

Abstract
Previous in vivo studies established that inactivated Francisella tularensis immune complexes (mAb-iFt) are a more protective vaccine against lethal tularemia than iFt alone. Subsequent in vitro studies revealed enhanced DC maturation marker expression with mAb-iFt stimulation. The goal of this study was to determine the mechanism of enhanced DC maturation. Multiparameter analysis of surface marker expression and cytokine secretion demonstrates a requirement for FcγR signaling in enhanced DC maturation. MyD88 was also found to be essential for heightened DC maturation, implicating MyD88-dependent TLRs in DC maturation. Upon further study, we discovered that TLR2 drives cytokine secretion, but surprisingly TLR9 is required for DC maturation marker upregulation. These studies reveal a separation of DC cytokine and maturation marker induction pathways and demonstrate that FcγR-TLR/MyD88 synergy underlies the enhanced dendritic cell maturation in response to the mAb-iFt vaccine.
Keywords: dendritic cell maturation, flow cytometry, immune complex, Francisella tularensis, TLR 2, TLR 9, MyD88, Fcγ Receptors
Graphical Abstract
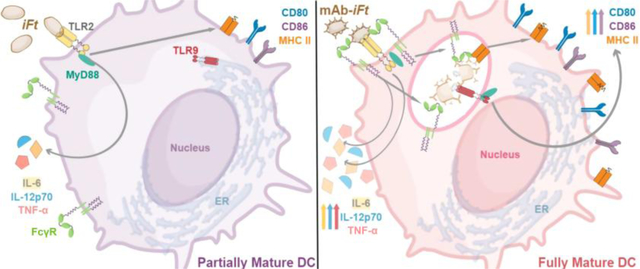
1. Introduction*
According to the World Health Organization, vaccines prevent 2 – 3 million deaths per year, however there are many infectious diseases for which there are no vaccine, including tularemia [1]. Respiratory tularemia, which can be fatal in up to 60% of cases, is caused by the intracellular bacterium Francisella tularensis (Ft) [2]. We have previously demonstrated that opsonizing the inactivated Ft with an IgG monoclonal antibody enhances immunity and protection against mucosal challenge with Ft [3]. This vaccine approach is also known as an immune complex. Immune complexes have been shown to be potent activators of immunity both in the context of chronic inflammatory/autoimmune diseases (i.e. atherosclerosis, multiple sclerosis, lupus, rheumatoid arthritis, etc.) as well as vaccine responses [4–8]. It is therefore important to study how immune complexes elicit immune reactions in order to design effective vaccines with minimal side-effects.
Dendritic cells (DCs) are key players in eliciting adaptive immune cell activation; they link the innate and adaptive arms of immunity by presenting antigen and secreting factors including cytokines. Because activation of T cells relies heavily on their interaction with DCs, the type of adaptive response generated is largely dependent on the DC maturation status [9]. At steady state, most DCs are considered immature; they are highly phagocytic and express very low levels of co-stimulatory molecules (e.g. CD40, CD80, CD83, & CD86). At this stage, MHC-restricted antigen presentation to T cells without co-stimulatory molecules results in tolerogenic responses. To elicit an immunogenic response, DCs must recognize an antigen as foreign through various pattern recognition receptors (PRRs). Signaling through these receptors will trigger induction of DC maturation which includes: increased proteolytic processing, decreased phagocytic capacity; increased surface expression of major histocompatibility complex class II (MHC II) and co-stimulatory molecules; and migration of DCs to lymph nodes where they can present antigen to T cells [10, 11]. Studies using mAb-iFt-stimulated DCs/macrophages have shown increased maturation marker expression, secretion of IL-1β & TNF-03B1, and increased presentation to T cells [12–14]. We therefore sought to define the cellular/molecular mechanism of mAb-iFt-induced DC maturation.
Fc receptors are one type of receptor which assist DC recognition of mAb-iFt; they bind the constant region (Fc) of antibodies thus, allowing the DC to bind and take up immune complexes. mAb-iFt, contains mouse IgG2a so our focus is on Fc receptors which bind IgG. There are three different classes of receptors which bind IgG: FcγRs, FcRn, and TRIM21 [15]. FcγRs are the only surface-expressed IgG receptor and therefore, likely the first of the three to bind IgG immune complexes. In mice there are four FcγRs: FcγRI, IIb, III, and IV; all of which, except FcγRIIb, are considered activating receptors based on the presence of an Immunoreceptor Tyrosine-based Activation Motif (ITAM) within the receptors’ associated γ-chain. FcγRIIb, does not associate with Fc receptor γ-chain and instead contains an Immunoreceptor Tyrosine-based Inhibition Motif (ITIM) within its cytoplasmic tail. Because the activating receptors contain an ITAM, their signaling cascades are very similar to other receptors containing ITAMs (e.g. B cell receptor). Upon ligation and crosslinking of an activating FcγR, tyrosine residues in the ITAM are phosphorylated by an active Src family kinase. This phosphorylation creates a docking site for active spleen tyrosine kinase (Syk) which subsequently phosphorylates downstream signaling mediators such as phosphatidylinositol-3kinase (PI3K) [15]. It is important to note however, that mAb-iFt could be recognized by receptors other than FcγRs.
Another receptor family involved in DC recognition of antigen is toll-like receptors (TLRs). TLRs are part of the pattern recognition receptor family which recognize repeating patterns within pathogens as foreign. The recognition of pathogens through TLRs triggers signaling cascades that include adaptor proteins such as myeloid differentiation primary response 88 (MyD88), kinases like interleukin-1 receptor-associated kinases (IRAKs), et cetera which ultimately lead to activation of transcription factors such as nuclear factor kappa-light-chain-enhances of activated B cells (NF-κB). The signaling cascades triggered by TLR ligation can elicit DC maturation, migration, and therefore an enhanced ability to trigger immunogenic responses in adaptive immune cells. Studies have shown that macrophages stimulated with mAb-iFt have increased IL-6 and IL-1β secretion because iFt can be recognized/bound by TLR2 [14, 16]. It is therefore likely that TLR ligation of mAb-iFt plays a role in DCs maturation.
In this study, we demonstrate a requirement for both MyD88 and FcR γ-chain ITAM signaling in the maturation of mAb-iFt-stimulated bone marrow-derived DCs (BMDCs). Fc receptor γ-chain deficient and WT cells treated with a FcR γ-chain signaling inhibitor have decreased maturation in mAb-iFt stimulated cells. In addition, we see massive reduction in the maturation of mAb-iFt-stimulated cells lacking MyD88. We explored this further and found, surprisingly, that mAb-iFt-stimulated BMDCs deficient in TLR2 have no changes in CD80, CD86, or MHC II expression when compared to the WT BMDCs. They do, however, have changes in IL-6, IL-2p70, and TNF-α secretion. Ultimately, we found that TLR9 is responsible for mAb-iFt-induced expression of DC maturation markers. These studies reveal a separation of DC cytokine and maturation marker induction pathways, demonstrating that both aspects need to be examined when studying DC maturation. In addition, the requirement for both FcR γ-chain ITAM signaling and TLR2/9 in mAb-iFt-induced BMDC maturation reveals a FcγR-TLR/MyD88 synergy that underlies enhanced dendritic cell maturation in response to the mAb-iFt vaccine.
2. Materials and Methods
2.1. Mice
Wildtype C57BL/6 mice were bred under pathogen free conditions in Albany Medical College’s Animal Resource Facility. In the few instances where our own WT mice were not available for use (Figure S1), C57BL/6 mice were ordered from Taconic Biosciences (Rensselaer, NY). TLR2−/− (B6.129-Tlr2tm1Kir/J), TLR4−/− (B6(Cg)-Tlr4tm1.2Karp/J), and MyD88−/− (B6.129P2(SJL)-Myd88tm1.1Defr/J) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). FcR-γ−/− (B6.129P2-Fcer1gtm1Rav N12) mice were obtained from Taconic Biosciences. Mice were provided with water and food ad lib. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC).
2.2. Reagents
Anti-F. tularensis LPS monoclonal antibody (mouse IgG2a) was purchased from Fitzgerald Industries (Acton, MA). Lipopolysaccharide from E. coli O111:B4 (LPS) was purchased from Millipore Sigma, (Burlington, MA). Poly(I:C), a TLR3 ligand; ODN 1826, a TLR9 agonist; ODN 2088, a TLR9 antagonist; and R406, a Syk inhibitor, were purchased from InvivoGen (San Diego, CA). Bio-Plex® kits were purchased from Bio-Rad (Hercules, CA). eFluor 450 conjugated anti-MHC Class II (I-A/I-E) Monoclonal Antibody (clone M5/114.15.2) was purchased from eBioscience (Waltham, MA). PE conjugated anti-CD80 Antibody (clone 16–10A1) and APC conjugated anti-CD86Antibody (clone Gl-1) were purchased from BioLegend (San Diego, CA). αTLR2 was purchased from InvivoGen. αFcγRI clone # 290322 was purchased from R&D Systems. αFcγRII/III (Mouse BD Fc Block) clone 2.4G2 was purchased from BD Biosciences (San Jose, CA). Mouse IgG2a Isotype Control clone UPC-10 for making mAb-beads was purchased from Millipore Sigma.
2.3. Buffers
ACK ammonium-chloride-potassium lysis buffer was composed of: 150mM NH4CL, 10mM KHCO3, 0.1mM EDTA. BMDC culture media included: 10% heat inactivated FBS (HyClone, Marlborough, MA), 1% 100mM sodium pyruvate (Millipore Sigma), 1% 200mM L-glutamine (Gibco), 1% MEM non-essential amino acids (Millipore Sigma), 1%, penicillin-streptomycin 100× solution (HyClone), 0.05mM 2-mercaptoethanol (Bio-Rad), RPMI 1640, and 50ng/mL recombinant mouse Flt3 ligand (R&D Systems, Minneapolis, MN).
2.4. Bone Marrow-derived Dendritic Cells
Femurs were removed from FcR γ-chain−/− (FcR-γ−/−), MyD88−/−, TLR2−/−, or wild-type C57BL/6 mice and crushed using a pestle and mortar containing RPMI1640 (Gibco, Gaithersburg, MD). Debris was removed via 70μm nylon cell strainer, and the cells recovered by centrifugation (200 × g, 5 min.). The resulting cell pellet was re-suspended in ACK lysis buffer, centrifuged, and re-suspended in BMDC culture media; the media was replaced after 3 days. At 6 days, cells were mostly non-adherent; adherent and settled cells were lifted via scraping. All cells (adherent and non-adherent) were then re-suspended in fresh BMDC culture media, plated in 6 well plates (1 × 106 cells/well), and incubated overnight before stimulation.
2.5. Generation of Stimuli
iFt:
Inactivation of Francisella tularensis (Ft) and generation of mAb-iFt complexes were done essentially as described [13]. Briefly, Ft were grown on chocolate agar plates (BD Biosciences), and a single colony was picked and expanded in Mueller Hinton Broth (MHB) (BD Biosciences) for 24 hours. The concentration of the bacteria was estimated by optical density (OD) at 610nm and Ft was once again expanded in MHB overnight. The resulting culture was washed twice in PBS and fixed in 2% methanol-free formaldehyde (MFF) (Millipore Sigma). This inactivated Ft (iFt) was washed in PBS and re-suspended at a concentration between 4 × 109 and 4 × 1010 bacteria/mL. Fixation Inactivation of Ft was confirmed by lack of growth on chocolate agar plates for 7 days.
mAb-iFt:
To generate mAb-iFt, 4 × 109 iFt were opsonized with 10 μg of anti-F. tularensis LPS monoclonal antibody in 1 mL PBS at 4°C overnight. mAb-iFt immune complexes were then washed twice in PBS, aliquoted at a concentration of 4 × 109 bacteria/mL, and stored at −20°C until use.
BSA- & mAb-beads:
Glass beads (2μm in diameter) were coated with either bovine serum albumin or mouse IgG2a isotype control via poly-L-lysine-dimethyl pimelimidate linkers as previously described [17].
2.6. BMDC stimulation
BMDCs (1 × 106) were stimulated with culture media, iFt (50/cell), mAb-iFt (50/cell), LPS (2.25ng/mL), BSA- or mAb-beads (5 – 30 per cell), Poly(I:C) (5μg/mL), or ODN 1826 (5μM) for 24 hours. For Syk inhibition studies, BMDCs were pretreated with 0.1, 0.3, 1, or 3μM of R406 for 30 minutes before stimulation. For αTLR2 treatment (10μg/mL) and TLR9 inhibition (10μM ODN 2088), BMDCs were pretreated for 1 hour before stimulation. For FcγR blocking, αFcγRI clone # 290322 and/or αFcγRII/III (Mouse BD Fc Block) clone 2.4G2 were added to the cells as a pre-treatment was for 20 minutes prior to stimulation.
2.7. Flow cytometry
Cells (5 × 105) were harvested, counted, and placed into wells of a u-bottom 96-well non-treated plate with 0.2mg of IgG from human serum (Millipore Sigma) for 10 minutes on ice. Cells were then stained (MHC Class II, CD80, and CD86), fixed, re-suspended in 400μl of PBS-BSA, and analyzed using FlowJo software. Cell debris was removed before gating on single cells (Figure 1A). Unstained cells were then used to draw the positive/negative gates on the resultant histograms (i.e. Figure 1B) Due to the differences in scatter generated by the beads, voltages had to be altered for the bead-containing samples; BSA-beads served as a negative control for gating. Therefore, when depicting the results from these experiments, MFI ratios were used for comparisons (i.e. mAb-beads/BSA-beads & mAb-iFt/iFt). Cells incubated in media alone had a basal level of marker expression which was not present in unstained cells (and therefore not autofluorescence).
Figure 1.

Stimulation with mAb-iFt enhances BMDC maturation. WT BMDCs were stimulated with LPS (2.25ng/mL), iFt (MOI 50), or mAb-iFt (MOI 50) for 24 hours. Supernatants were collected for cytokine analysis (D) and cells were stained for CD80, CD86, and MHC II (A – C). (A) After removal of doublets and debris, unstained cells were used to gate on CD80+, CD86+, or MHC II+ cell populations. Histograms representative of one experiment are shown. (B) Geometric mean fluorescent intensities (MFI) for CD80, CD86, or MHC II positive cells from the histograms in (A) were combined from three separate experiments and plotted as mean ± SEM. Further characterization of the MHC II+ cells was then conducted by plotting CD80 vs CD86 as shown in (C). Dot plots are representative of one experiment whereas data from three separate experiments is shown in the bar graphs as mean ± SEM. (D) Cytokine analysis of BMDC supernatants was completed via Bio-Rad multiplex kit. Concentrations of IL-6, IL-12p70, and TNF-α from three different experiments are shown as mean ± SEM. All bar graphs were analyzed with ordinary one-way ANOVA with aTukey’s post-test after Log10 transforming the data to account for heteroskedasticity. *p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 compared to media unless noted with a bar.
2.8. Cytokine Analysis
BMDCs were stimulated as described above. At 24 hours, the supernatants were collected and stored at −20°C for cytokine analysis. Cytokine release was quantified using the Bio-Plex® multiplex assay per manufacturers’ instructions with the exception that all assay volumes were reduced by half. Washes were completed with 100μl of 1× wash buffer.
2.9. Fluorescent Microscopy
In order to confirm that the mAb-beads bound BMDCs and were internalized, we labeled the plasma membranes of 2 × 106 BMDCs with PKH67 (Millipore Sigma) (2 ½ minutes in the dark, room temperature). The labeled cells were then washed and subsequently incubated with either BSA- or mAb-beads for 10 minutes at either 37°C or 4°C. Once allowed to take up the beads, cells were fixed with 4% paraformaldehyde for 20 minutes at room temperature, washed, and mounted onto slides for visualization. Some cells were DAPI stained prior to mounting. Internalized targets were detected as beads surrounded by PKH67 stained membrane.
2.10. Data Analysis
All data shown are in raw format to illustrate differences between groups/treatments. However, due to heteroskedasticity, all data was Log10 transformed prior to statistical analysis. In some cases where there were zeros as values, the number 1 was added to all data prior to Log10 transformation. Statistical analysis and post-tests are specifically indicated within each figure legend for clarity.
3. Results
3.1. Targeting iFt to FcγRs via mAb-iFt results in enhanced DC maturation.
We previously reported that BMDCs stimulated with the immune complex, mAb-iFt, have higher expression of maturation markers/costimulatory receptors (i.e. CD80, CD83, CD86, and MHC II) compared to cells stimulated with inactivated Francisella tularensis (iFt) alone [12]. We wanted to not only confirm that mAb-iFt-induced maturation was enhanced compared to iFt stimulation, but also know whether increased maturation marker expression corresponded with increased cytokine secretion. Before stimulating the BMDCs, however, we wanted to analyze the phenotype and heterogeneity of our BMDCs. We followed the staining and gating strategy of Xu, et. al. to look for the presence of plasmacytoid DCs (pDCs), CD24+ conventional DCs (cDCs), and/or CD11b+ cDCs (Figure S1) [18]. This analysis revealed that our BMDCs are a heterogeneous population which contains about 30% pDCs, and a variety of cDCs (25% CD24+ and 61% CD11+). Next, WT BMDCs were stimulated with iFt, mAb-iFt, or LPS for 24 hours before supernatants were collected and cells stained for maturation markers CD80, CD86, and MHC II. After removal of debris and selecting for single cells, the unstained cells were used to draw gates for CD80+, CD86+, and MHC II+ populations individually. With this strategy, shown in Figures 1 A & B, we found that BMDCs exposed to media alone (blue-shaded peaks) have a basal level of marker expression quite different than the unstained population (gray-shaded peaks). The other stimulants (iFt, mAb-iFt, and LPS) had further increases in each of these markers when compared to media alone. More specifically, iFt stimulation resulted in a trend toward higher expression of CD80 & MHC II, and significantly higher levels of CD86 compared to the cells incubated in media alone. mAb-iFt- stimulated cells had significantly higher expression of all markers when compared to media. The CD86 and MHC II levels seen in the mAb-iFt-stimulated cells also trended toward increased expression over the iFt alone, whereas the CD80 expression was significantly increased over the iFt-stimulated cells. Our positive control in this instance, LPS-stimulation, was significantly increased over the media alone group in every instance; soluble mAb did not increase expression of CD80, CD86, or MHC II (Figure S2). In addition, it is important to note that when we observed any increase in CD80, CD86, or MHC II, the entire peak on the histogram(s) of Fig. 1A shifted to a higher MFI rather than a specific DC population becoming enriched for CD80, CD86, or MHC II. This would imply that the BMDCs increase their maturation marker expression when stimulated regardless of DC subtype (plasmacytoid vs. conventional). Overall, these MFI bar graphs confirmed our previous studies; MFI trends support the idea that BMDCs stimulated with mAb-iFt have enhanced maturation.
We then questioned whether the MHC II+ cells were also positive for CD80 and/or CD86. The answer to this question would make a large impact on the outcome of the overall immune response; if cells which upregulate MHC II expression do not have increased CD80/CD86 expression, a tolerant reaction would occur. Both MHC II and CD80/CD86 should be expressed on the cells if they are to trigger a protective immune response against iFt. In the previous study, we had shown a protective response in mice immunized with mAb-iFt. Thus, we predicted to see an increase in CD80 and/or CD86, however we wanted to confirm this in our in vitro BMDC system. We assessed MHC II+ single cells for both CD80 & CD86 expression 24 hours after the stimulation described above. As shown in the dot plots and the bar graphs of Figure 1C, both the proportion and the frequency of cells expressing both CD80 and CD86 in MHC II+ BMDCs was significantly increased compared to media, regardless of the stimulant used. There were, no significant differences found between the various stimulants when assessing the triple positive population (MHC II+CD80+CD86+), however there is an increasing trend seen where iFt induces an increase over media, mAb-iFt stimulation further increases the triple positive population, and LPS stimulation induces largest triple positive population.
Surface expression of T cell ligands/coreceptors, however, is not the only requirement for activation of naïve T cells; cytokines are also needed to activate and inform the differentiation of T cells (e.g. TH1 vs TH2). Therefore, we analyzed the amount of secreted IL-6, IL-12p70, & TNF-α in the supernatants of cells in panels A – C using Bio-Plex® multiplex immunoassay kits. Similar to the MFI analysis, iFt-stimulated cells had a slight increase in cytokine expression over media, whereas LPS- and mAb-iFt-stimulated cells secreted greater amounts of IL-6, IL-12p70, and TNF-α (Figure 1D). Because cytokine secretion of IL-6, IL-12p70, and TNF-α as well as maturation markers CD80, CD86, and MHC II expression were all greater in mAb-iFt-stimulated cells than iFt-stimulated, we concluded that targeting iFt to FcγRs in the form of an immune complex enhances multiple aspects of BMDC maturation.
3.2. Fc receptor gamma chain (FcR-γ) is required for mAb-iFt-induced BMDC maturation.
Our previous in vivo studies with mAb-iFt demonstrated a requirement for Fc receptor gamma chain (FcR-γ) [3]. We therefore hypothesized that FcR-γ signaling is needed for the increase in DC maturation observed in Figure 1. Thus, we stimulated both WT and FcR-γ knockout (FcR-γ−/−) BMDCs with iFt or mAb-iFt for 24 hours before collecting supernatants and staining for maturation markers. The resulting data (Figure 2A & B) showed both heteroskedasticity and high variability in the effect of mAb-iFt on maturation when compared to iFt alone. Specifically, the difference between mAb-iFt-stimulated and iFt-stimulated MFIs shown in Figure 2A, is much smaller than previously noted in Figure 1. It is important to note here that each individual experiment pooled into the figure shown (n = 3) included duplicates or triplicates which, when combined, exhibited very low variability. In addition, each experiment exhibited the same trends. Which leads us to believe that the variability seen is due to natural variation in different batches of iFt and mAb-iFt. We consequently Log10 transformed the data prior to statistical analysis to account for the heteroskedasticity. MFI and cytokine secretion data display notable decreases when comparing mAb-iFt-stimulated FcR-γ−/− cells (gray bars) to WT cells (white bars). This is not the case with iFt. Instead, FcR-γ−/− cells stimulated with iFt had small, insignificant increases in CD80 and CD86 expression and no changes in MHC II or cytokine secretion (Figure 2). Interestingly, we observed a dramatic increase in CD80 and CD86 expression in FcR-γ−/− BMDCs stimulated with LPS (Figure 2A) whereas cytokine secretion had a downward trend, especially in IL-12p70 where the decrease was significant (Figure 2B). This was not entirely unexpected as Wei, et. al. showed a decrease in IL-6 and TNF-α secretion in LPS-stimulated bone marrow-derived macrophages, a cell type similar to BMDCs [19]. In contrast, surface expression of the maturation markers CD80 and CD86 were increased in response to LPS, suggesting that maturation markers and cytokine secretion may be independently regulated in BMDCs. The observation of slight increases in CD80 and 86 MFIs in iFt-stimulated knockout cells was also unexpected and will require further study in the future, as we do not know the cause of this result. Nevertheless, the fact that mAb-iFt-stimulated FcR-γ−/− cells had reductions in BMDC maturation while the iFt-stimulated knockout cells did not, reveals that maturation of mAb-iFt-stimulated BMDCs requires FcR-γ while iFt alone does not.
Figure 2. BMDCs deficient in FcR-γ or Syk signaling have decreased maturation compared to WT when stimulated with mAb-iFt.

(A & B) WT (white bars) and FcR-γ−/− (gray bars) BMDCs and their corresponding supernatants were stimulated and studied as described in Figure 1. (C & D) BMDCs were pretreated with R406 for 30 minutes and subsequently stimulated with media or mAb-iFt (50 iFt/cell) for 24 hours. Supernatants were collected for cytokine analysis (D) and cells were stained for CD80, CD86, & MHC II (C). R406 was present for the entire duration of the experiment. Data are combined from three experiments where bars represent mean ± SEM. Transformed data from panels A & B were analyzed by two-way ANOVA with Sidak’s post-test (WT vs. knockout). Transformed data from mAb-iFt stimulated cells of panel C were analyzed with an ordinary one-way ANOVA with a Dunnett’s multiple comparison test (everything compared to 0μM R406). Cytokine concentrations in panel D were transformed and then analyzed via two-way ANOVA with Sidak’s post-test (WT vs. knockout).*p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, & **** p ≤ 0.0001.
It is important to note that FcR-γ−/− mice have been documented to have markedly reduced surface expression of Fc receptors [20]. Also, the inhibitory receptor, FcγRIIb, does not utilize γ-chain. It is therefore possible that the mAb-iFt cytokine results in Figure 2B & D are due to an increase in mAb-iFt binding to the inhibitory receptor and subsequent skewing toward an inhibitory response in the DCs. In addition, the changes in LPS-induced maturation in FcR-γ−/− BMDCs (Figure 2A & B) were somewhat unexpected as stated above. Therefore, we decided to examine the role of activating FcγRs in BMDC maturation without altering the binding profile of the various stimuli. To accomplish this, we utilized a pharmacological inhibitor of FcR-γ signaling. As mentioned in the introduction, ITAM signaling of FcR-γ relies on phosphorylation by src family kinases. Syk is then recruited and activated by the phospho-ITAM in order to propagate the signaling cascade to other downstream effectors [15]. Inhibition of Syk would consequently result in abrogation of FcR-γ signaling. We therefore pretreated BMDCs with either media or varying doses of the Syk inhibitor R406 to assess the role of FcR-γ in another way. After pre-treatment, cells were incubated with media or mAb-iFt (in the continued presence of inhibitor) for 24 hours prior to maturation marker staining and cytokine analysis. As Figure 2C depicts, increasing amounts of R406 result in decreased CD80 and CD86 MFIs in mAb-iFt-stimulated cells. However, MHC II MFI does not change with the amount of R406 suggesting that MHC II is not affected by FcR-γ signaling. Cytokine analyses also show decreases in the inhibitor pretreated BMDCs that are stimulated with mAb-iFt, in fact the decrease observed in CD86 was significant. In the case of LPS stimulation, R406 pre-treatment significantly decreases CD80 and CD86 expression (Figure S3) confirming the decreases observed in Figures 2 A & B. Cytokine data from LPS-stimulated cells, however, exhibited increases when cells were pretreated with R406. Together with the FcR-γ−/− results, these data suggest that enhanced maturation of mAb-iFt-stimulated BMDCs seen in Figure 1 relies on FcR-γ ITAM signaling. However, it is important to note that FcR-γ is utilized by several receptors other than FcγRs such as some activating Fc receptors (e.g. FcεRI & FcαRI), TLR4, IL-3 receptor, TREM, Dectin-2, and others. [19, 21, 22] Thus, we used FcγR-selective blocking antibodies to determine their contribution to BMDC activation/maturation. WT BMDCs were first incubated with 10μg/mL of either an anti-FcγRI antibody, anti-FcγRIIb/III antibody, or both. Blocking antibodies remained present for the entirety of the experiment. αFcγRI or αFcγRIIb/III alone, or the two combined had no significant effect on the maturation status of BMDCs incubated with media alone. However, incubation with αFcγRIIb/III alone or in combination with αFcγRI decreased maturation marker expression and cytokine production in BMDCs stimulated with mAb-iFt (Figure 3). This implicates FcγRIIb/III signaling in mAb-iFt-induced maturation. As FcγRIIb is inhibitory, the αFcγRII/III is likely blocking BMDC maturation via the activating FcγRIII. Thus, we conclude that FcγRIII mediates mAb-iFt-induced maturation in our BMDCs. This conclusion is in line with previous literature stating that FcγRIII in mouse cells is biased to binding immune complexes [23, 24].
Figure 3. Blocking mAb-iFt binding to FcγRs with specific antibodies reduces BMDC maturation induced by mAb-iFt stimulation.

WT BMDCs were pretreated with 10μg/mL of anti-FcγRI, anti-FcγRII/III, or both antibodies for 20 minutes and subsequently stimulated with media or mAb-iFt (50 iFt/cell) for 24 hours. Supernatants were collected for cytokine analysis (B) and cells were stained for CD80, CD86, & MHC II (A). Blocking antibodies were present for the entire duration of the experiment. Data are combined from two experiments where bars represent mean ± SEM. MFI data (A) were Log10 transformed and subsequently analyzed via two way ANOVA with Dunnett’s multiple comparisons test (Media pre-treatment vs Antibody pre-treatment). Cytokine concentrations in panel B were also transformed and analyzed the same way. *p ≤ 0.05 & ** p ≤ 0.01.
3.3. Monoclonal antibody-coated beads (mAb-Beads) do not elicit BMDC maturation.
The fact that the Syk inhibitor R406 significantly reduces mAb-iFt-stimulated BMDC maturation (Fig 2), and that blocking FcγRIIb/III decreases both maturation and cytokine secretion (Fig 3), implicates FcγR signaling in BMDC maturation. To determine whether FcγR signaling alone is sufficient for mAb-iFt-enhanced DC maturation, we tested the ability of mAb-coated beads (lacking other ligands) to stimulate BMDCs. Glass beads (2 μm) were coated with either mouse IgG2a (mAb-beads) or bovine serum albumin (BSA-beads). mAb-beads are used routinely by the Lennartz laboratory to study FcR-mediated phagocytosis in bone marrow-derived macrophages; BSA-beads serve as a non-binding, non-internalized control [17]. By using mouse IgG2a, mAb-Beads should engage the same FcγR(s) as mAb-iFt but will lack the signaling contributed by the iFt. To account for any mechanical effects the glass beads may have on the BMDCs, we will use BSA-coated beads which neither bind nor are internalized by bone marrow-derived macrophages [25]. After confirming mAb-bead binding/internalization via fluorescent microscopy (data not shown), BMDCs were stimulated for 24 hours with iFt, mAb-iFt, BSA-beads, or mAb-Beads. Then supernatants were collected for cytokine analysis and cells were stained for maturation markers. As the presence of beads significantly shifted the scatter pattern of the BMDCs, it was necessary to use different voltages for the bead-containing samples, making direct comparison between BMDCs stimulated with mAb-iFt to those stimulated with beads impossible. To allow for comparisons between the two target types (mAb-iFt vs. mAb-beads), we normalized the mAb-iFt and mAb-bead MFIs to their controls (iFt and BSA-beads, respectively) and report the ratios (Fig 4B & C). For all measures, the bead ratios were lower than their respective iFt measures, trending lower for CD80 and MHCII but significantly lower for CD86 (Fig 4B). Release of all the measured cytokines by mAb beads was significantly lower than its’ mAb-iFt counterpart (Fig 4C). Together with the data from Figure 1, we conclude that BMDC maturation requires input from both iFt and IgG, with the IgG input likely coming from FcγRIII signaling.
Figure 4. Monoclonal antibody-coated beads (mAb-Beads) do not elicit BMDC maturation.

WT BMDCs were stimulated with media, iFt (50/cell), mAb-iFt (50 iFt/cell), BSA-beads, or mAb-beads (5 – 30 beads/cell) for 24 hours. Supernatants were collected for cytokine analysis (B) and cells were stained for CD80, CD86, and MHC class II (A). Data are combined from two experiments where bars represent mean ± SEM. Data were Log10 transformed to account for heteroskedasticity and then analyzed by unpaired two-tailed t-test (compared to media). *p ≤ 0.05 & ** p ≤ 0.01.
3.4. TLR2 plays a role in BMDC cytokine secretion, but does not influence surface maturation marker expression induced by mAb-iFt stimulation.
We have shown thus far that mAb-iFt stimulation of BMDC results in an enhanced maturation status which requires FcR-γ signaling (Figures 1 – 3). However, stimulating DCs with mAb-Beads results in little maturation (Figure 4). One explanation for this could be the presence of toll-like receptor (TLR) ligands on mAb-iFt. Many studies have shown evidence for FcγR crosstalk/synergy with TLR, and in the context of mAb-iFt FcγRs have been shown to synergize with TLR2 in eliciting cytokine secretion from macrophages [14, 16, 26, 27]. We therefore postulated that TLR2 could play a role in mAb-iFt-enhanced DC maturation. We stimulated WT and TLR2 knockout (TLR2−/−) cells with LPS, iFt, or mAb-iFt as previously described. Surprisingly, we found that mAb-iFt stimulated cells had no significant changes to their maturation marker MFIs despite the absence of TLR2 (Figure 5A). This came as a surprise to us, as TLR2 has been shown to play a role in mAb-iFt stimulation of macrophages [14, 16]. iFt-stimulated TLR2−/− BMDCs, on the other hand, had significantly diminished MFIs across the board as expected and LPS-stimulated cells had unchanged MFIs.
Figure 5. TLR2 deficient BMDCs stimulated with mAb-iFt have decreases in cytokine secretion, but no changes in maturation marker expression.

WT (white bars) or TLR2−/− (gray bars) BMDCs were stimulated as described in Figure 1. Maturation marker expression was determined by fluorescent staining (A), while supernatants were analyzed via multiplex for cytokine concentrations (B). Data are combined from three experiments where bars represent mean ± SEM. Data were Log10 transformed to account for heteroskedasticity prior to two-way analysis of variance with Sidak’s post-test (WT vs. knockout). *p ≤ 0.05, ** p ≤ 0.01, & *** p ≤ 0.001.
Cytokine data from the TLR2−/− cells revealed that secretion of IL-6, IL-12p70, and TNF-α was diminished in the knockout cells compared to WT regardless of stimuli (Figure 5B). This corroborated the MFI data of iFt-stimulated cells, and supported the results observed by other groups in mAb-iFt-stimulated macrophages. The IL-6 and IL-12p70 results for LPS, however, were a bit unexpected; in the presence of αTLR2, LPS-induced IL-6 secretion was reduced and IL-12p70 was significantly reduced. One possible explanation for our results could be LPS contamination with a TLR2 ligand, as has been previously reported [28]. We therefore pretreated our BMDCs with an anti-TLR-2 antibody (αTLR2) which blocks ligand from binding to assess the role of TLR2 in BMDC maturation and cytokine secretion. We first verified the function of the antibody by stimulating BMDCs with Pam3CSK4 after pre-treatment with or without αTLR2 (data not shown). We then tested the αTLR2 in our system. Specifically, BMDCs were pretreated with αTLR2 and subsequently stimulated with LPS, iFt, or mAb-iFt. After 24 hours of stimulation, maturation markers were quantified by flow cytometry. Surprisingly, only mAb-iFt-induced maturation was inhibited by αTLR2 pre-treatment (Figure S4). This was contrary to our results using TLR2−/− cells, but Fc-FcγR interactions could be one explanation for this discrepancy. The αTLR2 is an IgG2a antibody and can therefore bind to FcγRs present on the BMDC surface. This could, in turn, block the ability of mAb-iFt to interact with FcγRs on the BMDC surface. Regardless, the MFI and cytokine secretion data together reveal a partial role for TLR2 in mAb-iFt-induced BMDC maturation, specifically the secretion of proinflammatory cytokines.
3.5. Myeloid differentiation primary response 88 (MyD88) deficient BMDCs do not mature when stimulated with mAb-iFt.
TLR2 deficient cells revealed the receptor’s role in proinflammatory cytokine secretion (Figure 5). These results, however, did not explain the MFI differences observed between iFt and mAb-iFt stimulation. We hypothesized that another TLR could be responsible for this difference. Myeloid differentiation primary response 88 (MyD88) is an adaptor protein involved in all TLR signaling with the exception of TLR3. Because of MyD88’s heavy involvement in TLR signaling, comparing the response of WT to MyD88−/− cells would allow us to assess whether any TLR, other than TLR3, plays a role in mAb-iFt-induced BMDC maturation. BMDCs were stimulated with Poly(I:C) – a TLR3 agonist, LPS – a TLR4 agonist, iFt, or mAb-iFt for 24 hours before the collection of supernatants and staining of cells for maturation markers. LPS, being a TLR4 ligand, has a significant decrease in CD80, CD86, and MHC II MFIs when MyD88 is absent (gray bars). Poly(I:C)-treated cells, on the other hand, were unaffected by an absence of MyD88, due to Poly(I:C)’s reliance on TLR3 for activation. iFt is known to be a TLR2 agonist, and so MyD88−/− cells had a dramatic drop in MFIs across the board. This is also the case for mAb-iFt stimulated knockout cells; MyD88−/− cells had significant decreases in CD80 and CD86 MFIs and a substantial decrease MHC II MFI in comparison to WT cells (Figure 6A). Cytokine secretion of WT and MyD88−/− had similar trends; secretion was significantly decreased across the board in MyD88−/− cells when compared to WT cells with the exception of Poly(I:C) stimulation, which was unaffected (Figure 6B). Together these data show that MyD88 is critical for mAb-iFt-induced BMDC maturation.
Figure 6. MyD88 deficient BMDCs do not mature when stimulated with mAb-iFt.

(A) WT (white bars) or MyD88−/− (gray bars) BMDCs were stimulated with iFt (50 iFt/cell), mAb-iFt (50 iFt/cell), LPS (2.25ng/mL), or Poly(I:C) (10μg/mL) for 24 hours before being stained for maturation markers. (B) Cytokines analysis was completed on the BMDC supernatants via multiplex. Data are combined from three experiments where bars represent mean ± SEM and Log10 transformed to account for heteroskedasticity prior to analysis via two-way ANOVA with Sidak’s post-test (WT vs. knockout). *p ≤ 0.05, *** p ≤ 0.001, & **** p ≤ 0.0001.
3.6. The TLR9 antagonist, ODN 2088, inhibits enhanced maturation marker expression in mAb-iFt-stimulated BMDCs.
As shown above, MyD88 is required for BMDC marker expression and cytokine secretion (Figure 6), but TLR2 is not required for maturation marker expression (Figure 5). We therefore postulated that a TLR other than TLR2 or TLR3 was required for the increase in maturation marker expression. We initially thought to examine other surface-expressed TLRs however, studies conducted with TLR4−/− BMDCs showed no effect on mAb-iFt-induced maturation (Figure S5). The only other surface-expressed TLR that could play a role (TLR5) binds flagellin, which Ft does not possess. We therefore turned to endosomal TLRs.
TLR9 is known to be activated by ligation of unmethylated CpG DNA motifs which is readily found in bacterial DNA, making TLR9 a logical candidate for our next studies. To examine TLR9’s role in BMDC maturation, we utilized a TLR9 antagonist to inhibit signaling. BMDCs were pretreated with culture media or the antagonist, ODN 2088, for 1 hour prior to stimulation with LPS, iFt, mAb-iFt, or ODN 1826 - a TLR9 agonist. As expected, MFIs of LPS-treated BMDCs remained unchanged regardless of pretreatment. TLR9 agonist-treated cells displayed decreased maturation marker MFIs in the antagonist pretreated cells (gray bars). The iFt-stimulated cells had some subtle differences between media and antagonist pretreated cells, none of which were large enough to imply a major role for TLR9 in iFt-induced BMDC maturation. Cells stimulated with mAb-iFt, on the other hand, had significant decreases in CD80, CD86, and MHC II MFIs when cells were pretreated with antagonist, illustrating a role for TLR9 in upregulation of maturation marker expression (Figure 7A).
Figure 7. mAb-iFt-stimulated BMDCs have reduced maturation marker expression when TLR9 signaling is blocked.

(A) WT BMDCs were pretreated with either media (white bars) or the TLR9 antagonist ODN 2088 (10μM, gray bars) for 1 hour before being stimulated with iFt (50 iFt/cell), mAb-iFt (50 iFt/cell), LPS (2.25ng/mL), or the TLR9 ligand ODN 1826 (5μM) for 24 hours. BMDCs were subsequently stained for maturation markers (A) and supernatants analyzed by multiplex for cytokines (B). Data are combined from three experiments where bars represent mean ± SEM. Data were Log10 transformed to account for heteroskedasticity and subsequently analyzed via two-way ANOVA with Sidak’s post-test (media vs. antagonist). *p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, & **** p ≤ 0.0001.
We then analyzed cytokine secretion in supernatants of the cells from panel A. As expected, cells stimulated with ODN 1826 (agonist) had a massive decrease in cytokine secretion when pretreated with TLR9 antagonist whereas cells treated with LPS or iFt did not. Surprisingly, cells stimulated with mAb-iFt had practically no change in cytokine secretion when pretreated with the antagonist, with the exception of CD80. LPS and ODN 1826 stimulation results in panel A confirm the effectiveness and specificity of the antagonist pretreatment; thus, we concluded that TLR9 plays a central role in mAb-iFt-induced BMDC maturation marker expression, but not in cytokine secretion (Figure 6B).
4. Discussion
Our earlier studies with mAb-iFt demonstrated that intranasally administered mAb-iFt enhances protection against Ft challenge when compared to iFt. This enhanced protection required the constant region of IgG within mAb-iFt, FcR-γ, and FcRn [3]. Additional studies in vitro subsequently revealed increased binding, internalization, and presentation of mAb-iFt compared to iFt [12]. More recently, in vivo studies showed increased number and frequency of activated lung DCs, as well as frequency and number of gamma interferon-secreting effector memory CD4+ T cells in mAb-iFt-immunized mice [29]. Together these studies illustrate an importance for DC maturation in generating a protective immune response against Ft. Thus, the goal of this study was to determine the mechanism of maturation in mAb-iFt-stimulated DCs. As Figure 1 shows, mAb-iFt-stimulated BMDCs exhibit higher maturation marker expression (though only CD80 was significantly increased when comparing iFt to mAb-iFt stimulation) and cytokine secretion compared to iFt-stimulated cells. Based on previous studies, from our laboratory and others [3, 12, 14, 16, 29], we hypothesized that the enhanced maturation would rely on both FcγRs and TLR2. Subsequently, both our BMDC maturation marker expression and cytokine secretion results show that enhanced maturation of mAb-iFt-stimulated BMDCs relies not only on signaling through FcR-γ, FcγRs, and TLR2, but also signaling through MyD88 and TLR9.
To determine the mechanism(s) of mAb-iFt-stimulated DC maturation, the degree of DC maturation needed to be assayed. It is important to note that DC maturation consists of several changes over time: phagocytic capacity decreases, proteolytic processing increases, expression of T cell costimulatory markers and MHC molecules increase, cytokine secretion increases, and migration to lymph nodes commences [10, 30, 31]. In this study, we chose to measure both maturation marker expression and cytokine secretion to assay DC maturation status. Many studies assay solely maturation marker expression levels to determine DC maturation. However, we decided to include cytokine secretion because numerous studies have shown that cytokines have an immense impact on T cell activation, for example, differential skewing of T helper phenotype with specific cytokine stimulation (e.g. IL-12 skews to TH1 phenotype) [32–34]. Somewhat surprisingly, we show that when using mAb-iFt-stimulated TLR2−/− DCs and TLR9 antagonist pretreated DCs, changes in maturation marker expression do not equate to changes in proinflammatory cytokine secretion (Figures 5 & 7). In addition, studies have shown that not just cytokines, but the combination of costimulatory receptors with cytokines impact T cell skewing [35]. Together this demonstrates the importance of including cytokine secretion assays in addition to maturation marker studies in determining the maturation status of DCs. Furthermore, we find it intriguing that mAb-iFt-elicited cytokine secretion is dependent on the presence of TLR2 (Figure 5), whereas maturation marker expression relies on TLR9 signaling (Figure 7). However, both cytokine and maturation marker increases are dependent on the presence of MyD88 (Figure 6). We surmise that perhaps the adaptor protein MyD88-adaptor-like (Mal a.k.a. TIRAP) could play a role in this discrepancy. Mal has been found not only to form a bridge between MyD88 and certain TLRs, but to also bind various phosphatidylinositol- or phosphatidylserine-rich regions of membrane, and conduct signaling functions independent of MyD88 [36–38]. It would be interesting to conduct follow-up studies on how TLR2 and TLR9 signaling differ in the context of mAb-iFt stimulation by focusing on the role Mal/TIRAP plays (or doesn’t play) for each receptor.
The ability of DCs to present antigen to T cells is critical for effective immune activation and protection. Previous studies from our laboratory have shown, in vitro, that mAb-iFt stimulation of DCs results in more effective T cell activation. These same studies also discovered enhanced persistence of Ft antigen within DCs compared to stimulation with iFt alone [12]. In addition, Iglesias, et al. found that mAb-iFt binding to FcγRs leads to enhanced internalization, whereas iFt alone does not engage Fc receptors. Engagement of FcγRs by mAb-iFt (shown in Figure 4) likely leads to altered intracellular trafficking compared to iFt alone, which could be an explanation for the differences previously observed in antigen persistence. Our studies herein could further imply differential trafficking as a possible mechanism for enhanced persistence. In addition to a need for FcγR engagement and signaling, which we show in Figures 2 – 4, mAb-iFt maturation was shown to also require TLR9 (Figure 7) to increase maturation marker expression. At steady state, TLR9 localizes in the endoplasmic reticulum in an inactive form. Upon cell stimulation, inactive TLR9 migrates to endo-lysosomal compartments where it becomes activated [39, 40]. The requirement for TLR9 in mAb-iFt-induced maturation therefore suggests that mAb-iFt traffics specifically to compartments which contain TLR9. iFt stimulation of TLR9 antagonist pretreated DCs, however, did not show any maturation deficiencies, suggesting that iFt either does not engage TLR9 or does not traffic to compartments containing TLR9. Unmethylated CpG motifs are a known TLR9 ligand which are readily found in bacterial DNA including that of Ft; it is thus more likely that iFt does not traffic to a TLR9-containing compartment. It has also been shown that B cell receptor signaling through phospholipase D can recruit TLR9-containing endosomes to the autophagosome where internalized BCR was located [41]. BCR signaling is nearly homologous to that of FcγRs due to the presence of ITAMs in the invariant CD79B and CD79A molecules that associate with the BCR, suggesting that engagement of FcγRs by mAb-iFt could lead to differential trafficking. Further support of this idea comes from studies from Means, et. al. This study shows that FcγRIIA in humans cooperates with TLR9, in fact FcγRIIA is required for the uptake of autoantibody immune complexes in patients with systemic lupus erythematosus and subsequent trafficking to TLR9-containing lysosomes [42]. It is thought that the mouse FcγRIII is the orthologue to human FcγRIIA [43]. It is therefore likely that the engagement of FcγRs by mAb-iFt, specifically FcγRIII as illustrated in Figure 3, could lead to specific delivery of mAb-iFt to the compartment(s) containing active TLR9. This trafficking discrepancy and subsequent engagement of TLR9 could then lead to variances in the: level and amount of antigen degradation, amount and type of peptide loaded into MHC molecules, and antigen retention time. These variances can ultimately influence the amount and effectiveness of T cell activation. Alternatively, requirement for TLR9 with mAb-iFt, but not iFt, could be due to simple probability. We know from previous studies that mAb-iFt is taken up both in greater amounts and more rapidly than iFt alone. Because there would be more mAb-iFt within a given cell than iFt, there is simply a higher probability that mAb-iFt would engage TLR9 than iFt. If true, then mAb-iFt might elicit more T cell activation because the DCs containing the complexes are more mature. This could result from more efficient (i.e. faster and more) internalization of mAb-iFt and/or differential intracellular trafficking when compared to iFt alone. However, the intracellular trafficking routes of each stimuli will need to be studied to determine which explanation holds true. In addition, future studies should investigate whether mAb-iFt engagement of FcγRs leads to the recruitment of TLR9.
All our mAb-iFt studies to date, including this one, suggest a need for FcγR ITAM signaling in mAb-iFt elicited protection, antigen presentation, or DC maturation [3, 12]. It is important to note that iFt alone did not require FcR-γ to elicit an increase in maturation marker expression or cytokine secretion (Figure 2), demonstrating that the receptor most likely binds the antibody portion of mAb-iFt and not iFt itself. It is therefore likely that the receptor which utilizes FcR-γ within our system is a FcγR. We did confirm involvement of FcγRIII by using the 2,4G2 blocking antibody as a pretreatment (Figure 3). Furthermore, there is ample evidence for synergy/crosstalk between ITAM-containing receptors, like FcγRs, and TLRs in the literature [16, 26, 27, 44]. This data, along with our current study, suggests that FcγR/TLR synergy is also likely in the context of mAb-iFt. A review on FcR/TLR synergy by Lennartz & Drake discusses various ways in which synergy/crosstalk could occur such as signaling intermediates, transcriptional regulation, and post-transcriptional regulation [26]. Particularly relevant to this study are: tyrosine kinases (i.e. Syk & BTK); transcriptional regulation through NF-κB; and post-transcriptional regulation through micro-RNAs and/or long non-coding RNAs (lnc-RNAs). Our studies herein further emphasize the importance of this synergy by showing mAb-iFt-stimulated DC maturation relies on both MyD88 (Figure 5) and ITAM signaling (Figure 2). Future studies should therefore examine the mechanisms of synergy more in depth. Examination of the effects Syk/BTK, different NF-κB subunits and micro-RNA/lnc-RNA exert on mAb-iFt-elicited DC maturation would provide invaluable information on how this synergy occurs.
Supplementary Material
mAb-iFt-stimulation enhances dendritic cell maturation when compared to iFt alone.
mAb-iFt-stimulation requires both Fc receptor γ-chain and MyD88 signaling pathways.
TLR2 and TLR9 signaling are required for full mAb-iFt-induced DC maturation.
Enhanced DC cytokine secretion & marker expression utilize different TLR pathways.
Cytokine secretion & maturation marker expression require TLR2 & 9, respectively.
Acknowledgments
Raju Sunagar for instruction on making mAb-iFt.
Michelle Lennartz, Ph.D. and Cheryl Hanes for microscopy assistance and making the BSA-Beads and mAb-Beads.
Paul Feustel for help with statistical analyses.
Jim Drake, Ph.D. and Michelle Lennartz, Ph.D. for editing and proof-reading assistance.
This work was supported by the National Institutes of Health National Institute of Allergy and Infectious Disease (NIAID) grant numbers: 2P01AI05632008A1, 1R01AI10013801A1, and 1R21AI13973301.
Footnotes
Abbreviations: Ft – Francisella tularensis; iFt – methanol-free formaldehyde-inactivated Francisella tularensis; mAb-iFt – immune complex containing inactivated Francisella tularensis and monoclonal antibody against Francisella tularensis lipopolysaccharide; BMDCs – Bone marrow-derived dendritic cells; FcγR – Fc gamma receptor I (aka CD64); FcR-γ – Fc receptor gamma chain
References
- [1].Infographics: #VaccinesWork, World Health Organization, Online, 2019. [Google Scholar]
- [2].Jia Q, Horwitz MA, Live Attenuated Tularemia Vaccines for Protection Against Respiratory Challenge With Virulent F. tularensis subsp. tularensis, Frontiers in cellular and infection microbiology, 8 (2018) 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rawool DB, Bitsaktsis C, Li Y, Gosselin DR, Lin Y, Kurkure NV, Metzger DW, Gosselin EJ, Utilization of Fc receptors as a mucosal vaccine strategy against an intracellular bacterium, Francisella tularensis, Journal of immunology (Baltimore, Md. : 1950), 180 (2008) 5548–5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rhoads JP, Lukens JR, Wilhelm AJ, Moore JL, Mendez-Fernandez Y, Kanneganti TD, Major AS, Oxidized Low-Density Lipoprotein Immune Complex Priming of the Nlrp3 Inflammasome Involves TLR and FcgammaR Cooperation and Is Dependent on CARD9, Journal of immunology (Baltimore, Md. : 1950), 198 (2017) 2105–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Arazi A, Neumann AU, Modeling immune complex-mediated autoimmune inflammation, Journal of theoretical biology, 267 (2010) 426–436. [DOI] [PubMed] [Google Scholar]
- [6].Aibara N, Ichinose K, Baba M, Nakajima H, Satoh K, Atarashi R, Kishikawa N, Nishida N, Kawakami A, Kuroda N, Ohyama K, Proteomic approach to profiling immune complex antigens in cerebrospinal fluid samples from patients with central nervous system autoimmune diseases, Clinica chimica acta; international journal of clinical chemistry, 484 (2018) 26–31. [DOI] [PubMed] [Google Scholar]
- [7].Maamary J, Wang TT, Tan GS, Palese P, Ravetch JV, Increasing the breadth and potency of response to the seasonal influenza virus vaccine by immune complex immunization, Proceedings of the National Academy of Sciences of the United States of America, 114 (2017) 10172–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pepponi I, Stylianou E, van Dolleweerd C, Diogo GR, Paul MJ, Drake PM, Ma JK, Reljic R, Immune-complex mimics as a molecular platform for adjuvant-free vaccine delivery, PloS one, 8 (2013) e60855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Subauste CS, de Waal Malefyt R, Fuh F, Role of CD80 (B7.1) and CD86 (B7.2) in the immune response to an intracellular pathogen, Journal of immunology (Baltimore, Md. : 1950), 160 (1998) 1831–1840. [PubMed] [Google Scholar]
- [10].Dalod M, Chelbi R, Malissen B, Lawrence T, Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming, The EMBO journal, 33 (2014) 1104–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Clatworthy MR, Aronin CE, Mathews RJ, Morgan NY, Smith KG, Germain RN, Immune complexes stimulate CCR7-dependent dendritic cell migration to lymph nodes, Nature medicine, 20 (2014) 1458–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Iglesias BV, Bitsaktsis C, Pham G, Drake JR, Hazlett KR, Porter K, Gosselin EJ, Multiple mechanisms mediate enhanced immunity generated by mAb-inactivated F. tularensis immunogen, Immunology and cell biology, 91 (2013) 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bitsaktsis C, Babadjanova Z, Gosselin EJ, In vivo mechanisms involved in enhanced protection utilizing an Fc receptor-targeted mucosal vaccine platform in a bacterial vaccine and challenge model, Infection and immunity, 83 (2015) 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Duffy EB, Periasamy S, Hunt D, Drake JR, Harton JA, FcgammaR mediates TLR2-and Syk-dependent NLRP3 inflammasome activation by inactivated Francisella tularensis LVS immune complexes, Journal of leukocyte biology, 100 (2016) 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rosales C, Fcgamma Receptor Heterogeneity in Leukocyte Functional Responses, Frontiers in immunology, 8 (2017) 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hunt D, Drake LA, Drake JR, Murine macrophage TLR2-FcgammaR synergy via FcgammaR licensing of IL-6 cytokine mRNA ribosome binding and translation, PloS one, 13 (2018) e0200764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hanes CM, D’Amico AE, Ueyama T, Wong AC, Zhang X, Hynes WF, Barroso MM, Cady NC, Trebak M, Saito N, Lennartz MR, Golgi-Associated Protein Kinase C-epsilon Is Delivered to Phagocytic Cups: Role of Phosphatidylinositol 4-Phosphate, Journal of immunology (Baltimore, Md. : 1950), 199 (2017) 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH, Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking, Journal of immunology (Baltimore, Md. : 1950), 179 (2007) 7577–7584. [DOI] [PubMed] [Google Scholar]
- [19].Wei ZM, Wang Z, Wan XJ, Li XJ, Li YX, Bai Y, Yang X, Yang Y, Jiao SC, Liu ZF, FcRgamma deficiency improves survival in experimental sepsis by down-regulating TLR4 signaling pathway, Immunologic research, 67 (2019) 77–83. [DOI] [PubMed] [Google Scholar]
- [20].van Vugt MJ, Heijnen IA, Capel PJ, Park SY, Ra C, Saito T, Verbeek JS, van de Winkel JG, FcR gamma-chain is essential for both surface expression and function of human Fc gamma RI (CD64) in vivo, Blood, 87 (1996) 3593–3599. [PubMed] [Google Scholar]
- [21].Hida S, Yamasaki S, Sakamoto Y, Takamoto M, Obata K, Takai T, Karasuyama H, Sugane K, Saito T, Taki S, Fc receptor gamma-chain, a constitutive component of the IL-3 receptor, is required for IL-3-induced IL-4 production in basophils, Nature immunology, 10 (2009) 214–222. [DOI] [PubMed] [Google Scholar]
- [22].Brandsma AM, Hogarth PM, Nimmerjahn F, Leusen JH, Clarifying the Confusion between Cytokine and Fc Receptor “Common Gamma Chain”, Immunity, 45 (2016) 225–226. [DOI] [PubMed] [Google Scholar]
- [23].Nimmerjahn F, Ravetch JV, Fc-receptors as regulators of immunity, Advances in immunology, 96 (2007) 179–204. [DOI] [PubMed] [Google Scholar]
- [24].Nimmerjahn F, Activating and inhibitory FcgammaRs in autoimmune disorders, Springer seminars in immunopathology, 28 (2006) 305–319. [DOI] [PubMed] [Google Scholar]
- [25].Larsen EC, Ueyama T, Brannock PM, Shirai Y, Saito N, Larsson C, Loegering D, Weber PB, Lennartz MR, A role for PKC-epsilon in Fc gammaR-mediated phagocytosis by RAW 264.7 cells, The Journal of cell biology, 159 (2002) 939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lennartz M, Drake J, Molecular mechanisms of macrophage Toll-like receptor-Fc receptor synergy, F1000Research, 7 (2018) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Krupa A, Fudala R, Florence JM, Tucker T, Allen TC, Standiford TJ, Luchowski R, Fol M, Rahman M, Gryczynski Z, Gryczynski I, Kurdowska AK, Bruton’s tyrosine kinase mediates FcgammaRIIa/Toll-like receptor-4 receptor crosstalk in human neutrophils, American journal of respiratory cell and molecular biology, 48 (2013) 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Muroi M, Ohnishi T, Azumi-Mayuzumi S, Tanamoto K, Lipopolysaccharide-mimetic activities of a Toll-like receptor 2-stimulatory substance(s) in enterobacterial lipopolysaccharide preparations, Infection and immunity, 71 (2003) 3221–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bitsaktsis C, Babadjanova Z, Gosselin EJ, In Vivo Mechanisms Involved in Enhanced Protection Utilizing an FcR-Targeted Mucosal Vaccine Platform in a Bacterial Vaccine and Challenge Model, Infection and immunity, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Alloatti A, Kotsias F, Magalhaes JG, Amigorena S, Dendritic cell maturation and cross-presentation: timing matters!, Immunological reviews, 272 (2016) 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Granucci F, Ferrero E, Foti M, Aggujaro D, Vettoretto K, Ricciardi-Castagnoli P, Early events in dendritic cell maturation induced by LPS, Microbes and infection, 1 (1999) 1079–1084. [DOI] [PubMed] [Google Scholar]
- [32].Bourque J, Hawiger D, Immunomodulatory Bonds of the Partnership between Dendritic Cells and T Cells, Critical reviews in immunology, 38 (2018) 379–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mohamadzadeh M, Olson S, Kalina WV, Ruthel G, Demmin GL, Warfield KL, Bavari S, Klaenhammer TR, Lactobacilli activate human dendritic cells that skew T cells toward T helper 1 polarization, Proceedings of the National Academy of Sciences of the United States of America, 102 (2005) 2880–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dumitriu IE, Dunbar DR, Howie SE, Sethi T, Gregory CD, Human dendritic cells produce TGF-beta 1 under the influence of lung carcinoma cells and prime the differentiation of CD4+CD25+Foxp3+ regulatory T cells, Journal of immunology (Baltimore, Md. : 1950), 182 (2009) 2795–2807. [DOI] [PubMed] [Google Scholar]
- [35].Lichtenegger FS, Mueller K, Otte B, Beck B, Hiddemann W, Schendel DJ, Subklewe M, CD86 and IL-12p70 are key players for T helper 1 polarization and natural killer cell activation by Toll-like receptor-induced dendritic cells, PloS one, 7 (2012) e44266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bernard NJ, O’Neill LA, Mal, more than a bridge to MyD88, IUBMB life, 65 (2013) 777–786. [DOI] [PubMed] [Google Scholar]
- [37].Bonham KS, Orzalli MH, Hayashi K, Wolf AI, Glanemann C, Weninger W, Iwasaki A, Knipe DM, Kagan JC, A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction, Cell, 156 (2014) 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dowling JK, Tate MD, Rosli S, Bourke NM, Bitto N, Lauterbach MA, Cheung S, Ve T, Kobe B, Golenbock D, Mansell A, The Single Nucleotide Polymorphism Mal-D96N Mice Provide New Insights into Functionality of Mal in TLR Immune Responses, Journal of immunology (Baltimore, Md. : 1950), (2019). [DOI] [PubMed] [Google Scholar]
- [39].Lee BL, Moon JE, Shu JH, Yuan L, Newman ZR, Schekman R, Barton GM, UNC93B1 mediates differential trafficking of endosomal TLRs, eLife, 2 (2013) e00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Leifer CA, Medvedev AE, Molecular mechanisms of regulation of Toll-like receptor signaling, Journal of leukocyte biology, 100 (2016) 927–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chaturvedi A, Dorward D, Pierce SK, The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens, Immunity, 28 (2008) 799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD, Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9, The Journal of clinical investigation, 115 (2005) 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nimmerjahn F, Ravetch JV, Fcgamma receptors as regulators of immune responses, Nature reviews. Immunology, 8 (2008) 34–47. [DOI] [PubMed] [Google Scholar]
- [44].Hirsch I, Janovec V, Stranska R, Bendriss-Vermare N, Cross Talk between Inhibitory Immunoreceptor Tyrosine-Based Activation Motif-Signaling and Toll-Like Receptor Pathways in Macrophages and Dendritic Cells, Frontiers in immunology, 8 (2017) 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.