

Abstract
Split reporters based on fluorescent proteins and luciferases have emerged as valuable tools for measuring interactions in biological systems. Relatedly, biosensors that transduce measured input signals into outputs that influence the host system are key components of engineered gene circuits for synthetic biology applications. While small-molecule-based imaging agents are widely used in biological studies, and small-molecule-based drugs and chemical probes can target a range of biological processes, a general method for generating a target small molecule in a biological system based on a measured input signal is lacking. Here, we develop a proximity-dependent split esterase that selectively unmasks ester-protected small molecules in an interaction-dependent manner. Exploiting the versatility of an ester-protected small-molecule output, we demonstrate fluorescent, chemiluminescent, and pharmacological probe generation, each created by masking key alcohol functional groups on a target small molecule. We show that the split esterase system can be used in combination with ester-masked fluorescent or luminescent probes to measure protein–protein interactions and protein–protein interaction inhibitor engagement. We demonstrate that the esterase-based reporter system is compatible with other commonly used split reporter imaging systems for the simultaneous detection of multiple protein–protein interactions. Finally, we develop a system for selective small-molecule-dependent cell killing by unmasking a cytotoxic molecule using an inducible split esterase. Presaging utility in future synthetic biology-based therapeutic applications, we also show that the system can be used for intercellular cell killing via a bystander effect, where one activated cell unmasks a cytotoxic molecule and kills cells physically adjacent to the activated cells. Collectively, this work illustrates that the split esterase system is a valuable new addition to the split protein toolbox, with particularly exciting potential in synthetic biology applications.
Short abstract
In this work we develop a split esterase biosensor technology that unmasks ester-masked small molecules, triggered by measured input signals, as a new approach for biosensing and synthetic biology.
Introduction
Protein–protein interactions (PPIs) are critical regulators of diverse cellular processes1−4 and are increasingly recognized as viable therapeutic targets for the treatment of multiple disease states.5−7 Synthetic biology-based biosensor systems that drive cell fate changes based on measured PPIs are increasingly critical components of engineered gene circuits. For example, split proteases can be used to engineer cell receptors8,9 and drive gene expression, and split Cas910 can be used to control gene editing, both based on fused sensor domains triggered by a PPI. Our group has developed proximity-dependent split T7 RNAPs as a versatile strategy for encoding PPIs in RNA signals for applications in biosensing, cell engineering, and directed evolution.11−14 In a similar approach, a bioluminescence resonance energy transfer-based system that enables transcriptional activation with improved PPI specificity was recently engineered.15 While methods to generate genetic output responses based on PPI inputs are continuing to improve, a simple and general method to create a small-molecule output based on measured PPIs is lacking, despite the versatility of small molecules as biological indicators and mediators.
Aside from exploiting PPIs for engineering purposes, methods for detecting the interactions between proteins and the disruption of those interactions due to therapeutic target engagement in live cells are critical for understanding PPIs.16 One of the most widely used approaches to measure PPIs in live cells is protein fragment complementation assays (PCAs), which involves fusing interacting proteins to complementary fragments of a split protein reporter.17−19 Interactions between the fused proteins drive assembly of the split reporter, which in turn generates an output signal. In the context of analysis, a variety of reporters have been developed, including luciferases,20 fluorescent proteins,21 and horseradish peroxidase,22 which produce photons, fluorescence, or reactive molecule outputs, respectively.
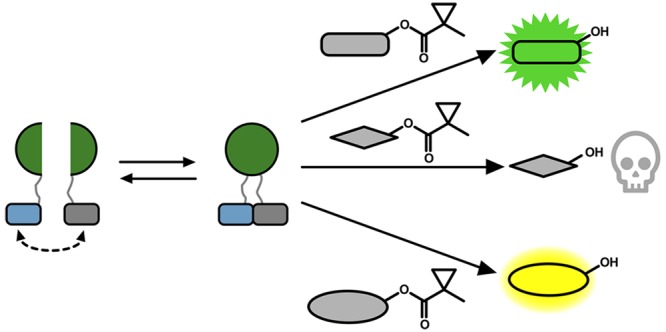
We envisioned a new split reporter system that would be capable of unmasking small molecules in a PPI-dependent manner. Small molecules provide substantial flexibility as an output signal for both analysis and generating bioactive molecules for synthetic biology purposes. Selective enzyme/substrate pairs that are orthogonal to endogenous cellular machinery have found utility for neuronal imaging,23 cell-specific pharmacology,24−26 and imaging of cellular interactions.27,28 For example, porcine liver esterase (PLE) was discovered to be able to process 1-methylcyclopropyl (CM)-masked phenol substrates when expressed in human cells, whereas endogenous esterases are not capable of processing this bulky protecting group.23 We reasoned that selective enzyme/substrate pairs may also be amenable to the creation of biosensor platforms by identifying proximity-dependent split sites of the enzymes that can ultimately unmask small molecules based on PPI-driven enzyme assembly. While it is conceptually possible to use the pre-existing split β-lactamase29 with β-lactam prodrugs30 or split β-galactosidase31 with galactoside prodrugs,32,33 this has not yet been demonstrated. We envisioned using a selective esterase–ester pair for this strategy. Exogenous esterases have previously been expressed in mammalian cells for prodrug metabolism,34 analysis of calcium signaling,35 cell-type specific pharmacology,23,36 and neuronal imaging.23 In addition, ester masking strategies for a variety of functional groups, including CM-masked phenol substrates, are well-established.23,37−40 On the basis of this work, we aimed to engineer a selective split esterase–ester substrate pair that would be capable of unmasking small molecules in a PPI-dependent manner (Figure 1a).
Figure 1.
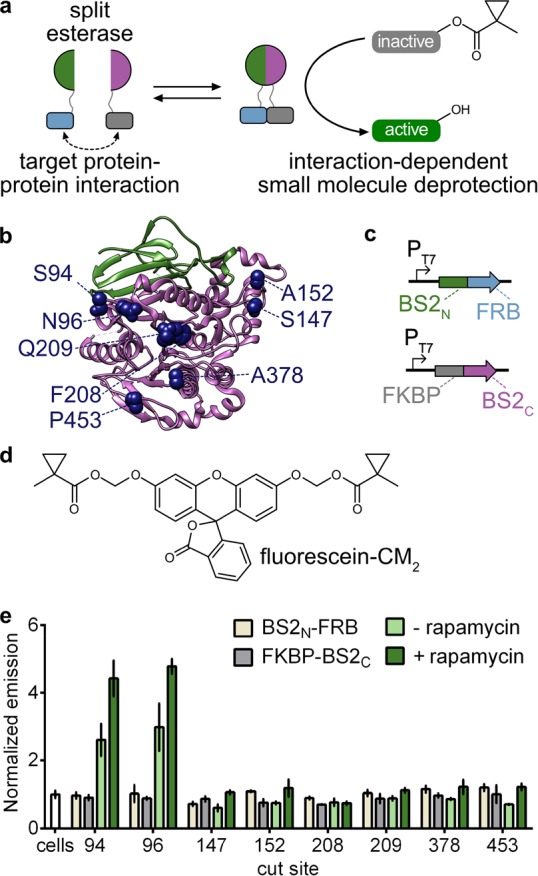
Development of a split esterase sensor. (a) Schematic of a PPI-driven split esterase assembly to unmask 1-methylcyclopropyl (CM)-masked molecules. Interacting proteins are fused to inactive fragments of BS2 esterase. Interaction between the fusion proteins results in assembly of the esterase which cleaves an inactive small molecule to generate an active molecule and output signal. (b) Mapping the cut sites onto a homologous B. subtilis esterase structure (PDB 1QE3). The BS2N fragment (green) and BS2C (magenta) from the lead split site, S94, are shown. Split sites occur after the designated amino acids. (c) Vector system to identify PPI-dependent esterase fragments in E. coli. (d) Chemical structure of masked fluorophore fluorescein-CM2. (e) Fluorescence output of split esterase fragments. E. coli expressing BS2N-fused FRB (tan), FKBP-fused BS2C (gray), or both in the absence (light green) or presence (green) of rapamycin were incubated with fluorescein-CM2 for 4 h and then analyzed for fluorescence. Error bars are the standard deviation for n = 3 replicates.
In this work, we develop a split BS2 esterase system using a fluorescence-based screen to identify interaction-dependent esterase fragments in Escherichia coli. We then demonstrate that the split esterase is capable of detecting interactions between multiple PPIs in mammalian cells, including small-molecule-induced dimerization domains, leucine zipper peptides, and medically relevant proteins of the B cell lymphoma (Bcl-2) family of apoptotic regulatory proteins and Bcl-2 homology 3 (BH3)-only interacting domains. We validate that the split esterase system is capable of measuring time-dependent engagement of a small-molecule PPI inhibitor in live cells. Moreover, we go on to multiplex our split esterase reporter with existing PCA technologies to simultaneously monitor two PPIs in both the intracellular and extracellular environment. In addition to demonstrating outputs from fluorescent and chemiluminescent probes, we go on to show that the split esterase can generate a bioactive molecule output to control cellular cytotoxicity, highlighting the versatility of small-molecule signals. Taken together, these results establish the split esterase as a versatile new addition to the PCA toolbox.
Results
Development of a Proximity-Dependent Split Esterase
To develop a split esterase reporter, we aimed to leverage the fluorescein α-cyclopropyl ester (fluorescein-CM2) fluorogenic molecule to rapidly screen esterase cut sites for PPI-dependent esterase activity in E. coli. We selected BS2 esterase from Bacillus subtilis as our exogenous esterase, which, like PLE, acts on sterically hindered esters. BS2, however, can be efficiently expressed in E. coli and has been successfully used in plate-based screens with high enzymatic activity.41 First, to confirm BS2-mediated unmasking of fluorescein-CM2, we incubated fluorescein-CM2 with E. coli expressing BS2, PLE, or a negative control protease. Both BS2 and PLE showed an enhanced fluorescent signal due to fluorescein-CM2 unmasking (Figure S2a), but BS2 showed significantly greater activity, likely due to improved expression in E. coli. More importantly, when expressed in mammalian cells, both BS2 and PLE show a robust fluorescent signal (Figure S2b–d). On the basis of these results, we moved forward with BS2 as our target for split esterase development.
To develop a split esterase, we screened eight potential split sites on BS2, each located on surface-exposed loop regions (Figure 1b). We fused the split esterase fragments via flexible linkers to the rapamycin dimerization domains, FRB and FKBP,42 coexpressed the fragments in E. coli, and measured the activity on fluorescein-CM2 in the absence and presence of rapamycin (Figure 1c,d). Splitting of BS2 at two of the eight sites, positions 94 and 96, produced robust enzymatic activity, which was enhanced in the presence of rapamycin (Figure 1e). We selected cut site 94 (resulting in a 10.5 kDa N-terminal fragment and a 43.6 kDa C-terminal fragment) as our lead and further validated esterase assembly with an abscisic-acid-inducible dimerization system by fusing the fragments to the ABI and PYL proteins.43 Similarly, esterase assembly was enhanced with the addition of the small-molecule dimerization trigger in E. coli (Figure S3). Given the performance of the system, we imaged lysate from E. coli cotransformed with N-terminal split BS2 (BS2N)-fused FRB and FKBP-fused C-terminal split BS2 (BS2C) as a proof-of-principle experiment toward a cell-free synthetic enzymatic detection system.44 Indeed, the split esterase assembly was robust enough to detect fluorescein-CM2 cleavage by eye and to discern the presence of rapamycin (Figure S4).
Monitoring Small-Molecule-Activated PPIs in Mammalian Cells with Split BS2 Esterase
Given that the split esterase performed well in E. coli, we next sought to determine if the system could function in mammalian cells. We first fused the split esterase fragments to tightly binding leucine zipper domains ZA and ZB45 (BS2N-ZA and ZB-BS2C) to optimize the deployment of the reporter in mammalian cells, including vector concentrations and experimental timing (Figure S5). Once we obtained optimized conditions, we then tested the rapamycin-dependent dimerization system in mammalian cells. A fluorescent signal, and therefore BS2 activity, was only observed in the presence of rapamycin in both imaging and plate-reader-based cell assays (Figure 2 and Figure S6). While esterase activity was observed in the absence of rapamycin in E. coli, esterase assembly was fully PPI-dependent in mammalian cells. We hypothesized that this discrepancy was due to a substantial overexpression of esterase fragments in E. coli, as compared with the relatively lower concentrations achieved in mammalian cells, which enables purely PPI-dependent assembly. Similar to the rapamycin system, we also observed robust PPI-dependent BS2 activity in mammalian cells with the ABA-inducible dimerization system (Figure S7).
Figure 2.
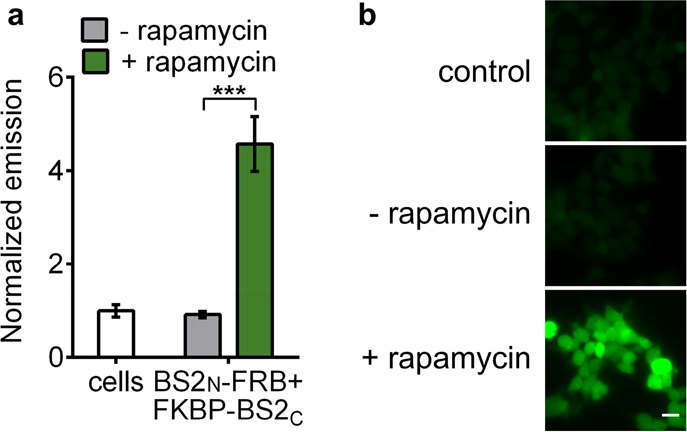
Split BS2 can detect small-molecule-activated PPIs. (a) HEK293T cells cotransfected with BS2N-fused FRB and FKBP-fused BS2C or HEK293T control cells (white) were incubated with rapamycin (green) or a DMSO control (gray). After 24 h, fluorescein-CM2 was applied, and the cells were analyzed by a plate reader for fluorescence. (b) HEK293T cells were treated identically to conditions in part a and analyzed by fluorescence microscopy. Error bars are the standard deviation for n = 4 replicates. Unpaired t test; ***P < 0.0001. Scale bars shown are 20 μm.
Monitoring Bcl-2 Family PPIs and Inhibitor Engagement
Having established small-molecule-dependent activation of split BS2 in mammalian cells, we next examined whether the system can detect therapeutically relevant PPIs. We selected the Bcl-2 family of apoptotic regulatory proteins to detect both the interaction network with BH3 domains and their pharmacological engagement.46 We generated fusions of BS2C to the antiapoptotic proteins Bcl-2 and Mcl-1 and cloned fusions of BS2N to the BH3 binding domains of tBID, which binds Bcl-2 and Mcl-1, NOXA, a selective Mcl-1 ligand, or deadBID, a control peptide without the necessary BH3 domain (Figure 3a). Cotransfection of BS2N-fused deadBID with either of the antiapoptotic protein-fused BS2C showed no significant signal over nontransfected cells incubated with fluorescein-CM2 as measured by either imaging or plate reader assays (Figure 3b,c and Figure S8). As expected, Mcl-1-fused BS2C showed significant esterase assembly when combined with either tBID- or NOXA-fused BS2N, while Bcl-2-fused BS2C only showed robust esterase activity in combination with tBID-fused BS2N. Collectively, these results indicate that split BS2 is capable of measuring biologically relevant PPIs in mammalian cells in a manner that recapitulates the well-studied selectivity profiles of these PPI targets.
Figure 3.

Split BS2 can detect Bcl-2 family PPIs and inhibitor engagement. (a) Vector system to test Bcl-2 split esterase PPI detection. (b) HEK293T cells cotransfected with the plasmids shown in part a were incubated with fluorescein-CM2 and analyzed for fluorescence by a plate reader. The normalized emission for interactions between deadBID/Bcl-2 protein (gray), tBID/Bcl-2 protein (green), and NOXA/Bcl-2 protein is shown. HEK293T control cells (white) were similarly analyzed. (c) HEK293T cells cotransfected and incubated with fluorescein-CM2 as in part b were analyzed by fluorescence microscopy. (d) HEK293T cells cotransfected with BS2N-fused tBID and Bcl-2-fused BS2C were treated with DMSO (time = 0) or ABT199 for 0–3 h (green) followed by incubation with fluorescein-CM2 and analyzed for fluorescence by a plate reader. HEK293T control cells (white) were similarly analyzed. (e) Vector system to simultaneously detect two PPIs and Bcl-2 inhibition. (f) HEK293T cells were cotransfected with the split esterase plasmids or split Nluc plasmids shown in part e. The two cell populations were mixed and treated with ABT-199 or a DMSO control. After 24 h, the cells were incubated with fluorescein-CM2 and analyzed (green). Immediately after analysis, furimazine was added, and the cells were reimaged (orange). The Bcl-2/tBID interaction was selectively blocked and detected with the esterase reporter (left) or Nluc reporter (right). HEK293T control cells were similarly analyzed. Error bars are the standard deviation for n = 4 (b), n = 6 replicates (d), and n = 8 replicates (f). Unpaired t test; *P < 0.01, ***P < 0.0001. Scale bars shown are 20 μm.
We next aimed to measure pharmacological engagement of PPI inhibitors, a common use of PCA systems, which would also allow us to test the time-dependency of split BS2 disassembly. We selected the FDA approved Bcl-2 inhibitor, ABT-199 (Venetoclax),47 and monitored tBID-fused BS2N and Bcl-2-fused BS2C disassembly over time (Figure 3d and Figure S9a,b). Treatment resulted in significantly decreased esterase activity within 1 h with no appreciable esterase activity detected after 6 h. We compared this to Nanobit, a structurally optimized split NanoLuc (Nluc) luciferase reporter, which has been used to monitor numerous PPIs and their modulation by small molecules (Figure S9c).48 While both systems reported on the ABT-199-mediated blockade of tBID-Bcl-2 assembly with approximately 10-fold reduction in signal on comparable time scales, a high level of signal remained with Nanobit. Nonetheless, these results confirm that split BS2 performs similarly to an established PPI inhibitor screening system, such as the state-of-the-art Nluc system.
Since both Nluc and split BS2 reporters measure pharmacological engagement in mammalian cells, and the two PCA systems should be orthogonal to one another, we reasoned that we could multiplex the reporters to simultaneously detect two PPIs and Bcl-2 inhibition (Figure 3e). We cotransfected one cell population with BS2N-fused tBID and Bcl-2-fused BS2C and another population with Nluc11S-fused tBID and Mcl-1-fused Nluc114. The two cell populations were then mixed and treated with ABT-199. As expected, only the Bcl-2 and tBID interaction was blocked, as esterase activity (as measured by fluorescence) was significantly decreased in the presence of the inhibitor while Nanobit activity (as measured by luminescence) remained constant (Figure 3f). We also swapped the fusion partners on Nluc and split BS2, with Nanobit reporting on the Bcl-2/tBID interaction and split BS2 reporting on the Mcl-1/tBID interaction, and observed a decrease in luminescence, but not fluorescence, with ABT-199 treatment.
Generation of a Chemiluminescent Signal Output for Detecting Extracellular PPIs
On the basis of the ability of the split esterase to sensitively and selectively monitor intracellular PPIs in mammalian cells, we next aimed to detect an extracellular PPI. Such an extracellular system could be useful for investigating both ligand- and receptor-mediated dimerization of transmembrane cell-surface receptors, such as receptor tyrosine kinases or G protein-coupled receptors. Additionally, measuring extracellular interactions also presented us with an opportunity to develop another signal output of split BS2. Specifically, we aimed to develop a masked chemiluminescent molecule that directly generates photons in an activity-dependent manner by split BS2. To this end, we first synthesized Chemilum-CM (Figure 4a, Figure S10 and Notes S1 and S2), a methylcyclopropyl ester-masked pro-chemiluminescent substrate based on a previously reported scaffold.49,50 We reasoned that esterase activity on Chemilum-CM would release the chemiluminescent form of the probe, which spontaneously reacts to generate a photon and emit steady-state luminescence. To confirm that Chemilum-CM can act cooperatively with BS2 to generate luminescence, we incubated Chemilum-CM with HEK293T cells transfected with a glycophosphatidyl inositol (GPI)-anchored51 full-length BS2 esterase and observed a robust luminescence signal (Figure S11a). We next examined whether the split esterase could provide a sensitive and fast readout on extracellular PPIs. HEK293T cells cotransfected with GPI-anchored BS2N-fused FRB and FKBP-fused BS2C were cultured in the presence and absence of rapamycin over time and subsequently incubated with Chemilum-CM (Figure 4b,c). Luminescence increased with rapamycin concentration and was observed 30 min post-rapamycin-addition and increased over time, suggesting that split esterase assembly is occurring at the cell membrane (Figure 4c,d). Luminescence also corresponded to the amount of split esterase used for transfection (Figure S11b). The BS2/Chemilum-CM system therefore functioned as a “synthetic luciferase”, allowing us to perform side-by-side direct comparisons between split BS2 and Nluc.
Figure 4.

Multiplexed PPI analysis with split BS2. (a) Chemilum-CM is unmasked by esterase activity and generates a photon. (b) Vector system to monitor extracellular PPIs. (c) HEK293T cells were cotransfected with plasmids shown in part b. Rapamycin (blue) or a DMSO control (gray) were added to cells for 0–6 h. Media was replaced with Chemilum-CM and analyzed for luminescence. HEK293T control cells (white) were similarly analyzed. (d) HEK293T cells cotransfected as in part c or HEK293T control cells (triangle) were incubated with rapamycin (blue) or a DMSO control (gray). After 6 h, the cells were analyzed with Chemilum-CM as in part c. (e) Vector system to simultaneously monitor extracellular and intracellular PPIs. (f) HEK293T cells were transfected with all four plasmids shown in part d. Rapamycin or a DMSO control was added to the cells for 24 h. Media was replaced with Chemilum-CM (10 μM) and analyzed for luminescence (blue). The cells were then rinsed, incubated with furimazine, and analyzed for bioluminescence (orange). Error bars are the standard deviation for n = 4 replicates. Unpaired t test; *P < 0.01, **P < 0.001, ***P < 0.0001.
We next tested whether we could monitor intracellular and extracellular PPIs simultaneously, using the selective luminescent signals generated from Nluc/luciferin and BS2/Chemilum-CM2. We used BS2N-ZA and ZB-BS2C with GPI anchors to monitor extracellular interactions (Figure 4e) and used ZBneg,45 a triple mutant of ZB with weakened affinity for ZA, as a negative BS2C fusion control. To monitor intracellular PPIs, we used Nluc11S-fused FRB and FKBP-fused Nluc114. All four plasmids were simultaneously transfected into mammalian cells, followed by treatment with rapamycin or DMSO carrier as a control. After 24 h, we added Chemilum-CM2, measured luminescence, and observed robust esterase activity only in cells expressing the ZA/ZB interaction fusions (Figure 4f). Cells expressing the ZBneg fusion did not exhibit significant chemiluminescence over untransfected control cells, indicating that esterase assembly was interaction-dependent, even on the cell surface. We then rinsed the cells, administered the Nanobit substrate, furimazine, and again measured luminescence. As expected, cells that were treated with rapamycin showed significantly enhanced luminescence activity over DMSO-treated cells. While luminescence was also observed in DMSO-treated cells, the signal was comparable to cells expressing just Nluc11S-fused FRB or mismatched PPI controls and, moreover, is a previously reported artifact attributed to the background of the N-terminal fragment of Nluc (Figure S12).48 Collectively, these results confirm not only that split BS2 functions as a proximity-dependent split reporter at the cell surface but also that the BS2/Chemilum-CM system is fully compatible with commonly used split luciferase reporters.
Proximity-Dependent Uncaging of Bioactive Molecules
Finally, we tested the utility of the split esterase to unmask a pharmacological agent in a proximity-dependent manner. The prodrug irinotecan (CPT-11) is converted to the cytotoxic active metabolite SN-38, a 1000-fold more potent topoisomerase-1 inhibitor, by carboxylesterases.34,52,53 Irinotecan is currently in clinical trials for neuroblastoma,54 colon cancer,55 and other solid malignancies.56−60 New strategies to more effectively activate irinotecan by modified rabbit and human carboxylesterases are in development.34,52 We reasoned that our new split esterase system could potentially be used to activate irinotecan analogues in an orthogonal and programmable manner. We therefore synthesized a new methyl-cyclopropyl-masked version of irinotecan to generate SN-38-CM2 (Figure 5a, Note S3). Incubation of SN-38-CM2 with BS2 in vitro showed >95% conversion to SN-38 within 5 min (Figure S13).
Figure 5.

Small-molecule-induced intra- and intercellular cell death. (a) Chemical structure of masked chemotherapeutic SN-38-CM2. (b) MDA-MB-231 luciferase cells were cotransfected with GPI-anchored BS2N-fused FRB and FKBP-fused BS2C. Rapamycin (blue square) or a DMSO control (gray square) was added to cells for 12 h prior to addition of SN-38-CM2. After 6 h, the cells were rinsed, cultured for 40 h, and imaged with d-luciferin. MDA-MB-231 luciferase control cells (gray dashed circle) were similarly analyzed. (c) MDA-MB-231 cells were cotransfected with GPI-anchored BS2N-fused FRB and FKBP-fused BS2C and then coplated with MDA-MB-453 luciferase cells. Rapamycin (blue square) or a DMSO control (gray square) and SN-38-CM2 were added to cells and imaged with d-luciferin as in part b. (d) Schematic of the coculture cytotoxicity assay. Split esterase or control MDA-MB-231 cells were plated in the center of a 3.5 cm2 dish (dashed center circle). MDA-MB-453 luciferase cells were plated around the split BS2 or control cells. PPI-mediated cleavage of SN-38-CM2 by split BS2 results in an active molecule to induce killing of neighboring cells. (e) Representative bioluminescence images of cocultures after treatment as in part d. (f) Quantification of light emission from bioluminescence images of cocultures. Photon counts along a 2 cm line in the direction of most observed cell killing from the hydrogel embedded cells (hydrogel) to the edge of each dish are plotted. (g) Commonly used PCA systems and their corresponding outputs. Error bars are the standard deviation for n = 4 replicates (b, c) and standard error of the mean for n = 8 replicates. Unpaired t test; *P < 0.01, **P < 0.001, ***P < 0.0001.
To test the PPI-dependent activity of the BS2/SN-38-CM2 system, we incubated SN-38-CM2 with MDA-MB-231 cells stably expressing luciferase and cotransfected with GPI-anchored BS2N-fused FRB and FKBP-fused BS2C in the presence or absence of rapamycin. After incubation with the masked pharmacological agent over time, cells were rinsed and cultured for 40 h prior to measuring cell viability via luciferase assay. Increased cytotoxicity was observed in cells treated with rapamycin over DMSO-treated cells and was comparable to activity observed with BS2 (Figure 5b, Figure S14). In addition, DMSO-treated cells and untransfected control cells showed no significant cytotoxicity with up to 1 μM SN-38-CM2. The reduced cell survival observed at higher concentrations of SN-38-CM2 for control conditions is likely due to slow background hydrolysis of the CM motif as shorter incubation times do not show any significant changes to cell proliferation (Figure S14). If background hydrolysis unmasks only 2–5% of SN-38-CM2 at the highest concentration, significant cell toxicity would be expected on the basis of reported IC50 values with comparable treatment times.61 While careful tuning of the SN-38-CM2 incubation is necessary to ensure maximum PPI-dependent esterase-mediated cell death, our experiments suggest that further optimization of the BS2/SN-38-CM2 system could effectively result in complete reduction of cell survival rate.
Since the split esterase/SN-38-CM2 system induced robust cytotoxicity in cells expressing the esterase system, we aimed to detect intercellularly mediated cell death. Such an approach, if successful, could lead to “sentinel” cells that respond to activation signals and release cytotoxic molecules that kill adjacent cells only when activated. To test this idea, we first coplated MDA-MB-231 cells cotransfected with GPI-anchored BS2N-fused FRB and FKBP-fused BS2C alongside MDA-MB-453 luciferase cells. We incubated the cells in the presence or absence of rapamycin and SN-38-CM2 and then imaged with d-luciferin. Indeed, we observed rapamycin-dependent cytotoxicity of the second cell population (Figure 5c).
To further visualize intercellular-mediated cell death based on the relative location of a second cell population using a bystander effect, we used MDA-MB-231 cells cotransfected with GPI-anchored BS2N-fused FRB and FKBP-fused BS2C suspended in a hydrogel, plated in the middle of a 3.5 cm2 tissue culture dish in the presence or absence of rapamycin with SN-38-CM2 (Figure 5d). After the matrix solidified, MDA-MB-453 luciferase expressing cells were plated around the hydrogel. The cells were then rinsed and assayed for cytotoxicity via light emission after 40 h. We then quantified luminescence from the hydrogel to the edge of the plate where the most cell killing was observed (Figure 5e,f, Figure S15). The reporter cells nearest the rapamycin-treated split BS2 cells showed significant cytotoxicity indicated by reduced light emission. Light emission was comparable between control cells and split BS2 cells in the absence of rapamycin. These results confirm that cells engineered with the split esterase reporter system can generate a cytotoxic molecule and kill adjacent cells in a small-molecule-dependent manner.
Conclusions
In this work, we developed a strategy for proximity-dependent uncaging of small molecules using an engineered esterase biosensor system. This approach comprises split versions of BS2 esterase that assemble to form a functional esterase when brought into contact by fused PPIs, which subsequently activates ester-masked small molecules. By utilizing small-molecule products as the output of BS2 PPI detection, we show that we can generate fluorescent, chemiluminescent, and pharmacological output signals, thereby adding a versatile new addition to the PCA toolbox (Figure 5g). The split esterase platform can enable a range of new applications in mammalian cells, including both PPI detection and synthetic biology. For in vitro applications with split BS2, current efforts have shown that the individual fragments express poorly in E. coli, precluding adequate protein purification (Figure S16). Optimization of structural stability after enzyme fragmentation has previously been reported,48 and we are currently investigating evolution strategies62,63 to improve stability and solubility of split BS2.
The small-molecule output made possible by BS2 can be used to generate fluorescent or luminescent signals and is compatible with other common PCA detection tools such as luciferase for analysis purposes. By modulating the properties of the ester-masked imaging agents, it should be possible to rapidly alter the dynamic range and sensitivity of BS2 imaging. Indeed, cell permeability, pharmacokinetics, and emission maximum have all been investigated for d-luciferin, enabling improved tissue distribution, sensitivity, and dynamic range in bioluminescence imaging.64,65
While Chemilum-CM enabled sensitive detection of extracellular PPIs, it can currently only detect full-length BS2 intracellularly, suggesting that improved cellular uptake is necessary to detect intracellular split BS2 (Figure S17). Chemiluminescent probes with improved cellular uptake and increased quantum yields have recently been reported,66,67 and these strategies should readily extend to our chemiluminescent scaffold. Fluorophores have also been extensively designed for enhanced brightness and photostability for cellular imaging,68−70 and we are currently investigating ester-masking strategies for new imaging scaffolds. For in vivo imaging, masked red-shifted fluorophores should be easily accessible, as well as masked versions of other imaging modalities such as magnetic resonance-based imaging reagents.
While the CM-masked substrates were not efficiently hydrolyzed by endogenous esterases in these studies or in a panel of cell lines previously tested (including HeLa, CHO, and neuronal cells),23 cell types that express more hydrolytic enzymes could be a concern for this strategy. When fluorescein-CM2 was incubated with different cell lines, including metastatic cancer cells, immune cells, and liver cells, varying endogenous activity on the probe was observed (Figures S18 and S19). We are therefore currently investigating new esters with increased steric bulk for improved biorthogonality to native esterases across a wider range of cellular environments and contexts.
In principle, any bioactive molecule that relies on an alcohol position for function can be masked and therefore unmasked by BS2 in an activity-dependent manner, which we demonstrated in this work through controlled cellular cytotoxicity of SN-38. Aside from activating killing in the cells expressing the biosensor, we also show that split BS2 in combination with an ester-masked pharmacological agent induced intercellular cell killing based on the proximity of the second cell population to the esterase expressing cells. While robust cytotoxicity is also observed in cells expressing split BS2, this could be mitigated against by expressing known resistance proteins to SN-3871,72 and would enable a constant esterase source for adjacent cell killing. We are currently pursuing integrating split BS2 with cell engineering approaches, such as chimeric antigen receptor T cell therapy, to integrate activity-dependent small-molecule activation at cancer sites. We also anticipate additional new synthetic biology opportunities for cell engineering and cell signaling-based prodrug release with PPI-dependent split esterase small-molecule activation.
Acknowledgments
This work was supported by the Yhim Foundation, the National Institute of General Medical Sciences (R35 GM119840), and the National Institute of Mental Health (RF1MH114102) of the National Institutes of Health to B.C.D. Chemical synthesis and characterization was supported by the National Science Foundation (CHE 1653474). K.K. was supported by a Chemical Biology Training Grant from the National Institutes of Health (T32 GM008720).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00567.
Experimental details on ester synthesis, split esterase development, imaging, and cell killing assays (PDF)
Author Contributions
K.A.J., K.K., M.W.B., J.C.L., and B.C.D. conceived of the experimental design. K.A.J. and K.K. performed experiments. M.W.B. synthesized and characterized SN-38-CM2. W.A. and A.R.L. synthesized and characterized Chemilum-CM. K.A.J. and B.C.D. wrote the paper with input from all coauthors.
The authors declare the following competing financial interest(s): A.R.L. declares a financial stake in BioLum Sciences, LLC.
Supplementary Material
References
- Braun P. Interactome mapping for analysis of complex phenotypes: Insights from benchmarking binary interaction assays. Proteomics 2012, 12 (10), 1499–1518. 10.1002/pmic.201100598. [DOI] [PubMed] [Google Scholar]
- Jones S.; Thornton J. M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (1), 13–20. 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof T. C. The synaptic cycle - A cascade of protein-protein interactions. Nature 1995, 375 (6533), 645–653. 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- Kolch W. Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000, 351, 289–305. 10.1042/0264-6021:3510289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin M. R.; Tang Y. Y.; Wells J. A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21 (9), 1102–1114. 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran X.; Gestwicki J. E. Inhibitors of protein-protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. 10.1016/j.cbpa.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J. A.; McClendon C. L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450 (7172), 1001–1009. 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Daringer N. M.; Dudek R. M.; Schwarz K. A.; Leonard J. N. Modular extracellular sensor architecture for engineering mammalian cell-based devices. ACS Synth. Biol. 2014, 3 (12), 892–902. 10.1021/sb400128g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink T.; Lonzaric J.; Praznik A.; Plaper T.; Merljak E.; Leben K.; Jerala N.; Lebar T.; Strmsek Z.; Lapenta F.; Bencina M.; Jerala R. Design of fast proteolysis-based signaling and logic circuits in mammalian cells. Nat. Chem. Biol. 2019, 15 (2), 115–122. 10.1038/s41589-018-0181-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B.; Volz S. E.; Zhang F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015, 33 (2), 139–142. 10.1038/nbt.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J. Y.; Zinkus-Boltz J.; Dickinson B. C. Evolution of a split RNA polymerase as a versatile biosensor platform. Nat. Chem. Biol. 2017, 13 (4), 432–438. 10.1038/nchembio.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J. Y.; Dewey J. A.; Hadji A.; LaBelle J. L.; Dickinson B. C. RNA polymerase tags to monitor multidimensional protein-protein interactions reveal pharmacological engagement of Bcl-2 proteins. J. Am. Chem. Soc. 2017, 139 (34), 11964–11972. 10.1021/jacs.7b06152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J. Y.; Kentala K.; Dickinson B. C. Multidimensional control of Cas9 by evolved RNA polymerase-based biosensors. ACS Chem. Biol. 2018, 13 (2), 431–437. 10.1021/acschembio.7b00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J.; Disare M.; Dickinson B. C. Evolution of c-terminal modification tolerance in full-length and split T7 RNA polymerase biosensors. ChemBioChem 2019, 20 (12), 1547–1553. 10.1002/cbic.201900325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C. K.; Cho K. F.; Kim M. W.; Ting A. Y. Luciferase-LOV BRET enables versatile and specific transcriptional readout of cellular protein-protein interactions. eLife 2019, 8, 21. 10.7554/eLife.43826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piehler J. New methodologies for measuring protein interactions in vivo and in vitro. Curr. Opin. Struct. Biol. 2005, 15 (1), 4–14. 10.1016/j.sbi.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Shekhawat S. S.; Ghosh I. Split-protein systems: beyond binary protein-protein interactions. Curr. Opin. Chem. Biol. 2011, 15 (6), 789–797. 10.1016/j.cbpa.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola T. K. Visualization of molecular interactions using bimolecular fluorescence complementation analysis: Characteristics of protein fragment complementation. Chem. Soc. Rev. 2009, 38 (10), 2876–2886. 10.1039/b909638h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy I.; Michnick S. W. Mapping biochemical networks with protein fragment complementation assays. Methods Mol. Biol. 2015, 1278, 467–481. 10.1007/978-1-4939-2425-7_31. [DOI] [PubMed] [Google Scholar]
- Azad T.; Tashakor A.; Hosseinkhani S. Split-luciferase complementary assay: Applications, recent developments, and future perspectives. Anal. Bioanal. Chem. 2014, 406 (23), 5541–5560. 10.1007/s00216-014-7980-8. [DOI] [PubMed] [Google Scholar]
- Kodama Y.; Hu C. D. Bimolecular fluorescence complementation (BiFC): A 5-year update and future perspectives. BioTechniques 2012, 53 (5), 285–298. 10.2144/000113943. [DOI] [PubMed] [Google Scholar]
- Martell J. D.; Yamagata M.; Deerinck T. J.; Phan S.; Kwa C. G.; Ellisman M. H.; Sanes J. R.; Ting A. Y. A split horseradish peroxidase for the detection of intercellular protein-protein interactions and sensitive visualization of synapses. Nat. Biotechnol. 2016, 34 (7), 774–780. 10.1038/nbt.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L.; Yang Y. L.; Wysocki L. M.; Arnold A. C.; Hu A.; Ravichandran B.; Sternson S. M.; Looger L. L.; Lavis L. D. Selective esterase-ester pair for targeting small molecules with cellular specificity. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (13), 4756–4761. 10.1073/pnas.1111943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay M. P.; Anderson R. F.; Ferry D. M.; Wilson W. R.; Denny W. A. Synthesis and evaluation of nitroheterocyclic carbamate prodrugs for use with nitroreductase-mediated gene-directed enzyme prodrug therapy. J. Med. Chem. 2003, 46 (25), 5533–5545. 10.1021/jm030308b. [DOI] [PubMed] [Google Scholar]
- Gruber T. D.; Krishnamurthy C.; Grimm J. B.; Tadross M. R.; Wysocki L. M.; Gartner Z. J.; Lavis L. D. Cell-specific chemical delivery using a selective nitroreductase-nitroaryl pair. ACS Chem. Biol. 2018, 13 (10), 2888–2896. 10.1021/acschembio.8b00524. [DOI] [PubMed] [Google Scholar]
- Hay M. P.; Wilson W. R.; Denny W. A. Nitroarylmethylcarbamate prodrugs of doxorubicin for use with nitroreductase gene-directed enzyme prodrug therapy. Bioorg. Med. Chem. 2005, 13 (12), 4043–4055. 10.1016/j.bmc.2005.03.055. [DOI] [PubMed] [Google Scholar]
- Porterfield W. B.; Jones K. A.; McCutcheon D. C.; Prescher J. A. A “caged” luciferin for imaging cell-cell contacts. J. Am. Chem. Soc. 2015, 137 (27), 8656–8659. 10.1021/jacs.5b02774. [DOI] [PubMed] [Google Scholar]
- Sellmyer M. A.; Bronsart L.; Imoto H.; Contag C. H.; Wandless T. J.; Prescher J. A. Visualizing cellular interactions with a generalized proximity reporter. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (21), 8567–8572. 10.1073/pnas.1218336110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau A.; Primeau M.; Trudeau L. E.; Michnick S. W. Beta-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein-protein interactions. Nat. Biotechnol. 2002, 20 (6), 619–622. 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
- Phelan R. M.; Ostermeier M.; Townsend C. A. Design and synthesis of a beta-lactamase activated 5-fluorouracil prodrug. Bioorg. Med. Chem. Lett. 2009, 19 (4), 1261–1263. 10.1016/j.bmcl.2008.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi F.; Charlton C. A.; Blau H. M. Monitoring protein-protein interactions in intact eukaryotic cells by beta-galactosidase complementation. Proc. Natl. Acad. Sci. U. S. A. 1997, 94 (16), 8405–8410. 10.1073/pnas.94.16.8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A.; Tekumalla V.; Krishnan A.; Pal-Bhadra M.; Bhadra U. Development of pyrrolo 2,1-c 1,4 -benzodiazepine beta-galactoside prodrugs for selective therapy of cancer by ADEPT and PMT. ChemMedChem 2008, 3 (5), 794–802. 10.1002/cmdc.200700328. [DOI] [PubMed] [Google Scholar]
- Leenders R. G. G.; Damen E. W. P.; Bijsterveld E. J. A.; Scheeren H. W.; Houba P. H. J.; van der Meulen-Muileman I. H.; Boven E.; Haisma H. J. Novel anthracycline-spacer-beta-glucuronide, -beta-glucoside, and -beta-galactoside prodrugs for application in selective chemotherapy. Bioorg. Med. Chem. 1999, 7 (8), 1597–1610. 10.1016/S0968-0896(99)00095-4. [DOI] [PubMed] [Google Scholar]
- Wierdl M.; Morton C. L.; Weeks J. K.; Danks M. K.; Harris L. C.; Potter P. M. Sensitization of human tumor cells to CPT-11 via adenoviral-mediated delivery of a rabbit liver carboxylesterase. Cancer Res. 2001, 61 (13), 5078–5082. [PubMed] [Google Scholar]
- Rehberg M.; Lepier A.; Solchenberger B.; Osten P.; Blum R. A new non-disruptive strategy to target calcium indicator dyes to the endoplasmic reticulum. Cell Calcium 2008, 44 (4), 386–399. 10.1016/j.ceca.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Yang Y. L.; Lee P.; Sternson S. M. Cell type-specific pharmacology of NMDA receptors using masked MK801. eLife 2015, 4, 12. 10.7554/eLife.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont K.; Webster R.; Gardner I.; Dack K. Design of ester prodrugs to enhance oral absorption of poorly permeable compounds: Challenges to the discovery scientist. Curr. Drug Metab. 2003, 4 (6), 461–485. 10.2174/1389200033489253. [DOI] [PubMed] [Google Scholar]
- Lavis L. D. Ester bonds in prodrugs. ACS Chem. Biol. 2008, 3 (4), 203–206. 10.1021/cb800065s. [DOI] [PubMed] [Google Scholar]
- Azzolini M.; Mattarei A.; La Spina M.; Marotta E.; Zoratti M.; Paradisi C.; Biasutto L. Synthesis and evaluation as prodrugs of hydrophilic carbamate ester analogues of resveratrol. Mol. Pharmaceutics 2015, 12 (9), 3441–3454. 10.1021/acs.molpharmaceut.5b00464. [DOI] [PubMed] [Google Scholar]
- Landowski C. P.; Song X. Q.; Lorenzi P. L.; Hilfinger J. M.; Amidon G. L. Floxuridine amino acid ester prodrugs: Enhancing Caco-2 permeability and resistance to glycosidic bond metabolism. Pharm. Res. 2005, 22 (9), 1510–1518. 10.1007/s11095-005-6156-9. [DOI] [PubMed] [Google Scholar]
- Schmidt M.; Henke E.; Heinze B.; Kourist R.; Hidalgo A.; Bornscheuer U. T. A versatile esterase from Bacillus subtilis: Cloning, expression, characterization, and its application in biocatalysis. Biotechnol. J. 2007, 2 (2), 249–253. 10.1002/biot.200600174. [DOI] [PubMed] [Google Scholar]
- Choi J. W.; Chen J.; Schreiber S. L.; Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996, 273 (5272), 239–242. 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- Liang F. S.; Ho W. Q.; Crabtree G. R. Engineering the ABA plant stress pathway for regulation of induced proximity. Sci. Signaling 2011, 4 (164), rs2 10.1126/scisignal.2001449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgman C. E.; Jewett M. C. Cell-free synthetic biology: Thinking outside the cell. Metab. Eng. 2012, 14 (3), 261–269. 10.1016/j.ymben.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh I.; Hamilton A. D.; Regan L. Antiparallel leucine zipper-directed protein reassembly: Application to the green fluorescent protein. J. Am. Chem. Soc. 2000, 122 (23), 5658–5659. 10.1021/ja994421w. [DOI] [Google Scholar]
- Czabotar P. E.; Lessene G.; Strasser A.; Adams J. M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15 (1), 49–63. 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- Cang S. D.; Iragavarapu C.; Savooji J.; Song Y. P.; Liu D. L. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8, 129. 10.1186/s13045-015-0224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon A. S.; Schwinn M. K.; Hall M. P.; Zimmerman K.; Otto P.; Lubben T. H.; Butler B. L.; Binkowski B. F.; Machleidt T.; Kirkland T. A.; Wood M. G.; Eggers C. T.; Encell L. P.; Wood K. V. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 2016, 11 (2), 400–408. 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- Lippert A. R. Unlocking the potential of chemiluminescence imaging. ACS Cent. Sci. 2017, 3 (4), 269–271. 10.1021/acscentsci.7b00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green O.; Eilon T.; Hananya N.; Gutkin S.; Bauer C. R.; Shabat D. Opening a gateway for chemiluminescence cell imaging: Distinctive methodology for design of bright chemiluminescent dioxetane probes. ACS Cent. Sci. 2017, 3 (4), 349–358. 10.1021/acscentsci.7b00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Grenier V.; Hong W.; Muller V. R.; Miller E. W. Fluorogenic targeting of voltage-sensitive dyes to neurons. J. Am. Chem. Soc. 2017, 139 (48), 17334–17340. 10.1021/jacs.7b07047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutova M.; Goldstein L.; Metz M.; Hovsepyan A.; Tsurkan L. G.; Tirughana R.; Tsaturyan L.; Annala A. J.; Synold T. W.; Wan Z. S.; Seeger R.; Anderson C.; Moats R. A.; Potter P. M.; Aboody K. S. Optimization of a neural stem-cell-mediated carboxylesterase/irinotecan gene therapy for metastatic neuroblastoma. Mol. Ther. Oncolytics 2017, 4, 67–76. 10.1016/j.omto.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierdl M.; Tsurkan L.; Hatfield M. J.; Potter P. M. Tumour-selective targeting of drug metabolizing enzymes to treat metastatic cancer. Br. J. Pharmacol. 2016, 173 (19), 2811–2818. 10.1111/bph.13553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBois S. G.; Mosse Y. P.; Fox E.; Kudgus R. A.; Reid J. M.; McGovern R.; Groshen S.; Bagatell R.; Maris J. M.; Twist C. J.; Goldsmith K.; Granger M. M.; Weiss B.; Park J. R.; Macy M. E.; Cohn S. L.; Yanik G.; Wagner L. M.; Hawkins R.; Courtier J.; Lai H.; Goodarzian F.; Shimada H.; Boucher N.; Czarnecki S.; Luo C. Q.; Tsao-Wei D.; Matthay K. K.; Marachelian A. Phase II trial of alisertib in combination with irinotecan and Temozolomide for patients with relapsed or refractory neuroblastoma. Clin. Cancer Res. 2018, 24 (24), 6142–6149. 10.1158/1078-0432.CCR-18-1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata N.; Maeda H.; Ishibashi K.; Hirata K.; Makiyama A.; Iwamoto S.; Takemoto H.; Imasato M.; Yoshida Y.; Munemoto Y.; Tanaka C.; Morita Y.; Hotta Y.; Toyofuku A.; Nagasaka T.; Morita S.; Sakamoto J.; Mishima H. Multicenter open-label randomized phase II study of second-line panitumumab and irinotecan with or without fluoropyrimidines in patients with KRAS wild-type metastatic colorectal cancer (PACIFIC study). Med. Oncol. 2019, 36 (6), 46. 10.1007/s12032-019-1254-4. [DOI] [PubMed] [Google Scholar]
- Nogami N.; Hotta K.; Segawa Y.; Takigawa N.; Hosokawa S.; Oze I.; Fujii M.; Ichihara E.; Shibayama T.; Tada A.; Hamada N.; Uno M.; Tamaoki A.; Kuyama S.; Ikeda G.; Osawa M.; Takata S.; Tabata M.; Tanimoto M.; Kiura K. Phase II study of irinotecan and amrubicin in patients with relapsed non-small cell lung cancer: Okayama Lung Cancer Study Group Trial 0402. Acta Oncol. 2012, 51 (6), 768–773. 10.3109/0284186X.2011.648342. [DOI] [PubMed] [Google Scholar]
- Murakami S.; Saito H.; Kondo T.; Ito H.; Oshita F.; Yamada K. Phase II study of nedaplatin and irinotecan as adjuvant chemotherapy for completely resected non-small cell lung cancer. Cancer Chemother. Pharmacol. 2018, 81 (1), 81–87. 10.1007/s00280-017-3460-0. [DOI] [PubMed] [Google Scholar]
- Wills B.; Cardona A. F.; Rojas L.; Ruiz-Patino A.; Arrieta O.; Reguart N.; Carranza H.; Vargas C.; Otero J.; Corrales L.; Martin C.; Cuello M.; Pino L. E.; Rolfo C.; Rosell R.; Zatarain-Barron Z. L.; Latin-Amer Consortium Invest L. Survival outcomes according to TIMP1 and EGFR expression in heavily treated patients with advanced non-small cell lung cancer who received biweekly irinotecan plus bevacizumab. Anticancer Res. 2017, 37 (11), 6429–6436. 10.21873/anticanres.12097. [DOI] [PubMed] [Google Scholar]
- Vo K. T.; Michlitsch J. G.; Shah A. T.; Long-Boyle J.; Kim M. O.; Gustafson C. Phase 1 multicenter trial to assess the maximum tolerated dose, safety, pharmacokinetics, and pharmacodynamics of pazopanib in combination with irinotecan and Temozolomide (PAZIT) for children and young adults with advanced sarcoma. J. Clin. Oncol. 2018, 36 (15), 2. 10.1200/JCO.2018.36.15_suppl.TPS10576. [DOI] [Google Scholar]
- Palmerini E.; Jones R. L.; Setola E.; Picci P.; Marchesi E.; Luksch R.; Grignani G.; Cesari M.; Longhi A.; Abate M. E.; Paioli A.; Szucs Z.; D’Ambrosio L.; Scotlandi K.; Fagioli F.; Asaftei S.; Ferrari S. Irinotecan and Temozolomide in recurrent Ewing sarcoma: An analysis in 51 adult and pediatric patients. Acta Oncol. 2018, 57 (7), 958–964. 10.1080/0284186X.2018.1449250. [DOI] [PubMed] [Google Scholar]
- Ciomei M.; Croci V.; Stellari F.; Amboldi N.; Giavarini R.; Pesenti E. Antitumor activity of edotecarin in breast carcinoma models. Cancer Chemother. Pharmacol. 2007, 60 (2), 229–235. 10.1007/s00280-006-0365-8. [DOI] [PubMed] [Google Scholar]
- Cabantous S.; Waldo G. S. In vivo and in vitro protein solubility assays using split GFP. Nat. Methods 2006, 3 (10), 845–854. 10.1038/nmeth932. [DOI] [PubMed] [Google Scholar]
- Wang T. N.; Badran A. H.; Huang T. P.; Liu D. R. Continuous directed evolution of proteins with improved soluble expression. Nat. Chem. Biol. 2018, 14 (10), 972–980. 10.1038/s41589-018-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M. S.; Chaurette J. P.; Adams S. T.; Reddy G. R.; Paley M. A.; Aronin N.; Prescher J. A.; Miller S. C. A synthetic luciferin improves bioluminescence imaging in live mice. Nat. Methods 2014, 11 (4), 393–395. 10.1038/nmeth.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchimaru T.; Iwano S.; Kiyama M.; Mitsumata S.; Kadonosono T.; Niwa H.; Maki S.; Kizaka-Kondoh S. A luciferin analogue generating near-infrared bioluminescence achieves highly sensitive deep-tissue imaging. Nat. Commun. 2016, 7, 11856. 10.1038/ncomms11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hananya N.; Shabat D. Recent advances and challenges in luminescent imaging: Bright outlook for chemiluminescence of dioxetanes in water. ACS Cent. Sci. 2019, 5 (6), 949–959. 10.1021/acscentsci.9b00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan L. S.; Gerberich J.; Cao J.; An W. W.; Jenkins B. A.; Mason R. P.; Lippert A. R. Kinetics-based measurement of hypoxia in living cells and animals using an acetoxymethyl ester chemiluminescent probe. ACS Sens 2019, 4 (5), 1391–1398. 10.1021/acssensors.9b00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; Muthusamy A. K.; Liang Y. J.; Brown T. A.; Lemon W. C.; Patel R.; Lu R. W.; Macklin J. J.; Keller P. J.; Ji N.; Lavis L. D. A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 2017, 14 (10), 987–994. 10.1038/nmeth.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavis L. D.; Raines R. T. Bright building blocks for chemical biology. ACS Chem. Biol. 2014, 9 (4), 855–866. 10.1021/cb500078u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun J. V.; Petersson E. J.; Chenoweth D. M. Rational design and facile synthesis of a highly tunable quinoline-based fluorescent small-molecule scaffold for live cell imaging. J. Am. Chem. Soc. 2018, 140 (30), 9486–9493. 10.1021/jacs.8b03738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandu H.; Aluzaite K.; Fogh L.; Thrane S. W.; Noer J. B.; Proszek J.; Do K. N.; Hansen S. N.; Damsgaard B.; Nielsen S. L.; Stougaard M.; Knudsen B. R.; Moreira J.; Hamerlik P.; Gajjar M.; Smid M.; Martens J.; Foekens J.; Pommier Y.; Brunner N.; Schrohl A. S.; Stenvang J. Molecular characterization of irinotecan (SN-38) resistant human breast cancer cell lines. BMC Cancer 2016, 16, 34. 10.1186/s12885-016-2071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata S.; Oka M.; Shiozawa K.; Tsukamoto K.; Nakatomi K.; Soda H.; Fukuda M.; Ikegami Y.; Sugahara K.; Yamada Y.; Kamihira S.; Doyle L. A.; Ross D. D.; Kohno S. Breast cancer resistance protein directly confers SN-38 resistance of lung cancer cells. Biochem. Biophys. Res. Commun. 2001, 280 (5), 1216–1223. 10.1006/bbrc.2001.4267. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.