

Abstract
Objective
Higher blood malondialdehyde (biomarker of lipid peroxidation) levels in the first hours of traumatic brain injury (TBI) have been found in patients with a worst prognosis. The objective of this study was to determine whether serum malondialdehyde levels during the first week of severe TBI could be used as mortality biomarkers. This was a multicenter, prospective and observational study performed in six Spanish Intensive Care Units. We included patients with severe TBI (defined as Glasgow Coma Scale < 9), and with Injury Severity Score in non-cranial aspects < 9. We determined serum malondialdehyde concentrations at days 1, 4 and 8 of TBI. We stablished 30-day mortality as the end-point study.
Results
We found that serum malondialdehyde concentrations at days 1 (p < 0.001), 4 (p < 0.001), and 8 (p < 0.001) of TBI were higher in non-survivor (n = 34) than in survivor (n = 90) patients. We found an area under curve of serum malondialdehyde concentrations at days 1, 4, and 8 of TBI to predict 30-day mortality of 77% (p < 0.001), 87% (p < 0.001) and 84% (p < 0.001) respectively. Thus, the new and most relevant findings of our study were serum malondialdehyde levels during the first week of TBI could be used as mortality biomarkers.
Keywords: Malondialdehyde, Brain trauma, Patients, Mortality, Injury
Introduction
Traumatic brain injury (TBI) leads to many deaths, and also to many disabilities and consumption of resources [1]. In TBI could appears a secondary brain injury during the following hours or days after TBI due to the neuroinflammatory response and the generation of free radicals [2–5].
Under physiologic conditions, the production of reactive oxygen species (ROS) in brain tissue is balanced by the action of antioxidant agents. However, in TBI appears an increase in the production of ROS and this leads to lipid peroxidation. The degradation of cellular membrane phospholipids during this lipid peroxidation leads to the production of Malondialdehyde, which is an end-product of lipid peroxidation [6, 7], comes to extracellular space and afterwards to the blood; and its determination has been used to estimate lipid oxidation [6, 7].
Previous studies have reported higher blood levels of malondialdehyde in TBI patients than in controls subjects [8–15]. In addition, higher blood malondialdehyde levels in the first hours of TBI in patients with a worst prognosis have been found [13–16]. Thus, the objective of this study was to determine serum malondialdehyde levels during the first week of a severe TBI and to analyze whether those levels during the first week are difference between survivor and non-survivor patients, and whether could be used as biomarkers of mortality.
Main text
Methods
Design and subjects
We performed this prospective and observational in 6 Intensive Care Units of Spain. The Institutional Ethic Review Board of each hospital approved the study: Hospital Universitario Nuestra Señora de Candelaria of Santa Cruz de Tenerife, Hospital Clínico Universitario of Valencia, Hospital General of La Palma, Hospital Insular of Las Palmas de Gran Canaria, Hospital Universitario de Canarias of La Laguna, and Hospital Universitario Dr. Negrín of Las Palmas de Gran Canaria. Besides, legal guardians of patients signed the informed consent form to study participation.
Patients with severe TBI, defined as Glasgow Coma Scale (GCS) [17] < 9 points, and with Injury Severity Score (ISS) [18] in non-cranial aspects < 9 points were included. We excluded patients with malignant or inflammatory disease, age < 18 years, pregnancy or comfort measures only.
In a previous study by our team were determined serum malondialdehyde concentrations in the day of TBI in some of those patients [13]. In our current research, we determine serum concentrations of malondialdehyde in the day 1, 4 and 8 of TBI in 118 patients.
Clinical and demographic variables
We recollected Acute Physiology and Chronic Health Evaluation II (APACHE II) score [19] and brain lesions by computer tomography (CT) Marshall classification [20], and other clinical and demographic variables as in our previous study [13]. We established 30-day mortality as the end-point of the study.
Collection of blood samples and determination of serum malondialdehyde levels
Collection of blood samples were obtained on days 1, 4 and 8 of TBI and were processed as in our previous study [13]. Determination of serum malondialdehyde levels was carried out with thiobarbituric acid-reactive substance (TBARS) method of Kikugawa et al. [21] with some modifications as in our previous study [13].
Statistical methods
We carried out statistical analyses similarly as in our previous study [13], using Wilcoxon–Mann–Whitney test and Chi square test to compare variables between patient groups, multiple logistic regression analysis to determine the association between serum malondialdehyde levels and 30 day-mortality, and Spearman’ coefficient to determine the association between continuous variables. In addition, to determine the capacity of serum malondialdehyde levels at day 1, 4 and 8 of TBI for 30-day mortality, receiver operating characteristic (ROC) analyses were performed.
Results
In Table 1 appears demographic and clinical variables of non-surviving (n = 34) and surviving patients (n = 90). Non-surviving TBI patients showed higher female rate, APACHE-II score and age, and lower GCS than survivors. Besides, CT findings were different in surviving and non-surviving patients. We found that non-survivor in respect to survivor patients showed higher serum malondialdehyde concentrations at days 1 (p < 0.001), 4 (p < 0.001), and 8 (p < 0.001) of TBI (Fig. 1).
Table 1.
Clinical and biochemical characteristics of 30-day survivor and non-survivor trauma brain injury patients
| Non-survivors (n = 34) | Survivors (n = 90) | p Value | |
|---|---|---|---|
| Computer tomography classification—n (%) | 0.01 | ||
| Type 1 | 0 | 0 | |
| Type 2 | 5 (14.7) | 25 (27.8) | |
| Type 3 | 6 (17.6) | 15 (16.7) | |
| Type 4 | 9 (26.5) | 13 (14.4) | |
| Type 5 | 6 (17.6) | 32 (35.6) | |
| Type 6 | 8 (23.5) | 5 (5.6) | |
| CT with high risk of death (types 3, 4, 6)—n (%) | 23 (67.6) | 33 (36.7) | 0.002 |
| Gender female—n (%) | 13 (38.2) | 15 (16.7) | 0.02 |
| Age (years)—median (p 25–75) | 65 (55–75) | 46 (28–62) | < 0.001 |
| Temperature (°C)—median (p 25–75) | 36.0 (35.0–37.0) | 37.0 (36.0–37.3) | 0.07 |
| Creatinine (mg/dl)—median (p 25–75) | 0.80 (0.70–1.10) | 0.80 (0.70–1.00) | 0.50 |
| Glycemia (g/dL)—median (p 25–75) | 160 (125–191) | 139 (121–167) | 0.11 |
| Sodium (mEq/L)—median (p 25–75) | 141 (136–147) | 140 (138–143) | 0.41 |
| Bilirubin (mg/dl)—median (p 25–75) | 0.70 (0.53–1.05) | 0.60 (0.40–0.80) | 0.06 |
| Lactic acid (mmol/L) median (p 25–75) | 2.30 (1.25–4.58) | 1.75 (1.10–2.50) | 0.08 |
| PaO2 (mmHg)—median (p 25–75) | 142 (97–195) | 148 (110–242) | 0.45 |
| PaO2/FI02 ratio—median (p 25–75) | 294 (167–395) | 336 (246–400) | 0.11 |
| Leukocytes—median*103/mm3 (p 25–75) | 14.9 (9.7–21.6) | 13.9 (10.1–19.0) | 0.47 |
| Fibrinogen (mg/dl)—median (p 25–75) | 348 (300–475) | 371 (286–471) | 0.70 |
| aPTT (seconds)—median (p 25–75) | 29 (25–37) | 28 (25–31) | 0.25 |
| INR—median (p 25–75) | 1.12 (1.03–1.48) | 1.11 (1.00–1.24) | 0.19 |
| Platelets—median*103/mm3 (p 25–75) | 172 (125–232) | 182 (135–238) | 0.49 |
| Hemoglobin (g/dL)—median (p 25–75) | 11.9 (10.0–13.7) | 11.2 (10.0–13.0) | 0.73 |
| ISS—median (ppe 25–75) | 25 (25–25) | 25 (25–34) | 0.28 |
| Glasgow Coma Scale score—median (p 25–75) | 4 (3–7) | 7 (5–8) | < 0.001 |
| APACHE–II score—median (p 25–75) | 25 (23–28) | 18 (14–22) | < 0.001 |
| ICP (mmHg)—median (p 25–75) | 25 (11–30) | 15 (14–20) | 0.36 |
| CPP (mmHg)—median (p 25–75) | 61 (52–70) | 68 (57–70) | 0.60 |
| Malondialdehyde (nmol/mL)—median (p 25–75) | 2.03 (1.36–4.12) | 1.35 (1.05–1.77) | < 0.001 |
PaO2 pressure of arterial oxygen/fraction inspired oxygen, FIO2 pressure of arterial oxygen/fraction inspired oxygen, aPTT activated partial thromboplastin time, INR international normalized ratio, ISS Injury Severity Score, GCS Glasgow Coma Scale, APACHE II Acute Physiology and Chronic Health Evaluation, ICP intracranial pressure, CPP cerebral perfusion pressure, p 25–75 percentile 25th–75th
Fig. 1.
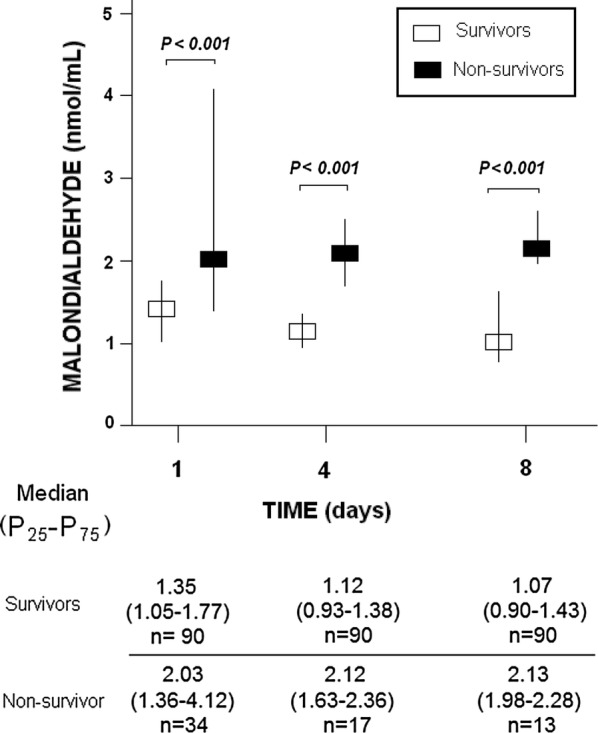
Serum malondialdehyde levels at day 1, 4 and 8 of trauma brain injury in 30-day survivor and non-survivor patients
We found in ROC curve analyses an area under curve of serum malondialdehyde concentrations at days 1, 4, and 8 of TBI to predict 30-day mortality of 77% (p < 0.001), 87% (p < 0.001) and 84% (p < 0.001) respectively (Table 2).
Table 2.
Receiver operation characteristic analysis using serum malondialdehyde levels at day 1, 4 and 8 of trauma brain injury as predictor of mortality at 30 days
| Day 1 | Day 4 | Day 8 | |
|---|---|---|---|
| Cut-off of malondialdehyde (mmol/mL) | > 1.96 | > 1.83 | > 1.83 |
| AUC, 95% CI, and p value |
0.77 (0.68–0.84) p < 0.001 |
0.87 (0.79–0.93) p < 0.001 |
0.84 (0.75–0.90) p < 0.001 |
| Sensitivity and 95% CI | 56% (38%–73%) | 77% (50%–93%) | 85% (55%–98%) |
| Specificity and 95% CI | 84% (75%–91%) | 92% (85%–97%) | 86% (77%–92%) |
| Positive likelihood ratio and 95% CI | 3.6 (2.0–6.3) | 9.8 (4.6–21.8) | 5.9 (3.4–10.2) |
| Negative likelihood ratio and 95% CI | 0.5 (0.4–0.8) | 0.4 (0.2–0.7) | 0.2 (0.1–0.6) |
| Positive predicted value and 95% CI | 58% (44%–71%) | 65% (44%–81%) | 46% (33%–60%) |
| Negative predicted value and 95% CI | 84% (78%–88%) | 93% (88%–96%) | 98% (92%–99%) |
AUC area under curve, CI confidence intervals
We found in multiple logistic regression analysis an association between serum malondialdehyde concentrations and mortality controlling for sex, CT, age and CGS (OR = 3.91; 95% CI 1.61–9.50; p = 0.003). CT findings were including in the regression analysis as CT findings with low risk of death (including CT types 2 and 5) and with high risk of death (including CT types 3, 4 and 6). That classification was based in the fact that we found a mortality rate of 16.7% (5/30) in patients with CT type 2, 28.6% (6/21) with type 3, 40.9% (9/22) with type 4, 15.8% (6/38) with type 5 and 61.5% (8/13) with type 6.
We found an association of serum melatonin levels with GCS (rho = − 0.21; p = 0.02), but not with age (rho = 0.11; p = 0.24). In addition, we found higher serum melatonin levels in female than in male (p = 0.01).
Discussion
The new and most relevant findings of our study were that non-survivor TBI patients showed higher serum malondialdehyde levels during the first week of TBI that survivors, and that those levels during the first week of TBI could be used as mortality biomarkers.
Previously there were reported higher circulating levels of malondialdehyde in TBI patients than in controls subjects [8–15], and higher malondialdehyde levels on the first hours of TBI in patients with worst prognosis [13–16]. Higher malondialdehyde levels in erythrocytes obtained within 24 h of TBI in non-survivor than in survivor TBI patients have been found in a study by Nayak et al. [14]. Higher malondialdehyde levels in cerebrospinal fluid obtained by lumbar puncture during the first day of TBI in non-survivor than in survivor TBI patients have been found in the study by Kasprzak et al. [15]. Concentrations of malondialdehyde in blood from jugular bulb blood samples and from peripheral venous blood samples were obtained from 30 TBI patients at admission, and at 6, 12, 24 and 48 h after TBI in a study by Paolin et al. [16]; and malondialdehyde levels in jugular bulb blood samples at 6 and 12 h after TBI were higher in patients with poorer than in patients with a good 6-month neurological outcome [16]. Higher serum malondialdehyde levels on the first day of TBI obtained from a central venous catheter in non-survivor than in survivor TBI patients have been found in another study by our team [13]. Thus, the finding of our current study that serum malondialdehyde levels obtained from a central venous catheter during the first week of a severe TBI were higher in survivor than in non-survivor patients is a novel finding.
Another interesting finding of our study was the significant negative association between serum melatonin levels and GCS; thus, patients with higher clinical impairment showed higher oxidative damage.
The antioxidant administration of different agents, such as melatonin [24, 25] or memantine [26], has reduced the increase of brain tissue malondialdehyde levels in animal models of TBI. In addition, the administration of different antioxidant, such as alpha-phenyl-tert-butyl-nitrone or melatonin, has reduced the neurological deficit in animal models of TBI [27]. In a randomized clinical trial of TBI patients of small sample size (18 patients with and 14 without the administration of amantadine), the patient group receiving amantadine showed lower serum malondialdehyde levels and mortality rate (6% vs 43%) [28].
We believed that those higher serum malondialdehyde levels during the first week after TBI in non-survivor patients found in our study reflects a higher ROS production and a higher lipid peroxidation in non-survivor TBI patients. That higher production of ROS production in non-surviving TBI patients could play a rol on microvascular regulation loss and vasogenic edema formation, cellular dysfunction, and patient death [22, 23]. Thus, the administration of antioxidant agents could be a therapeutic way to explore in those patients.
Conclusions
The new and most relevant findings of our study were that non-survivor TBI patients showed higher serum malondialdehyde levels during the first week of TBI that survivors, and that those levels during the first week of TBI could be used as mortality biomarkers.
Limitations
Some limitations should be recognized in our study. First, the determination of other compounds of oxidant and antioxidant states was not made. Second, we have only determined malondialdehyde levels in blood from a central venous catheter, and not in cerebrospinal fluid and in blood from jugular bulb. However, in some studies was found a worst prognosis in TBI patients with high levels of malondialdehyde on the first hours after TBI in cerebrospinal fluid [15] and in blood from jugular bulb [16].
Acknowledgements
This study was supported by a Grant (OA18/011) from Fundación DISA a la Investigación Médica 2017 (Santa Cruz de Tenerife. Spain). Funding did not influence in the study design, the collection, analysis, and interpretation of data, the manuscript writing, and the decision to submit it for publication.
Abbreviations
- ICU
Intensive Care Unit
- PaO2
pressure of arterial oxygen/fraction inspired oxygen
- FIO2
pressure of arterial oxygen/fraction inspired oxygen
- INR
international normalized ratio
- aPTT
activated partial thromboplastin time
- ICP
intracranial pressure
- CPP
cerebral perfusion pressure
- ISS
Injury Severity Score
- GCS
Glasgow Coma Scale
- SOFA
Sepsis-related Organ Failure Assessment
- APACHE
Acute Physiology and Chronic Health Evaluation
Authors’ contributions
LL conceived, designed and coordinated the study, participated in acquisition and interpretation of data, and drafted the manuscript. MMM, LR, MA, JJC, JSV, and VGM participated in acquisition of data. PAG participated in blood determination levels. AJ participated in the interpretation of data. All authors revised the manuscript critically for important intellectual content, made the final approval of the version to be published, and were agree to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Funding
This study was supported by a Grant (OA18/011) from Fundación DISA a la Investigación Médica 2017 (Santa Cruz de Tenerife. Spain). Funding did not influence in the study design, the collection, analysis, and interpretation of data, the manuscript writing, and the decision to submit it for publication.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
We performed this prospective and observational in 6 Intensive Care Units of Spain. The Institutional Ethic Review Board of each hospital approved the study: Hospital Universitario Nuestra Señora de Candelaria of Santa Cruz de Tenerife, Hospital Clínico Universitario of Valencia, Hospital General of La Palma, Hospital Insular of Las Palmas de Gran Canaria, Hospital Universitario de Canarias of La Laguna, and Hospital Universitario Dr. Negrín of Las Palmas de Gran Canaria. Besides, legal guardians of patients signed the informed consent form to study participation.
Consent to publish
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Leonardo Lorente, Email: lorentemartin@msn.com.
María M. Martín, Email: mar.martinvelasco@gmail.com
Pedro Abreu-González, Email: pabreu@ull.es.
Luis Ramos, Email: lramosgomez@gmail.com.
Juan J. Cáceres, Email: juanjose.caceresagra@gobiernodecanarias.org
Mónica Argueso, Email: moni_begasa@hotmail.com.
Jordi Solé-Violán, Email: jsolvio@gobiernodecanarias.org.
Alejandro Jiménez, Email: ajimenezsosa@gmail.com.
Victor García-Marín, Email: vicgarmar666@gmail.com.
References
- 1.Brain Trauma Foundation. American Association of Neurological Surgeons. Congress of Neurological Surgeons Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2007;24(Suppl 1):S1–S106. doi: 10.1089/neu.2007.9999. [DOI] [PubMed] [Google Scholar]
- 2.Ikeda Y, Long DM. The molecular basis of brain injury and brain edema: the role of oxygen free radicals. Neurosurgery. 1990;27:1–11. doi: 10.1227/00006123-199007000-00001. [DOI] [PubMed] [Google Scholar]
- 3.McCall JM, Braughler JM, Hall ED. Lipid peroxidation and the role of oxygen radicals in CNS injury. Acta Anaesthesiol Belg. 1987;38:373–379. [PubMed] [Google Scholar]
- 4.Warner DS, Sheng H, Batinić-Haberle I. Oxidants, antioxidants and the ischemic brain. J Exp Biol. 2004;207:3221–3231. doi: 10.1242/jeb.01022. [DOI] [PubMed] [Google Scholar]
- 5.Hall ED. Lipid antioxidants in acute central nervous system injury. Ann Emerg Med. 1993;22:1022–1027. doi: 10.1016/S0196-0644(05)82745-3. [DOI] [PubMed] [Google Scholar]
- 6.Draper HH, Hadley M. Malondialdehyde determination as index of lipid peroxidation. Methods Enzymol. 1990;186:421–431. doi: 10.1016/0076-6879(90)86135-I. [DOI] [PubMed] [Google Scholar]
- 7.Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A. Biomarkers of oxidative damage in human disease. Clin Chem. 2006;52:601–623. doi: 10.1373/clinchem.2005.061408. [DOI] [PubMed] [Google Scholar]
- 8.Hu S, Zheng L, Chen B, Xie J, Yang C. The role of the leukocytes in pathogenesis of secondary brain injury. Human Yi Ke Da Xue Xue Bao. 1999;24:56–58. [PubMed] [Google Scholar]
- 9.Hohl A, Gullo-Jda S, Silva CC, Bertotti MM, Felisberto F, Nunes JC, de Souza B, Petronilho F, Soares FM, Prediger RD, Dal-Pizzol F, Linhares MN, Walz R. Plasma levels of oxidative stress biomarkers and hospital mortality in severe head injury: a multivariate analysis. J Crit Care. 2012;27(523):e11–e19. doi: 10.1016/j.jcrc.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Cristofori L, Tavazzi B, Gambin R, Vagnozzi R, Vivenza C, Amorini AM, Di Pierro D, Fazzina G, Lazzarino G. Early onset of lipid peroxidation after human traumatic brain injury: a fatal limitation for the free radical scavenger pharmacological therapy? J Investig Med. 2001;49:450–458. doi: 10.2310/6650.2001.33790. [DOI] [PubMed] [Google Scholar]
- 11.Wang HC, Lin YJ, Shih FY, Chang HW, Su YJ, Cheng BC, Su CM, Tsai NW, Chang YT, Kwan AL, Lu CH. The role of serial oxidative stress levels in acute traumatic brain injury and as predictors of outcome. World Neurosurg. 2016;87:463–470. doi: 10.1016/j.wneu.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 12.Lin WM, Chen MH, Wang HC, Lu CH, Chen PC, Chen HL, Tsai NW, Su YJ, Li SH, Kung CT, Chiu TM, Weng HH, Lin WC. Association between peripheral oxidative stress and white matter damage in acute traumatic brain injury. Biomed Res Int. 2014;2014:340936. doi: 10.1155/2014/340936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lorente L, Martín MM, Abreu-González P, Ramos L, Argueso M, Cáceres JJ, Solé-Violán J, Lorenzo JM, Molina I, Jiménez A. Association between serum malondialdehyde levels and mortality in patients with severe brain trauma injury. J Neurotrauma. 2015;32:1–6. doi: 10.1089/neu.2014.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nayak C, Nayak D, Bhat S, Raja A, Rao A. Relationship between neurological outcome and early oxidative changes in erythrocytes in head injury patients. Clin Chem Lab Med. 2007;45:629–633. doi: 10.1515/CCLM.2007.123. [DOI] [PubMed] [Google Scholar]
- 15.Kasprzak HA, Woźniak A, Drewa G, Woźniak B. Enhanced lipid peroxidation processes in patients after brain contusion. J Neurotrauma. 2001;18:793–797. doi: 10.1089/089771501316919157. [DOI] [PubMed] [Google Scholar]
- 16.Paolin A, Nardin L, Gaetani P, Rodriguez Y, Baena R, Pansarasa O, Marzatico F. Oxidative damage after severe head injury and its relationship to neurological outcome. Neurosurgery. 2002;51:949–954. doi: 10.1097/00006123-200210000-00018. [DOI] [PubMed] [Google Scholar]
- 17.Teasdale G, Jennett B. Assessement of coma and impaired conciousness. A practical scale. Lancet. 1974;2:81–88. doi: 10.1016/s0140-6736(74)91639-0. [DOI] [PubMed] [Google Scholar]
- 18.Baker SP, O’Neill B, Haddon W, Jr, Long WB. The injury severity score: a method for describing patients with multiple injuries and evaluating emergency care. J Trauma. 1974;14:187–196. doi: 10.1097/00005373-197403000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13:818–829. doi: 10.1097/00003246-198510000-00009. [DOI] [PubMed] [Google Scholar]
- 20.Marshall LF, Marshall SB, Klauber MR, Van Berkum Clark M, Eisenberg H, Jane JA, Luerssen TG, Marmarou A, Foulkes MA. The diagnosis of head injury requires a classification based on computed axial tomography. J Neurotrauma. 1992;9(Suppl 1):S287–S292. [PubMed] [Google Scholar]
- 21.Kikugawa K, Kojima T, Yamaki S, Kosugi H. Interpretation of the thiobarbituric acid reactivity of rat liver and brain homogenates in the presence of ferric ion and ethylediaminotetraacetic acid. Anal Biochem. 1992;202:249–255. doi: 10.1016/0003-2697(92)90102-D. [DOI] [PubMed] [Google Scholar]
- 22.Maldonado MD, Murillo-Cabezas F, Terron MP, Flores LJ, Tan DX, Manchester LC, Reiter RJ. The potential of melatonin in reducing morbidity-mortality after craniocerebral trauma. J Pineal Res. 2007;42:1–11. doi: 10.1111/j.1600-079X.2006.00376.x. [DOI] [PubMed] [Google Scholar]
- 23.Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerman M, Cirak B, Ozguner MF, Dagtekin A, Sutcu R, Altuntas I, Delibas N. Does melatonin protect or treat brain damage from traumatic oxidative stress? Exp Brain Res. 2005;163:406–410. doi: 10.1007/s00221-005-2338-2. [DOI] [PubMed] [Google Scholar]
- 25.Horakova L, Onrejickova O, Barchrrata K, Vajdova M. Preventive effect of several antioxidants after oxidative stress on rat brain homogenates. Gen Physiol Biophys. 2000;19:195–205. [PubMed] [Google Scholar]
- 26.Ozsüer H, Görgülü A, Kiriş T, Cobanoğlu S. The effects of memantine on lipid peroxidation following closed-head trauma in rats. Neurosurg Rev. 2005;28:143–147. doi: 10.1007/s10143-004-0374-1. [DOI] [PubMed] [Google Scholar]
- 27.Messenge C, Margail I, Verrechia C, Allix M. Protective effect of melatonin in a model of traumatic brain injury in mice. J Pineal Res. 1998;25:41–46. doi: 10.1111/j.1600-079X.1998.tb00384.x. [DOI] [PubMed] [Google Scholar]
- 28.Saniova B, Drobny M, Lehotsky J, Sulaj M, Schudichova J. Biochemical and clinical improvement of cytotoxic state by amantadine sulphate. Cell Mol Neurobiol. 2006;26:1475–1482. doi: 10.1007/s10571-006-9033-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.