

SUMMARY
We have developed a cancer model of gliomas in human cerebral organoids that allows direct observation of tumor initiation as well as continuous microscopic observations. We used CRISPR/Cas9 technology to target an HRasG12V-IRES-tdTomato construct by homologous recombination into the TP53 locus. Results show that transformed cells rapidly become invasive and destroy surrounding organoid structures, overwhelming the entire organoid. Tumor cells in the organoids can be orthotopically xenografted into immunodeficient NOD/SCID IL2RG−/− animals, exhibiting an invasive phenotype. Organoid-generated putative tumor cells show gene expression profiles consistent with mesenchymal subtype human glioblastoma. We further demonstrate that human-organoid-derived tumor cell lines or primary human-patient-derived glioblastoma cell lines can be transplanted into human cerebral organoids to establish invasive tumor-like structures. Our results show potential for the use of organoids as a platform to test human cancer phenotypes that recapitulate key aspects of malignancy.
In Brief
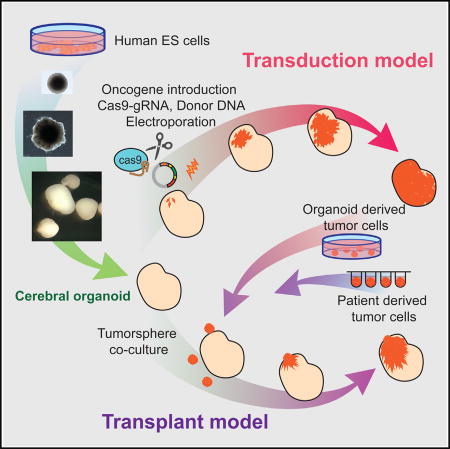
Ogawa et al. show that human cerebral organoids can be used as a platform for tumor formation. CRISPR/Cas9 manipulation of oncogenes/tumor suppressors initiates tumorigenesis in cerebral organoids, allowing microscopic observation of tumor development. Additionally, human cerebral organoids can be used as a platform for tumor cell transplantation.
INTRODUCTION
Cancer models are essential in our understanding of the nature of neoplastic disease, as well as more practical and translational aspects, such as their use as screening platforms to evaluate the effectiveness of candidate therapeutic modalities. Both require that models capture the essential features of naturally occurring disease and translate these into outcomes that are equivalent to those in natural tumors. In recent years, cancer research has shifted from mouse genetic models to patient-derived xenograft (PDX) models of cancer that are xenografted onto immunodeficient mice for tumor propagation. This has been largely fueled by the fact that results derived from mouse genetic models of experimental therapeutics are not always useful predictors of outcomes in human clinical trials (Hackam and Redelmeier, 2006; Shanks et al., 2009; Van Der Worp et al., 2010). The usefulness of these PDX models, however, is often handicapped by limited donor availability. Furthermore, propagation of these PDX models is only faithful to normal tumor behavior for a limited number of mouse passages, after which selection within the mouse leaves an indelible genetic and/or epigenetic signature (Ben-David et al., 2017). In practice, this means that selection pressure for propagation in mice may make tumors less representative of the properties associated with the original tumor. Furthermore, as PDX tumor models are patient derived, one has little control over the type of oncogenic mutations or epigenetic states at play within a particular tumor.
To complement the use of PDX models in a genetically defined manner, we have combined recent technological advances in human organoid technology and CRISPR genome engineering to generate a genetically defined model of human glioblastoma (GBM). Here, we demonstrate the formation of human cerebral organoids in which a small number of cells undergo CRISPR-mediated homologous recombination to simultaneously disrupt the TP53 tumor suppressor locus and express the oncogenic HRasG12V. Modified cells proliferate, show invasive phenotypes within organoids, and can be transplanted into immunodeficient mice. These tumors, when transplanted into mice, possess proliferative hallmarks of tumorigenesis, are invasive, and exhibit disease pathology from which mice succumb. Furthermore, human-patient-derived tumor samples can be transplanted into organoids. This technology allows recreation of the putative initiating genetic events and observation of the natural history of tumor initiation of human gliomas, a process normally invisible in human patients.
RESULTS
Generation and Transformation of Human Cerebral Organoids
To generate human cerebral organoids, we adapted the culture method of Lancaster and Knoblich (2014). Organoids were grown for a period of 4 months and allowed to mature. During the development of organoids, neural rosettes initially appear and then subsequently form cortical structures accompanied by the expression of cortical layer markers CTIP2 and SATB2 (Figure S1). At this phase, the number of neural stem cells also decrease and become more sparse. Neural stem cells become sparse, as evidenced by a decrease in the SOX2-positive population; nevertheless, cerebral organoids, as established by the Lancaster and Knoblich (2014) protocol, do not achieve complete postmitotic maturity and never completely lose the neural stem cell population. Given that and our previous experience with Ras and TP53 and the short incubation time, we opted to use this combination as a proof of principle (Marumoto et al., 2009). We co-injected the two plasmids close to the surface of the 4-month-old organoids, where the cortical structure is formed, and electroporated. The two injected plasmids consist of the pSpCas9(BB)-2A-GFP-expressing plasmid designed by the Zhang lab (Ran et al., 2013) with a TP53 single-guide RNA (sgRNA). The sgRNA was designed to target a region within the human TP53 locus inside exon 4, which is within the transcriptional activation domain of TP53 and upstream of its DNA-binding domain. The specificity of sgRNA was tested by transfection to 293T cells (Figure S2); insertions or deletions are likely to lead to a truncated TP53 gene, as it will be frame-shifted or contain an internal deletion. A second construct is a donor plasmid containing an expression cassette for an oncogenic cytomegalovirus (CMV) promoter-driven HRasG12V that will integrate disrupting the open reading frame of TP53 by homologous recombination. Immediately downstream 3′ of HRasG12V, we introduced an IRES-tdTomato cassette as a fluorescent marker. The tdTomato allows monitoring of tumor cells with the oncogenic cassette. Integration within the human TP53 locus is promoted by the inclusion of two adjacent homology arms in the donor plasmid. These homology arms are flanked by the same sgRNA-targeted cognate sequence of the TP53-targeted locus. In this manner, the CRISPR/Cas9 expression will linearize the co-transfected donor construct in addition to cutting the genomic loci of both TP53 alleles (Figure 1A). It will generate a linear donor construct, an oncogenic cassette flanked with homology arms targeting the same TP53 exon 4 locus at its 5′ and 3′ ends. The simultaneous cuts within the genomic locus and the targeting plasmid maximize the likelihood of integrating the RAS expression cassette in the TP53 locus, leading to loss of TP53 activity.
Figure 1. Generation and Transformation of Human Cerebral Organoids Using the CRISPR/Cas9 System.

(A) Schematic representation of the CRISPR/Cas9-mediated homologous recombination strategy. CRISPR/Cas9 TP53 sgRNA plasmid was co-electroporated with HRasG12V-expressing homologous recombination donor vector (HRasG12V-tdTomato p53 exon HA), which is also targeted and cleaved by the TP53 sgRNA (orange). Two homologous regions (HA1 and HA2, purple) mediate integration within the TP53 gene locus.
(B) Co-localization of CRISPR/Cas9 (GFP) and HRasG12V (tdTomato) observed 2 weeks after transfection.
(C) Ki-67 immunostaining shows HRasG12V-transduced cells are highly proliferative compared to controls.
(D) Time-lapse imaging of HRasG12V-transduced cells showing invasion from 4 weeks on after electroporation. (a) High-magnification view of the invasive edges of a 13-week transduced organoid.
Scale bars: 100 mm in (B), (C), and (D-a) and 1 mm in (D).
Two weeks after electroporation, organoids containing cells expressing both GFP from the CRISPR/Cas9 construct and tdTomato from the HRasG12V cassette can be observed (Figure 1B). It is in this population where the oncogenic recombination events are expected to appear. At 2 weeks post-electroporation, individual cells expressing tdTomato are observable. These cells do not overtly increase in number for another week, but then they start growing and dividing through the entire organoid until completely taking over the organoid. As shown in Figure 1D, time-lapse microscopic imaging allows observation of tumor development from the moment it is electroporated.
The control un-transduced organoids, at 11 weeks, display a sparse but visible labeling of the Ki-67 proliferative marker. This is in marked contrast with the lower panels in Figure 1C, which show that HRasG12V-transduced and TP53-disrupted cells that carry the tdTomato marker greatly increase the Ki-67 population within the organoid. To quantify the difference, we analyzed Ki-67-positive cells along the 400-mm superficial layers of the organoids, which is where tdTomato-positive cells are localized. In control organoids, 3.0% of the cells are still proliferative and Ki-67 positive, whereas in the transduced organoids, 18.6% of the cells are Ki-67 positive (p = 0.014), of which 16.9% are tdTomato positive and, hence, tumor-contributed (Figures S3A and S3B). This is consistent with the presence of an oncogenic event and a proliferative, invasive cell population (Figure 1D). Furthermore, these cells express the transcription factors SOX2 and OLIG2, two brain tumor stem cell markers associated with neural stem/progenitor cells. The tdTomato-positive population diffusely infiltrates into the organoid and are SOX2/OLIG2 double-positive for these stem cell markers (Figures S3C and S3D). These cells were 70.5% positive for SOX2, compared to 35.0% in controls (p = 0.029). Similarly, OLIG2 was expressed in 20.8% of the transduced cells and only in 7.8% (p = 0.0089) of the control cells (Figure S3E).
Tumor Progression and Invasion in Organoids
To quantify tumor proliferation and invasion, we dissociated organoids at 8 weeks after electroporation and quantified by flow cytometry the tdTomato-positive population. Results show that, at 8 weeks post-electroporation, tumorigenic cells encompass only 5.7% of the organoid. This is in marked contrast from 16 weeks post-electroporation, when 86.8% of the organoid is composed of tumor. Normal cerebral organoids will form smooth spheroid structures, but tumor-dominated organoids, as shown in an organoid 16 weeks post-electroporation, will show marked buds never seen with normal organoids. This invasive phenotype is exacerbated and generates projections of tumor mass beyond the boundaries of the organoid. This is reminiscent of invasive edges in human tumors, a behavior never observed in normal organoid cultures. This behavior is similarly absent in the early stages of tumor growth (Figure 2A).
Figure 2. Molecular Architecture of Invasive Tumor in Organoids.

(A) Organoid images and FACS profile of tdTomato+ transduced cells in organoids at 8 and 16 weeks after electroporation. FACS analysis shows that tdTomato+ cells are 5.7% of the organoid at 8 weeks, whereas at 16 weeks, they are (86.8%) of the organoid. Arrowheads show abnormal budding regions within the organoid (arrowhead).
(B–E) Immunostaining at 16 weeks for post-electroporation organoids. Transduced cells exhibit large numbers of OLIG2+ (B), GFAP+ (C), and SOX2+ (D), and cells are highly proliferative as seen in Ki-67 expression (E).
(F) RNA-seq analysis of FACS-sorted tdTomato+ (POS) and tdTomato− (NON) fractions. Samples were classified using the Verhaak four GBM subtypes: mesenchymal (orange), proneural (purple), classical (green), and neural (gray) subtypes. Upregulated (red) or dowregulated (blue) genes of each subtype are highlighted along the left side. Relative expression is plotted for each organoid sample (red indicates up, and blue indicates down). A two-tailed Welch’s t test was used to determine significance of differential enrichment/depletion between POS and NON organoids of average log-normalized fragments per kilobase of transcript per million mapped reads (FPKM) values (Figure S4).
(G) Fraction of POS and NON genes in each of the eight Verhaak gene sets (binomial test; NS, not significant).
Scale bars: 1 mm in (A) and (B) and 200 mm in (C)–(E).
See also Figure S4.
To verify the glioma stem cell identity within the putative tumor cells, we analyzed histologically the tumor-invaded organoids. The tumor organoids showed increased expression of OLIG2 (Figure 2B), glial fibrillary acidic protein (GFAP) (Figure 2C), and SOX2 (Figure 2D) markers of GBM stem cells, as well as marked increase in proliferation, as evidenced by very extensive Ki-67 labeling within the tdTomato-positive electroporated cell population (Figure 2E).
Molecular Signature of Tumor Organoids
To determine the molecular identity of the organoid-generated tumors, we characterized the expression profiles of the tumor cells within organoids. We sorted tdTomato-positive and -negative cell fractions of organoids for four independent batches of electroporated organoids. Transcriptome data were then evaluated against the Verhaak subtypes of clinical samples of GBM (Verhaak et al., 2010). To do this, we clustered the original 840 Verhaak diagnostic genes by their reported centroid signatures. Eight sets of genes that were up- or downregulated in classical, neural, proneural, and mesenchymal GBM subtypes defined the tumors. We then analyzed the expression of these genes across our tdTomato-positive and -negative fractions (Figure 2F) and tested the overlap ratios of up- and downregulated genes in each fraction for the eight Verhaak gene sets (Figure 2G). A comparison of the average normalized expression of genes in each of the eight Verhaak gene sets showed significant differences between tdTomato-positive and -negative fractions. Specifically, tdTomato-positive fractions tended to express genes that were upregulated in mesenchymal subtypes, while negative fractions, on average, upregulated genes that were more like neural stem cells, presumably representing normal stem cells in organoids. We conclude that the HRasG12V TP53−/− population was mesenchymal, consistent with previous results with mouse brain tumors (Friedmann-Morvinski et al., 2012; Marumoto et al., 2009). Thus, human organoid tumors fall within categories of defined subtypes of patient-derived GBM.
Transgene Characterization of Tumors in Organoids
Tumors were dissociated and grew well in 2D conditions if dishes were coated with Matrigel but grew as spheres if grown directly on plastic dishes (Figure 3A). The formation of neurospheres is consistent with the presence of a stem cell population. We characterized the tumor explants grown in 2D culture and observed simultaneous expression of GFAP, an astrocytic or neural stem cells marker, and SATB2, a marker for upper-layer cortical neurons (Figure 3B). Similarly, we observed cells that co-expressed the neural stem cell marker SOX2 with NeuN, a marker of differentiated neurons. We also observed cells expressing OLIG2, a marker of oligodendrocyte progenitors, which was present in cells expressing TUJ1, a marker for immature neurons. The abnormal simultaneous expression of stem cell markers, line-age-specific progenitor markers, and differentiated markers in gliomas following the transduction of oncogenes was also previously noted in human gliomas and also in our mouse glioma model (Friedmann-Morvinski et al., 2012; Galli et al., 2004).
Figure 3. Tumors from Organoids Show Tumorigenic Behavior and Invasiveness In Vivo.

(A) Organoid tumor explants were propagated either on Matrigel for 2D culture or as neurospheres.
(B) Tumor explants on 2D culture shows co-expression of GFAP and SATB2 (left), NeuN and SOX2 (middle), and OLIG2 with TuJ1 (right).
(C) CRISPR-mediated integration analysis. Southern probe design for tdTomato and the endogenous TP53 site.
(D) Sanger sequencing of the integration loci.
(E) Gross appearance and fluorescence signal of xenografted mouse brain. The dotted area shows a slightly pinkish appearance from tdTomato expression. Right panel indicates the fluorescence signal with a red fluorescent protein (RFP) filter.
(F) Kaplan-Meier survival curve of xenografted mice with mean survivals of 90 and 110 days.
(G) H&E staining of xenografted mouse brain coronal sections. Highly invasive phenotypes of the tumors.
(H) H&E staining shows a high degree of microscopic invasiveness. The tumor is diffuse (arrow) and perivascular (arrowhead).
(I–K) Higher magnification shows perivascular invasion (I; arrowhead indicates blood vessel), nuclear pleomorphism (J; arrowhead indicates giant cell, and arrows indicate hyperchromatic nuclei), and necrotic areas (K; arrow).
(L) Immunostaining with anti-CD31 antibody visualizes the endothelium and microvascular angiogenesis (upper panel). Organoid-derived tumors show high Ki-67 and are positive for SOX2 and GFAP.
Scale bars: 100 mm in (A), 20 mm in (B), and 100 mm in (H)–(L).
To ascertain that our design of the oncogenic driver was working as intended, we performed Southern blot analysis using a tdTomato probe and a TP53 probe (Figure 3C). In the case of successful targeting of the TP53 locus with our oncogenic cassette, the TP53 probe would be split into 2 fragments of 3,095 bp and 7,610 bp with a Hind III genomic DNA digest. An untargeted allele would give a single signal of 2,678 bp. Similarly, the tdTomato probe would give no signal if untargeted, but would give a single 7,610-bp band if successfully integrated. The Southern blot analysis shows that, as expected, we found two bands at 3,095 bp and 7,610 bp with the TP53 probe, as well as a single 7,610-bp signal with the tdTomato probe, thus confirming successful targeting by CRISPR/Cas9-mediated homologous recombination. To substantiate this result, we performed PCR amplification on the integration junctions and, following Sanger sequencing, confirmed the integration site with an A/T insertion within the exon 4 of TP53 immediately adjacent to the CRISPR/Cas9 cleavage site, which frameshifts TP53, rendering it inactive. The second allele revealed the integration of the HRasG12V-IRES-tdTomato cassette, which inactivatesTP53 (Figure 3D). These results confirm the design and consequent expression of the oncogene with the marker, tdTomato, and disruption of TP53.
Tumors in Organoids Are Tumorigenic and Invasive In Vivo
We tested the oncogenic potential of these cells in vivo. About 3 × 105 organoid-derived tumor cells were stereotaxically injected unilaterally in the hippocampus of immunodeficient NOD/SCID/IL2RGKO mice, which started to die within months. The mean survival times were of 90 and 100 days as shown in the Kaplan-Meier curves (Figure 3F). tdTomato-positive tumors were clearly visible (Figure 3E). H&E staining shows extensive microscopic invasiveness and nuclear pleomorphism, as previously described for human GBM. As with human GBM, the organoid-derived tumors frequently spread along blood vessels as shown (Figures 3H and 3I, arrowheads). Tumors are hyper-cellular with large nucleated giant cells, bizarre multinucleated cells (Figure 3J; arrowhead), and occasional areas of small cells with hyperchromatic nuclei (Figure 3J, arrow), and they also show some necrotic areas (Figure 3K). Large areas with foci of tumor cells show extensive Ki-67 expression. Tumors were highly angiogenetic, as assessed by high levels of CD31 staining, and showed markers of GBM neural stem cells (SOX2 and GFAP) (Figure 3L). These observations prove that organoid-derived GBMs have full oncogenic potential and characteristics of human tumors.
Tumor Organoids Are Serially Transplantable
We generated organoid-derived GBM cell-culture spheres and co-cultured these with new intact mature organoids. Tumor-spheres spontaneously attached onto the non-transformed organoids and started to grow on them (Figure 4A). Tumor cells started to invade the organoid, and by day 24, the tumor co-culture had invaded ~30% of the original organoid. The invasive cells were highly proliferative, as shown by Ki-67 staining, and expressed the stem cell marker SOX2 and GFAP at high levels.
Figure 4. Organoids as Platforms for Tumor Transplantation.

(A) Tumorsphere of the transformed organoid-derived cells attaches and spreads through an untransduced mature organoid showing an invasive phenotype (arrowheads). Tumor cells are highly proliferative (Ki-67) and show high expression of SOX2 and are GFAP positive as they invade the organoid.
(B) Kaplan-Meier survival curve of mice injected with SK2176 and SK429 (n = 3; mean survival for SK2176 xenografts: 34 days). SK429 xenografts show a lack of lethality in mice.
(C and D) SK2176 cell-line-generated tumorspheres attach and invade mature cerebral organoids over the course of 21 days (C, arrowheads). SK429 and SK2176 both express proliferation marker Ki-67, but only SK2176 displayed an invasive phenotype (D).
Scale bars: 1 mm in (A, top panel) and (C) and 100 mm in (A, lower panel) and (D).
To test whether tumor characteristics of existing patient-derived cell lines could be tested in the context of cerebral organoids, we tested two patient-derived GBM cell lines, SK2176 and SK429. We labeled both cell lines with tdTomato and then injected these into NSG mouse brains. SK429 failed to cause any disease in mice within 6 months, whereas SK2176 killed mice in less than 40 days when xenografted orthotopically (Figure 4B). When tested within the organoid context, SK429 grew on the surface but did not invade the parenchyma of the organoids. SK2176, on the other hand, invaded and proliferated within the organoid parenchyma readily (Figures 4C and 4D). Thus, invasiveness in organoids, at least in these two cases, correlates with lethality in mice. Although both cell lines were highly proliferative, as shown by Ki-67 staining, only SK2176 displayed an invasive phenotype. Thus, the results shown suggest a possibility of testing human primary tumor explant properties in cerebral organoids.
DISCUSSION
Tumors can be induced in human cerebral organoids by introducing CRISPR/Cas9 and sgRNAs in combination with the activated oncogene HRasG12V and simultaneous disruption of the tumor suppressor, TP53. Use of human cerebral organoids and the genetic manipulation will allow us to observe the earliest steps of tumorigenesis in a human context with a defined genetic manipulation. Here, we only introduced two genes for a proof of principle; one could conceivably introduce additional genes or larger gene expression cassettes to construct relevant organoid models. Although RAS activation and TP53 deletion are not a common mutation in GBMs, the extensive characterization of experimental tumors with this combination, and our previous mouse models, made a direct comparison of mouse and organoid models possible. The tumors generated in human cerebral organoids are invasive and, over the course of a few weeks, destroy all normal organoid tissue and substitute it with tumor tissue. Xenograft models of organoid-derived tumor cells exhibited uncontrolled invasive growth that led to morbidity and death of the injected mice, thus demonstrating that organoid-generated tumors exhibit essential features of tumorigenesis. This was also confirmed at the molecular level by the expression profiles of these cells, which match those previously observed in human GBMs (Verhaak et al., 2010). The most commonly observed subtype belonged to the mesenchymal secondary GBM, which is consistent with our previously generated lentiviral mouse model (Friedmann-Morvinski et al., 2012; Marumoto et al., 2009).
In addition to being transplantable from organoids to mice, organoid-derived tumors were transplantable from organoid to organoid. The organoids, in addition to being platforms for testing the role of tumor suppressors and oncogenes, can also be used to support the growth of primary human brain tumor explants, which proliferate well on organoids when co-cultured and allowed to attach. Though the number of cell lines we have used is limited, it is interesting to note that a primary patient-derived cell line—which, when orthotopically injected, failed to kill mice—also failed to invade into the parenchyma of our organoids. In contrast, a second primary isolate and an established tumor cell line that readily kills mice clearly invaded the interior of organoids. Future experiments with greater numbers of primary isolates and cell lines will likely clarify whether the invasive behavior of primary patient-derived isolates and cell lines in organoids are predictive of invasiveness and/or lethality in mice and, by extension, in human patients. We think that organoid tests could possibly be good complements to PDX models in mice. It will be interesting to compare these two systems and see what information each system could provide toward predicting behaviors of tumors in patients. A recent study shows through genomic analyses that PDX models appear to only recapitulate normal human tumor genetic alterations for a very limited number of mouse passages (Ben-David et al., 2017). On the other hand, another recent study of breast cancer organoids shows that their organoid system maintains original tumor genomic polymorphisms (Sachs et al., 2018). This leads to the possibility that organoids might be better preserving human mutational profiles than PDX models. Future whole-genome sequencing studies of organoid-generated and propagated tumors should address whether the niche within the organoid model has properties preserving genomic stability. A still unexplored possibility is whether tumor cells from a different histogenesis, such as breast cancers and lung cancers, which frequently metastasize to the brain, could also be studied in brain organoids.
Cerebral organoid models, although promising, lack some essential cell types that are certainly important for tumor pathogenesis. Among these are a lack of endothelial cells. For example, we and others have observed that GBMs have a preference for tissue invasion along existing vasculature tracking along existing blood vessels (Figure 4D) (Watkins et al., 2014). Thus, the absence of endothelial cells could limit glioma natural history reconstruction in organoids. In the future, this might be addressed by co-culture with endothelial and mesenchymal progenitors to produce more realistic cerebral organoids (Ibrahim and Richardson, 2017; Lauschke et al., 2017). Other important components absent are hematopoietic-stem-cell-derived microglial cells that mediate brain inflammatory and injury responses. These could conceivably be incorporated by co-culture or by providing hematopoietic progenitors to give rise to microglial cells (Douvaras et al., 2017; Takata et al., 2017). In addition to issues of missing cell types, there are outstanding issues with human cerebral organoids. An unresolved issue is the known variability in organoid generation. As compared to gut organoids, cerebral organoid generation is more variable (Camp et al., 2015; Quadrato et al., 2017). Brain organoids frequently also contained some amount of non-CNS differentiated tissues, which could conceivably complicate interpretation (Figure S1B). This common occurrence has been identified in single-cell studies as mesodermal precursors and an unidentified proliferative precursor population (Quadrato et al., 2017). In addition, human cerebral organoids do not fully differentiate. Of the human cerebral cortex’s six layers, cerebral organoids can only produce the deepest layers. Although the normal adult human brain is largely postmitotic, human cerebral organoids have significant numbers of SOX2+ stem/progenitor cells, even when fully mature (Figure S1A).
Despite these current limitations, it is clear that human pluripotent-cell-derived cerebral organoids can support tumorigenesis. The tumors generated are similar to those generated in mice by other methods using the same oncogenes/tumor suppressors and are transplantable like patient-derived tumors. Furthermore, human cerebral organoids support both proliferation and invasive behaviors using patient-derived primary tumor explants as well as established cell lines with unknown mutagenic genetic/epigenetic history. We propose that human cerebral organoids offer an interesting platform to explore ex vivo the natural history of cancer.
EXPERIMENTAL PROCEDURES
All animal experiments were performed in accordance with a Salk Institute IACUC-approved animal protocol. Similarly, human-originated materials were approved by the Salk Institute IRB.
Plasmid Construction and Cell Culture
sgRNAs targeted to TP53 exon 4 were designed using CRISPRdirect (Naito et al., 2015) (http://crispr.dbcls.jp) and cloned into the PX459 plasmid (Ran et al., 2013). The donor plasmid, the HRasG12V IRES tdTomato fragment, was cloned into pcDNA3.1(+) with 200-bp homology arms synthesized as gBlock fragments (Integrated DNA Technologies).
Cerebral organoids were generated from the human embryonic stem cell (ESC) line H9 using the protocol previously described (Lancaster and Knoblich, 2014).
Organoid Electroporation
4-month-old cerebral organoids were co-electroporated using a 3-mm-diameter tweezer electrode and the Electro Square Porator (ECM 830) from BTX Harvard Apparatus. Electroporated organoids were quickly placed back into the cerebral organoid differentiation medium and then incubated in an orbital shaker in an incubator with 5% CO2 at 37°C and kept shaking at 80 rpm.
Cell Sorting and RNA Sequencing
Transduced organoids were dissociated to create single-cell suspensions for cell sorting. Cells were sorted into TRIzol, and RNA was prepared for sequencing using Illumina TruSeq RNA chemistry. RNA-sequencing (RNA-seq) counts were generated with the STAR aligner on the hg19 human genome.
Mouse Orthotopic Xenografts
The transduced organoid-derived cell lines or human-patient-derived tumor cell lines were dissociated with TrypLE. Single-cell suspensions in cerebral organoid differentiation medium were stereotaxically injected (3 × 105 cells per animal) into the hippocampus of NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice, as previously described (Marumoto et al., 2009).
Sphere Co-culture with Intact Organoids
Transduced organoid-derived cell lines were cultured on non-coated plastic dishes in cerebral organoid differentiation medium to induce sphere formation. Human-patient-derived glioblastoma cell lines were transferred into low-attachment 96-well plates in cerebral organoid differentiation medium at a density of 18,000 cells per well. 72 hr after tumorsphere formation, these were transferred into 6-well plates and mixed with intact human cerebral organoids to initiate invasion (10 spheres with 5 organoids per well) and incubated in an orbital shaker in an incubator for the remainder of the experiment.
Statistical Analysis
Data are presented as mean ± SEM and were analyzed using paired two-tailed Student’s t test to determine significance. For statistical significance, p values < 0.05 were considered significant.
Further details and an outline of resources used in this work can be found in the Supplemental Experimental Procedures.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-sequencing data reported in this paper is GenBank: GSE109982.
Supplementary Material
Highlights.
Human cerebral organoids can be used as a model for tumor formation
Oncogene manipulation by CRISPR/Cas9 initiates tumorigenesis in cerebral organoids
Time-lapse microscopic imaging allows observation of tumor development in organoids
Tumor cells derived from organoids display an invasive phenotype in xenografted mice
Acknowledgments
We thank Dr. Nissi Varki for her evaluation of tumor histopathology. We thank Dr. Santosh Kesari for providing patient-derived GBM cell lines. I.M.V. is an American Cancer Society Professor of Molecular Biology and holds the Irwin and Joan Jacobs Chair in Exemplary Life Science. This work was supported, in part, by a grant from the NIH (R01 CA095613); a Cancer Center Core grant (P30 CA014195-38); Ipsen; the H.N. and Frances C. Berger Foundation; the Leona M. and Harry B. Helmsley Charitable Trust grant #2017-PG-MED001, the Glenn Center for Aging Research and the Razavi Newman Integrative Genomics and Bioinformatics Core Facility, with funding from NIH-NCI CCSG: P30 014195; the Waitt Advanced Biophotonics Core Facility of the Salk Institute, with funding from NIH-NCI CCSG: P30 014195, NINDS Neuroscience Core grant NS072031, and the Waitt Foundation; the Stem Cell Core Facility of the Salk Institute, with funding from the Helmsley Charitable Trust; the Flow Cytometry Core Facility of the Salk Institute, with funding from NIH-NCI CCSG: P30 014195; and the NGS Core Facility of the Salk Institute, with funding from NIH-NCI CCSG: P30 014195, and the Chapman Foundation.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.105.
AUTHOR CONTRIBUTIONS
Conceptualization, J.O., G.M.P., and I.M.V.; Methodology, J.O.; Formal Analysis, J.O. and M.N.S.; Investigation, J.O.; Data Curation, M.N.S.; Resources, M.N.S.; Writing – Original Draft, J.O., G.M.P., and I.M.V.; Writing – Review & Editing, J.O., G.M.P., M.N.S., and I.M.V.; Visualization, J.O. and M.N.S.; Supervision, I.M.V.; Funding Acquisition, I.M.V.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Ben-David U, Ha G, Tseng Y-Y, Greenwald NF, Oh C, Shih J, McFarland JM, Wong B, Boehm JS, Beroukhim R, Golub TR. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017;49:1567–1575. doi: 10.1038/ng.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Bräuninger M, Lewitus E, Sykes A, Hevers W, Lancaster M, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA. 2015;112:15672–15677. doi: 10.1073/pnas.1520760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douvaras P, Sun B, Wang M, Kruglikov I, Lallos G, Zimmer M, Terrenoire C, Zhang B, Gandy S, Schadt E, et al. Directed differentiation of human pluripotent stem cells to microglia. Stem Cell Reports. 2017;8:1516–1524. doi: 10.1016/j.stemcr.2017.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, Ellisman MH, Verma IM. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338:1080–1084. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- Hackam DG, Redelmeier DA. Translation of research evidence from animals to humans. JAMA. 2006;296:1731–1732. doi: 10.1001/jama.296.14.1731. [DOI] [PubMed] [Google Scholar]
- Ibrahim M, Richardson MK. Beyond organoids: in vitro vasculogenesis and angiogenesis using cells from mammals and zebrafish. Reprod. Toxicol. 2017;73:292–311. doi: 10.1016/j.reprotox.2017.07.002. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014;9:2329–2340. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauschke K, Frederiksen L, Hall VJ. Paving the way toward complex blood-brain barrier models using pluripotent stem cells. Stem Cells Dev. 2017;26:857–874. doi: 10.1089/scd.2017.0003. [DOI] [PubMed] [Google Scholar]
- Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, Verma IM. Development of a novel mouse glioma model using lentiviral vectors. Nat. Med. 2009;15:110–116. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y, Hino K, Bono H, Ui-Tei K. CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics. 2015;31:1120–1123. doi: 10.1093/bioinformatics/btu743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Min Yang S, Berger DR, Maria N, Scholvin J, Goldman M, Kinney JP, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545:48–53. doi: 10.1038/nature22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, Balgobind AV, Wind K, Gracanin A, Begthel H, et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172:373–386.e10. doi: 10.1016/j.cell.2017.11.010. [DOI] [PubMed] [Google Scholar]
- Shanks N, Greek R, Greek J. Are animal models predictive for humans? Philos. Ethics Humanit. Med. 2009;4:2. doi: 10.1186/1747-5341-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata K, Kozaki T, Lee CZW, Thion MS, Otsuka M, Lim S, Utami KH, Fidan K, Park DS, Malleret B, et al. Induced-pluripotent-stem-cell-derived primitive macrophages provide a platform for modelling tissue-resident macrophage differentiation and function. Immunity. 2017;47:183–198.e6. doi: 10.1016/j.immuni.2017.06.017. [DOI] [PubMed] [Google Scholar]
- Van Der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O’Collins V, Macleod MR. Can animal models of disease reliably inform human studies? PLoS Med. 2010;7:e1000245. doi: 10.1371/journal.pmed.1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins S, Robel S, Kimbrough IF, Robert SM, Ellis-Davies G, Sontheimer H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014;5:5196. doi: 10.1038/ncomms5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-sequencing data reported in this paper is GenBank: GSE109982.