

Abstract
The contribution of mosaic alterations to tumors of the nervous system and to non-malignant neurological diseases has been unmasked thanks to the development of Next Generation Sequencing (NGS) technologies. We report here the case of a young patient without any remarkable familial medical history who was first referred at 7 years of age, for an autism spectrum disorder (ASD) of Asperger type, not associated with macrocephaly. The patient subsequently presented at 10 years of age with multiple nodular lesions located within the trigeminal, facial and acoustic nerve ganglia and at the L3 level. Histological examination of this latter lesion revealed a glioneuronal hamartoma, exhibiting heterogeneous PTEN immunoreactivity, astrocyte and endothelial cell nuclei expressing PTEN, but not ganglion cells. NGS performed on the hamartoma allowed the detection of a PTEN pathogenic variant in 30% of the reads. The presence of this variant in the DNA extracted from blood and buccal swabs in 3.5 and 11% of the NGS reads, respectively, confirmed the mosaic state of the PTEN variant. The anatomical distribution of the lesions suggests that the mutational event affecting PTEN occurred in neural crest progenitors, thus explaining the absence of macrocephaly. This report shows that mosaic alteration of PTEN may result in multiple central and peripheral nervous system hamartomas and that the presence of such alteration should be considered in patients with multiple nervous system masses, even in the absence of cardinal features of PTEN hamartoma tumor syndrome, especially macrocephaly.
Keywords: PTEN, Mosaics, Hamartomas, Central nervous system, Neural crest derivatives
Introduction
Since 2009, the development of Next Generation Sequencing (NGS) technologies allowing whole exome and genome sequencing has unmasked the mutability of the human genome with an estimate of 1,58 coding variation occurring de novo per exome, at the pre-zygotic level [1]. The rate of de novo variations occurring at the post-zygotic level, resulting in mosaicism, and their contribution to human diseases are probably underestimated [2]. Mosaic causal alterations in central nervous system (CNS) tumors have been described in several genes such as NF2 in meningiomas and ependymomas [3], and TP53 in choroid plexus tumors [4, 5] and in a case of neuroblastoma [6]. Several recent studies have also pointed to the role of somatic mutations in non-malignant neurological diseases of childhood, such as malformations of cortical development, epilepsy or autism spectrum disorders [7]. Mosaic alterations of PTEN, corresponding either to nucleotide variations, genomic rearrangements or 10q23 microdeletions encompassing the PTEN locus, have already been reported in several patients exhibiting syndromic features pathognomonic of PTEN hamartoma tumor syndrome (PHTS), such as macrocephaly, Lhermitte-Duclos Disease, mucosal papillomatous lesions, hamartomatous polyposis and thyroid goiter [8–11]. In one patient, the father of an index case with PHTS, clinical expression was restricted to macrocephaly [8]. Germline mosaic alterations of the PTEN locus, associated in trans with inherited PTEN variants, have also been reported in a distinct clinical presentation corresponding to segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome due to PTEN nullizygosity [12, 13] and for review see ref. [14]. We report herein the case of a young patient who presented with several brain and spinal cord lesions, resulting from a mosaic PTEN alteration restricted to discrete neural subpopulations.
Case presentation
The patient was an 11-year-old male, without any remarkable familial medical history. He was born at term with normal growth parameters (3100 g (15.8th centile), 53 cm (91st centile), OFC (33 cm 6th centile). He was able to walk unaided at 16 months of age. Physiotherapy was performed for slight hypotonia and moderate global coordination disorder. He developed normal language skills but presented with a mild social communication disorder and a learning disability without any cognitive impairment. He was first referred to the department of genetics at 7 years of age, for an autism spectrum disorder (ASD) of Asperger type, according to the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM-IV). Physical examination at this age was normal; growth parameters were in the normal range and, more notably, there was no macrocephaly (+1SD). Skin examination revealed a small congenital retro-auricular hamartoma. Blood karyotype was normal and screening for fragile X syndrome and metabolic disorders was negative. At ten years of age, the patient complained of headaches and presented painful limping and lower limb asymmetry.
Magnetic resonance imaging (MRI) revealed intracranial extra-cerebral and spinal intra-dural masses, T1-hypointense, T2-hyperintense with contrast enhancement after gadolinium injection. These nodular lesions were located within the ganglion of the trigeminal, facial and acoustic nerves (Fig. 1a and b). An extramedullary intradural nodule with similar imaging characteristics was detected at the L3 level (Fig. 1c). A diagnosis of neurofibromatosis type II and schwannoma predisposition syndrome was initially considered but screening of NF2, INI1, SMARCB1, and LZTR1 on the patient’s blood using NGS did not reveal any detectable germline alteration. The L3 lesion was surgically removed. Six months post-operatively, control MRI showed stable volumes of the cranial lesions. It also revealed a cerebellar cortical lesion consisting in “focal micropolygyria” of the right hemisphere (Fig. 1d), differing from Lhermitte-Duclos disease in which the cerebellar cortex appears broadened on MRI.
Fig. 1.
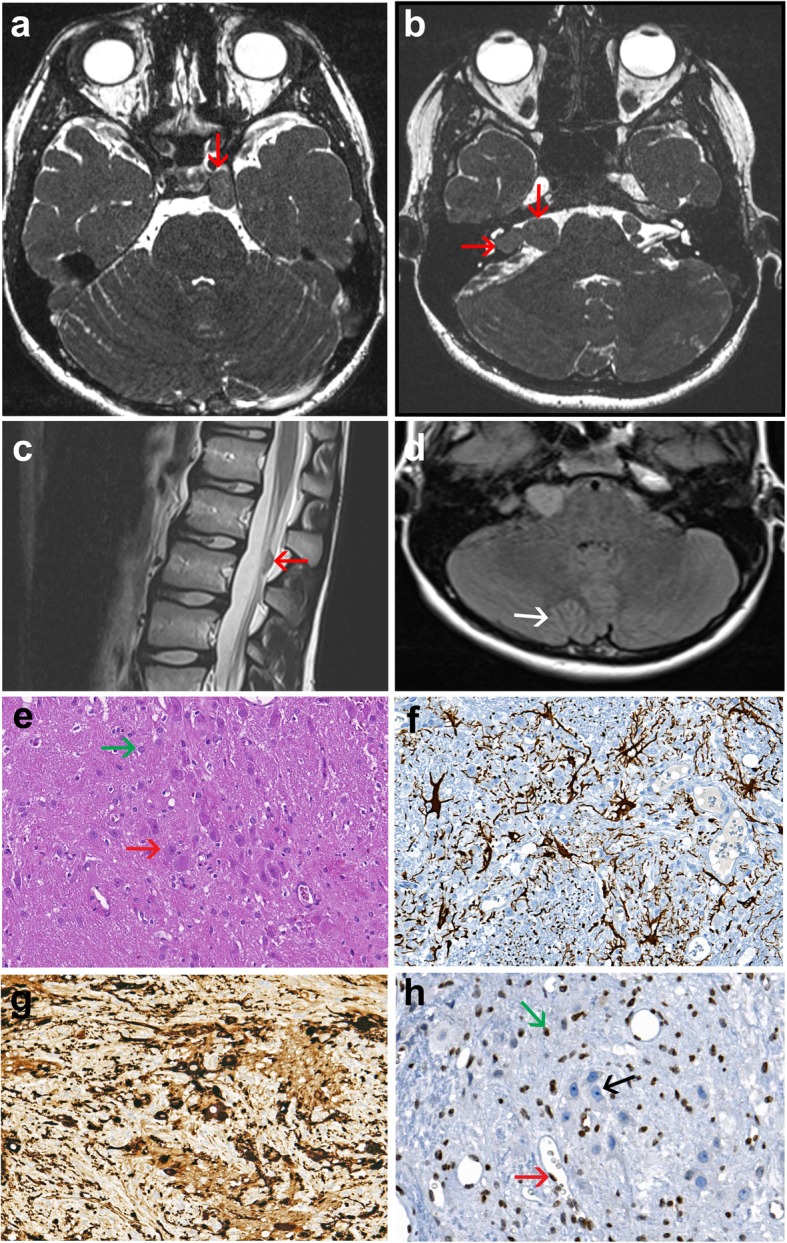
Imaging characteristics of the brain and spinal lesions; pathological hallmarks of the spinal lesion. a-d, MRI of the case. Axial T2-weighted images show the well circumscribed lesion located within the cavernous sinus (a), at the level of the ganglion of the trigeminal nerve measuring 20 × 9 mm close to the not invaded internal carotid (red arrow) and associated with bilateral asymmetric lesions (b), measuring 14 × 12 mm in the interpedoncular fossa and 11 × 10 mm in the cerebellopontine angle within the ganglia of cranial nerves VII and VIII respectively (red arrows) and a nodule at the L3 level (c). T2 weighted axial plane of the cerebellum displaying exaggerated foliation of the right hemispheric cerebellar cortex (white arrow) located close to the vermis (d). e-h, histopathology of the resected L3 lesion. Histological view shows all but disorganized components of a spinal ganglion (e), including often dysmorphic ganglion cells (red arrow) and astrocytes (green arrow) lying in a fibrillar background [OM × 200]. Immunohistochemistry displays multiple GFAP positive astrocytes [OM × 200] (f), and numerous MAP2 positive ganglion cells [OM × 200] (g). PTEN immunolabeling reveals immunoreactive astrocytes (green arrow), endothelial (red arrow) and satellite cell nuclei, whereas positivity of ganglion cell (black arrow) nuclei is lost [OM × 100] (h). (OM: original magnification)
Histological examination of the well circumscribed L3 lesion measuring 1 cm in width revealed the presence of a heterogeneous lesion resembling a disorganized ganglion. It was composed of enlarged dysmorphic ganglion cells, either dispersed or arranged in small clusters, intermixed with protoplasmic astrocytes, spindle cells as well as small round cells lying in a fibrillar network (Fig. 1e). Neither necrosis nor mitoses were identified. Immunohistochemical study indicated that astrocytes expressed GFAP (Fig. 1f), and only scarce Olig2-positive cells were observed, indicative of defective astrocytic maturation. SOX10 and PS100 were positive in the vast majority of cells, corresponding to neural crest cells. Ganglion cells were strongly immunoreactive for MAP2 (Fig. 1g), chromogranin and NeuN. The fibrillar background was synaptophysin- and neurofilament-positive corresponding to disordered assembly of axons. CD34 immunolabelled endothelial cells only. The proliferative marker Ki67 was negative. Based on these findings, the final neuropathological diagnosis of glioneuronal hamartoma was established.
PTEN immunoreactivity appeared heterogeneous, with astrocyte and endothelial cell nuclei expressing PTEN, but not ganglion cells (Fig. 1h). To interpret the latter result, PTEN immunolabeling was performed on several adult samples including one frontal cortex sample, two sympathetic ganglia and one dorsal root ganglion, used as controls. Despite repeated immunolabelling with PTEN antibody, no immunoreactivity was detected in ganglion cells. NGS performed on DNA extracted from the L3 lesion (see Additional file 1) revealed in 30% of the reads a PTEN pathogenic variant within exon 8 (c.970dup; p.(Asp324Glyfs*3); NM_000314.6). No other PTEN alteration, corresponding either to a second pathogenic nucleotide variant or a deletion of the PTEN locus, was detected in the L3 lesion. This allelic imbalance was not suggestive of a germline heterozygous PTEN alteration, which usually yields a percentage of mutant reads close to or above 50%, when associated with loss of heterozygosity, and led us to suspect a mosaic alteration of PTEN. NGS performed at high depth (>500X) on DNA extracted from blood and buccal swabs found this variation in 3.5 and 11% of the reads, respectively, confirming the mosaic state of the PTEN variant. In order to quantify the variant allelic fraction present in the different tissues, using an independent method, we performed a targeted analysis based on QMPSF (Quantitative Multiplex PCR of Short fluorescent Fragments) and dye-labeled primers specific to PTEN exon 8. This analysis confirmed the presence of the PTEN variant in 28, 6 and 12% of the DNA extracted from the hamartoma, blood and buccal swabs, respectively (see Additional file 1). Although we could not formally exclude the fact that the detection of the PTEN variant in a small fraction of blood DNA corresponded to circulating DNA originating from the hamartoma, its detection in buccal swabs makes this hypothesis unlikely. Therefore the final diagnosis was a glioneuronal hamartoma resulting from a mosaic PTEN alteration and we extrapolated that the brain lesions also corresponded to glioneuronal hamartoma, as they were located within the Gasser, as well as within Corti and Scarpa ganglia.
Discussion and conclusions
The PTEN protein is a phosphatase functioning as a key negative regulator of the PI3/AKT/mTOR cascade. Germline alterations of PTEN, cause PHTS, encompassing Cowden syndrome (CS; OMIM 158350), Bannayan-Riley-Ruvalcaba syndrome (BRRS; OMIM 153480) and Proteus and Proteus-like syndrome [for review see ref. [15, 16]. Marked macrocephaly (usually > + 3SD to +6SD) in young children, is very specific to PHTS, when compared to other syndromes with ASD, and is a required major criterion for the diagnosis of PTHS [15–17]. In Cowden mouse models, it has been shown that macrocephaly results from the increased proliferation and the decreased apoptosis of neural stem cells in the ventricular zones [18]. The absence of significant macrocephaly in our patient did not lead us to consider a diagnosis of PTHS and strongly suggested that radial glial cells and intermediate progenitors did not harbor the deleterious PTEN variant at embryonic stages. Despite the absence of macrocephaly, we nevertheless postulate that the Asperger syndrome or mild ASD present in this patient was probably also related to the mosaic PTEN variant.
As regards the development of glioneuronal hamartomas in the cranial and spinal ganglia, the mutational event in our patient very likely occurred at the end of the third post-conception week (day 18- day 21). At this time, the neural crest which arises from neuroepithelial cells adjacent to the edges of the neural groove forms two longitudinal strips on both sides of the embryonic midline and, shortly after, becomes segmented to form cranial nerve and spinal cord ganglia from the sixth post-conception week. This hypothesis is consistent with previous reports in which it has been stated that deleterious somatic mutations must occur early during development to have phenotypic effects, even though the pathogenic consequences may not be observed before childhood or adulthood [2]. It should be noticed that the NGS analysis of PTEN in the hamartoma did not show loss of heterozygosity. This in agreement with a previous study reporting that, in PTEN variation carriers, hamartomas may develop without loss of the wild-type allele [19].
There is no obvious hypothesis that could explain the detection in blood of a PTEN variant which occurred in neural crest progenitors. Nevertheless, recent data have revealed that neural crest cell progenitors are in fact multipotent, harbor mesenchymal potential [20] and that some hematopoietic mesenchymal stem cells derive from the neural crest [21]. The higher variant allelic fraction observed in buccal swabs, as compared to the blood, might be explained by the contribution of the neural crest to many ectomesenchymal derivatives in the cranial region.
In conclusion, this report underlines that mosaic alteration of PTEN is sufficient to cause multiple central and peripheral nervous system hamartomas and that the presence of such alteration should be considered in patients with multiple nervous system masses, even in the absence of cardinal features of PHTS, especially macrocephaly.
Additional file
Acknowledgements
We are grateful to Sophie Coutant and Myriam Vezain for bioinformatic analyses of NGS data, to Beatrice Parfait for the analysis of genes involved in schwannoma predisposition syndromes and to Nikki Sabourin-Gibbs for help in editing the manuscript.
Abbreviations
- ASD
Autism spectrum disorder
- CNS
Central nervous system
- MRI
Magnetic resonance imaging
- NGS
Next Generation Sequencing
- PHTS
PTEN hamartoma tumor syndrome
Authors’ contributions
TF and AL designed the study; AG, VG, A-MC, FD, SR and MB collected the clinical data. FM and AL performed and interpreted the pathological analyses. AH performed and interpreted the neuroimaging. SV and JB performed the genetic analyses and CH, SB-D and TF analyzed the results. AG, AL and TF wrote the manuscript. All authors approved the manuscript.
Availability of data and materials
Most data generated or analyzed during this study are included in this article. Additional datasets are available from the corresponding author on request.
Ethics approval and consent to participate
Genetic analyses were performed after appropriate informed consent was obtained and the patient’s parents provided informed consent for publication.
Competing interests
The authors have no competing interest to declare.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s40478-019-0841-0.
References
- 1.Goldmann JM, Wong WS, Pinelli M, Farrah T, Bodian D, Stittrich AB, et al. Parent-of-origin-specific signatures of de novo mutations. Nat Genet. 2016;48(8):935–939. doi: 10.1038/ng.3597. [DOI] [PubMed] [Google Scholar]
- 2.Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. 2015;31(7):382–392. doi: 10.1016/j.tig.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans DG, Sainio M, Baser ME. Neurofibromatosis type 2. J Med Genet. 2000;37(12):897–904. doi: 10.1136/jmg.37.12.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renaux-Petel M, Charbonnier F, Théry JC, Fermey P, Lienard G, Bou J, et al. Contribution of de novo and mosaic TP53 mutations to li-Fraumeni syndrome. J med genet. 2018;55(3):173–180. doi: 10.1136/jmedgenet-2017-104976. [DOI] [PubMed] [Google Scholar]
- 5.Trubicka J, Filipek I, Iwanowski P, Rydzanicz M, Grajkowska W, Piekutowska-Abramczuk D, et al. Constitutional mosaicism of a de novo TP53 mutation in a patient with bilateral choroid plexus carcinoma. Cancer Genet. 2017;216-217:79–85. doi: 10.1016/j.cancergen.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Behjati Sam, Maschietto Mariana, Williams Richard D., Side Lucy, Hubank Mike, West Rebecca, Pearson Katie, Sebire Neil, Tarpey Patrick, Futreal Andrew, Brooks Tony, Stratton Michael R., Anderson John. A Pathogenic Mosaic TP53 Mutation in Two Germ Layers Detected by Next Generation Sequencing. PLoS ONE. 2014;9(5):e96531. doi: 10.1371/journal.pone.0096531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodin Rachel E., Walsh Christopher A. Somatic Mutation in Pediatric Neurological Diseases. Pediatric Neurology. 2018;87:20–22. doi: 10.1016/j.pediatrneurol.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gammon A, Jasperson K, Pilarski R, Prior T, Kuwada S. PTEN mosaicism with features of Cowden syndrome. Clin Genet. 2013;84(6):593–595. doi: 10.1111/cge.12078. [DOI] [PubMed] [Google Scholar]
- 9.Golas MM, Auber B, Ripperger T, Pabst B, Schmidt G, Morlot M, et al. Looking for the hidden mutation: Bannayan-Riley-Ruvalcaba syndrome caused by constitutional and mosaic 10q23 microdeletions involving PTEN and BMPR1A. Am J Med Genet A. 2019;179(7):1383–1389. doi: 10.1002/ajmg.a.61166. [DOI] [PubMed] [Google Scholar]
- 10.Pritchard CC, Smith C, Marushchak T, Koehler K, Holmes H, Raskind W, et al. A mosaic PTEN mutation causing Cowden syndrome identified by deep sequencing. Genet Med. 2013;15(12):1004–1007. doi: 10.1038/gim.2013.51. [DOI] [PubMed] [Google Scholar]
- 11.Salo-Mullen EE, Shia J, Brownell I, Allen P, Girotra M, Robson ME, et al. Mosaic partial deletion of the PTEN gene in a patient with Cowden syndrome. Familial Cancer. 2014;13(3):459–467. doi: 10.1007/s10689-014-9709-4. [DOI] [PubMed] [Google Scholar]
- 12.Caux F, Plauchu H, Chibon F, Faivre L, Fain O, Vabres P, et al. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur J hum genet. 2007;15(7):767–73. 3. doi: 10.1038/sj.ejhg.5201823. [DOI] [PubMed] [Google Scholar]
- 13.Zhou X.-P. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Human Molecular Genetics. 2000;9(5):765–768. doi: 10.1093/hmg/9.5.765. [DOI] [PubMed] [Google Scholar]
- 14.Nathan N, Keppler-Noreuil KM, Biesecker LG, Moss J, Darling TN. Mosaic disorders of the PI3K/PTEN/AKT/TSC/mTORC1 signaling pathway. Dermatol Clin. 2017;35(1):51–60. doi: 10.1016/j.det.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen-Kiss E, Beinkampen S, Adler B, Frazier T, Prior T, Erdman S, et al. A retrospective chart review of the features of PTEN hamartoma tumour syndrome in children. J Med Genet. 2017;54(7):471–478. doi: 10.1136/jmedgenet-2016-104484. [DOI] [PubMed] [Google Scholar]
- 16.Yehia L, Eng 65 YEARS OF THE DOUBLE HELIX: one gene, many endocrine and metabolic syndromes: PTEN-opathies and precision medicine. Endocr Relat Cancer. 2018;25(8):T121–T140. doi: 10.1530/ERC-18-0162. [DOI] [PubMed] [Google Scholar]
- 17.Ciaccio Claudia, Saletti Veronica, D'Arrigo Stefano, Esposito Silvia, Alfei Enrico, Moroni Isabella, Tonduti Davide, Chiapparini Luisa, Pantaleoni Chiara, Milani Donatella. Clinical spectrum of PTEN mutation in pediatric patients. A bicenter experience. European Journal of Medical Genetics. 2019;62(12):103596. doi: 10.1016/j.ejmg.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 18.Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor. Science. 2001;294(5549):2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- 19.Reifenberger J, Rauch L, Beckmann MW, Megahed M, Ruzicka T, Reifenberger G. Cowden's disease: clinical and molecular genetic findings in a patient with a novel PTEN germline mutation. Br J Dermatol. 2003;148(5):1040–1046. doi: 10.1046/j.1365-2133.2003.05322.x. [DOI] [PubMed] [Google Scholar]
- 20.Ciaccio Claudia, Saletti Veronica, D'Arrigo Stefano, Esposito Silvia, Alfei Enrico, Moroni Isabella, Tonduti Davide, Chiapparini Luisa, Pantaleoni Chiara, Milani Donatella. Clinical spectrum of PTEN mutation in pediatric patients. A bicenter experience. European Journal of Medical Genetics. 2019;62(12):103596. doi: 10.1016/j.ejmg.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Isern J, García-García A, Martín AM, Arranz L, Martín-Pérez D, Torroja C et al (2014) The neural crest is a source of mesenchymal stem cells with specialized hematopoietic stem cell niche function. Elife. 3:e03696. doi: 10.7554/eLife.03696 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Most data generated or analyzed during this study are included in this article. Additional datasets are available from the corresponding author on request.