

Abstract
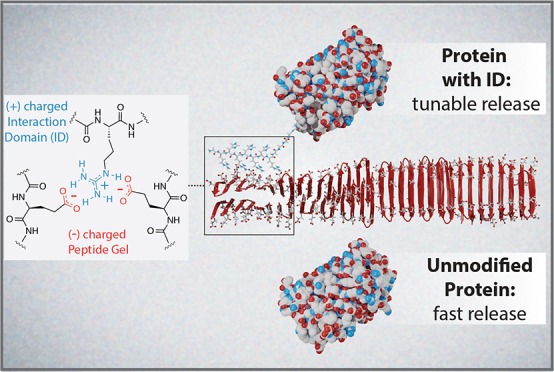
Protein biologics are an important class of drugs, but the necessity for frequent parenteral administration is a major limitation. Drug-delivery materials offer a potential solution, but protein-material adsorption can cause denaturation, which reduces their effectiveness. Here, we describe a new protein delivery platform that limits direct contact between globular protein domains and material matrix, yet from a single subcutaneous administration can be tuned for long-term drug release. The strategy utilizes complementary electrostatic interactions made between a suite of designed interaction domains (IDs), installed onto the terminus of a protein of interest, and a negatively charged self-assembled fibrillar hydrogel. These intermolecular interactions can be easily modulated by choice of ID to control material interaction and desorption energies, which allows regulation of protein release kinetics to fit desired release profiles. Molecular dynamics studies provided a molecular-level understanding of the mechanisms that govern release and identified optimal binding zones on the gel fibrils that facilitate strong ID–material interactions, which are crucial for sustained release of protein. This delivery platform can be easily loaded with cargo, is shear-thin syringe implantable, provides improved protein stability, is capable of a diverse range of in vitro release rates, and most importantly, can accomplish long-term control over in vivo protein delivery.
Short abstract
This study describes the use of positively charged interaction domains to control the rate of protein release from negatively charged peptide hydrogels for improving protein delivery.
Introduction
Protein therapeutics have become a significant source of new drugs due to their high affinity and selectivity.1 However, protein drugs cannot be taken orally and often require frequent parenteral administration for optimal efficacy, both of which can complicate treatment regimens. Thus, a delivery method that utilizes a single administered dosage of drug with long-term release would be a powerful clinical tool for improving treatment efficacy and patient compliance. Many different materials-based protein delivery platforms have been developed,1−3 but various challenges remain that limit further application.4 For example, the formulation process of encapsulating protein can lack precise control over the amount of drug loaded, and undesirable interactions between proteins and organic cosolvents, cross-linking agents, or the materials themselves can lead to denaturation and/or immunogenicity.
Hydrogels made from either synthetic or natural polymers are promising materials for improving protein delivery.2,5 Their aqueous interiors are often compatible with long-term protein encapsulation, but significant material optimization is typically necessary to tune the rate of drug release. One strategy to control delivery is to engineer binding interactions between the protein and ligands displayed within the gel matrix, where stronger interactions lead to slower rates of release. Ligands such as nucleic acids, peptides, or other proteins have been employed for these affinity-controlled systems.6−14 However, since protein–ligand binding is often specific, unique materials must be engineered to deliver each protein of interest. The design of a general affinity-based platform that could be used to deliver a larger array of proteins would have utility. In principle, the primary interactions that govern protein–ligand binding, such as H-bonding, hydrophobics, and electrostatics, could be used to engineer such a system. From a design standpoint, electrostatic interactions are attractive offering tunable affinity and plasticity in their geometric requirements.15−26 However, the folded conformations of many proteins are marginally stable, and encapsulation of a charged protein into a material network of opposite charge can cause nonspecific protein–matrix interactions that results in protein aggregation or denaturation. Additionally, engineering precise charge complementarity between the protein and matrix can be challenging due to the varied distribution of charged residues on protein surfaces and the heterogeneous spatial display of charge within polymers.27,28
Reported herein is the design of an electrostatics-driven delivery system that offers tunable release of proteins. This platform is comprised of two components, a highly negatively charged fibrillar hydrogel network and a family of designed positively charged interaction domains (IDs) that can be fused to the termini of, in principle, any negatively charged protein. The ID is responsible for sequestering the protein to the gel fibrillar scaffold and controlling its eventual release. Importantly, this strategy ensures that the globular domain of a gel-encapsulated protein is noninteracting with the material matrix via charge repulsion, and matrix binding is largely facilitated by the ID (Figure 1). Furthermore, since the suite of different IDs affords a high degree of control over release rates, it eliminates the need for re-engineering the hydrogel for delivering different proteins.
Figure 1.
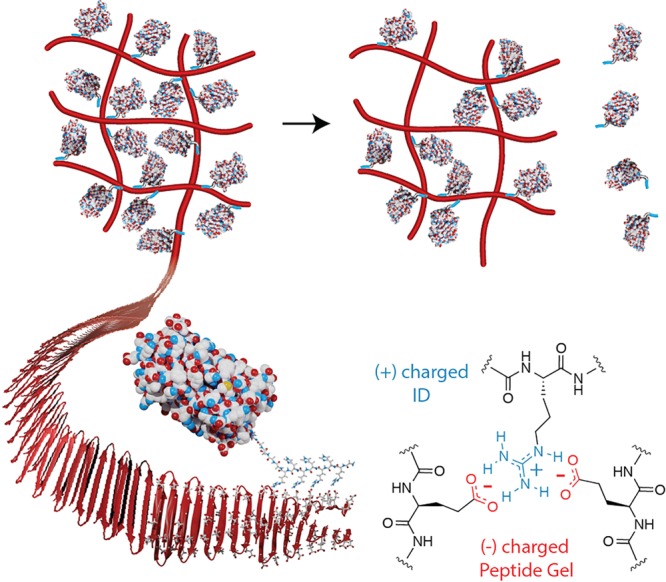
Controlling protein release from a negatively charged peptide hydrogel using a cationic interaction domain (ID).
Results and Discussion
Design of the Interaction Domains and Complement Hydrogel Scaffold
The design of our delivery system was inspired by the additive nature of biomolecular electrostatic interactions that impact macromolecular folding,29,30 association,31−33 and ultimate function. Extensive biophysical studies in proteins demonstrated that salt bridges can help stabilize the folded native state. The strength of these interactions is dependent on both the type of residues involved and their location within the protein.30,34−36 With this in mind, we designed a family of interaction domains with the potential to make a varying number of weak, medium, and strong interactions with the fibers of the gel network to control release. The general design of the ID utilizes a linear sequence (RH)nR-(GGSGS)2- that contains a varying number of (RH) repeats. The alternating sequence is designed to place all of the guanidinium side chains of arginine on the same face of an extended β-strand conformation to maximize potential interactions with the gel’s β-sheet rich fibers (Figure 1). Arginine was chosen to be the driver of ID association to the fibers based on its high pKa and known ability to engage in multiple binding orientations with carboxylates.37 Histidine was incorporated at the alternate position to aid solubilization, as opposed to a noncharged hydrophobic residue which would result in an amphiphilic sequence prone to aggregation. We also envisioned that for the longer IDs, the histidines could act as an intrinsic purification handle eliminating the need for a separate poly-histidine domain. Lastly, IDs are appended to a protein of interest with a simple glycine/serine linker. ID amino acid composition, sequence, and length influence the type and strength of electrostatic interactions made between the ID and fiber, which in turn, dictates protein release kinetics.
The hydrogel component of the delivery system is prepared from AcVES3, a self-assembling peptide. AcVES3 contains strands of alternating hydrophobic and hydrophilic residues that flank a central tetrapeptide capable of promoting a type II’ β-turn (Figure 1, Table 1). Sequences of this type are known to assemble into highly homogeneous β-hairpin-rich fibers.38−46 Hydrophilic residues are displayed on the solvent-exposed surface of the fibers, while the hydrophobic residues are buried within the core of a bilayer.47 To test our hypothesis that IDs can control protein release from the gel, we first designed a library of enhanced green fluorescent protein (EGFP) analogues containing different IDs at their N-termini (Table 1, Figure S1). EGFP, which has the β-barrel structure found in all fluorescent proteins, is an excellent model because its spectral properties allow for easy quantification and assessment of activity.48 Importantly, EGFP is negatively charged (pI ≈ 5.6) and allows us to test one of our design principles, which asserts that matching the electrostatic potential of a protein’s globular domain with the gel scaffold should limit their direct interaction.
Table 1. Electrostatic Components for Proteins and AcVES3.
| protein | basic A.A. in ID | calc pI |
|---|---|---|
| EGFP | 5.6 | |
| For EGFP/mRuby3, N-Terminal IDn = G(RH)nR(GGSGS)2- | ||
| ID1-EGFP | 2 Arg, 1 His | 5.9 |
| ID2-EGFP | 3 Arg, 2 His | 6.1 |
| ID3-EGFP | 4 Arg, 3 His | 6.3 |
| ID4-EGFP | 5 Arg, 4 His | 6.5 |
| ID5-EGFP | 6 Arg, 5 His | 6.7 |
| ID6-EGFP | 7 Arg, 6 His | 7.0 |
| H6-EGFP | 6 His | 6.0 |
| H12-EGFP | 12 His | 6.2 |
| NH6-EGFP | 3 Arg, 2 Lys, 6 His | 6.3 |
| mRuby3 | 5.8 | |
| ID5-mRuby3 | 6 Arg, 5 His | 8.4 |
| For IFNα, C-Terminal IDn = -(SGSGG)2(RH)nR | ||
| IFNα | 5.4 | |
| IFNα-ID3 | 4 Arg, 3 His | 7.0 |
| IFNα-ID5 | 6 Arg, 5 His | 8.4 |
| AcVES3 peptide hydrogel | net charge | |
|---|---|---|
| Ac-VEVSVSVEVDPLPTEVSVEVEV-NH2 | –5 | |
In Vitro Release of ID-EGFP from AcVES3 Hydrogel
The library of EGFP analogues was generated by bacterial expression (Figure S1) and AcVES3, used for hydrogelation, was synthesized by solid phase peptide synthesis (Figures S2 and S3). The conformation of the peptide within the empty gel is rich in β-sheet structure, as observed by CD, and TEM indicates a highly homogeneous fibrous network (Figure S4). Precise amounts of protein can be easily loaded directly into the AcVES3 gel network during hydrogel formation with 100% efficiency (Video S1). This is in contrast to preformed gels that must be loaded by permeation. Direct encapsulation of the ID proteins (∼1 mg/mL) does not affect the rheological properties of the AcVES3 hydrogel, as time-sweep measurements for ID-EGFP-loaded gels display shear-thin recovery properties that are identical to gel alone (Figures 2A and S5). The unaltered behavior of the material is further exemplified by the consistency of the storage moduli before and after shear-thinning across the different protein-loaded gels (Figure 2B), similar kinetics of gel formation at early time points (Figure S5), and the observance of similar fibers by TEM (Figure S4). We also assessed the rheological behavior at higher protein concentrations (3 and 5 mg/mL) and observed similar shear-thin recovery properties (Figure S5). Since the IDs do not affect peptide self-assembly or hydrogelation under these conditions, we postulate that the electrostatic interactions between the ID and material must be formed later in the assembly process, after a highly charged fiber surface is generated. The ability of the protein-loaded gels to form rigid materials and reheal after shearing is important because it demonstrates that they can be administered by syringe injection for in vivo delivery.
Figure 2.
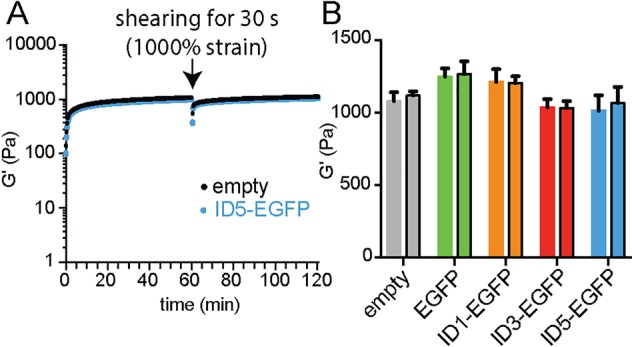
Shear-thin recovery properties of AcVES3 hydrogels with and without protein. (A) Comparison of time-sweep experiments for empty hydrogel and ID5-EGFP-containing gel. Other ID-EGFP analogues behave in a similar manner, as shown in Figure S5. (B) Storage modulus of gels containing protein before and after (bars outlined with black) shearing compared to empty hydrogel.
We then showed that the IDs could achieve a diverse range of in vitro release profiles (Figure 3A). As expected, unmodified EGFP, which is negatively charged, undergoes burst release within 2 days from the like-charged gel network. ID1-EGFP is released at a rate similar to EGFP, with ID2-EGFP showing only a modest increase in retention. In contrast, the ID3–6 analogues offer excellent prolonged control over release. After 36 days, ∼50% of ID3-EGFP was released, whereas only about 5% of ID6-EGFP had been released. Intermediate amounts (∼20%) of ID4- and ID5-EGFP are released over the same time period. Further, experiments performed in the presence of a high concentration of salt greatly expedited release indicating that electrostatics is the primary interaction governing matrix binding and release (Figure S6). The slow release at longer times (>2 weeks) for ID3–6 might be due to ID proteins located deep within the gel undergoing multiple desorption and resorption events en route to exiting the matrix.
Figure 3.
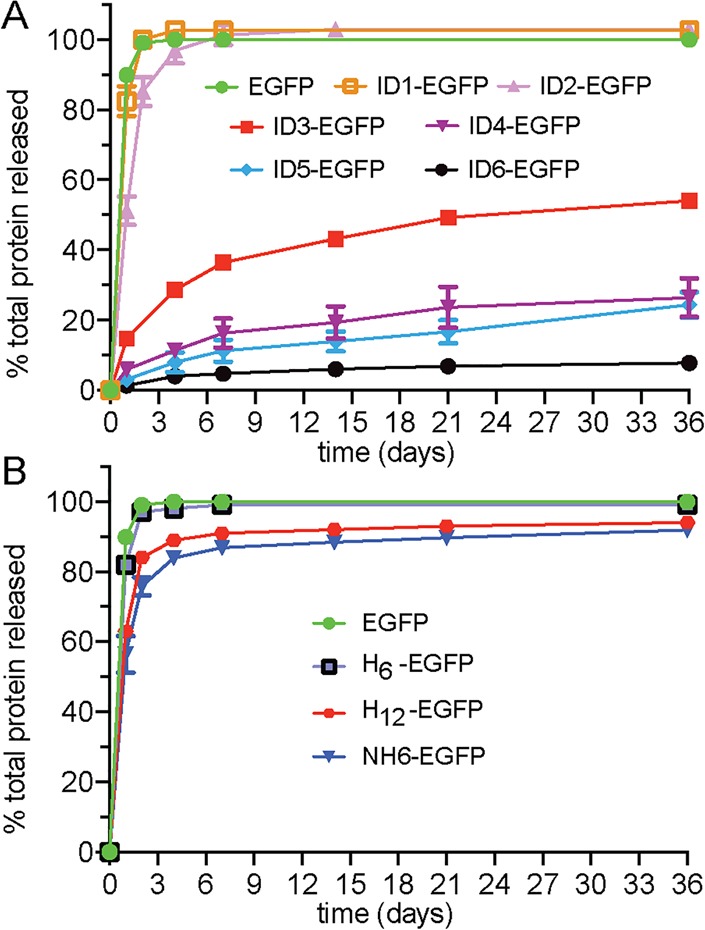
Release of EGFP versus analogues containing (A) IDs and (B) control domains.
In addition to studying the ID-appended proteins, we also investigated the release of EGFP-containing control domains to confirm that amino acid composition and local sequence arrangement of ID residues are important design determinants for release (Table 1 and Figure S1). Controls H6 and H12 contain 6 and 12 histidine residues, respectively, appended to the N-terminus of EGFP. The NH6 control domain originates from a common fusion used in protein expression, which contains a variety of basic residues distributed throughout its sequence. While the IDs afforded a range of release rates, the polyhistidine controls offered little ability to retain protein within the material (Figure 3B). This demonstrates the importance of arginine within the ID design and suggests that charge alone is not sufficient for controlling release. In fact, if charge content was the only defining factor controlling release rates, then the isoelectric point (pI) of any N-terminally modified protein should be an absolute predictor for release rates. Although there is good correlation within the ID1–6 family, the release profiles of ID3-EGFP and the NH6-EGFP control are quite different, despite having similar pIs (∼6.3). The control data also show that ID length is not a sole determinant of release behavior. The H12-fusion offers no control over EGFP release compared to ID5, despite having the same number of residues outside their linker domains. These results indicate that in addition to charge content and length, the composition and sequential arrangement of residues within the ID are important design factors.
Proteins released from the gel retain their activity. Fluorescence spectra of the ID-EFGPs before gel encapsulation and after gel release were identical (Figure S7). Further, qualitative evidence of active protein remaining in the materials is demonstrated by the visual examination of the gels over time (Figure S8). Conversely, EGFP encapsulated into cationic peptide hydrogels led to weak, brittle gels, suggesting that the oppositely charged EGFP interferes with peptide self-assembly (Figure S9). Furthermore, the EGFP that is released is denatured and displays attenuated fluorescence. This highlights the delicate nature of protein encapsulation and that denaturation can be mitigated through the installation of an ID.
Fine-Tuning Release and Time-Staggered Delivery
As shown in Figure 3A, there is a significant gap between the release profiles of ID2- and ID3-EGFP. This region of delivery space can be accessed using combinations of different IDs within the family. The expected release of protein can be determined by fitting each of the release curves in Figure 3A to a first-order kinetics model, then performing linear combinations of the fitted data. To address this previously neglected delivery profile, the release curves for ID2- and ID3-EGFP were fit and linearly combined at a ratio of 4:6 (ID2-EGFP/ID3-EGFP, Figure 4A). The predicted protein release behavior closely matched the experimental release data. Although this is only a simple test case, more complex combinations should be readily possible.
Figure 4.
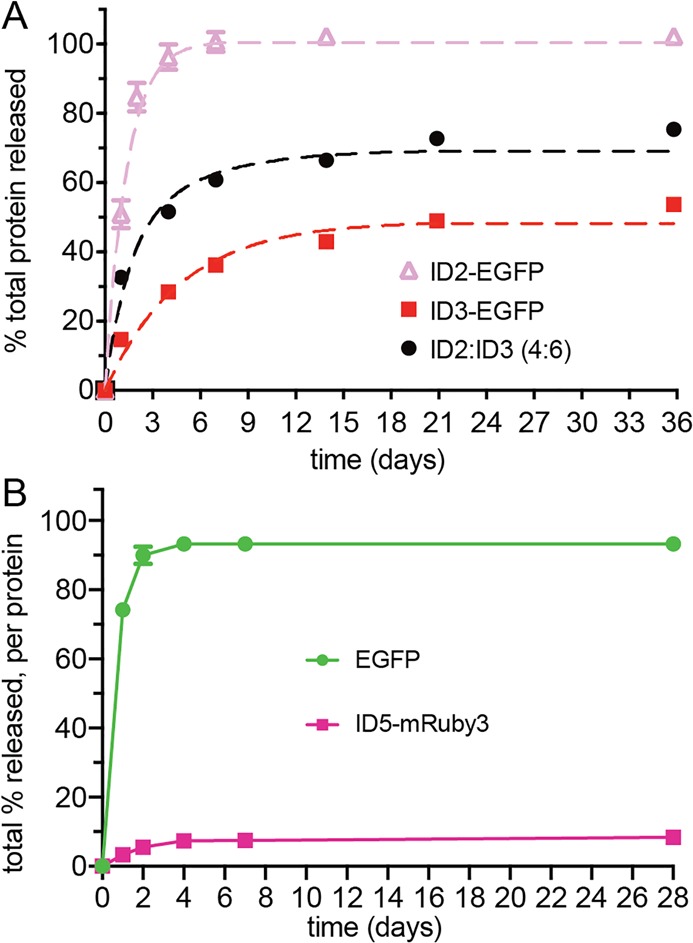
(A) Release profiles for mixtures of EGFP containing different interaction domains can be predicted, as shown with a 4:6 ratio of ID2-EGFP/ID3-EGFP. First, release data for each pure ID protein was fit to first-order kinetics (dashed pink and red lines), obtained by numerically integrating the equation [Pu]t = k1 [Pb] –k–1 [Pu], where t is time, Pu is unbound protein, Pb is bound protein, and k1 and k–1 are adsorption/desorption rate constants. Fits are linearly combined to predict the release of the mixed system (black dashed line), which agrees well with the experimental data (black circle). (B) Co-encapsulation and staggered release of distinct proteins.
In addition to fine-tuning the release of one specific protein of interest, our system can also be used to stagger the release of multiple proteins. Combination therapies can significantly improve treatment outcomes, with sequential delivery of different drugs often leading to improved efficacy over monotherapy or simultaneous codelivery.49−52 To model this treatment paradigm, we generated ID5-mRuby3, the ID5 variant of the monomeric red fluorescent protein mRuby3 (Table 1, Figure S1)53 and coencapsulated it into an AcVES3 hydrogel with EGFP. These fluorescent proteins have a difference in absorbance wavelength maxima of 70 nm, allowing for their precise concentration determination within the same sample. Figure 4B shows that independent control over the release of multiple proteins from a single gel is possible. EGFP was released within several days, followed by slow sustained delivery of the ID5-containing protein. This staggered release is shown qualitatively in Figure S10 through the visual monitoring of fluorescence over time.
Installing IDs onto the C-Terminus of Interferon-α for Controlled Cytokine Delivery
After demonstrating control over the release of model fluorescent proteins, we sought to expand the utility of the ID delivery platform toward the release of a clinically relevant protein, interferon-α (IFNα). Type I interferons, including IFNα, are important cytokines that are involved in the innate immune response and have been used for the treatment of viral infections and as cancer immunotherapies.54−56 IFNα is negatively charged (pI ≈ 5.4) similar to EGFP and mRuby3, which should lead to fast release of the unmodified protein from the negatively charged AcVES3 gel. Unlike the fluorescent proteins, however, IFNα is α-helical and contains multiple intramolecular disulfide bonds. We designed the IFNα analogues with the IDs at the C-terminus (Table 1), to further test the generality of ID placement. Since protein termini can be involved in binding interactions, having flexibility with respect to ID location is crucial. Also, putting the ID at the C-terminus opens the possibility that this strategy can be applied to proteins that utilize N-terminal signal peptides for secretory or protein folding purposes during the expression and maturation process.
ID proteins were cloned into the pPAL7 vector containing IFNα.57 After expression, proteins were purified/cleaved using Profinity eXact resin (Figure S11). As mentioned earlier, the longer IDs might be useful purification handles. As a proof of concept, IFNα-ID5 was first purified to homogeneity by metal affinity utilizing the ID domain prior to incubation with the eXact resin (Figure S11). This showcases the potential dual functionality of this domain to both control release and act as an intrinsic purification handle. Once purified, CD experiments were performed that showed that the ID does not interfere with the protein’s helical structure (Figure S12). We then performed concentration-dependent studies of IFNα and the ID analogues in the presence of HL116 cells.57 When stimulated with IFNα, HL116 expresses luciferase that can report on protein activity levels.58 Importantly, the IFNα-ID proteins had similar picomolar activity as the native IFNα, indicating that the ID does not hinder binding to the IFNα receptor and activation of cell-signaling pathways (Figure 5A). In vitro release experiments show that the IDs can effectively control delivery of IFNα (Figure 5B). As seen with the EGFP studies, the unmodified IFNα was mostly released within a few days, while IFNα-ID5 had a much slower release (<20% after 2 weeks). IFNα-ID3 offered intermediate release to unmodified and ID5-functionalized protein as expected. IFNα-ID protein released from the gel retained its ability to stimulate HL116 cells (Figure 5C). These results further validate the capability of IDs to modulate protein release and demonstrate that they can be used for proteins of different structure (α-helical or β-sheet) when incorporated at either protein termini. Although we anticipate that the IDs will be compatible with many negatively charged proteins, there will certainly be outliers.
Figure 5.

(A) Dose response for HL116 cell stimulation for IFNα and two ID analogues. (B) Release profiles of IFNα and its analogues. (C) HL116 stimulation activity of released IFNα-ID5 compared to soluble IFNα (500 pM) and empty gel (no lum = no luminescence).
Molecular-Level Insight into ID Binding, Mobility, and Release
The release data suggest that ID composition, sequence, and length influence release kinetics. To gain molecular-level insight into how the IDs physically engage the hydrogel’s fibrils, which affects binding and subsequent release, molecular dynamics (MD) simulations were performed. AcVES3 assembles into fibrils with two opposing hydrophilic faces, each containing distinct strips of glutamate-derived negative charge that traverse the fibril long axis (Figure S13). Microsecond simulations performed for each ID show that these charged strips on the fiber are the main orchestrators of the ID–fibril interaction. As designed, simulations suggest that the IDs bind to the fibrils in an extended conformation to maximize the electrostatic interactions between the arginine and glutamate residues. Conversely, when ID6 was forced to bind a fibril in a nonextended compact conformation, the calculated interaction energy was more than 35 kcal/mol less favorable than the extended conformation (Figure S14).59,60 Interaction energies were determined for the entire family of IDs and showed that the longer IDs have greater binding potentials (Figure 6A). Interestingly, the interaction energies are much greater than what might be expected for typical electrostatic interactions in proteins (normally on the order of single digit kcal/mol)29,30 and are more comparable to the energies observed for the complexation of oppositely charged peptide homopolymers.61,62 Additional experiments were performed to assess ID mobility once bound to the fibril. Here, an opposing force to ID binding was applied to mimic the induced repulsion between an appended negatively charged protein and the like-charged fiber. These simulations show that shorter IDs were much more mobile (Figure 6B). Video S2 shows the dynamics of ID1 versus ID6 over 100 ns, with the shorter ID being more conformationally flexible and traveling a longer distance along the fiber. Here, the mobility was also quantitated by measuring the displacement of the IDs’ center of mass relative to its initial docking site as a function of time (Figure 6C). Figure 6D shows snapshots of the two IDs from Video S2 before and after the simulation; ID6 remains more centrally bound to the fibril’s high-density charged regions, as compared to ID1. To assess the energetic requirements for ID desorption from the gel matrix, the release curves for the ID-EGFP analogues (Figure 3A) were analyzed to derive activation energies using the Arrhenius rate law (Figure 6E). The longer IDs required more energy for protein release, and the observed trend shows an inverse correlation to the calculated interaction energies for ID binding (Figure 6A).
Figure 6.

(A) Computationally determined interaction energies for IDs. (B) Rate of movement for IDs during simulations. (C) Change in the center of mass and (D) visual snapshots before and after 100 ns simulation for ID1 and ID6, demonstrating differences in mobility. (E) Calculated desorption energies for ID-EGFP determined by fitting the Arrhenius rate law (k = Ae(−Ea/RT)) to the release curves in Figure 3A, where k is the rate constant, A is the Arrhenius pre-exponential factor, Ea is the activation (desorption) energy, R is the gas constant, and T is the temperature.
Further analysis of the MD trajectories indicates that a substantial variety of interactions are made between the ID arginines and the glutamate side chains of the fiber, ranging from weak transient interactions to strong multidentate interactions that firmly anchor the ID onto the fiber. These binding interactions were assigned into one of three groups, namely, weak (type I), intermediate (type II), and strong (type III). This was accomplished across all ID family members by dividing each of their respective 100 ns trajectories into 5 ns sections and then computing the interaction energy for all of the arginine residues within that time frame. All arginine–glutamate intermolecular contacts <5.0 Å were binned into their respective types (I, II, or III) based on their computed energy. Within each type, the mobility of each arginine residue was determined, as well as the average number of glutamate side chains the arginine guanidinium group could make contact with during the simulation. Arginine residues that made weak interactions (type I) had an average interaction energy of −2.8 kcal/mol, traveled the greatest distance over the course of the simulation period (>4 Å), and were bound to at most one glutamate (on average 0.50). The intermediate interactions (type II) had more favorable binding energies (−6.2 kcal/mol), which were less mobile (traveled ∼3.3 Å) and could make contacts with at least one glutamate (average 0.98). The strongest contacts (type III) had an average interaction energy of −12 kcal/mol, which is roughly on par with the energetics of poly-arginine/poly-glutamate scaffolds reported previously.61 Type III arginine residues migrated very little during the simulation (<3 Å) and made contacts with multiple glutamates at a given time (average 1.56). Various molecular binding geometries were observed within each type; Figure 7A shows examples based on the statistics compiled from the simulations.
Figure 7.

Molecular insight into the ID and fiber interaction. (A) Potential binding modes of arginine, classified as type I, II, or III. (B) Histogram showing the probability of forming a second arginine type III interaction as a function of sequential position when an existing type III interaction is made at position i. (C) Graphical representation of an ID binding to a fiber containing four repeat units of a self-assembled AcVES3 peptide. The right-most panel contains the entire sequence of AcVES3. The ID shown at top contains the minimal sequence necessary to make type III interactions with arginine located at positions i, and i + 6.
We next investigated whether a particular ID could make multiple type III interactions, and if so, we sought to determine whether there was a positional dependence of arginine residues that allowed for multiple interactions. Figure 7B shows the probability of finding an arginine engaged in a type III interaction (i + n) residues away from an arginine at sequence position (i) that is also engaged in a type III interaction. The data indicate that arginine residues occupying (i, i + 6) sequential arrangements are more likely to engage in simultaneous type III interactions. Having discovered this positional dependence within the ID, we turned our attention to the model of the fibril to search for repetitive complementary glutamate-based motifs that could support the formation of multiple type III interactions. Each face of a given hydrogel fibril contains two strips of negative charge resulting from the solvent-exposed glutamate side chains (Figure S13). This molecular arrangement is depicted in the cartoon shown in Figure 6C. Optimal binding zones were identified every 5 Å along one of the charged strips, where each zone contains three proximal glutamates. The greater density of glutamate residues here increases the probability of the ID forming type III interactions. Within the ID, the average measured distance between the Cα carbons of two bound (i, i + 6) arginine residues engaged in a type III interaction and the distance between two binding zones along the fiber are both ∼20 Å. Alternative arrangements of arginine residues within the ID are less capable of binding to optimal zones simultaneously. For example, arginines at (i, i + 2) positions are 6.8 Å apart, a distance that does not fully complement the 5 Å repeat between zones. The importance of the (i, i + 6) arrangement is exemplified by the observed change in ID release behavior transitioning from ID2 to ID3 (Figures 3A and 6A,E). ID2 shows almost no ability to control release as compared to ID3, with the latter being the first interaction domain in the series capable of displaying arginine side chains in an (i, i + 6) arrangement. In addition to making more interactions of all types, the domains beyond ID3 have an increased probability of forming pairs of type III interactions. Thus, the collective assessment of ID behavior through experimental release and computational analysis suggests ID–gel interaction energies, ID motility, and activation energies for ID desorption all regulate the release of protein.
Control over in Vivo Protein Release Using IDs
Finally, we used our system to locally deliver protein in vivo with temporal control, using mRuby3 as a model. At the outset, we acknowledged that a peptide hydrogel containing l-amino acids will be digested by secreted proteases and phagocytosed by macrophages in subcutaneous tissue, both of which would reduce the overall delivery lifetime of the material. In an effort to hinder these processes, we conducted in vivo experiments using hydrogels consisting of either AcVES3 (also termed l-AcVES3) or its enantiomer d-AcVES3 (Figure S3). We hypothesized that the D-gel, which contains mainly d-amino acids, would be more proteolytically stable than l-AcVES3 and would prolong the protein delivery period. The L- and D-gels form similar fibrous nanostructures with identical mechanical properties (Figures S4 and S5), and we observe the same in vitro release profiles with the mRuby3 analogues (Figure S15). This suggests that release is dictated by the general arrangement of charge displayed on the gel fibers, not by side chain stereochemistry. Therefore, any observed differences in release rates in vivo should be attributed to differences in peptide gel stability.
In the first set of in vivo experiments, mice were injected with l-AcVES3 gel containing either mRuby3 or ID5-mRuby3 in their left and right dorsal flanks, respectively (Figures 8A and S16A). mRuby3 was rapidly released within 1 day, and ID5-mRuby3 was largely retained within the material, indicating the ID was effective in slowing protein release. Analogous experiments comparing release of the mRuby3 analogues using d-AcVES3 hydrogels produced the same results (Figure S16B). Next, mice were injected on their left and right dorsal flanks with ID5-mRuby3 encapsulated in either l-AcVES3 or d-AcVES3 gels, respectively, and release was measured over 28 days (Figures 8B and S16C). Release from the l-AcVES3 gel occurred over 10–14 days, demonstrating that the ID can enable extended release of protein, while release from the d-AcVES3 gel was sustained through 21 days. Four of the five mice had detectable levels of ID5-mRuby3 in the d-AcVES3 gel after 28 days (Figure S16C). These results demonstrate the ID operates within an in vivo setting, and that the stereochemical character of the gel can enhance the delivery lifetime. To investigate whether biodegradation of the hydrogel material was indeed responsible for longer delivery, histology was conducted on the tissue surrounding both left and right flank injection sites. The left flank tissue shows subcutaneous macrophage infiltration (for which the nuclei are stained purple) and the absence of the L-gel (Figure 8C). In contrast, the right flank tissue shows remaining D-gel surrounded by macrophages, presumably still working to clear the material through phagocytosis (Figure 8D). As shown in Figure 8B, after 28 days the right flank tissue was still fluorescent, which signifies the presence of ID5-mRuby3 within the remaining gel. The current limiting factor for in vivo delivery using our ID-system is thus the clearance of AcVES3 gel, not the release kinetics of the ID, and we are currently exploring approaches to further extend the material’s lifetime. Nonetheless, the ID delivery approach has the potential to release protein over the course of 3 weeks from a single administration, a marked improvement over the often-daily parenteral administration typically needed for protein drugs.
Figure 8.

(A) In vivo comparison of mRuby3 and ID5-mRuby3 released from l-AcVES3 hydrogels. (B) Comparison of l-AcVES3 and d-AcVES3 hydrogels for ID5-mRuby3 release. (C) Histological analysis of injection site subcutis for l-AcVES3 and (D) d-AcVES3 for the mouse in panel (B) at 28 days post injection. Engorged macrophages are present throughout the injection site for l-AcVES3 gel with complete clearance of the material, whereas significant amounts of d-AcVES3 gel remained (marked by *) with macrophages at the periphery. Scale bars = 100 μm.
Conclusions
We have developed a modular strategy to control protein release from a biocompatible peptide hydrogel that mitigates gel-based protein denaturation and the need for gel re-engineering for delivering different proteins of interest. A suite of IDs for controlling release was designed for convenient incorporation at either termini of proteins during expression. Individual or combinations of different IDs can be used to finely tune the release profiles for a given protein. Furthermore, our system also allows for the time-staggered delivery of different proteins, which should be beneficial in drug combination regimens. The presence of the IDs did not alter the material properties of the hydrogel nor did they affect the structural or functional properties of the proteins examined in this study. MD simulations afforded molecular-level insight into the behavior of the ID–gel interaction, which helped elucidate the mechanism that dictates sustained protein release. Simulations also suggest opportunities for future ID design to further fine-tune delivery. Most importantly, the ID delivery approach was validated in vivo, with sustained release of protein for over 3 weeks using a hydrogel composed of mainly d-amino acids. This platform represents a promising strategy to deliver negatively charged proteins and complements existing affinity-controlled delivery methods.
Acknowledgments
This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health (1ZIABC011313). We would like to thank Dr. Dominic Esposito for kindly providing the initial unmodified fluorescent protein plasmids and the empty pDonr/pDest cloning vectors, Dr. Mark Walter for kindly providing the initial unmodified pPAL7-IFNα plasmid and the HL116 cells, Dr. Joseph Kalen for providing experimental guidance with the in vivo imaging experiments, and Lisa Riffle for performing the in vivo injections of protein-loaded hydrogels. We would also like to thank Dr. Scott Walsh for guidance with the HiFi assembly protocol, Dr. Ziqiu Wang for assistance with the TEM experiments, and Dr. Rachel Abrams for useful comments during the preparation of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00501.
Author Present Address
▽ (Y.Y.) Tokyo University of Pharmacy and Life Sciences, Tokyo, Japan.
Author Present Address
° (S.T.) Geisel School of Medicine, Dartmouth College, Hanover, NH.
Author Present Address
◆ (C.A.) Charles River Laboratories, Frederick, MD.
The authors declare no competing financial interest.
Supplementary Material
References
- Mitragotri S.; Burke P. A.; Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat. Rev. Drug Discovery 2014, 13 (9), 655–672. 10.1038/nrd4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakulska M. M.; Miersch S.; Shoichet M. S. Designer protein delivery: From natural to engineered affinity-controlled release systems. Science 2016, 351 (6279), aac4750 10.1126/science.aac4750. [DOI] [PubMed] [Google Scholar]
- Yu M.; Wu J.; Shi J.; Farokhzad O. C. Nanotechnology for protein delivery: Overview and perspectives. J. Controlled Release 2016, 240, 24–37. 10.1016/j.jconrel.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiskoot W.; Randolph T. W.; Volkin D. B.; Middaugh C. R.; Schöneich C.; Winter G.; Friess W.; Crommelin D. J. A.; Carpenter J. F. Protein instability and immunogenicity: Roadblocks to clinical application of injectable protein delivery systems for sustained release. J. Pharm. Sci. 2012, 101 (3), 946–954. 10.1002/jps.23018. [DOI] [PubMed] [Google Scholar]
- Vermonden T.; Censi R.; Hennink W. E. Hydrogels for protein delivery. Chem. Rev. 2012, 112 (5), 2853–2888. 10.1021/cr200157d. [DOI] [PubMed] [Google Scholar]
- Martino M. M.; Briquez P. S.; Ranga A.; Lutolf M. P.; Hubbell J. A. Heparin-binding domain of fibrin(ogen) binds growth factors and promotes tissue repair when incorporated within a synthetic matrix. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (12), 4563–4568. 10.1073/pnas.1221602110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulic K.; Shoichet M. S. Tunable growth factor delivery from injectable hydrogels for tissue engineering. J. Am. Chem. Soc. 2012, 134 (2), 882–885. 10.1021/ja210638x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.-C.; Anseth K. S. Controlling affinity binding with peptide-functionalized poly(ethylene glycol) hydrogels. Adv. Funct. Mater. 2009, 19 (14), 2325–2331. 10.1002/adfm.200900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belair D. G.; Khalil A. S.; Miller M. J.; Murphy W. L. Serum-Dependence of Affinity-Mediated VEGF Release from Biomimetic Microspheres. Biomacromolecules 2014, 15 (6), 2038–2048. 10.1021/bm500177c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon O.; Powell C.; Solorio L. D.; Krebs M. D.; Alsberg E. Affinity-based growth factor delivery using biodegradable, photocrosslinked heparin-alginate hydrogels. J. Controlled Release 2011, 154 (3), 258–266. 10.1016/j.jconrel.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakulska M. M.; Vulic K.; Shoichet M. S. Affinity-based release of chondroitinase ABC from a modified methylcellulose hydrogel. J. Controlled Release 2013, 171 (1), 11–16. 10.1016/j.jconrel.2013.06.029. [DOI] [PubMed] [Google Scholar]
- Wylie R. G.; Ahsan S.; Aizawa Y.; Maxwell K. L.; Morshead C. M.; Shoichet M. S. Spatially controlled simultaneous patterning of multiple growth factors in three-dimensional hydrogels. Nat. Mater. 2011, 10 (10), 799–806. 10.1038/nmat3101. [DOI] [PubMed] [Google Scholar]
- Battig M. R.; Soontornworajit B.; Wang Y. Programmable Release of Multiple Protein Drugs from Aptamer-Functionalized Hydrogels via Nucleic Acid Hybridization. J. Am. Chem. Soc. 2012, 134 (30), 12410–12413. 10.1021/ja305238a. [DOI] [PubMed] [Google Scholar]
- Fettis M. M.; Wei Y.; Restuccia A.; Kurian J. J.; Wallet S. M.; Hudalla G. A. Microgels with tunable affinity-controlled protein release via desolvation of self-assembled peptide nanofibers. J. Mater. Chem. B 2016, 4 (18), 3054–3064. 10.1039/C5TB02446C. [DOI] [PubMed] [Google Scholar]
- Wang X.; Shi C.; Zhang L.; Bodman A.; Guo D.; Wang L.; Hall W. A.; Wilkens S.; Luo J. Affinity-controlled protein encapsulation into sub-30 nm telodendrimer nanocarriers by multivalent and synergistic interactions. Biomaterials 2016, 101, 258–271. 10.1016/j.biomaterials.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ageitos J. M.; Pulgar A.; Csaba N.; Garcia-Fuentes M. Study of nanostructured fibroin/dextran matrixes for controlled protein release. Eur. Polym. J. 2019, 114, 197–205. 10.1016/j.eurpolymj.2019.02.028. [DOI] [Google Scholar]
- Koshy S. T.; Zhang D. K. Y.; Grolman J. M.; Stafford A. G.; Mooney D. J. Injectable nanocomposite cryogels for versatile protein drug delivery. Acta Biomater. 2018, 65, 36–43. 10.1016/j.actbio.2017.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B.; Song J.; Zhang S.; Anderson M. A.; Ao Y.; Yang C.-Y.; Deming T. J.; Sofroniew M. V. Sustained local delivery of bioactive nerve growth factor in the central nervous system via tunable diblock copolypeptide hydrogel depots. Biomaterials 2012, 33 (35), 9105–9116. 10.1016/j.biomaterials.2012.08.060. [DOI] [PubMed] [Google Scholar]
- Pakulska M. M.; Elliott Donaghue I.; Obermeyer J. M.; Tuladhar A.; McLaughlin C. K.; Shendruk T. N.; Shoichet M. S. Encapsulation-free controlled release: Electrostatic adsorption eliminates the need for protein encapsulation in PLGA nanoparticles. Sci. Adv. 2016, 2 (5), e1600519 10.1126/sciadv.1600519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu B. B.; Hagerman S. R.; Jamieson K.; Veselinovic J.; O’Neill N.; Holler E.; Ljubimova J. Y.; Hammond P. T. Multilayer films assembled from naturally-derived materials for controlled protein release. Biomacromolecules 2014, 15 (6), 2049–2057. 10.1021/bm5001839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young S.; Wong M.; Tabata Y.; Mikos A. G. Gelatin as a delivery vehicle for the controlled release of bioactive molecules. J. Controlled Release 2005, 109 (1–3), 256–274. 10.1016/j.jconrel.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Holland T. A.; Tabata Y.; Mikos A. G. Dual growth factor delivery from degradable oligo(poly(ethylene glycol) fumarate) hydrogel scaffolds for cartilage tissue engineering. J. Controlled Release 2005, 101 (1), 111–125. 10.1016/j.jconrel.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Freeman I.; Kedem A.; Cohen S. The effect of sulfation of alginate hydrogels on the specific binding and controlled release of heparin-binding proteins. Biomaterials 2008, 29 (22), 3260–3268. 10.1016/j.biomaterials.2008.04.025. [DOI] [PubMed] [Google Scholar]
- Freeman I.; Cohen S. The influence of the sequential delivery of angiogenic factors from affinity-binding alginate scaffolds on vascularization. Biomaterials 2009, 30 (11), 2122–2131. 10.1016/j.biomaterials.2008.12.057. [DOI] [PubMed] [Google Scholar]
- Nagy-Smith K.; Yamada Y.; Schneider J. P. Protein release from highly charged peptide hydrogel networks. J. Mater. Chem. B 2016, 4 (11), 1999–2007. 10.1039/C5TB02137E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco M. C.; Pochan D. J.; Wagner N. J.; Schneider J. P. The effect of protein structure on their controlled release from an injectable peptide hydrogel. Biomaterials 2010, 31 (36), 9527–9534. 10.1016/j.biomaterials.2010.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada A.; Nakamura H. Nature of the charge distribution in proteins. Nature 1981, 293 (5835), 757–758. 10.1038/293757a0. [DOI] [PubMed] [Google Scholar]
- Barlow D. J.; Thornton J. M. Ion-pairs in proteins. J. Mol. Biol. 1983, 168 (4), 867–885. 10.1016/S0022-2836(83)80079-5. [DOI] [PubMed] [Google Scholar]
- Perutz M. Electrostatic effects in proteins. Science 1978, 201 (4362), 1187–1191. 10.1126/science.694508. [DOI] [PubMed] [Google Scholar]
- Horovitz A.; Serrano L.; Avron B.; Bycroft M.; Fersht A. R. Strength and co-operativity of contributions of surface salt bridges to protein stability. J. Mol. Biol. 1990, 216 (4), 1031–1044. 10.1016/S0022-2836(99)80018-7. [DOI] [PubMed] [Google Scholar]
- Battiste J. L.; Mao H.; Rao N. S.; Tan R.; Muhandiram D. R.; Kay L. E.; Frankel A. D.; Williamson J. R. α Helix-RNA Major Groove Recognition in an HIV-1 Rev Peptide-RRE RNA Complex. Science 1996, 273 (5281), 1547–1551. 10.1126/science.273.5281.1547. [DOI] [PubMed] [Google Scholar]
- Schreiber G.; Fersht A. R. Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 1996, 3 (5), 427–431. 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- Ma J.-B.; Ye K.; Patel D. J. Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature 2004, 429 (6989), 318–322. 10.1038/nature02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D. E.; Becktel W. J.; Dahlquist F. W. pH-Induced denaturation of proteins: a single salt bridge contributes 3–5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry 1990, 29 (9), 2403–2408. 10.1021/bi00461a025. [DOI] [PubMed] [Google Scholar]
- Sun D. P.; Sauer U.; Nicholson H.; Matthews B. W. Contributions of engineered surface salt bridges to the stability of T4 lysozyme determined by directed mutagenesis. Biochemistry 1991, 30 (29), 7142–7153. 10.1021/bi00243a015. [DOI] [PubMed] [Google Scholar]
- Tissot A. C.; Vuilleumier S.; Fersht A. R. Importance of two buried salt bridges in the stability and folding pathway of barnase. Biochemistry 1996, 35 (21), 6786–6794. 10.1021/bi952930e. [DOI] [PubMed] [Google Scholar]
- Donald J. E.; Kulp D. W.; DeGrado W. F. Salt bridges: Geometrically specific, designable interactions. Proteins: Struct., Funct., Genet. 2011, 79 (3), 898–915. 10.1002/prot.22927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J. P.; Pochan D. J.; Ozbas B.; Rajagopal K.; Pakstis L.; Kretsinger J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002, 124 (50), 15030–15037. 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- Haines-Butterick L.; Rajagopal K.; Branco M.; Salick D.; Rughani R.; Pilarz M.; Lamm M. S.; Pochan D. J.; Schneider J. P. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (19), 7791–7796. 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giano M. C.; Pochan D. J.; Schneider J. P. Controlled biodegradation of self-assembling beta-hairpin peptide hydrogels by proteolysis with matrix metalloproteinase-13. Biomaterials 2011, 32 (27), 6471–6477. 10.1016/j.biomaterials.2011.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga A. S.; Sinthuvanich C.; Gaspar D.; Franquelim H. G.; Castanho M.; Schneider J. P. Arginine-rich self-assembling peptides as potent antibacterial gels. Biomaterials 2012, 33 (35), 8907–8916. 10.1016/j.biomaterials.2012.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micklitsch C. M.; Medina S. H.; Yucel T.; Nagy-Smith K. J.; Pochan D. J.; Schneider J. P. Influence of hydrophobic face amino acids on the hydrogelation of beta-hairpin peptide amphiphiles. Macromolecules 2015, 48 (5), 1281–1288. 10.1021/ma5024796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonmez C.; Nagy K. J.; Schneider J. P. Design of self-assembling peptide hydrogelators amenable to bacterial expression. Biomaterials 2015, 37, 62–72. 10.1016/j.biomaterials.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. J.; Brat G. A.; Medina S. H.; Tong D. D.; Huang Y.; Grahammer J.; Furtmuller G. J.; Oh B. C.; Nagy-Smith K. J.; Walczak P.; Brandacher G.; Schneider J. P. A multiphase transitioning peptide hydrogel for suturing ultrasmall vessels. Nat. Nanotechnol. 2016, 11 (1), 95–102. 10.1038/nnano.2015.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinthuvanich C.; Nagy-Smith K. J.; Walsh S. T. R.; Schneider J. P. Triggered formation of anionic hydrogels from self-assembling acidic peptide amphiphiles. Macromolecules 2017, 50 (15), 5643–5651. 10.1021/acs.macromol.7b01056. [DOI] [Google Scholar]
- Shi J.; Fichman G.; Schneider J. P. Enzymatic control of the conformational landscape of self-assembling peptides. Angew. Chem., Int. Ed. 2018, 57 (35), 11188–11192. 10.1002/anie.201803983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy-Smith K.; Moore E.; Schneider J.; Tycko R. Molecular structure of monomorphic peptide fibrils within a kinetically trapped hydrogel network. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (32), 9816–9821. 10.1073/pnas.1509313112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez E. A.; Campbell R. E.; Lin J. Y.; Lin M. Z.; Miyawaki A.; Palmer A. E.; Shu X.; Zhang J.; Tsien R. Y. The growing and glowing toolbox of fluorescent and photoactive proteins. Trends Biochem. Sci. 2017, 42 (2), 111–129. 10.1016/j.tibs.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson T. P.; Peters M. C.; Ennett A. B.; Mooney D. J. Polymeric system for dual growth factor delivery. Nat. Biotechnol. 2001, 19, 1029–1034. 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- Simmons C. A.; Alsberg E.; Hsiong S.; Kim W. J.; Mooney D. J. Dual growth factor delivery and controlled scaffold degradation enhance in vivo bone formation by transplanted bone marrow stromal cells. Bone 2004, 35 (2), 562–569. 10.1016/j.bone.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Kumar V. A.; Taylor N. L.; Shi S.; Wickremasinghe N. C.; D’Souza R. N.; Hartgerink J. D. Self-assembling multidomain peptides tailor biological responses through biphasic release. Biomaterials 2015, 52, 71–78. 10.1016/j.biomaterials.2015.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder P.; Baxa U.; Walsh S. T. R.; Schneider J. P. Design of a multicompartment hydrogel that facilitates time-resolved delivery of combination therapy and synergized killing of glioblastoma. Angew. Chem., Int. Ed. 2018, 57 (46), 15040–15044. 10.1002/anie.201806483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajar B. T.; Wang E. S.; Lam A. J.; Kim B. B.; Jacobs C. L.; Howe E. S.; Davidson M. W.; Lin M. Z.; Chu J. Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci. Rep. 2016, 6, 20889. 10.1038/srep20889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Navajas J. M.; Lee J.; David M.; Raz E. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 2012, 12 (2), 125–135. 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller L.; Aigner P.; Stoiber D. Type I interferons and natural killer cell regulation in cancer. Front. Immunol. 2017, 8 (304), 304. 10.3389/fimmu.2017.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli G.; Scagnolari C.; Moschella F.; Proietti E. Twenty-five years of type I interferon-based treatment: a critical analysis of its therapeutic use. Cytokine Growth Factor Rev. 2015, 26 (2), 121–31. 10.1016/j.cytogfr.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruganti S.; Accavitti-Loper M. A.; Walter M. R. Production and characterization of thirteen human type-I interferon-α subtypes. Protein Expression Purif. 2014, 103, 75–83. 10.1016/j.pep.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uze G.; Di Marco S.; Mouchel-Vielh E.; Monneron D.; Bandu M. T.; Horisberger M. A.; Dorques A.; Lutfalla G.; Mogensen K. E. Domains of interaction between alpha interferon and its receptor components. J. Mol. Biol. 1994, 243 (2), 245–257. 10.1006/jmbi.1994.1651. [DOI] [PubMed] [Google Scholar]
- Aqvist J.; Medina C.; Samuelsson J. E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng., Des. Sel. 1994, 7 (3), 385–91. 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- Panel N.; Sun Y. J.; Fuentes E. J.; Simonson T. A simple PB/LIE free energy function accurately predicts the peptide binding specificity of the Tiam1 PDZ domain. Front. Mol. Biosci. 2017, 4 (65), 65. 10.3389/fmolb.2017.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrauskas V.; Maximowitsch E.; Matulis D. Thermodynamics of ion pair formations between charged poly(amino acid)s. J. Phys. Chem. B 2015, 119 (37), 12164–12171. 10.1021/acs.jpcb.5b05767. [DOI] [PubMed] [Google Scholar]
- Robison A. D.; Sun S.; Poyton M. F.; Johnson G. A.; Pellois J. P.; Jungwirth P.; Vazdar M.; Cremer P. S. Polyarginine interacts more strongly and cooperatively than polylysine with phospholipid bilayers. J. Phys. Chem. B 2016, 120 (35), 9287–9296. 10.1021/acs.jpcb.6b05604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.