. doi: 10.1016/0003-2697(77)90692-3. [DOI] [PubMed] [Google Scholar]
- 28.Sannaningaiah D., Subbaiah G.K., Kempaiah K. Pharmacology of spider venom toxins. Toxin Rev. 2014;33:206–220. doi: 10.3109/15569543.2014.954134. [DOI] [Google Scholar]
- 29.Uzawa S. Uber die phosphomonoesterase und die phosphodiesterase. J. Biochem. 1932;15:19–28. doi: 10.1093/oxfordjournals.jbchem.a125167. [DOI] [Google Scholar]
- 30.Aird S.D. Ophidian envenomation strategies and the role of purines. Toxicon. 2002;40:335–393. doi: 10.1016/S0041-0101(01)00232-X. [DOI] [PubMed] [Google Scholar]
- 31.Aird S.D. Taxonomic distribution and quantitative analysis of free purine and pyrimidine nucleosides in snake venoms. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2005;140:109–126. doi: 10.1016/j.cbpc.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 32.Hargreaves M.B., Stoggall S.M., Collis M.G. Evidence that the adenosine receptor mediating relaxation in dog lateral saphenous vein and guinea-pig aorta is of the A2b subtype. Br. J. Pharmacol. 1991;102:198. [Google Scholar]
- 33.Sobrevia L., Yudilevich D.L., Mann G.E. Activation of A2-purinoceptors by adenosine stimulates L-arginine transport (system y+) and nitric oxide synthesis in human fetal endothelial cells. J. Physiol. 1997;499:135–140. doi: 10.1113/jphysiol.1997.sp021916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uzair B., Khan B.A., Sharif N., Shabbir F., Menaa F. Phosphodiesterases (PDEs) from Snake Venoms: Therapeutic Applications. Protein Pept. Lett. 2018;25:612–618. doi: 10.2174/0929866525666180628160616. [DOI] [PubMed] [Google Scholar]
- 35.Iwanaga S., Suzuki T. Enzymes in snake venom. In: Lec C.Y., editor. Snake Venoms. Volume 19. Springer; New York, NY, USA: 1979. pp. 61–158. [Google Scholar]
- 36.Pollack S.E., Tetsuo U., David S.A. Snake venom phosphodiesterase: A zinc metalloenzyme. J. Protein. 1983;2:1–12. doi: 10.1007/BF01025165. [DOI] [Google Scholar]
- 37.Levy Z., Bdolah A. Multiple molecular forms of snake venom phosphodiesterase from Vipera palastinae. Toxicon. 1976;14:389–391. doi: 10.1016/0041-0101(76)90086-6. [DOI] [PubMed] [Google Scholar]
- 38.Ferrè F., Clote P. DiANNA 1.1: An extension of the DiANNA web server for ternary cysteine classification. Nucleic Acids Res. 2006;34(Suppl. 2):W182–W185. doi: 10.1093/nar/gkl189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perron S., Mackessy S.P., Hyslop R.M. Purification and characterization of exonuclease from rattlesnake venom. Acad. Sci. 1993;25:21–22. [Google Scholar]
- 40.Mackessy S.P. Ph.D. Thesis. Washington State University; Pullman, WA, USA: 1989. Venoms as Trophic Adaptations: An Ultrastructural Investigation of the Venom Apparatus and Biochemical Characterization of the Proteolytic Enzymes of the Northern Pacific Rattlesnake Crotalus Viridis Oreganus. [Google Scholar]
- 41.Xue Z., Xu D., Wang Y., Zhang Y. ThreaDom: Extracting protein domain boundary information from multiple threading alignments. Bioinformatics. 2013;29:i247–i256. doi: 10.1093/bioinformatics/btt209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker J.M., editor. The Proteomics Protocols Handbook. Humana Press; Totowa, NJ, USA: 2005. pp. 571–607. [Google Scholar]
- 43.Trummal K., Aaspõllu A., Tõnismägi K., Samel M., Subbi J., Siigur J., Siigur E. Phosphodiesterase from Vipera lebetina venom—Structure and characterization. Biochimie. 2014;106:48–55. doi: 10.1016/j.biochi.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 44.Mitra J., Bhattacharyya D. Phosphodiesterase from Daboia russelli russelli venom: Purification, partial characterization and inhibition of platelet aggregation. Toxicon. 2014;88:1–10. doi: 10.1016/j.toxicon.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Razzell W.E., Khorana H.G. Studies on polynucleotides. III. Enzymic degradation; substrate specificity and properties of snake venom phosphodiesterase. J. Biol. Chem. 1959;234:2105–2113. [PubMed] [Google Scholar]
- 46.Peng L., Xu X., Shen D., Zhang Y., Song J., Yan X., Guo M. Purification and partial characterization of a novel phosphodiesterase from the venom of Trimeresurus stejnegeri: Inhibition of platelet aggregation. Biochimie. 2011;93:1601–1609. doi: 10.1016/j.biochi.2011.05.027. [DOI] [PubMed] [Google Scholar]
- 47.Santoro M.L., Vaquero T.S., Paes Leme A.F., Serrano S.M. NPP-BJ, a nucleotide pyrophosphatase/phosphodiesterase from Bothrops jararaca snake venom, inhibits platelet aggregation. Toxicon. 2009;54:499–512. doi: 10.1016/j.toxicon.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 48.Gupta R., Jung E., Brunak S. Prediction of N-glycosylation Sites in Human Proteins. [(accessed on 12 December 2018)];2004 Available online: http://www.cbs.dtu.dk/services/NetNGlyc/
- 49.Sulkowski E., Bjork W., Laskowski M. A specific and nonspecific alkaline monophosphatase in the venom of Bothrops atrox and their occurrence in the purified venom phosphodiesterase. J. Biol. Chem. 1963;238:2477–2486. [PubMed] [Google Scholar]
- 50.Laskowski R.A., MacArthur M.W., Thornton J.M. PROCHECK: Validation of protein structure coordinates. In: Rossmann M.G., Arnold E., editors. International Tables of Crystallography: Volume F: Crystallography of Biological Macromolecules. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2001. pp. 722–725. [Google Scholar]
- 51.Webb B., Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci. 2016;86:3. doi: 10.1002/cpps.20. [DOI] [PubMed] [Google Scholar]
- 52.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L., et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gorelik A., Randriamihaja A., Illes K., Nagar B. Structural basis for nucleotide recognition by the ectoenzyme CD203c. FEBS J. 2018;285:2481–2494. doi: 10.1111/febs.14489. [DOI] [PubMed] [Google Scholar]
- 54.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 55.Colovos C., Yeates T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bowie J., Luthy R., Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 57.Lüthy R., Bowie J.U., Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 58.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. doi: 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- 60.Hospital A., Andrio P., Fenollosa C., Cicin-Sain D., Orozco M., Gelpí J.L. MDWeb and MDMoby: An integrated web-based platform for molecular dynamics simulations. Bioinformatics. 2012;28:1278–1279. doi: 10.1093/bioinformatics/bts139. [DOI] [PubMed] [Google Scholar]
- 61.Jansen S., Perrakis A., Ulens C., Winkler C., Andries M., Joosten R.P., Van Acker M., Luyten F.P., Moolenaar W.H., Bollen M. Structure of NPP1, an Ectonucleotide pyrophosphatase/phosphodiesterase involved in tissue calcification. Structure. 2012;20:1948–1959. doi: 10.1016/j.str.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 62.Zalatan J.G., Fenn T.D., Brunger A.T., Herschlag D. Structural and functional comparisons of nucleotide pyrophosphatase/phosphodiesterase and alkaline phosphatase: Implications for mechanism and evolution. Biochemistry. 2006;45:9788–9803. doi: 10.1021/bi060847t. [DOI] [PubMed] [Google Scholar]
- 63.Stein A.J., Bain G., Prodanovich P., Santini A.M., Darlington J., Stelzer N.M.P., Sidhu R.S., Schaub J., Goulet L., Lonergan D., et al. Structural Basis for Inhibition of Human Autotaxin by Four Potent Compounds with Distinct Modes of Binding. Mol. Pharmacol. 2015;88:982–992. doi: 10.1124/mol.115.100404. [DOI] [PubMed] [Google Scholar]
- 64.Rokyta D.R., Margres M.J., Calvin K. Post-transcriptional Mechanisms Contribute Little to Phenotypic Variation in Snake Venoms. G3 Genes Genomes Genet. 2015;5:2375–2382. doi: 10.1534/g3.115.020578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hausmann J., Kamtekar S., Christodoulou E., Day J.E., Wu T., Fulkerson Z., Albers H.M., Van Meeteren L.A., Houben A.J.S., Van Zeijl L., et al. Structural basis for substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Boil. 2011;18:198–204. doi: 10.1038/nsmb.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Touw W.G., Baakman C., Black J., te Beek T.A., Krieger E., Joosten R.P., Vriend G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015;43:D364–D368. doi: 10.1093/nar/gku1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Philipps G.R. Purification and characterization of phosphodiesterase I from Bothrops atrox. Biochim. Biophys. Acta. 1976;432:237–244. doi: 10.1016/0005-2787(76)90165-9. [DOI] [PubMed] [Google Scholar]
- 68.Boman H.G. On the specificity of the snake venom phosphodiesterase. Ann. N. Y. Acad. Sci. 1959;81:800–803. doi: 10.1111/j.1749-6632.1959.tb49363.x. [DOI] [PubMed] [Google Scholar]
- 69.Ke H., Wang H., Ye M. Structural Insight into the Substrate Specificity of Phosphodiesterases. Pharmacol. Itch. 2011;204:121–134. doi: 10.1007/978-3-642-17969-3_4. [DOI] [PubMed] [Google Scholar]
- 70.Bendtsen J.D., Nielsen H., von Heijne G., Brunak S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 71.Von Heijne G. On the hydrophobic nature of signal sequences. Eur. J. Biochem. 1981;116:419–422. doi: 10.1111/j.1432-1033.1981.tb05351.x. [DOI] [PubMed] [Google Scholar]
- 72.Lively M.O., Walsh K.A. Hen oviduct signal peptidase is an integral membrane protein. J. Biol. Chem. 1983;258:9488–9495. [PubMed] [Google Scholar]
- 73.Ullah A., Mariutti R.B., Masood R., Caruso I.P., Costa G.H., de Freita C.M., Santos C.R., Zanphorlin L.M., Mutton M.J.R., Murakami M.T., et al. Crystal structure of mature 2S albumin from Moringa oleifera seeds. Biochem. Biophys. Res. Commun. 2015;468:365–371. doi: 10.1016/j.bbrc.2015.10.087. [DOI] [PubMed] [Google Scholar]
- 74.Khan A.R., James M.N. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Edgar R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hofmann K., Baron M.D. Institute for Animal Health Ash Road Pirbright, Surrey GU24 0, 2017 (NF U.K.)
- 77.Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schneider T.D., Stephens R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marchler-Bauer A., Bryant S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–W331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wass M.N., Kelley L.A., Sternberg M.J. 3DLigandSite: Predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010;38:W469–W473. doi: 10.1093/nar/gkq406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.De Castro E., Sigrist C.J.A., Gattiker A., Bulliard V., Langendijk-Genevaux P.S., Gasteiger E., Bairoch N., Hulo A. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34:W362–W365. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yaseen A., Li Y. Dinosolve: A protein disulfide bonding prediction server using context-based features to enhance prediction accuracy. BMC Bioinform. 2013;14:S9. doi: 10.1186/1471-2105-14-S13-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theor. Comput. 2015;8:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Anandakrishnan R., Aguilar B., Onufriev A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulation. Nucleic Acids Res. 2012;40:W537–W541. doi: 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Darden T., York D., Pedersen L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 86.DeLano W.L. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 87.Roy A., Kucukural A., Zhang Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dolinsky T.J., Nielsen J.E., McCammon J.A., Baker N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ullah A., Masood R., Hayat Z., Hafeez A. Determining the Structures of the Snake and Spider Toxins by X-Rays. Methods Mol. Biol. 2020;2068:163–172. doi: 10.1007/978-1-4939-9845-6_8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.