

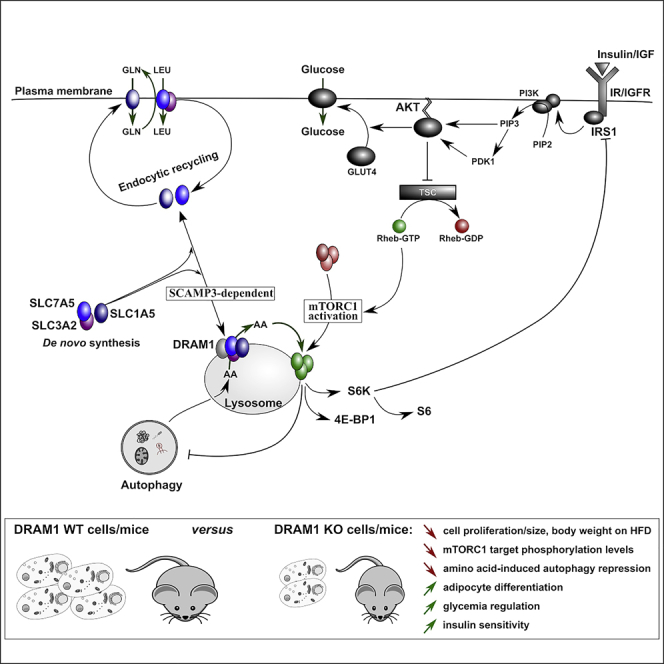
Summary
Sensing nutrient availability is essential for appropriate cellular growth, and mTORC1 is a major regulator of this process. Mechanisms causing mTORC1 activation are, however, complex and diverse. We report here an additional important step in the activation of mTORC1, which regulates the efflux of amino acids from lysosomes into the cytoplasm. This process requires DRAM-1, which binds the membrane carrier protein SCAMP3 and the amino acid transporters SLC1A5 and LAT1, directing them to lysosomes and permitting efficient mTORC1 activation. Consequently, we show that loss of DRAM-1 also impacts pathways regulated by mTORC1, including insulin signaling, glycemic balance, and adipocyte differentiation. Interestingly, although DRAM-1 can promote autophagy, this effect on mTORC1 is autophagy independent, and autophagy only becomes important for mTORC1 activation when DRAM-1 is deleted. These findings provide important insights into mTORC1 activation and highlight the importance of DRAM-1 in growth control, metabolic homeostasis, and differentiation.
Keywords: mTOR, DRAM-1, autophagy, amino acid transporters, SCAMP3, insulin signaling, and adipocyte differentiation
Graphical Abstract
Highlights
-
•
DRAM-1 is required for efficient activation of the nutrient-sensing complex mTORC1
-
•
DRAM-1 and SCAMP3 direct newly synthesized amino acid transporters to lysosomes
-
•
DRAM-1 drives lysosomal amino acid efflux to promote mTORC1 activation
-
•
Loss of DRAM-1 increases insulin sensitivity and enhances adipocyte differentiation
mTORC1 is a nutrient-sensing complex that affects many cellular processes. Beaumatin et al. show that DRAM-1 and SCAMP3 are required for efficient activation of mTORC1. Consequently, they show that loss of DRAM-1 impairs cell growth and amino-acid-induced autophagy repression while promoting insulin sensitivity, glycemic regulation, and adipocyte differentiation.
Introduction
Cell growth and homeostasis are critically dependent on nutrient availability for biosynthesis. Under fed conditions, cells utilize external nutrients; in periods of starvation, cells activate macroautophagy (hereafter autophagy) that produces biosynthetic precursors and energy through the lysosomal breakdown of internal macromolecules (Dunn, 1990, Takeshige et al., 1992). The interplay between use of external and internal precursors is tightly controlled by mechanisms that sense and respond to nutrient availability. A major factor in this regard is the mTORC1 complex, which is activated by amino acids and contains the serine/threonine kinase mTOR (Goberdhan et al., 2016). When activated, mTOR phosphorylates substrates that promote protein translation, such as ribosomal protein S6 kinase-1 (S6K1) and eukaryotic translation initiation factor 4 binding protein 1 (4E-BP1) (Hara et al., 1998). At the same time, mTOR phosphorylates Unc-51-like autophagy-activating kinase 1 (ULK1), causing inhibition of autophagy (Jung et al., 2009). Reciprocal effects occur under starved conditions when mTORC1 is repressed (Egan et al., 2011, Gwinn et al., 2008). Changes in the activation of mTORC1 also affect insulin sensitivity by modulating the activation of the serine/threonine kinase AKT downstream of insulin receptors. This then influences the ability of cells to respond to changes in serum glucose and also the differentiation and deposition of adipose tissue.
Due to its impact on multiple cellular processes, the activation of mTORC1 is highly regulated, and several mechanisms have been described (Goberdhan et al., 2016). Many studies have shown that mTORC1 senses amino acids at the lysosome, and in nutrient-replete conditions, mTORC1 is localized at lysosome membranes—an interaction that is lost upon starvation (Sancak et al., 2010). In this context, mTORC1 is thought to initiate amino acids sensing from within the lysosome by what has been termed an “inside-out” mechanism involving the vacuolar ATPase (Zoncu et al., 2011). It has also been reported, however, that amino acids and in particular leucine—the main activating amino acid for mTORC1—are sensed in the cytoplasm via leucyl-tRNA synthetase and SESTRIN-2 (Han et al., 2012, Parmigiani et al., 2014, Wolfson et al., 2016). In this study, we report that DRAM-1 is a component of the nutrient-sensing system that links lysosomal and cytoplasmic sensing by facilitating amino acid transport across the lysosomal membrane. Moreover, we show that loss of DRAM-1 leads to an inhibition of mTORC1 activation, which affects metabolic signaling pathways and organismal health.
Results
DRAM1 Promotes mTORC1 Activation Independent of Autophagy
We previously identified DRAM-1 as a lysosomal membrane protein that promotes autophagy downstream of p53 (Crighton et al., 2006). Because autophagy and mTORC1 activity are connected (Tan and Miyamoto, 2016), we questioned whether DRAM-1 also affects mTORC1. To examine this, we starved and re-fed Saos2-TetOn-DRAM1 cells that had been incubated in either the absence or presence of doxycycline (Dox) to induce DRAM-1 levels. Analysis of S6K1 phosphorylation, an established readout of mTORC1 activity (Burnett et al., 1998), upon starvation in Earle’s balanced salt solution (EBSS) followed by addition of amino acids, clearly showed that higher levels of DRAM-1 facilitated mTORC1 activation at all time points analyzed (Figure 1A). To test whether this effect was a facet of endogenous DRAM-1, we isolated mouse embryonic fibroblasts (MEFs) from embryonic day 13.5 (E13.5) Dram-1flox/flox mice and infected these cells with a retrovirus expressing Cre recombinase or “empty” retroviral vector as control (Figure S1A). Starvation of these cells for 3 h followed by addition of leucine for 20 min caused a marked increase in S6K1 phosphorylation that was essentially absent in Dram-1−/− cells (Figure 1B). Decreased phosphorylation of another mTORC1 substrate, 4E-BP1, was also observed, which together with decreased S6K1 phosphorylation indicates a critical role for DRAM1 in the activation of mTORC1 (Figure 1B). Importantly, these effects were not observed upon Cre infection of wild-type MEFs (Figure S1B). Consistent with a defect in the ability to activate mTORC1, DRAM1-null cells have decreased growth rate and size when compared to controls (Figures 1C and 1D) and have a diminished ability to repress autophagy upon re-feeding—as assessed by lower levels of the lipidated form of LC3 (LC3-II), a marker of autophagosomes (Figures 1E and S1C; Klionsky et al., 2016).
Figure 1.

DRAM1 Stimulates Amino-Acid-Induced mTOR Activation
(A) Saos2 TetOn-DRAM1-myc-his-tagged cells were, where indicated, treated with doxycycline (Dox) for 24 h and then starved for 3 h in EBSS before treatment with EBSS containing essential amino acids for the indicated times. mTOR activation was evaluated by measuring phospho-S6 kinase levels by western blot. Total S6 kinase levels or actin were used as loading controls. DRAM1 expression was detected using an anti-Myc-tag antibody.
(B) Dram1flox/flox MEF expressing Cre recombinase (−/−) or a control vector (fl/fl) was starved for 3 h in EBSS prior to 20 min in EBSS containing 0.8 mM leucine. mTOR activation was evaluated by measuring phospho-S6 kinase, phospho-4E-BP1 levels by western blot. S6K, 4E-BP1, and ERK2 were used as loading controls.
(C) Flow cytometry analysis of cell size of Dram1flox/flox MEF expressing Cre recombinase (−/−) or a control vector (fl/fl) grown under control or starvation conditions for 3 h. FSC, forward scatter. Boxplot and whiskers: 1–99 percentile. Bar represents median. ∗p < 0.05.
(D) Cell proliferation of Dram1flox/flox MEF expressing Cre recombinase (−/−) or a control vector (fl/fl). Equal cell numbers were split on day 0 in complete DMEM. From day 2, cells were harvested daily and counted using Innovatis cell counter. Result shown is representative of 3 independent experiments. Data are mean ± SD. ∗p < 0.05.
(E) Dram1flox/flox MEF expressing Cre recombinase enzyme (−/−) or a control vector (fl/fl) were starved for 3 h in EBSS containing glutamine prior to DMEM treatment for the indicated times. Repression of autophagy by mTOR activation was assessed by western blot of LC3B (I and II) and phospho-S6 kinase. ERK2 was used as a loading control. Result shown is representative of 3 independent experiments.
See also Figures S1 and S2.
Due to DRAM-1’s previously described role in autophagy (Crighton et al., 2006), and because autophagy can increase amino acid levels and mTORC1 activity (Yu et al., 2010), we first considered that DRAM-1 may affect mTORC1 via its role in autophagy. As a result, we generated mice hemizygous for Dram-1 that also contain two floxed alleles of Atg7—an essential autophagy gene (Komatsu et al., 2005, Tanida et al., 1999). Intercrossing these mice resulted in animals with floxed Atg7 alleles, which were also either wild-type, hemizygous, or null for Dram-1 (Figure S2A). Examination of MEFs from these mice revealed, consistent with our previous observations, that loss of DRAM-1 severely impaired the ability to activate mTORC1, as assessed by S6K1 phosphorylation (Figure S2B). These cells also had a diminished ability to repress autophagy and a decreased growth rate (Figures S2C and S2D). Treatment with bafilomycin A1, an inhibitor of the lysosomal vacuolar ATPase (Bowman et al., 1988), also reduced leucine’s ability to activate mTORC1 in Dram1+/+ Atg7flox/flox cells (Figure S2E), underscoring the importance of lysosomal function in this response. However, and in contrast, infection of Dram1+/+ Atg7flox/flox cells with a retrovirus expressing Cre to delete Atg7 did not diminish mTORC1 activation (Figures S2F and S2G). In fact, loss of autophagy only reduced leucine-mediated mTORC1 activation when Dram-1 was also deleted (Figures S2G and S2H). This therefore shows that DRAM-1 has a role in mTORC1 activation that is independent of autophagy and that autophagy only serves as a back-up for mTORC1 activation when this DRAM-1/mTORC1 axis is impaired.
DRAM1 Promotes mTORC1 Activation by Binding the Amino Acid Transporters LAT1 and SLC1A5
To gain insight into DRAM-1’s role in mTORC1 activation, we searched for DRAM-1-interacting proteins among factors enriched from HeLa cells containing exogenous DRAM-1 linked to a tandem-affinity purification (TAP) tag (Gloeckner et al., 2007). Based on the frequency of peptide identification by mass spectrometry and our interest in proteins linked to nutrient sensing and autophagy, we were drawn to the amino acid transporters SLC3A2, SLC1A5, and SLC7A5 (Figure S3A). These transporters were previously linked to mTORC1 activation through their action at the plasma membrane, where SLC1A5 (also known as ASCT2) imports glutamine to facilitate the activity of a complex between SLC3A2 and SLC7A5 (also known, and hereafter collectively referred to, as LAT1 for large neutral amino acid transporter), which imports leucine and reciprocally exports glutamine (Nicklin et al., 2009). To confirm our TAP tag results in a different system, we performed immunoprecipitation in Saos2-TetOn-DRAM1 cells. Upon immunoprecipitation of DRAM-1, an interaction could clearly be seen with SLC1A5 (Figure 2A). An interaction could also be detected between DRAM-1 and both SLC7A5, indicating an interaction with LAT1 (Figure 2A). Moreover, immunoprecipitation of DRAM-1 mutants lacking regions from the N or C terminus revealed that LAT1 binds to DRAM-1’s C terminus (Figures S3B and S3C). To test whether the interaction with these transporters was inter-dependent, we repeated these immunoprecipitations in cells where we had knocked down either SLC7A5 or SLC1A5. This revealed that interaction between DRAM-1 and SLC7A5 was not diminished by knockdown of SLC1A5 and vice versa (Figures 2B and 2C), indicating that DRAM-1 interacts with the two amino acid transporters independently.
Figure 2.

DRAM1 Interacts with LAT1 and SLC1A5 Amino Acid Transporters to Stimulate mTOR Activation
(A) Saos2 TetOn-DRAM1 cells treated with or without doxycycline for 24 h before incubation in full DMEM or EBSS for 3 h. Cells were harvested and lysed prior to immunoprecipitation of DRAM1 using anti-Myc-tag antibody. Immunoblotting from total protein extracts (INPUT) or elutions (IP DRAM1) was undertaken to detect SLC7A5 (LAT1), SLC1A5, DRAM1 (Myc tag), and ERK2 (loading control). A representative result from at least 3 independent experiments is shown.
(B) Saos2 TetOn-DRAM1 cells expressing the indicated short hairpin RNA (shRNA) constructs were grown in the presence or absence of doxycycline for 24 h. Myc-tagged proteins were immunoprecipitated and samples analyzed by western blotting to detect DRAM-1 (Myc tag), SLC7A5, SLC1A5, or SLC3A2. ERK2 was used as a loading control.
(C) Quantifications of SLC7A5 and SLC1A5 bound to DRAM1 from 3 independent experiments. Data are mean ± SEM. ∗p < 0.05.
(D–F) Experiments were performed using Dram1flox/flox MEF expressing Cre recombinase (−/−) or a control vector (fl/fl).
(D) Cells were starved in EBSS with, where indicated, 2 mM glutamine for 3 h before incubation in EBSS containing 0.8 mM leucine for 20 min.
(E) Cells were starved in EBSS containing glutamine for 3 h before incubation for 20 min in EBSS containing 0.8 mM leucine, 10 mM D-phenylalanine, and 25 μg/mL cycloheximide as indicated.
(F) Cells were starved for 3 h in EBSS containing glutamine alone or supplemented with L-ɣ-glutamyl-p-nitroanilide (ɣ-GPNA) before treatment with EBSS containing 0.8 mM leucine alone or supplemented with 100 nM rapamycin for 20 min.
(D–F) mTOR activation was detected by measuring phospho-S6 kinase levels by western blotting. Total S6 kinase and ERK2 levels were used as loading controls.
See also Figure S3.
We were interested to know whether the effect of DRAM-1 on mTORC1 activation was related to SLC1A5 and LAT1 binding. Because pre-loading with glutamine can enhance the ability of LAT1 to import leucine (Nicklin et al., 2009), we first tested whether glutamine pre-loading could affect mTORC1 activation in Dram-1 flox/flox and Dram-1−/− cells. This clearly showed, in line with previous studies, that glutamine pre-loading enhances leucine’s effects on mTORC1, but again, little activation of mTORC1 was observed in DRAM1-null cells (Figure 2D). We next directly assessed the involvement of LAT1 and SLC1A5 in this system by respectively treating cells with D-phenylalanine (D-Phe) (Yanagida et al., 2001) and L-γ-glutamyl-p-nitroanilide (γ-GPNA) (Esslinger et al., 2005), two well-characterized inhibitors of leucine and glutamine uptake by these transporters. In both cases, the ability of glutamine pre-loading followed by addition of leucine to activate mTORC1 was abolished by these inhibitors, showing a clear role for these transporters in the activation of mTORC1 in these cells and underscoring DRAM-1’s role in this process (Figures 2E and 2F).
DRAM-1 Directs LAT1 and SLC1A5 to Lysosome Membranes to Invoke Amino Acid Efflux
Our findings caused us to consider that DRAM-1 loss may reduce mTORC1 activation by reducing the ability of SLC1A5/LAT1 to import leucine at the cell membrane. However, analysis of intracellular leucine by mass spectrometry in cells incubated in either EBSS or EBSS plus glutamine for 3 h followed by incubation in EBSS plus leucine indicated this was not the case (Figure 3A). Therefore, we considered that DRAM-1 may be promoting mTORC1 activation via SLC1A5/LAT1 at a different cellular location. As DRAM-1 is predominantly a lysosomal protein and because mTORC1 is localized at lysosomes when active, we considered that DRAM-1 may facilitate lysosomal localization of SLC1A5/LAT1 to lysosomes, enabling transfer of amino acids into this organelle. To test this, we performed three different experiments. First, we examined SLC1A5 localization by immunofluorescence in TetOn-DRAM1 cells, which showed that DRAM-1 expression causes a re-localization of a proportion of SLC1A5 to lysosomes (Figures 3B and 3C). Second, we generated a construct that expresses SLC3A2 linked C-terminally to mRFP and GFP (SLC3A2-RFP-GFP). In this context, based on the known membrane orientation of SLC3A2, GFP and mRFP should be located in the acidic lumen if this transporter is directed to the lysosomal membrane. As a result, because fluorescence from GFP, but not mRFP, is acid labile (Kimura et al., 2007), if DRAM-1 directs a proportion of these transporters to the lysosome, the relative fluorescent intensities between mRFP and GFP should change if DRAM-1 is deleted. Indeed, when analyzed by flow cytometry, the level of GFP relative to mRFP was indeed lower in Dram1flox/flox cells compared with DRAM1-null cells (Figure S4A). In addition, when analyzed by fluorescent microscopy, a significant reduction in the amount of this construct was observed at lysosomes in DRAM1-null cells compared to controls (Figures S4B–S4D). In contrast to these results, no DRAM1-dependent difference in GFP/mRFP intensity was observed using a construct in which SLC3A2 was fused N-terminally to red fluorescent protein (RFP) and GFP (RFP-GFP-SLC3A2), which results in the fluorophores being on the outside of the lysosome membrane (Figure S4A). In a final approach, we purified lysosomes using a construct expressing LAMP1-RFP-FLAG (Zoncu et al., 2011). This resulted in clear enrichment for SLC1A5/LAT1 as well as LC3-II and LAMP2 (known autolysosomal/lysosomal proteins) in the lysosomal fraction, and expression of DRAM-1 increased the amount of SLC1A5 and LAT1 in this cellular fraction (Figures 3D–3F and S4E). Collectively, these data show that DRAM-1 directs a proportion of SLC1A5 and LAT1 to the lysosome membrane.
Figure 3.

DRAM1 Drives LAT1 and SLC1A5 Amino Acid Transporters to Lysosomes, which Export Amino Acids and Induce mTOR Activation
(A) Dram1flox/flox MEFs expressing Cre recombinase (−/−) or a control vector (fl/fl) were starved for 3 h in EBSS with or without glutamine before treatment with 0.8 mM leucine in EBSS for 20 min. Intracellular leucine was determined by liquid chromatography-mass spectrometry (LC-MS) analysis.
(B) Saos TetOn-DRAM1-myc-his cells overexpressing V5-tagged SLC1A5 were grown for 24 h in the presence or absence of doxycycline (1 μg/mL). Cells were fixed and stained for LAMP2 and SLC1A5(V5-tag). Scale bars represent 50 μm.
(C) Quantification of colocalization coefficients of LAMP2 with SLC1A5-V5-tagged in Saos TetOn-DRAM1 cells treated or not with doxycycline for 24 h (average of at least 25 cells).
(D) Saos2 TetOn-DRAM1 cells expressing LAMP1-RFP-FLAG2× were induced with or without doxycycline for 24 h. Cells were then starved for 3 h in EBSS prior to 20-min treatment with 0.8 mM methyl-leucine ester. Lysosome-enriched fractions were analyzed by western blot for levels of LAMP2, SLC1A5, and SLC7A5.
(E and F) Quantification of 3 independent experiments was determined using ImageJ. Results represent the relative amounts of SLC7A5 (E) or SLC1A5 (F) normalized to LAMP2 and compared to non-induced starved DRAM1 cells. ∗p < 0.05.
(G) Intracellular leucine (Leu) or methyl leucine ester (CH3-Leu) were analyzed as described in (A) in cells that were starved in EBSS for 3 h before treatment with EBSS supplemented with 0.8 mM methyl-leucine ester for 20 min.
(H and I) Dram1flox/flox MEF expressing Cre recombinase (−/−) or a control vector (fl/fl) cells were starved in the absence or presence of glutamine for 3 h before treatment with EBSS supplemented with 0.8 mM leucine or 0.8 mM methyl leucine ester (H) or 0.8 mM methyl leucine ester and 10 mM D-phenylalanine (I) as indicated for 20 min. mTOR activation was detected by measuring phospho-S6 kinase levels by western blot. ERK2 was used as a loading control.
(J) Amino acid transporters SLC1A5 or SLC7A5 were downregulated using siRNAs in Dram1flox/flox MEFs expressing Cre recombinase (−/−) or a control vector (fl/fl). mTOR activation was detected by measuring phospho-S6 kinase levels by western blot after cells were starved 3 h in EBSS containing glutamine and treated with 0.8 mM methyl leucine ester for 20 min. ERK2 was used as a loading control.
(K) Metabolites from lysosomal fractions were analyzed for intralysosomal leucine content using high-performance liquid chromatography (HPLC)-MS. Results of 3 independent experiments are presented as the amount of lysosomal leucine measured for each condition and normalized to non-induced DRAM1 cells treated with methyl-leucine ester. ∗p < 0.05.
For micrographs, scale bars represent 50 μm. (A and G) Data are mean ± SD. (C–F and K) Data are mean ± SEM.
See also Figure S4.
If DRAM-1 brings a proportion of SLC1A5/LAT1 to the lysosomal membrane to facilitate leucine import, then DRAM-1 should not be required for mTORC1 activation in response to methyl-leucine—an analog of leucine that diffusees across membranes without the aid of transporters and accumulate in lysosomes, where it is de-esterified to yield leucine (Reeves, 1979). Analysis of methyl-leucine uptake by mass spectrometry showed it was indeed efficiently taken up by cells, and this, as observed with leucine in Figure 3A, was not affected by DRAM-1 deletion (Figure 3G). Surprisingly, however, mTORC1 activation by methyl-leucine was still dependent on DRAM-1 (Figure 3H). Moreover, methyl-leucine increased the lysosomal localization of SLC1A5 and SLC7A5 (Figures 3D–3F), and the activation of mTORC1 observed in Dram-1+/+ (Dram1fl/fl on figures) cells was blocked by inhibition of LAT1 with D-Phe or by small interfering RNA (siRNA)-mediated knockdown of SLC1A5 or SLC7A5 (Figures 3I, 3J, S4F, and S4G). These RNAi data also serve to corroborate our data using D-Phe and γ-GPNA in Figures 2E and 2F, underscoring the importance of these transporters in this response (Figures 2E, 2F, 3I, 3J, and S4G).
In light of these results, we considered instead that DRAM-1 may facilitate lysosomal localization of SLC1A5 and LAT1, enabling amino acid efflux from lysosomes, and it is this efflux that is required for mTORC1 activation. If this was correct, expression of DRAM-1 should not only increase the lysosomal localization of SLC1A5/LAT1, but this should also decrease intralysosomal levels of leucine and maybe also glutamine. To test this, we analyzed amino acid levels in purified lysosomal fractions, which clearly showed decreased levels of leucine and glutamine in DRAM1-expressing cells that had been treated with methyl-leucine (Figures 3K, S4E, and S4H). These findings are therefore consistent with a model in which DRAM-1 facilitates mTORC1 activation by directing a proportion of SLC1A5/LAT1 to the lysosomal membrane, where these transporters promote amino acid efflux in a manner independent of autophagy.
We were naturally interested to understand the mechanism by which DRAM-1 re-localizes SLC1A5/LAT1 to lysosomes. We found that the ability of DRAM-1 to re-localize SLC1A5 is blocked by short-term treatment with the translation inhibitors cycloheximide and anisomycin (Figure 4A). This did not, however, affect total SLC1A5 levels (Figure 4B), indicating that DRAM-1 somehow captures a proportion of newly synthesized transporter and restricts its traffic to the plasma membrane. With this theory in mind, we looked again at the other proteins we had found that bind to DRAM-1 (Figure S3A). Among these proteins was secretory carrier membrane protein 3 (SCAMP3), which is involved in membrane traffic events, including traffic to the plasma membrane. Subsequent studies not only confirmed that DRAM-1 binds SCAMP3 (Figure 4C) but also that DRAM-1 causes enhanced localization of SCAMP3 with DRAM-1 and SLC1A5 (Figure 4D). To test whether the lysosomal re-localization of SCAMP3 was mechanistically connected to the lysosomal re-localization of the amino acid transporters, we examined DRAM-1’s effects in cells where SCAMP3 had been silenced by RNAi. This completely impaired the ability of DRAM-1 to re-localize SLC1A5 to lysosomes and also abolished the ability to activate mTORC1 with leucine (Figures 4E–4G). These findings therefore support a hypothesis in which DRAM-1 binds and retains SCAMP3 and a proportion of newly synthesized SLC1A5/LAT1 at lysosomes.
Figure 4.

DRAM1 Interacts with SCAMP3 to Drive SLC1A5 and LAT1 to Lysosome Membranes
(A) Saos TetOn-DRAM1-myc-his cells overexpressing V5-tagged SLC1A5 were grown for 24 h in the presence or absence of doxycycline (1 μg/mL) and then treated for 4 h with cycloheximide (100 μg/mL) or anisomycin (10 μg/mL). Cells were fixed in 4% paraformaldehyde and stained for Lamp2 and V5.
(B) Saos2 TetOn-DRAM1-myc-his cells overexpressing V5-tagged SLC1A5 were, where indicated, treated with doxycycline for 24 h and then treated for 4 h with cycloheximide (100 μg/mL) or anisomycin (10 μg/mL). SLC1A5 expression was detected using an anti-V5 antibody: a short and long exposure is shown. DRAM-1 levels were detected using an anti-Myc tag antibody. Actin was used as a loading control
(C) Saos2 TetOn-DRAM-1 cells treated with or without doxycycline for 24 h were lysed prior to immunoprecipitation of DRAM-1 using anti-Myc-tag antibody. Immunoblotting from total protein extracts (INPUT) or elutions (IP DRAM-1) was undertaken to detect SCAMP3, DRAM-1 (Myc tag), and ERK2.
(D) Saos Tet-On-DRAM1-myc-his cells overexpressing V5-tagged SLC1A5 were grown for 24 h in the presence or absence of doxycycline (1 μg/mL). Cells were fixed in 4% paraformaldehyde and stained for SCAMP3, DRAM1 (Myc-tag), and SLC1A5 (V5-tag).
(E) SCAMP3 was downregulated using siRNA in Saos-2 TetOn-DRAM1-myc-his cells overexpressing V5-tagged SLC1A5, grown for 24 h in the presence or absence of doxycycline (1 μg/mL). Cells were fixed in 4% paraformaldehyde and stained for SLC1A5(V5-tag) and LAMP2.
(F) Colocalization coefficients were determined using Zeiss Zen Black software (average of at least 25 cells). Data are mean ± SEM.
(G) SCAMP3 was downregulated using siRNA in Saos TetOn-DRAM1-myc-his cells overexpressing V5-tagged SLC1A5, then, where indicated, treated with Dox for 24 h, and then starved for 3 h in EBSS before treatment with EBSS containing leucine for 20 min. mTORC1 activation was evaluated by measuring phospho-S6 kinase and phospho-S6 levels by western blot. Levels of total S6 kinase, S6, and actin were used as loading controls. DRAM1 expression was detected using an anti-Myc-tag antibody, and SCAMP3 knockdown was measured using an anti- SCAMP3 antibody.
DRAM-1 Controls Insulin Signaling
Changes in mTORC1 activity can have a negative impact on insulin signaling, because mTORC1 signaling drives negative feedback on insulin receptor substrate 1 (IRS-1) through inhibitory phosphorylation (Harrington et al., 2004, Tremblay and Marette, 2001, Um et al., 2004). This causes reduced phosphatidylinositol 3-kinase (PI3K) flux and a resultant decrease in the phosphorylation of AKT at T308—the site phosphorylated by PDK1 in response to insulin (Alessi et al., 1997). We were therefore interested to know whether DRAM-1 affects insulin signaling and found that DRAM1-null cells exhibit markedly increased and sustained AKT phosphorylation over a range of insulin concentrations (Figures 5A, 5B, and S5). In addition, and in line with our observations connecting DRAM-1 and mTORC1 activation, we found that the differential levels of AKT phosphorylation between Dram-1+/+ (fl/fl) and Dram-1−/− cells could be largely normalized by treatment with rapamycin, an inhibitor of mTORC1 (Brown et al., 1994, Sabatini et al., 1994), but was unaffected by deletion of Atg7, indicating an autophagy-independent link between DRAM-1, mTORC1, and insulin signaling (Figures 5C–5E). It is important to note that, in these experiments, the principal signal for mTORC1 activation, as measured by S6K phosphorylation, is excess insulin in insulin-treated cells. As a result, the comparative levels of S6K phosphorylation at T-389 between DRAM-1+/+ (fl/fl) and DRAM-1−/− cells is different than that observed when re-feeding amino acids is used as a stimulus for mTORC1, as shown in Figures 1, 2, 3, and 4.
Figure 5.

DRAM1 Promotes Insulin Resistance through mTOR Activation
(A) Dram1flox/flox MEFs expressing Cre recombinase (−/−) or a control vector (fl/fl) were grown overnight in serum-depleted DMEM prior to treatment with 5 nM insulin for 15 min. Western blots for phospho-AKT T308, phospho-S6 kinase, phospho-S6 ribosomal protein, and insulin receptor substrate 1 (IRS1) were performed to evaluate activation of the insulin pathway. ERK2 was used as a loading control.
(B) Similar experiments to those in (A) were performed to evaluate the kinetics of insulin pathway activation following different insulin exposure times.
(C) Cells were treated as in (A) except that, prior to insulin treatment, cells were incubated with or without 100 nM rapamycin for 4 h. Western blots for phospho-AKT T308, phospho-S6 kinase, phospho-S6 ribosomal protein, total AKT, total S6 kinase, and total S6 ribosomal protein were performed to evaluate outcomes of mTOR inhibition on cellular insulin sensitivities.
(D) Atg7flox/floxDram1−/− or Atg7flox/floxDram1+/+ MEFs were treated as in (C) in order to evaluate the role of autophagy in DRAM1-induced insulin resistance.
(E) Quantification of 3 independent experiments described in (D). Phospho-AKT T308 levels were normalized to total AKT levels. Data are mean ± SEM. ∗∗p < 0.01.
See also Figure S5.
Inspired by our observations that loss of DRAM-1 promotes insulin signaling, we wanted to know whether these effects cause changes in glucose or insulin tolerance in vivo. To test this, wild-type and Dram-1−/− mice were maintained on a high-fat diet (HFD) and subjected to a glucose tolerance test by oral gavage of glucose or to an insulin tolerance test following intraperitoneal injection of insulin. In line with enhanced insulin signaling in the absence of DRAM-1, this revealed that Dram-1−/− mice are able to mitigate hyperglycemia at a faster rate than wild-type animals (Figure 6A). In our insulin tolerance tests, we observed that wild-type and Dram-1−/− mice fed a HFD did not display differences in glycemia regulation when compared to mice fed a standard chow diet (Figures S6A and S6B). This lack of differences could be due to an excess of insulin injected, which in cells abolishes differences observed in AKT signaling (Figure S5). Interestingly, differences in insulin tolerance were revealed when mice were subjected to rapamycin injections, a treatment already described to further promote type 2 diabetes in mice. Indeed, and consistent with our previous observations, wild-type mice became almost insensitive to insulin upon mTOR inhibition and Dram-1−/− mice were not affected (Figures S6A and S6B). Over a longer period of time, this ability to tolerate and adjust hyperglycemic conditions at a faster rate was accompanied by reduced animal weight, lower levels of subcutaneous and epididymal fat, reduced liver weight, and lower serum levels of adiponectin—an indicator of the extent of adipose tissue in the whole animal (Figures 6B–6E and S6C). As before, treatment with rapamycin showed that these results were connected to the activity of mTORC1. Interestingly, analysis of fat from Dram-1−/− mice revealed no significant changes in adipocyte density or size (Figures S6D and S6E), indicating that DRAM-1 loss may instead be affecting the signaling and differentiation state within adipose tissue. In line with this and consistent with our previous observations, we observed that adipose tissue from Dram-1−/− mice, when compared to wild-type mice, had increased levels of phospho-AKT S473, a readout of the full activation of AKT (Figures 6F and S6I), and phospho-AKT-S473 and -T308 levels were similar in mice fed a HFD (Figures S6F, S6G, S6I, and S6J). Notably, and in accordance with our in vitro results (Figures 5C–5E), phosphorylation of AKT-T308 is only increased by rapamycin treatment in wild-type mice (Figures S6G and S6J).
Figure 6.

DRAM1-Null Mice Are Less Prone to HFD-Induced Glucose Intolerance
(A) Glucose tolerance test was performed on mice fed a high-fat diet for 22 weeks. Blood glucose levels were measured at different time points following glucose administration (10 mice per group).
(B) Mouse weights were measured weekly during the course of the HFD experiment. Results are mean ± SEM for each time point (10 mice per group).
(C and D) Subcutaneous (C) and epididymal adipose tissues (D) were weighed at the end of the HFD experiment.
(E) Plasma adiponectin levels were measured for each mouse using ELISA.
(F) AKT activation levels in epididymal adipose tissues were evaluated by immunohistochemistry (IHC) staining for phospho-AKT S473. Quantifications of at least 5 different mouse adipose tissues per group (5 pictures per mouse) were performed using Adobe Photoshop CS5.1 software.
(G and H) S6 kinase activation (G) and S636/639 IRS1 phosphorylation (H) in epididymal adipose tissues were evaluated by western blots for phospho-S6 kinase and phospho-S636/639 IRS1. Quantifications of 10 different mouse adipose tissues per group were performed using ImageJ. Where indicated, rapamycin (RAPA) was administrated (4 mg/kg) by intraperitoneal injection twice a week for the duration of the experiment.
All data are mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. n.s., non-significant.
See also Figure S6.
Interestingly, and consistent with previous studies (Laplante et al., 2012), adipocytes show no differences in phospho-S6K levels and phospho-IRS1 levels are significantly decreased in DRAM-1-null mice fed a HFD, when compared to similarly fed wild-type animals (Figures 6G, 6H, and S6H–S6K). Altogether, these results: (1) point to the existence of a compensatory mechanism that Dram-1−/− mice establish to overcome their innate defect in mTORC1 activation, (2) demonstrate that DRAM-1 is connected to the PI3K/AKT/mTOR pathway in mice, and (3) reveal a specific role of DRAM-1 during adipogenesis, as confirmed by the following results.
DRAM-1 Controls Adipocyte Differentiation
Our evidence suggested that DRAM-1 controls adipocyte signaling and differentiation and that this is associated with changes in mTORC1 activity. This idea is consistent with previous reports that have indicated that mTORC1 regulates this process (Martin et al., 2015). The role of mTORC1 is, however, reportedly complex, whereby the kinase complex promotes differentiation from precursor cells to pre-adipocytes but inhibits differentiation from pre-adipocytes to adipocytes (Laplante et al., 2012). As a result, although loss of DRAM-1 has an overall negative impact on the accumulation of adipose tissue (Figure 6), we were interested to understand how DRAM-1 might regulate the differentiation of adipocytes when cultured in isolation and, in particular, how DRAM-1 might affect the later stages of differentiation from pre-adipocytes to adipocytes. If this is again potentially connected to the regulation of mTORC1, we would predict DRAM-1, like mTORC1, would negatively affect this terminal stage. To test this, we utilized the 3T3-L1 cell line, which is a well-characterized model of differentiation from pre-adipocytes (Green and Kehinde, 1975). Our initial analysis supported our hypothesis when we found that DRAM-1 expression is increased in 3T3-L1 cells when induced to differentiate from pre-adipocytes to adipocytes (Figure S7A). We therefore decided to disrupt DRAM-1 in this line using CRISPR/Cas9 gene editing to assess the role of DRAM-1 in the later stages of adipocyte differentiation (Figure S7B). Similar to what is observed upon mTORC1 inhibition (Laplante et al., 2012), differentiation of these cells indeed showed that loss of DRAM-1 potentiates differentiation from pre-adipocytes to adipocytes, as assessed by increased oil red O staining (Figures 7A and 7B). This differentiation was accompanied by increased levels of adiponectin and peroxisome proliferator-activated receptor gamma (PPARγ)—another marker of terminal adipocyte differentiation (Figures 7C–7E). In addition, differentiation in DRAM-1-null cells was associated with increased AKT phosphorylation at T308 and decreased p-IRS1 levels, without changes in phospho-S6K (Figures 7D, 7F–7H, and S7C). This indicates that these effects are associated with differences in mTORC1 activity and insulin signaling as we had observed and are similar to those observed by Laplante et al. (2012) in their study looking at the effects of DEPTOR on differentiation from pre-adipocytes.
Figure 7.

DRAM1 Impairs 3T3-L1 Pre-adipocyte Differentiation
3T3-L1 pre-adipocytes infected with 2 different guide sequences targeting Dram1 or 2 non-targeting control guides (NTC) were left undifferentiated or differentiated for 8 days in differentiation media containing various amounts of troglitazone (0–4 μM).
(A) Cellular neutral lipid contents were shown following oil red O staining.
(B) Staining was quantified by eluting oil red O in isopropanol and measuring optical densities at a wavelength of 500 nm. Results are the mean staining of the 2 different Dram1 CRISPR cell lines for each condition normalized to the average staining of the 2 different NTC cell lines. Results are from 3 independent experiments.
(C) qPCR analysis showing Adiponectin expression levels in 3T3-L1 NTC or Dram1 CRISPR cells grown with regular or differentiation medium containing the indicated concentration of troglitazone.
(D) Pre-adipocyte differentiation efficiency was evaluated by western blots directed against phospho-AKT T308 and PPARɣ. AKT and ERK2 were used as loading controls.
(E) Quantification of PPARɣ levels measured from 3 independent experiments was performed using ImageJ.
(F) Quantification of phospho-AKT levels measured from 3 independent experiments was performed using ImageJ.
(G) mTOR activation was evaluated by western blotting for phospho-S636/639 IRS1 in Dram1 CRISPR 3T3-L1 cells or NTC CRISPR cells.
(H) Quantification of phospho-S636/639 IRS1 levels measured from 3 independent experiments was performed using ImageJ.
(B, C, E, F, and H) Data are mean ± SEM. ∗p < 0.05; ∗∗p < 0.01.
See also Figure S7.
When taken together, our results identify DRAM-1 as a critical component in mTORC1 activation by mediating amino acid efflux from lysosomes. As a consequence, loss of DRAM-1 impairs mTORC1 activity, leading to changes in insulin sensitivity and adipocyte differentiation, suggesting a key role for DRAM-1 in the control of obesity and diabetic control.
Discussion
We report here that DRAM-1 is involved in the activation of mTORC1. As mTORC1 activity represses autophagy (Rabanal-Ruiz et al., 2017), it could initially be considered that these findings are at odds with our previous report showing that DRAM-1 is a positive regulator of autophagy downstream of p53 (Crighton et al., 2006). One distinct difference in our studies, however, is that we showed that DRAM-1 positively affects autophagic flux when elevated by p53, which importantly also occurs in the context of p53-mediated activation of other target genes involved in an autophagic response (Kenzelmann Broz et al., 2013). In this study, we show that DRAM-1’s effects on mTORC1 occur in a different context—in either p53-null cells and/or in cells with basal (non-p53-stimulated) levels of DRAM-1. We acknowledge, however, that it could still be the case that the facilitation of autophagy by DRAM-1 could lead to an increase in intralysosomal amino acids and subsequent activation of mTORC1. However, we found that mTORC1 activation is an autophagy-independent function of DRAM-1 and that autophagy is only critically important for mTORC1 activation in the absence of DRAM-1. These results therefore highlight both context-specific effects/roles of DRAM-1 as well as the relative importance/contribution of internally derived amino acids for mTORC1 activation.
In light of these results, we searched for DRAM-1 binding factors that may be connected to mTORC1 activation. This led to the finding that the amino acid transporters LAT-1 and SLC1A5 bind DRAM-1 and that cells with depleted levels of LAT1 or SLC1A5 show markedly diminished amino-acid-induced mTORC1 activation. Initially, we considered that DRAM-1, a lysosomal protein, may bring these transporters to the lysosomal membrane to promote amino acid import into lysosomes—a step which at the time was undefined. We found, however, that this was not the case, and recent studies have now shown that a different lysosomal membrane protein, LAPTM4b, directs LAT-1 to the lysosomal membrane to facilitate amino acid import (Milkereit et al., 2015). In contrast to our initial thoughts, we found that DRAM-1, through its interaction with SCAMP3, directs LAT-1 and SLC1A5 to lysosomal membranes, where, instead of facilitating amino acid import, they facilitate amino acid export to the cytoplasm. The importance of this step is exemplified by the fact that increased DRAM-1 levels reduce intralysosomal leucine and that DRAM-1 is required for mTORC1 activation in cells that have been treated with esterified leucine, which can enter lysosomes freely, but upon cleavage to leucine requires transporter activity for lysosomal export (Reeves, 1979).
Recent studies have implicated SLC38A9 as a factor required for amino acid export across the lysosome membrane, leading to mTORC1 activation. Initially, SLC38A9 was considered an arginine sensor (Rebsamen et al., 2015, Wang et al., 2015), but recent in vitro assays have shown that SLC38A9 also has leucine transporter activity (Wyant et al., 2017). It is therefore natural to consider that there may be interplay between the activities of SLC38A9, LAT-1, and SLC1A5 in cells. For example, as LAT-1 is considered the principal leucine transporter at the plasma membrane, could it be possible that SLC38A9 is required for LAT-1 to export leucine? Or, conversely, are LAT-1 and SLC1A5 required for SLC38A9 function? Future studies in this area would certainly be insightful.
We also report that DRAM-1 is involved in several biological functions where mTORC1 has been shown to be a regulator. We show that DRAM-1 is important for insulin signaling and that, in vivo, this has implications for glucose tolerance in mice on a high-fat diet. Our finding, however, that loss of DRAM-1 promotes differentiation from pre-adipocytes to adipocytes at first hand seems incongruous with previous reports showing a positive role for mTORC1 in adipocyte differentiation. For example, it has been reported that treatment with rapamycin can inhibit adipocyte differentiation and knockout of TSC1/TSC2 can promote adipocyte differentiation (Yeh et al., 1995, Zhang et al., 2009). However, it is important to note that, unlike rapamycin, loss of DRAM-1 does not completely block mTORC1 but impairs its activation. In line with this “partial” inhibition of mTORC1, we found that DRAM-1 loss in pre-adipocytes does not affect the phosphorylation of S6K, but it does reduce phosphorylation of IRS-1 at S636/639—the site phosphorylated by mTORC1 (Tzatsos, 2009, Tzatsos and Kandror, 2006). This partial (or selective) inhibition of mTORC1 is not without precedent. Laplante et al. (2012) showed that overexpression of the mTOR inhibitor DEPTOR in pre-adipocytes also reduced IRS-1 phosphorylation while having no apparent effects on the phosphorylation of S6K. These changes in IRS-1 phosphorylation caused by loss of DRAM-1 or DEPTOR overexpression led to changes in AKT phosphorylation and upregulation of PPARγ—events that promote differentiation from pre-adipocytes to adipocytes. Despite the similarities between the role of DEPTOR and DRAM-1 on mTORC1 activity during differentiation to adipocytes, we have to acknowledge that the partial effects on mTORC1 activity caused by deletion of DRAM-1 may not be the only mechanism affected by DRAM-1 loss in this context. As yet unknown mechanisms may contribute to the differentiation process in a manner either dependent or independent of the regulation of mTORC1.
When taken together, our findings identify and characterize DRAM-1 as a critical component involved in mTORC1 activation. The impact of DRAM-1 loss not only impacts the biochemical readouts of mTORC1 activation but also markedly affects biological processes connected to mTORC1 and reveals potential roles for DRAM-1 in insulin sensitivity, obesity, and diabetic control.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies (anti-) | ||
| Myc tag | Millipore | cat# 05-724; RRID: AB_309938 |
| ERK2 | Santa Cruz biotechnology | cat# sc-154; RRID: AB_2141292 |
| Flag-Tag | Sigma-Aldrich | cat# 3165; RRID: AB_259529 |
| LAMP2 (aka CD107b) | BD PharMingen | cat# 555803; RRID: AB_396137 |
| SLC3A2 (aka CD98hc) | Santa Cruz biotechnology | cat# sc-9160; RRID: AB_638288 |
| SLC1A5 (aka ASCT2) | Cell Signaling | cat# 5345; RRID: AB_10621427 |
| SLC7A5 (aka LAT1) | Cell Signaling | cat# 5347; RRID: AB_10695104 |
| LC3B | Cell Signaling | cat# 2775; RRID: AB_515950 |
| Phospho S6 Ribosomal protein | Cell Signaling | cat# 4858; RRID: AB_516156 |
| S6 Ribosomal protein | Cell Signaling | cat# 2317; RRID: AB_2238583 |
| Phospho 4E-BP1 | Cell Signaling | cat# 2855; RRID: AB_560835 |
| 4E-BP1 | Cell Signaling | cat# 9644; RRID: AB_2097841 |
| Atg7 | Cell Signaling | cat# 8558; RRID: AB_10831194 |
| Phospho AKT (S473) | Cell Signaling | cat# 9271; RRID: AB_329825 |
| Phospho AKT (T308) | Cell Signaling | cat# 4056; RRID: AB_331163 |
| AKT | Cell Signaling | cat# 9272; RRID: AB_329827 |
| PPARγ | Cell Signaling | cat# 2430; RRID: AB_823599 |
| Anti-mouse IgG HRP-linked | Cell Signaling | cat# 7076S; RRID: AB_330924 |
| Anti-rabbit IgG HRP-linked | Cell Signaling | cat# 7074S; RRID: AB_2099233 |
| Phospho-IRS-1 | Cell Signaling | cat# 2388; RRIS: AB_330339 |
| Phospho p70 S6 Kinase | Cell Signaling | cat# 9234; RRID: AB_2269804 |
| p70 S6 Kinase | Cell Signaling | cat# 2708; RRID: AB_390722 |
| ARF6 | Cell Signaling | cat# 3546; RRID: AB_2058484 |
| V5-Tag | Cell Signaling | cat# 13202; RRID: AB_2687461 |
| LAMP2 Alexa Fluor 488 conjugate | Thermo Fisher Scientific | MA1-205-A488; RRID: AB_2688309 |
| Myc Tag Alexa Fluor 488 | Millipore | cat#16-224; RRID: AB_442399 |
| Scamp3 | Proteintech | 26888-1-AP; RRID: AB_2810962 |
| V5-Tag DyLight 650 conjugate | Thermo Fisher Scientific | MA5-15253-D650; RRID: AB_2537642 |
| VPS35 | Abcam | ab97545; RRID: AB_10696107 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-Phenylalanine | Sigma-Aldrich | cat# P1751 |
| L-Leucine methyl ester hydrochloride | Sigma-Aldrich | cat# L1002 |
| L-γ-glutamyl-p-nitoanilide | Sigma-Aldrich | cat# G6133 |
| L-Leucine | Sigma-Aldrich | cat# L8000 |
| cycloheximide | Sigma-Aldrich | cat# C4859 |
| puromycin | Sigma-Aldrich | cat# P9620 |
| polybrene | Sigma-Aldrich | cat# H9268 |
| doxycyclin | Sigma-Aldrich | cat# D9891 |
| Protein A Sepharose beads | Sigma-Aldrich | cat# P9424 |
| Anti-FLAG M2 Magnetic Beads | Sigma-Aldrich | cat# M8823 |
| bicinchonic acid solution | Sigma-Aldrich | cat# B9643 |
| Copper (II) sulfate solution | Sigma-Aldrich | cat# C2284 |
| troglitazone | Sigma-Aldrich | cat# T2573 |
| insulin | Sigma-Aldrich | cat# I9278 |
| IBMX | Sigma-Aldrich | cat# I5879 |
| dexamethasone | Sigma-Aldrich | cat# D4902 |
| Oil Red O solution | Sigma-Aldrich | cat# O1391 |
| Earle’s Balanced Salt Solution | Sigma-Aldrich | cat# E2888 |
| Fetal Bovin Serum | Thermo Fisher Scientific | cat# 10270-106 |
| penicillin/streptomycin | Thermo Fisher Scientific | cat# 15140-122 |
| glutamine | Thermo Fisher Scientific | cat# 25030-024 |
| accutase | Thermo Fisher Scientific | cat# A11105-01 |
| trypsin | Thermo Fisher Scientific | cat# 15090-046 |
| Dulbecco’s Modified Eagle Medium | Thermo Fisher Scientific | cat# 21969-035 |
| 50X MEM amino acid solution | Thermo Fisher Scientific | cat# 11130-036 |
| LysoTracker Deep Red | Thermo Fisher Scientific | cat# L12492 |
| HALT protease and phosphatase inhibitor cocktail | Thermo Fisher Scientific | cat# 78442 |
| Rapamycin | LC laboratories | cat# R-5000 |
| Bafilomycin A1 | LC laboratories | cat# B-1080 |
| Deposited Data | ||
| Full scans of western blot data and original microscopy images | This paper | Mendeley Data: https://doi.org/10.17632/37tgjx44g4.1 |
| Experimental Models: Cell Lines | ||
| Saos2 Tet-On DRAM1-myc tag | This lab | Crighton et al. 2006 |
| Phoenix-AMPHO | ATCC | cat# CRL-3213 |
| HEK293T | ATCC | cat# CRL-3216 |
| 3T3-L1 | ATCC | cat# CL-173 |
| DRAM1 fl/fl MEF | This article | N/A |
| ATG7 fl/fl MEF | This article | N/A |
| ATG7 fl/fl DRAM1 KO MEF | This article | N/A |
| HeLa | Beatson Institute stock (obtained from ATCC) | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice | Charles River | Strain Code: 027 |
| DRAM1fl/fl C57BL/6 mice | This article | N/A |
| DRAM1+/− C57BL/6 mice | This article | N/A |
| ATG7fl/fl C57BL/6 mice | Komatsu et al. 2005 | Komatsu et al. 2005 |
| Oligonucleotides | ||
| See table for primer sequences | N/A | |
| Recombinant DNA | ||
| pBabe-puro | Addgene | cat# 1764 |
| pLenti-puro | Addgene | cat# 39481 |
| pLX304-SLC3A2 | DNASU plasmid repository | cat# HsCD00440549 |
| pLX304-SLC1A5 | DNASU plasmid repository | cat# HsCD00436374 |
| pLZRS-SLC3A2-RFP-GFP | This article | N/A |
| pLZRS- RFP-GFP-SLC3A2 | This article | N/A |
| psPAX2 | Addgene | cat# 12260 |
| pMD2.G | Addgene | cat# 12259 |
| p LAMP1-mRFP-Flag2X | Addgene | cat# 34611 |
| pCMV6-mouse-DRAM1 | Origene | cat# MR220640 |
| pLentiCRISPR v2 | Addgene | cat# 52961 |
| pLentiCRISPR v2 mouse DRAM1 #A | This article | N/A |
| pLentiCRISPR v2 mouse DRAM1 #B | This article | N/A |
| pLentiCRISPR v2 NTC1 | This article | N/A |
| pLentiCRISPR v2 NTC2 | This article | N/A |
| si/shRNA | ||
| Renilla shRNA (Non-targeting control) | Mirimus | LPE shRen.713 |
| SLC1A5 shRNA | Mirimus | cat# SLC1A5_798 |
| SLC1A5 shRNA | Mirimus | cat# SLC1A5_1062 |
| SLC7A5 shRNA | Mirimus | cat# SLC7A5_1857 |
| SLC7A5 shRNA | Mirimus | cat# SLC7A5_2000 |
| ON-TARGETplus SLC1A5 smartpool siRNA | GE Dharmacon | cat# L-007429-00-0005 |
| ON-TARGETplus SLC7A5 smartpool siRNA | GE Dharmacon | cat# L-041166-01-0005 |
| ON-TARGETplus non-targeting controls siRNA | GE Dharmacon | cat# D-001810-01-05, D-001810-02-05, D-001810-03-05, D-001810-04-05 |
| Software and Algorithms | ||
| ImageJ64 | NIH | https://imagej.nih.gov/ij/ |
| Optimized CRISPR Design | Feng Zhang Lab (MIT) | http://zlab.bio/guide-design-resources |
| ZEN 2012 software | Zeiss | N/A |
| FlowJo software v.7.6.5 | FlowJo | N/A |
Lead Contact and Materials Availability
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Kevin M. Ryan (k.ryan@beatson.gla.ac.uk).
Reagents
D-Phenylalanine (Cat# P1751), L-Leucine methyl ester hydrochloride (Cat# L1002), L-γ-glutamyl-p-nitoanilide (γ-GPNA) (Cat# G6133), L-Leucine (Cat# L8000), cycloheximide (Cat# C4859), puromycin (Cat# P9620), polybrene (Cat# H9268), doxycycline (Cat# D9891), Protein A Sepharose beads (Cat# P9424), Anti-FLAG M2 Magnetic Beads (Cat# M8823), bicinchonic acid solution (BCA) (Cat# B9643), Copper (II) sulfate solution (Cat# C2284), troglitazone (Cat# T2573), insulin (Cat# I9278), IBMX (Cat# I5879), dexamethasone (Cat# D4902), Oil Red O solution (Cat# O1391), cOmplete protease inhibitor tablets (Cat# 4693124001) and Earle’s Balanced Salt Solution (EBSS) (Cat# E2888) were obtained from Sigma-Aldrich. Fetal Bovin Serum (FBS) (Cat# 10270-106), penicillin/streptomycin (Cat#15140-122), glutamine (Cat#25030-024), accutase (Cat#A11105-01), trypsin (Cat# 15090-046), Dulbecco’s Modified Eagle Medium (DMEM) (Cat# 21969-035), LysoTracker Deep Red (cat#L12492) and 50X MEM amino acid solution (Cat# 11130-036) were purchased from Thermo Fisher Scientific. Rapamycin (Cat# R-5000) and Bafilomycin A1 (Cat# B-1080) were obtained from LC laboratories.
Experimental Model and Subject Details
All in vivo experiments were performed using male C57BL/6 Dram-1 whole body knock-out or male C57BL/6 wild-type mice as approved by the Glasgow University Animal Welfare and Ethical Review Body and in accordance with UK Home Office guidelines. Mice were placed five per cage with free access to water and food. All experimental studies were started with animals at 10 weeks of age, were randomized and did not involve blinding.
For in vitro studies, E13.5 embryos from wild-type mice containing two floxed Dram-1 alleles or E13.5 embryos from intercross of DRAM1 hemizygous mice containing two floxed Atg7 alleles were used to generate Mouse Embryonic Fibroblasts (MEFs) with the indicated genotypes prior to immortalization by serial passages. Experiments were also conducted in Saos2 TetOn-DRAM1 cells (a derivative of Saos2 osteosarcoma cells, which can be obtained from ATCC (Cat# HTB-85) and the pre-adipocyte 3T3-L1 (ATCC, Cat# CL-173).
Cell culture
Cell lines were grown in DMEM supplemented by 10% FBS, 2 mM glutamine, 100 μg/mL streptomycin and 100 U/mL penicillin (complete DMEM) at 37°C 5% CO2. Saos2 TetOn-DRAM1 cells were either treated 24h with 1 μg/mL of doxycycline or left untreated prior to perform experiments. For amino acid-induced mTOR re-activation experiments, cells were washed twice in PBS, starved in EBSS containing or not 2 mM glutamine for 3 to 6 h and then incubated for the indicated times in EBSS containing specified treatments. 3T3-L1 pre-adipocyte cell line was obtained from ATCC.
Retrovirus/ Lentivirus production and infection
Retrovirus and lentivirus were produced using Phoenix-AMPHO cells (ATCC, Cat#: CRL-3213) and HEK293T cells respectively using calcium/phosphate transfection protocol. For lentivirus production, cells were transfected overnight with lentiviral, packaging and envelope plasmids (pPAX2 and pVSVG). While for retrovirus production, Phoenix-AMPHO cells were only transfected with retroviral constructs overnight. The following day, media were replaced by full DMEM containing 20% FBS for 48 h. Then media containing viruses were collected, filtered (0.45 μm), supplemented with 4 μg/ml Polybrene and transferred to the recipient cells. In the meantime, HEK293T (or Phoenix-AMPHO) cells were kept in DMEM containing 20%FBS for an additional 24 h in order to proceed to a second infection of recipient cells as described before. Finally, infected cells were selected with 1 μg/mL of puromycin for 10 days.
Constructs, shRNAs, siRNAs and CRISPR
SLC3A2-mRFP-eGFP and mRFP-eGFP-SLC3A2 were made by means of Gibson assembly using pLX304 SLC3A2 (DNASU plasmid repository) plasmid and the previously described mRFP-eGFP-LC3 construct (1). pLX304 SLC1A5 plasmid was obtained from DNASU plasmid repository. LAMP1-mRFP-Flag2X construct was obtained from Addgene (cat#34611) (2) and pCMV6-mouse-DRAM1 (Myc-DDK-tagged) was obtained from Origene (cat# MR220640). shRNA against Renilla (Non-targeting control), SLC1A5 and SLC7A5 were provided by Mirimus (cat#SLC1A5_798, SLC1A5_1062, SLC7A5_1857, SLC7A5_2000). ON-TARGETplus Mouse SLC1A5 smartpool siRNA (cat#L-056095-01-0005), ON-TARGETplus Mouse SLC7A5 smartpool siRNA (cat#L-041166-01-0005), ON-TARGETplus Human SCAMP3 siRNA (cat#L-013442-00-0005) and non-targeting controls (NTCs, cat#D-001810-01-05, D-001810-02-05, D-001810-03-05, D-001810-04-05) were obtained from GE Dharmacon.
The following sgRNA sequences were used in this study:
Mouse DRAM1 #A: CATCATCTCCTACGTGGTCG
Mouse DRAM1 #B: AGAGAGCACCGCGACCACGT
Non-targeting control #1 (NTC1): GTAGCGAACGTGTCCGGCGT (Wang et al., 2014)
Non-targeting control #2 (NTC2): GCTTGAGCACATACGCGAAT (Wang et al., 2014)
Protein extraction and Immunoblotting
Cells were washed in ice cold PBS and lysed in 1% Triton X-100, 0.1% SDS, 50 mM HEPES pH7.5, 150 mM NaCl, 100 mM NaF, 10 mM EDTA supplemented with HALT protease and phosphatase inhibitor cocktail (cat#87786,Thermo scientific). After 10 min centrifugation at 12000g at 4°C, the concentration of solubilised proteins was determined with the bicinchoninic acid assay (BCA). Protein samples were mixed with Laemmli loading buffer 4X (120 mM Tris pH6.8, 20% glycerol, 4% SDS, bromophenol blue) and subjected to Tris-Glycine SDS-PAGE. Then proteins were transferred onto PVDF membranes and immunoblotted using different antibodies using 1/1000 dilution: Anti-Myc tag (cat#05-724, Millipore), ERK2 (cat#sc-154, Santa Cruz biotechnology), SLC3A2 (cat#sc-9160, Santa Cruz biotechnology), LAMP2 (CD107b, cat#555803, BD PharMingen), SCAMP3 (cat# 26888-1-AP, Proteintech), Flag (cat#3165, Sigma-Aldrich). Anti- SLC1A5 (cat#5345), SLC7A5 (cat#5347), LC3B (cat#2775), Phospho-p70 S6 Kinase (Thr-389, cat#9234), p70 S6 Kinase (cat#2708), Phospho-S6 Ribosomal protein (S235/236, cat#4858), V5-Tag mAb (cat#13202), S6 Ribosomal protein (cat#2317), Phospho-4E-BP1 (Thr37/46, cat#2855), 4E-BP1 (cat#9644), Atg7 (cat#8558), Phospho-AKT (Thr308, cat#4056), AKT (cat#9272) PPARγ (cat#2430), IRS1 (cat#2382), Phospho-IRS1 (S636/639, cat#2388) and anti-mouse or anti-rabbit IgG HRP-linked (cat#7076S, cat#7074S, 1/5000) were from Cell Signaling Technology. Loading controls (ERK2 and actin) as well as total S6K, total AKT, total S6, total 4E-BP1 were probed following membrane stripping.
DRAM-1 immunoprecipitation
Saos2 TetOn-DRAM1 cells pre-treated with or without doxycycline were grown for 3-5 h in complete DMEM or EBSS. Cells were washed in ice cold PBS and lysed in 1.2% CHAPS, 150 mM NaCl, 50 mM HEPES pH 7.2 buffer supplemented by Complete protease inhibitors (Roche) for 20 minutes. Solubilised proteins were collected after centrifugation at 10000g for 10 minutes. 1 mg of total proteins, assessed by BCA, were mixed with 2 μg of anti-myc-tag antibody, incubated for 1 hour at 4°C prior to incubation overnight in presence of 50 μL of Protein A-Sepharose beads. Beads were washed 5 times using protein extraction buffer and elution was performed using Laemmli loading buffer. Samples were subjected to SDS-PAGE, transferred on PVDF membranes and immunoblotted against indicated proteins.
TAP-Tag experiment
For the TAP-Tag experiments, immunoprecipitated proteins were eluted from the sepharaose beads using 1% SDS and separated by SDS-PAGE, which was subsequently stained with Coomassie blue. Each gel lane was divided in 5 slices and digested according to a procedure previously described (McGarry et al., 2016). Briefly, the bands were excised and washed twice with 50 mM ammonium bicarbonate followed by 50 mM ammonium bicarbonate with 50% acetonitrile. Proteins were reduced using 10 mM DTT at 54°C for 30 minutes and alkylated with 55 mM iodoacetamide at room temperature for 45 minutes. Gel pieces were washed again with 50 mM ammonium bicarbonate and finally 50 mM ammonium bicarbonate with 50% acetonitrile. Gels were then dehydrated using 100% acetonitrile followed by drying in a speedvac. 5 μg/mL trypsin in 25 mM ammonium bicarbonate was added and incubated for 12 hours at 30°C. Tryptic peptides were extracted from gel pieces with two 50% v/v acetonitrile/water washes and evaporated to dryness prior to reconstituting in mobile phase for running.
For one of the replicates, the tryptic digest obtained was separated by nanoscale C18 reverse-phase liquid chromatography (EASY-nLC II Thermo Fisher Scientific) coupled to a Linear Trap Quadrupole (LTQ) Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Samples were loaded on a pre-column (C-18 Biosphere 5 μm, 120 Å–200 μm × 0.2 cm) for desalting, and subsequently eluted, at a flow of 0.6 μl/min, into an analytical column (C-18 Biosphere 5 μm, 120 Å–100 μm × 15 cm).
The eluting peptide solutions were electrosprayed into the mass spectrometer via a nanoelectrospray ion source. General mass spectrometric conditions were as follows: spray voltage, 2.1 kV, ion transfer tube temperature, 200°C. The mass spectrometer was operated in positive ion mode and used in data-dependent acquisition mode. A full scan (FT-MS) was acquired with resolution of 30,000 over mass range of 350–2000 amu, and the top ten most intense ions were selected for fragmentation using higher energy collision dissociation (HCD). Fragmentation spectra in HCD were acquired in the FTMS analyzer at a resolution of 7500 in centroid mode. Normalized collision energy used was 35. Former target ions selected for MS/MS were dynamically excluded for 60 s. The ‘lock mass’ function (lock mass = 445.120036 Da) was enabled for MS and MS/MS HCD scan modes.
For two other replicates, the tryptic digests were analyzed using an Ultimate 3000 nano-RP-LC (Dionex) coupled to a Q-Star XL (Applied Biosystems, Concorde, Canada). Samples were loaded onto a PepMap100 trap column at a flow of 20 μl/min of RP-LC buffer A (A: 5% ACN, 0.1% formic acid) and then separated using a PepMap C18 75-μm inner diameter × 15-cm analytical column. Analyst software was used for data-dependant acquisition. The basic survey scan (1 s) from 400 to 1200 Da was followed by four Enhanced Precursor Ion scans (1 s).
Data acquired with Q-star were exported from Analyst using the script Mascot.dll 1.6b23(Matrix Science, London, UK). Raw data obtained from Orbitrap were processed with Raw2Msm MGF files were searched using Mascot (Matrix Science), querying UniProt (UniProt Consortium, 2010) Homo sapiens plus an in-house database containing common proteomic contaminants and the sequence of Streptavidin-FLAG–DRAM-1. Mascot was searched assuming the digestion enzyme trypsin allowing for two miscleavages with a fragment ion mass tolerance of 0.1 Da and a parent ion tolerance of 35 ppm for Q-Star data and 10 ppm for Orbitrap data. The iodoacetamide derivative of cysteine was specified in Mascot as a fixed modification. Oxidation of methionine and Protein N-terminal acetylation were considered as variable modifications. Scaffold (version 4.3.2, Proteome Software) was used to summarize MS/MS based peptide and protein identifications.
Metabolite Extraction
Cells were rapidly washed 3 times with chilled PBS. Intracellular metabolites were extracted, in triplicate, by adding a volume equivalent to 1x106 cells/ml with extraction solution at 4°C (methanol, acetonitrile, and water 5:3:2) and incubating the plate on a rocking shaker for 5 minutes at 4°C. The intracellular extract was centrifuged at 16100 g for 10 min at 4°C and the supernatants were transferred into HPLC vials and stored at −80°C until LC-MS analysis.
LC-MS Analysis
A Q Exactive Orbitrap mass spectrometer (Thermo Scientific, Waltham, MA, USA) was used together with a Thermo Ultimate 3000 HPLC system. The HPLC setup consisted of a ZIC-pHILIC column (SeQuant, 150 × 2.1mm, 5 μm, Merck KGaA, Darmstadt, Germany), with a ZIC-pHILIC guard column (SeQuant, 20 × 2.1mm) and an initial mobile phase of 20% 20mM ammonium carbonate, pH 9.4, and 80% acetonitrile. Cell and media extracts (5 μl) were injected and metabolites were separated over a 15 minute mobile phase gradient, decreasing the acetonitrile content to 20%, at a flow rate of 200μL/min and a column temperature of 45°C. The total analysis time was 23 minutes. All metabolites were detected across a mass range of 75-1000 m/z using the Q Exactive mass spectrometer at a resolution of 35000 (at 200 m/z), with electrospray (ESI) ionization and polarity switching to enable both positive and negative ions to be determined in the same run. Temperature at the hESI source was 50°C and spray voltage was 4500V for positive mode and ˗3500V for negative mode. Sheath gas was 25 arbitrary units and auxiliary gas 15 arbitrary units for both ionization modes. A capillary temperature of 325°C was used for both positive and negative ionization modes. Lock masses were used and the mass accuracy obtained for all metabolites was below 5ppm. Data were acquired with Thermo Xcalibur software. The peak areas of different metabolites were determined using Thermo TraceFinder software where metabolites were identified by the exact mass of the singly charged ion and by known retention time on the HPLC column. Commercial standards of all metabolites detected had been analyzed previously on this LC-MS system with the pHILIC column. Intracellular metabolites were normalized to protein content of the cells, measured at the end of the experiment by the Lowry assay.
Confocal microscopy
Dram-1flox/flox MEF stably expressing Cre recombinase or a control vector and SLC3A2-mRFP-GFP or GFP-mRFP-SLC3A2 constructs were grown on glass coverslips for 48 h in regular DMEM and treated for 30 min in presence of LysoTracker Deep Red (L12492, ThermoFisher) followed by 3 washes of 10 min in regular DMEM media. Saos Tet-ON DRAM1-myc-his cells overexpressing V5-tagged SLC1A5 were grown on coverslips for 24hr in the presence or absence of doxycycline (1ug/ml), then treated for four h with cycloheximide (100ug/ml) or anisomycin (10ug/ml). Cells were fixed in 4% paraformaldehyde, permeabilized in PBS, 1%BSA, 0.2% Triton X-100 and stained for LAMP2 and V5 according to previously described protocol (Mrschtik et al., 2015). Coverslips were mounted on slides using DAKO mounting medium containing DAPI and z stack images were taken by mean of a ZEISS 710 confocal microscope with a × 63 objective. Quantification of lysosome volume and the proportion of lysosomes containing SLC3A2 was undertaken using Volocity 3D Imaging analysis software (Perkin-Elmer)
Lysosomal enrichment protocol
Saos2 TetOn-DRAM1 cells stably expressing LAMP1-mRFP-Flag2X construct were used for lysosomal fractionation assays according to the previously described protocol (Zoncu et al., 2011). Briefly, cells were either treated for 24h with doxycyclin or left untreated prior to starvation containing glutamine or fed treatments for 5 h. Cells were first mechanically disrupted in fractionation buffer (50 mM KCl, 90 mM K gluconate, 1mM EGTA, 5mM MgCl2, 50 mM sucrose, 5mM glucose, 2.5mM ATP, 20 mM HEPES pH7.4 supplemented by Complete protease inhibitors) before to centrifuge samples at 2000g at 4°C for 10 min to obtain a postnuclear supernatant fraction (PNS). PNS samples were then centrifuged 15 min at 20600g at 4°C prior to resuspension of pellets in fractionation buffer. These lysosomal enrich fractions were then incubated with 50 μL of anti-Flag magnetic beads for 3h to overnight prior extensive washes with fractionation buffer. Samples were differently treated according to the following experiment: For western blot analysis, proteins were eluted using Laemmli loading buffer. For amino acid lysosomal content assessments, beads were incubated in metabolites extraction buffer (aqueous solution of 50% methanol and 30% acetonitrile) for 10 min on ice, then samples were quickly centrifuged to pellet beads and to collect buffer containing lysosomal metabolites. Samples were analyzed by means of LC-MS as described above.
Cell proliferation
Cells were plated at a density of 10000 cells per well in 6-well plates for Dram-1flox/flox MEF expressing cre recombinase or a control vector while Atg7flox/flox Dram-1 knock-out or wild-type MEF were seeded at 20000 cells per well. Cell number was determined at the specified time after cells have been washed in PBS, trypsinized, re-suspended and counted using Casy Innovatis cell counter.
Flow cytometric analysis
Dram-1flox/flox MEF stably expressing cre recombinase or a control vector were infected with retroviral particles encoding SLC3A2-mRFP-GFP or GFP-mRFP-SLC3A2 constructs, followed by a week of antibiotic selection. Cells stably expressing the indicated construct were grown under control condition or starved for 3 h. Cells were then washed in PBS, harvested using Accutase and analyzed for GFP and mRFP mean fluorescent intensities (MFI). For each condition and cell lines, GFP MFI were normalized to mRFP MFI. Each ratio has been compared to the ratio from cells grown under control condition and expressing both DRAM1 and SLC3A2-mRFP-GFP.
In vitro insulin sensitivity
Cells were starved overnight from growth factors using DMEM containing glutamine, penicillin, streptomycin and 0.2% FBS. The following day cells were treated with or without 5 nM insulin for 15 minutes. Cells were washed in ice cold PBS and protein extract were made as described previously. Where indicated, cells were pre-incubated in media containing 100 nM rapamycin for 4 h prior to insulin treatments.
siRNA transfection
5 × 105 Dram-1flox/flox MEF cells stably expressing cre recombinase or a control vector were plated in 10 cm dishes. The following day cells are transfected with smart pool siRNAs using oligofectamine reagent. Briefly, 14.7 μL of 20 μM siRNA resuspended in 5 × siRNA Buffer (GE Dharmacon) are mixed with 1085 μL of Optimem. A mix of 24 μL oligofectamine with 66 μL Opti-mem was incubated for 5-10 minutes at room temperature prior addition with the first siRNA mix. After 20 minutes incubation at room temperature, the mix was added on cells for which media was previously replaced by 4.8 mL of Optimem. Cells were incubated overnight in presence of siRNAs before replacing media by regular growing media for 24 h. Cells were then plated according to the desired experiment.
Animal experiments
Mice were fed a western diet (cat# T-5TJN-1810842 from TestDiet® Limited) for the indicated time. Rapamycin was administrated (4mg/kg) by intraperitoneal injection twice a week for the duration of the experiment. Cohorts of 10 males for each group, and with the same age, were used to perform these experiments and mouse weights were measured weekly.
Mice were starved for 4 h prior to take basal glycemia readings from bleeding tails. Measurements were performed using Accu-check test strips and meter (Roche). Then mice were given either 3g/kg glucose by oral gavage or 0.5U/kg of insulin by intraperitoneal injections and blood glucose was assessed every 20 minutes for 2 h.
Immunohistochemistry
Mice were killed by CO2 mediated euthanasia before collecting different organs and tissues. Tissues were weighed prior to a minimum 24 h fixation step in 10% neutral-buffered formaldehyde. Paraffin-embedded sections were treated as previously described (Sakamaki et al., 2017). Briefly, sections were successively dipped into xylene (5 min), 100% ethanol (2 × 1 min), 70% ethanol (1 min) and deionized water for 5 min. Antigen retrieval was proceeded using Sodium citrate retrieval buffer pH 6 (cat#: TA-250-PM1X, Thermo Fisher Scientific) at 98°C for 25 min followed by 2 washes in Tris buffered saline and tween 20 (TBS-T). Sections were then blocked (Peroxidase-blocking solution, cat#: S2023, Dako) prior incubation overnight at 4°C with anti-Phospho AKT (cat#4060, Cell Signaling), Phospho-AKT-T308 (cat#4056, Cell signaling), or Phospho-S6k1-T389 (cat#orb6617, Biorbyt). Sections were then incubated with Labeled Polymer HRP anti-rabbit (cat#K4011, Dako). Incubation in DAB (3,3′-diaminobenzidine tetrahydrochloride, cat#3468, Dako) was used to reveal staining. Sections were mounted and observed with a Zeiss Axio Scope.A1 microscope (Zeiss) and ZEN 2012 software.
Plasma adiponectin levels
Plasma adiponectin levels were assessed using ELISA kit from Abcam (cat#ab108785) according to manufacturer’s protocols. Briefly, at the end of the HFD experiment, mice were killed by CO2-mediated euthanasia and blood samples were collected by direct cardiac puncture. Samples were stored at −20°C following 10 min centrifugation at 1500 x g and 4°C. Each sample was analyzed in duplicate using ELISA kits before to plot and to quantify the average of plasma adiponectin levels of each group.
3T3-L1 differentiation
1x105 cells were plated in each well of 6-well plates. Cells were grown with full DMEM media until they reached 100% confluence. Media was then replaced and cells were kept in full DMEM media for 2 additional days. Then they were incubated in full DMEM supplemented with 500 nM dexamethasone, 500 μM IBMX, 5 μg/mL insulin and various concentrations of troglitazone (0; 0.4; 4; 40 μM) for 2 days. The media was replaced by full DMEM supplemented only with 5 μg/mL insulin for 2 more days. Cells were then kept under full DMEM for an additional day before performing further experiments.
Oil Red O staining
Cells were washed in PBS prior to two successive incubations in 10% neutral-buffered formaldehyde for 10 and 60 min. Cells were rinsed twice in deionised water and dried following 5 min incubation in 60% ethanol. Neutral lipids were then stained using Oil Red O solution (Sigma) for 10 minutes. Excess of Oil Red O solution was eliminated by at least 5 washes with deionized water. Images were taken, plates were dried before solubilise staining using 100% isopropanol. Optical densities at 500 nm wavelength were read against a solution of 100% isopropanol.
RT-qPCR
RNA were extracted from cells using RNeasy ® Mini Kit (cat#74101, QIAGEN) and quantified using Nanodrop (ThermoFisher scientific). cDNAs were produced using High Capacity RNA-to-cDNA kit (Appliedbiosystems from Thermo Fisher Scientific) according to manufacturer’s protocol and qPCR were performed using DyNAmo HS SYBR Green qPCR Kit (cat#F-410, Thermo Fisher Scientific). PCR was performed on a C1000 Thermal Cycler (CFX96 Real time system, BioRad) as follows: 3 min at 95 °C, followed by 40 cycles of 20 s at 95 °C, 30 s at 58 °C, 20 s at 72 °C and a final 5 min at 72 °C. mRNA quantifications were calculated using ΔΔCt method.
Mouse primer sequences (5′ to 3′):
| DRAM1 fwd | CAGCCTTCATCATCTCCTACG |
| DRAM1 rev | ATGCAGAGAAGTTTATCATG |
| Adiponectin fwd | TGTTCCTCTTAATCCTGCCCA |
| Adiponectin rev | CCAACCTGCACAAGTTCCCTT |
| SLC1A5 fwd | GAAGAATGGTGTGGCCAAACAC |
| SLC1A5 rev | CTCTGAGCTCGGCATCTTGG |
| SLC7A5 fwd | CTTCGGCTCTGTCAATGGGT |
| SLC7A5 rev | TTCACCTTGATGGGACGCTC |
| Actin fwd | CTAAGGCCAACCGTGAAAAG |
| Actin rev | ACCAGAGGCATACAGGGACA |
| 18S fwd | GTAACCCGTTGAACCCCATT |
| 18S rev | CCATCCAATCGGTAGTAGCG |
Quantification and Statistical Analysis
All in vitro experiments were repeated at least three independent times and results are shown as mean ± SEM. All in vivo experiments are shown as mean ± SEM or SD. Statistical significances were determined by two-tailed unpaired Student’s t test for two group comparison and 2way ANOVA with Tukey or Dunnett for multiple group comparison. Results were considered statistically different when p values < 0.05 (∗); < 0.01 (∗∗); < 0.001 (∗∗∗). When required, no significance between results are mentioned as N.S.
Data and Code Availability
Full scans of western blot data and original microscopy images have been deposited in Mendeley Data (https://doi.org/10.17632/37tgjx44g4.1).
Acknowledgments
This work was supported by Cancer Research UK (A17196), Worldwide Cancer Research (16-1194), and INRA (UMR1419 NuMeA). We thank the Core Services and Advanced Technologies at the Cancer Research UK Beatson Institute (C596/A17196), with particular thanks to the Beatson Advanced Imaging Resource, Biological Services Unit, Histology and Proteomics Facility.
Author Contributions
F.B., J.S.L., and K.M.R. conceived the study and/or designed experiments. F.B., J.O., V.J.A.B., B.Z., J.-P.P., A.M.B., M.O., E.K., P.S.G., R.M., L.Y.L., C.N., J.L., and S.W.G.T. conducted and/or analyzed experiments. F.B. and K.M.R. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 3, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2019.07.021.
Supplemental Information
References
- Alessi D.R., James S.R., Downes C.P., Holmes A.B., Gaffney P.R., Reese C.B., Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Bowman E.J., Siebers A., Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA. 1988;85:7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E.J., Albers M.W., Shin T.B., Ichikawa K., Keith C.T., Lane W.S., Schreiber S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Burnett P.E., Barrow R.K., Cohen N.A., Snyder S.H., Sabatini D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crighton D., Wilkinson S., O’Prey J., Syed N., Smith P., Harrison P.R., Gasco M., Garrone O., Crook T., Ryan K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Dunn W.A., Jr. Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J. Cell Biol. 1990;110:1935–1945. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D.F., Shackelford D.B., Mihaylova M.M., Gelino S., Kohnz R.A., Mair W., Vasquez D.S., Joshi A., Gwinn D.M., Taylor R. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esslinger C.S., Cybulski K.A., Rhoderick J.F. Ngamma-aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg. Med. Chem. 2005;13:1111–1118. doi: 10.1016/j.bmc.2004.11.028. [DOI] [PubMed] [Google Scholar]
- Gloeckner C.J., Boldt K., Schumacher A., Roepman R., Ueffing M. A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics. 2007;7:4228–4234. doi: 10.1002/pmic.200700038. [DOI] [PubMed] [Google Scholar]
- Goberdhan D.C., Wilson C., Harris A.L. Amino acid sensing by mTORC1: intracellular transporters mark the spot. Cell Metab. 2016;23:580–589. doi: 10.1016/j.cmet.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green H., Kehinde O. An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell. 1975;5:19–27. doi: 10.1016/0092-8674(75)90087-2. [DOI] [PubMed] [Google Scholar]
- Gwinn D.M., Shackelford D.B., Egan D.F., Mihaylova M.M., Mery A., Vasquez D.S., Turk B.E., Shaw R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J.M., Jeong S.J., Park M.C., Kim G., Kwon N.H., Kim H.K., Ha S.H., Ryu S.H., Kim S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012;149:410–424. doi: 10.1016/j.cell.2012.02.044. [DOI] [PubMed] [Google Scholar]
- Hara K., Yonezawa K., Weng Q.P., Kozlowski M.T., Belham C., Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- Harrington L.S., Findlay G.M., Gray A., Tolkacheva T., Wigfield S., Rebholz H., Barnett J., Leslie N.R., Cheng S., Shepherd P.R. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C.H., Jun C.B., Ro S.H., Kim Y.M., Otto N.M., Cao J., Kundu M., Kim D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenzelmann Broz D., Spano Mello S., Bieging K.T., Jiang D., Dusek R.L., Brady C.A., Sidow A., Attardi L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016–1031. doi: 10.1101/gad.212282.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S., Noda T., Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C.M., Adams P.D., Adeli K. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M., Horvat S., Festuccia W.T., Birsoy K., Prevorsek Z., Efeyan A., Sabatini D.M. DEPTOR cell-autonomously promotes adipogenesis, and its expression is associated with obesity. Cell Metab. 2012;16:202–212. doi: 10.1016/j.cmet.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.K., Fitter S., Dutta A.K., Matthews M.P., Walkley C.R., Hall M.N., Ruegg M.A., Gronthos S., Zannettino A.C. Brief report: the differential roles of mTORC1 and mTORC2 in mesenchymal stem cell differentiation. Stem Cells. 2015;33:1359–1365. doi: 10.1002/stem.1931. [DOI] [PubMed] [Google Scholar]
- McGarry D.J., Shchepinova M.M., Lilla S., Hartley R.C., Olson M.F. A cell-permeable biscyclooctyne as a novel probe for the identification of protein sulfenic acids. ACS Chem. Biol. 2016;11:3300–3304. doi: 10.1021/acschembio.6b00742. [DOI] [PubMed] [Google Scholar]
- Milkereit R., Persaud A., Vanoaica L., Guetg A., Verrey F., Rotin D. LAPTM4b recruits the LAT1-4F2hc Leu transporter to lysosomes and promotes mTORC1 activation. Nat. Commun. 2015;6:7250. doi: 10.1038/ncomms8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrschtik M., O’Prey J., Lao L.Y., Long J.S., Beaumatin F., Strachan D., O’Prey M., Skommer J., Ryan K.M. DRAM-3 modulates autophagy and promotes cell survival in the absence of glucose. Cell Death Differ. 2015;22:1714–1726. doi: 10.1038/cdd.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P., Bergman P., Zhang B., Triantafellow E., Wang H., Nyfeler B., Yang H., Hild M., Kung C., Wilson C. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmigiani A., Nourbakhsh A., Ding B., Wang W., Kim Y.C., Akopiants K., Guan K.L., Karin M., Budanov A.V. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014;9:1281–1291. doi: 10.1016/j.celrep.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabanal-Ruiz Y., Otten E.G., Korolchuk V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017;61:565–584. doi: 10.1042/EBC20170027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsamen M., Pochini L., Stasyk T., de Araújo M.E., Galluccio M., Kandasamy R.K., Snijder B., Fauster A., Rudashevskaya E.L., Bruckner M. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature. 2015;519:477–481. doi: 10.1038/nature14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves J.P. Accumulation of amino acids by lysosomes incubated with amino acid methyl esters. J. Biol. Chem. 1979;254:8914–8921. [PubMed] [Google Scholar]
- Sabatini D.M., Erdjument-Bromage H., Lui M., Tempst P., Snyder S.H. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- Sakamaki J.I., Wilkinson S., Hahn M., Tasdemir N., O’Prey J., Clark W., Hedley A., Nixon C., Long J.S., New M. Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol. Cell. 2017;66:517–532.e9. doi: 10.1016/j.molcel.2017.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y., Bar-Peled L., Zoncu R., Markhard A.L., Nada S., Sabatini D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshige K., Baba M., Tsuboi S., Noda T., Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan V.P., Miyamoto S. Nutrient-sensing mTORC1: Integration of metabolic and autophagic signals. J. Mol. Cell. Cardiol. 2016;95:31–41. doi: 10.1016/j.yjmcc.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I., Mizushima N., Kiyooka M., Ohsumi M., Ueno T., Ohsumi Y., Kominami E. Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol. Biol. Cell. 1999;10:1367–1379. doi: 10.1091/mbc.10.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay F., Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J. Biol. Chem. 2001;276:38052–38060. doi: 10.1074/jbc.M106703200. [DOI] [PubMed] [Google Scholar]
- Tzatsos A. Raptor binds the SAIN (Shc and IRS-1 NPXY binding) domain of insulin receptor substrate-1 (IRS-1) and regulates the phosphorylation of IRS-1 at Ser-636/639 by mTOR. J. Biol. Chem. 2009;284:22525–22534. doi: 10.1074/jbc.M109.027748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzatsos A., Kandror K.V. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol. Cell. Biol. 2006;26:63–76. doi: 10.1128/MCB.26.1.63-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um S.H., Frigerio F., Watanabe M., Picard F., Joaquin M., Sticker M., Fumagalli S., Allegrini P.R., Kozma S.C., Auwerx J., Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Tsun Z.Y., Wolfson R.L., Shen K., Wyant G.A., Plovanich M.E., Yuan E.D., Jones T.D., Chantranupong L., Comb W. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science. 2015;347:188–194. doi: 10.1126/science.1257132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson R.L., Chantranupong L., Saxton R.A., Shen K., Scaria S.M., Cantor J.R., Sabatini D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351:43–48. doi: 10.1126/science.aab2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyant G.A., Abu-Remaileh M., Wolfson R.L., Chen W.W., Freinkman E., Danai L.V., Vander Heiden M.G., Sabatini D.M. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell. 2017;171:642–654.e12. doi: 10.1016/j.cell.2017.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagida O., Kanai Y., Chairoungdua A., Kim D.K., Segawa H., Nii T., Cha S.H., Matsuo H., Fukushima J., Fukasawa Y. Human L-type amino acid transporter 1 (LAT1): characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta. 2001;1514:291–302. doi: 10.1016/s0005-2736(01)00384-4. [DOI] [PubMed] [Google Scholar]
- Yeh W.C., Bierer B.E., McKnight S.L. Rapamycin inhibits clonal expansion and adipogenic differentiation of 3T3-L1 cells. Proc. Natl. Acad. Sci. USA. 1995;92:11086–11090. doi: 10.1073/pnas.92.24.11086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., McPhee C.K., Zheng L., Mardones G.A., Rong Y., Peng J., Mi N., Zhao Y., Liu Z., Wan F. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.H., Huang J., Düvel K., Boback B., Wu S., Squillace R.M., Wu C.L., Manning B.D. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS ONE. 2009;4:e6189. doi: 10.1371/journal.pone.0006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R., Bar-Peled L., Efeyan A., Wang S., Sancak Y., Sabatini D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Full scans of western blot data and original microscopy images have been deposited in Mendeley Data (https://doi.org/10.17632/37tgjx44g4.1).