

Dissecting the BAZ1B genetic circuitry in the neural crest brings out its critical role in shaping the modern human face.
Abstract
We undertook a functional dissection of chromatin remodeler BAZ1B in neural crest (NC) stem cells (NCSCs) from a uniquely informative cohort of typical and atypical patients harboring 7q11.23 copy number variants. Our results reveal a key contribution of BAZ1B to NCSC in vitro induction and migration, coupled with a crucial involvement in NC-specific transcriptional circuits and distal regulation. By intersecting our experimental data with new paleogenetic analyses comparing modern and archaic humans, we found a modern-specific enrichment for regulatory changes both in BAZ1B and its experimentally defined downstream targets, thereby providing the first empirical validation of the human self-domestication hypothesis and positioning BAZ1B as a master regulator of the modern human face. In so doing, we provide experimental evidence that the craniofacial and cognitive/behavioral phenotypes caused by alterations of the Williams-Beuren syndrome critical region can serve as a powerful entry point into the evolution of the modern human face and prosociality.
INTRODUCTION
Anatomically modern humans (AMHs) exhibit a suite of craniofacial and prosocial characteristics that are reminiscent of traits distinguishing domesticated species from their wild counterparts (1–3). This has led to the formulation of a self-domestication hypothesis according to which modern humans (3) went through a domestication process in the course of their evolution. Recent evidence, along with the well-warranted distinction between domestication and selective breeding (4), is also extending this notion to other species that might have undergone a self-domestication phase, such as cats, dogs, and bonobos (3). Thus, as self-domestication represents a special case of domestication, the most parsimonious hypothesis must posit the same core mechanisms to underlie both. For this reason, the self-domestication hypothesis also entails the prediction that key aspects of modern humans’ anatomy and cognition can be illuminated by studies of the so-called “domestication syndrome,” the core set of domestication-related traits that was recently proposed to result from mild neural crest (NC) deficits (5). However, both the neurocristopathic basis of domestication and its extension to the evolution of AMHs remain to be tested experimentally.
Williams-Beuren syndrome [WBS; OMIM (Online Mendelian Inheritance in Man) 194050] and Williams-Beuren region duplication syndrome (7dupASD; OMIM 609757), caused respectively by the hemideletion or hemiduplication of 28 genes at the 7q11.23 region [WBS critical region (WBSCR)], represent a paradigmatic pair of neurodevelopmental conditions whose NC-related craniofacial dysmorphisms and cognitive/behavioral traits (6, 7) bear directly on domestication-related traits relevant for AMHs (facial reduction and retraction, pronounced friendliness, and reduced reactive aggression) (fig. S1A). Structural variants in WBS genes, for example in the case of GTF2I and its paralogs, have been shown to underlie stereotypical hypersociability in domestic dogs and foxes (8, 9).
Among the WBSCR genes, we focus here on the chromatin regulator BAZ1B (also known as Williams syndrome transcription factor, WSTF), on the basis of the following lines of evidence that implicate it in domestication-relevant craniofacial features: (i) its established role in NC maintenance and migration in Xenopus laevis and the craniofacial defects observed in knockout mice (10, 11); (ii) the observation that its expression is affected by domestication-related events in canids (12); (iii) the first formulation of the neurocristopathic hypothesis of domestication, which included BAZ1B among the genes influencing NC development (5); (iv) the most comprehensive studies focusing on regions of the modern human genome associated with selective sweep signals compared to Neanderthals/Denisovans (hereafter “archaics”) (13, 14), one of which specifically included BAZ1B within the detected portions of the WBSCR; and (v) the thus far most detailed study systematically exploring high-frequency (HF) (>90%) changes in modern humans for which archaic humans carry the ancestral state, which found BAZ1B enriched for mutations in modern humans (most of which fall in the regulatory regions of the gene) (15).
Our previous work had established the largest cohort of 7q11.23 patient-derived induced pluripotent stem cell (iPSC) lines and revealed major disease-relevant transcriptional dysregulation that was already apparent at the pluripotent state and became further exacerbated upon differentiation (16). Here, we first harness this resource to dissect the impact of BAZ1B dosage on the NC of patients with WBS and 7dupASD, both in terms of function (i.e., NC migration and induction) and of transcriptional and chromatin dysregulation, thereby defining the BAZ1B dosage–dependent circuits controlling the NC. Next, we apply these experimentally determined BAZ1B-dependent circuits underlying craniofacial morphogenesis to interrogate the evidence from paleogenomic analyses, which were thus far only of a correlative nature. We find major convergence between the BAZ1B control and the genes harboring regulatory changes in the modern human lineage. Together, the definition of the role of BAZ1B dosage in craniofacial neurocristopathy and its application to domestication-relevant paleogenomics demonstrate a major contribution of BAZ1B to the modern human face and offer experimental validation for the prediction at the heart of NC-based accounts of (self-) domestication: that the modern human face acquired its shape as an instance of mild neurocristopathy.
RESULTS
Establishment and validation of an extensive cohort of patient-specific BAZ1B-interfered NC stem cell lines
To dissect the role of BAZ1B in the craniofacial dysmorphisms that characterize WBS and 7dupASD, we started from our previous characterization of WBS patient– and 7dupASD patient–specific iPSC lines and differentiated derivatives (16) and selected a cohort of 11 NC stem cell (NCSC) lines (four from patients with WBS, three from patients with 7dupASD, and four from control individuals), which also represent the largest cohort of patient-specific NCSCs described so far. Given the centrality of the cranial NC for the development of the face, we first validated the cranial identity of our NCSC cohort by transcriptomic profiling through a manually curated gene expression signature (fig. S2A), confirming their suitability for the study of craniofacial dysregulations. We then knocked down BAZ1B via RNA interference in all lines across the three genetic conditions, including also NCSCs derived from a particularly informative patient with atypical WBS (hereafter atWBS) bearing a partial deletion of the region that spares BAZ1B and six additional genes (Fig. 1A) (17). To establish a high-resolution gradient of BAZ1B dosages, we selected two distinct short hairpin RNA (shRNA) against BAZ1B (i.e., sh1 and sh2) along with a scrambled shRNA sequence (hereafter scr) as negative control, for a total of 32 NCSC lines. Knockdown (KD) efficiency was evaluated at the RNA level by quantitative polymerase chain reaction (qPCR) (Fig. 1B and fig. S1C), confirming the attainment of the desired gradient with an overall reduction of about 40% for sh1 and 70% for sh2, as well as reduction at the protein level, as detected by Western blot (fig. S1E).
Fig. 1. BAZ1B KD impairs migration and induction of patient-specific iPSC-derived NCSCs.
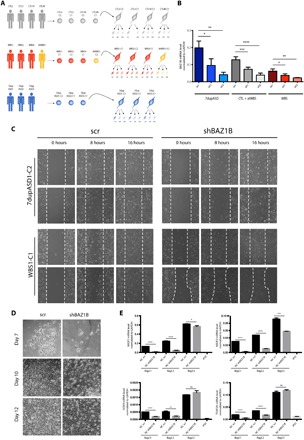
(A) Schematic representation of the KD strategy on our iPSC-derived NCSC cohort. (B) BAZ1B mRNA levels in all the interfered lines (scr, sh1, and sh2) as measured by qPCR. Data represent aggregates of samples with the same number of BAZ1B copies (7dup, CTL + atWBS, and WBS). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is used as a normalizer. (C) Eight- and 16-hour time points from the wound-healing assay analyses performed on a 7dupASD and a WBS NCSC line upon BAZ1B KD. Cells from the same line infected with the scr sh were used as references for the migration (n = 2). (D) Days 7, 10, and 12 of NC differentiation from embryoid bodies (EBs) of an scr-interfered iPSC line and its respective BAZ1B KD (n = 3). (E) mRNA levels of NC markers at day 12 of differentiation in three individual experimental replicates [bright-field images are reported in (D)]. An iPSC line is included as a negative control. Student’s t test was used (ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0. 0001).
BAZ1B dosage imbalance impairs NCSC migration and induction
NCSCs need to migrate to reach specific target regions in the developing embryo and give rise to distinct cell types and tissues, including craniofacial structures that are major areas of change in human evolution. Since BAZ1B KD was shown to affect the migration of the NC in X. laevis and to promote cancer cell invasion in different lung cancer cell lines (10, 18), we hypothesized that the BAZ1B dosage imbalances entailed in the 7q11.23 syndromes could result in a defective regulation of NCSC migration and might underlie the NC-related alterations typical of patients with WBS and 7dupASD. To test this, we compared the migration properties of patient-specific BAZ1B KD NCSC lines (sh2) to their respective control NCSC line (scr) by the well-established wound-healing assay. The 7dupASD NCSC KD lines took longer to fill the wound when compared to the respective control lines (scr), as indicated by images taken at 8 and 16 hours after a gap was created on the plate surface (Fig. 1C and fig. S1F). We instead observed an opposite behavior for the WBS BAZ1B KD lines, which were faster than the respective scr lines in closing the gap (Fig. 1C and fig. S1F). In contrast to the previous observations from X. laevis (10), we also observed a minor delay in NC induction as a consequence of BAZ1B KD (Fig. 1D and fig. S1D), by means of a differentiation protocol based on NC delamination from adherent embryoid bodies (EBs), which recapitulates the initial steps of NC generation (19). In particular, starting from 2 to 3 days after attachment of EBs, we observed a lower number of outgrowing cells in the KD line (Fig. 1D, days 7 and 10), coupled with an evidently higher cell mortality. Cells were eventually able to acquire the typical NC morphology, although lower differentiation efficiency was evident, as shown by images taken at day 12. In addition, the delay in NC formation was associated with a down-regulation of well-established critical regulators of NC migration and maintenance, including NR2F1, NR2F2, TFAP2A, and SOX9 (Fig. 1E). These results show that BAZ1B regulates the developing NC starting from its earliest migratory stages and that the symmetrically opposite 7q11.23 dosages alterations prime NCSCs to symmetrically opposite deficits upon BAZ1B interference. In turn, the central role of the NC in the development of facial morphology allows relating such findings to the symmetrically opposite craniofacial dysmorphisms of the two 7q11.23 syndromes.
BAZ1B interference disrupts key NC-specific transcriptional circuits
Having defined the functional impact of BAZ1B dosage on NC function, we predicted that a main molecular readout of its dosage imbalances would be at the level of transcriptional regulation, given its critical role as transcriptional regulator in different cell and animal models (20–22). To test this hypothesis and gain mechanistic insights into the specific BAZ1B dosage–dependent downstream circuits, we subjected 32 interfered NCSC lines to high-coverage RNA sequencing (RNA-seq) analysis. As shown in fig. S2A, a manually curated signature from an extensive literature review (23–28) validated the cranial identity of our NCSC lines, while clustering by Pearson correlation excluded the presence of any genotype- or hairpin-specific expression change. Confirming our previous observations in the two largest cohorts of iPSC lines (29), a principal component analysis (PCA) corroborated the significant impact of individual genetic backgrounds on transcriptional variability, with most “KD lines” clustering with their respective control “scr line.” This was consistent with the narrow range of experimentally interfered BAZ1B dosages and pointed to a selective BAZ1B dosage–dependent transcriptional vulnerability (fig. S2B).
To dissect it, we thus resorted to a combination of classical pairwise comparative analysis, contrasting shBAZ1B-interfered NCSC lines (sh1 + sh2) with their respective controls (scr), with a complementary regression analysis using BAZ1B expression levels as independent variables, subtracting the contribution of individual genetic backgrounds. This design increases robustness and sensitivity in the identification of genes that, across multiple genetic backgrounds and target gene dosages, might have a different baseline (scr) across individuals while still being robustly dysregulated upon BAZ1B interference.
The two analyses identified a total of 448 genes with false discovery rate (FDR) < 0.1 (1192 with P < 0.01 and FDR < 0.25) whose transcriptional levels followed BAZ1B dosage, in either a direct (202; 539 with P < 0.01 and FDR < 0.25) or an inverse (246; 653 with P < 0.01 and FDR < 0.25) fashion. In addition, genes identified in the regression analysis included around 90% of the differentially expressed genes (DEGs) (27 of 29, FDR < 0.1) found in the comparative analysis (Fig. 2A). Consistent with the differential efficiency of the two short hairpins, we found a globally stronger transcriptional impact for the group of samples targeted by sh2 (fig. S2C) and a milder but nevertheless clearly distinguishable effect of sh1, resulting in particularly informative gradient of dosages over the scr control lines.
Fig. 2. BAZ1B KD is responsible for transcriptional alterations in NC-related pathways.
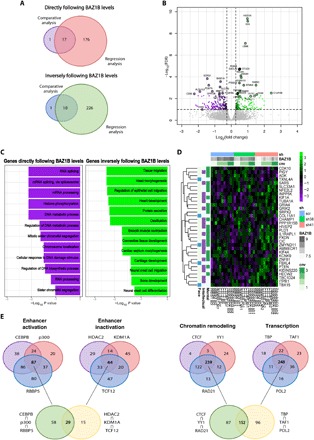
(A) Overlap between genes directly or inversely following BAZ1B levels identified in the pairwise comparative analysis (scr versus shBAZ1B) and in the regression analysis on BAZ1B-level sensitive genes on iPSC-derived NCSCs (FDR < 0.1). (B) Volcano plot reporting DEGs identified in the RNA-seq analysis on iPSC-derived NCSCs [fold change (FC) > 1.25; FDR < 0.1]. (C) Top most specific enrichments for GO biological processes among the DEGs in the RNA-seq analysis on iPSC-derived NCSCs. (D) Heat map representing DEGs that are dysregulated in genetic disorders involving mental retardation (“Mental”), intellectual disability (“Intellectual”), and/or facial dysmorphisms (“Face”) according to OMIM database classification. cnv, copy number variant. (E) Putative regulators of genes that follow BAZ1B levels identified by a master regulator analysis. Regulators were divided in four different groups based on their main functions.
Particularly noteworthy among the genes that we found correlated with BAZ1B levels were (i) crucial regulators of cranial NC, further highlighting a convergent BAZ1B dosage–dependent dysregulation of the foundational CUL3-centered regulatory axis orchestrating NC-mediated craniofacial morphogenesis (30), and (ii) genes associated with variation of human facial shape or causative of dysmorphic facial features and mild intellectual disability when mutated (Fig. 2B and table S1).
Gene Ontology (GO) analysis performed on genes directly following BAZ1B levels suggested specific enrichments in biological processes such as histone phosphorylation, chromosome localization, RNA processing, and splicing. Genes inversely following BAZ1B levels were instead enriched in categories particularly relevant for NC and NC-derivative functions, such as cell migration and cardiovascular and skeletal development (Fig. 2C). By querying the OMIM database, we found that several DEGs were associated with genetic disorders whose phenotypes include “mental retardation,” “intellectual disability,” and/or “facial dysmorphisms” (Fig. 2D), underscoring the pertinence of BAZ1B-dependent dysregulation across both the neurocristopathic and cognitive axes.
Last, a master regulator analysis identified candidate regulators of BAZ1B DEGs, including factors involved in enhancer marking [CEBPB, p300, RBBP5, HDAC2 (histone deacetylase 2), KDM1A, and TCF12], promoter activation [TBP (TATA box–binding protein), TAF1 (TBP-associated factor 1), and POL2 (polymerase 2)], and chromatin remodeling (CTCF, RAD21, and YY1) (Fig. 2E and fig. S2D), several of which are themselves causative genes of intellectual disability syndromes with neurocristopathic involvement, as in the case of our recently identified Gabriele–de Vries syndrome caused by YY1 haploinsufficiency (31, 32). Chromatin remodeling was indeed the most prominently enriched group within the overall domain of transcriptional regulation. Two master regulators are particularly noteworthy, as they are themselves regulated by BAZ1B dosage. The first is EGR1 (FDR < 0.1), which is itself among the genes inversely correlated with BAZ1B levels, which is implicated in cranial development (in animal models) (33, 34) and whose promoter has been recently shown to feature a bivalent state in human embryonic cranial NC (23, 35). The second is MXI1, identified as master regulator of genes directly following BAZ1B levels (FDR < 0.001), which is itself found among the genes inversely correlated with BAZ1B and is itself a regulator of BAZ1B, pointing to a cross-talk between the two (fig. S2C). Notably, two differentially expressed targets of MXI1, TGFB2 and NFIB, are also involved in intellectual disability and craniofacial defects (30, 36, 37).
BAZ1B regulates the NC epigenome in a dosage-dependent manner
The transcriptional readout and functional impact of BAZ1B dosage (at the level of NC induction and migration) established its role as a master controller of the NC. We thus predicted, on the basis of its molecular function, that BAZ1B would directly bind to key NC target genes and that for some of these, the binding would be dosage sensitive. These genes would be, in turn, the most likely direct targets to mediate the dosage-dependent transcriptional and functional phenotypes described above. To test this prediction, we set out to both identify BAZ1B direct targets and characterize their promoter and enhancer states, so as to mechanistically link their transcriptional dysregulation with BAZ1B dosage–dependent chromatin binding. Given the absence of chromatin immunoprecipitation (ChIP)–grade BAZ1B antibodies, to carry out our ChIP coupled with sequencing (ChIP-seq) on scr and KD lines, we first designed a tagging strategy to establish, by CRISPR-Cas9 editing, a series of in-frame 3xFLAG endogenously tagged BAZ1B alleles in representative iPSCs of the four genotypes (Fig. 3A and fig. S3, A and B). These were then differentiated to NCSCs (fig. S3C) and subjected to ChIP-seq with anti-FLAG antibody, enabling a faithful characterization of BAZ1B genome-wide occupancy across dosages (one tagged allele in WBS, two tagged alleles in atWBS and CTL, and two tagged alleles in the context of 1.5-fold dosages in 7dupASD).
Fig. 3. BAZ1B preferentially binds its targets at their enhancer regions and its KD causes a redistribution of enhancer histone marks.
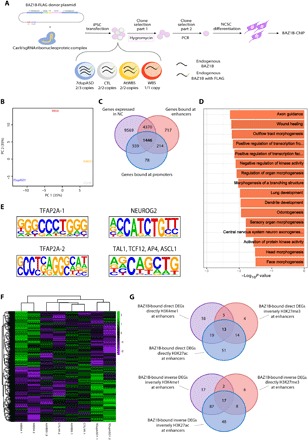
(A) Schematic representation of the strategy for CRISPR-Cas9–mediated tagging of endogenous BAZ1B. Briefly, iPSCs from the four genotypes were electroporated with the donor plasmid and the Cas9/single-guide RNA ribonucleoprotein complex; clones were selected via hygromycin and PCR, differentiated to NCSCs, and then subjected to ChIP-seq. (B) PCA showing the distribution of the four BAZ1B-tagged NCSC lines according to their chromatin profiles. (C) Overlap between genes expressed in our NCSC lines (purple) and genes bound by BAZ1B at their enhancer (red) or promoter (blue) regions. (D) Top most specific enrichments for GO biological processes among the genes that are bound by BAZ1B and expressed in our NCSC cohort. (E) Most represented BAZ1B DNA binding motifs identified by HOMER show high similarity to neural and NCSC-specific transcription factors motifs. (F) BAZ1B differentially bound regions according to its copy number (FDR < 0.1; n = 2). (G) Overlap between genes that are differentially expressed have their enhancers differentially marked concordantly (H3K27ac, H3K4me1, and H3K27me3) and are bound by BAZ1B at enhancers.
PCA shows a clear separation of the samples by BAZ1B copy number, with CTL and atWBS samples clustering more closely and WBS and 7dupASD samples clustering at opposing positions (Fig. 3B). To call NC-specific enhancer regions and promoter-enhancer associations, we exploited for chromatin annotation the unprecedented resolution afforded by the patients’ cohort with its underlying variability and proceeded to (i) select chromosomal regions featuring H3K4me1 and H3K27ac in at least two individuals; (ii) exclude regions marked with H3K4me3 in at least two individuals;(iii) eliminate regions bearing a transcription start site (TSS); and (iv) associate each putative enhancer to the closest TSS, identifying a total of 30,8470 putative enhancer regions. Notably, BAZ1B binds 75% of its targets at their enhancer regions (6747 genes), with the remaining 2297 targets bound at promoters (Fig. 3C). In addition, 40% of genes expressed in NC are bound by BAZ1B, either exclusively at enhancers (27.4%) or exclusively at promoters (3.5%) or at both regions (9%). This highlights its pervasiveness within the NC epigenome (Fig. 3C) and is also reflected in the key functional enrichments observed for the BAZ1B direct targets that are also expressed and that include “axon guidance,” “tube development,” “dendrite development,” “outflow tract morphogenesis,” “odontogenesis,” “wound healing,” and “endochondral bone morphogenesis” (Fig. 3D). Many of the phenomena captured by these GO categories (e.g., odontogenesis and endochondral bone morphogenesis) are linked to recent changes in the bone structure of modern (versus archaic) humans, with Homo sapiens having characteristically smaller teeth than its extinct relatives.
Last and consistent with the enrichments in NC-defining categories uncovered above, the analysis of BAZ1B bound regions revealed major convergence with the binding motifs of critical NC regulators, including two motifs similar to those of TFAP2A and NEUROG2, and one equally associated to TAL1, TCF12, AP4, and ASCL1 (Fig. 3E and text S1A). Thus, BAZ1B binding regions are enriched for target sites of major regulators of NC and its neural derivatives (38, 39), among which TFAP2A stands out given its core role in neural border formation and NC induction and differentiation (40) through the binding and stabilization of NC-specific enhancers, in concert with NR2F1, NR2F2, and EP300 (41).
Last, we identified 81 regions that are quantitatively bound by BAZ1B depending on its copy number (FDR < 0.1) (Fig. 3F), 153 regions differentially bound concordantly in WBS and 7dupASD compared to control and atWBS samples (FDR < 0.1) (fig. S4A), and 176 and 25 regions differentially bound preferentially in WBS (fig. S4B) and 7dupASD (FDR < 0.1) (fig. S4C), respectively.
Given the prominence of its binding to distal regulatory regions, we then set out to define the BAZ1B dosage–dependent impact on NCSC-specific enhancers by integrating H3K27ac, H3K4me1, H3K27me3, and H3K4me3 profiles. We thus performed a regression analysis on BAZ1B levels for the distribution of the three histone marks in the aforementioned regions and found H3K27ac to be the most affected, with 7254 genes differentially acetylated at their enhancers, followed by a differential distribution of the H3K4me1 (4048) and H3K27me3 (2136) marks (fig. S4D). This enabled the overlay of epigenomic and transcriptomic profiles, uncovering that among the 1192 DEGs identified in the regression RNA-seq analysis, 21.3% (257 of 1192) are associated to enhancers that are both bound by BAZ1B and differentially H3K27-acetylated in a manner concordant with BAZ1B levels (fig. S4E), with a stronger overlap for genes whose expression is inversely correlated with BAZ1B levels (160 versus 97). The same held for DEGs that have a concordant differential distribution of H3K4me1 mark at enhancers (123 versus 55), underscoring the consistency of the impact of BAZ1B dosage on distal regulation (fig. S4F). In contrast, a lower number of genes (36) showed a concordant differential distribution of the H3K27me3 mark and, at the same time, were bound by BAZ1B at enhancers (fig. S4G), indicating that BAZ1B preferentially affects active chromatin. From this integrative analysis, we could thus lastly identify a core set of 30 bona fide direct targets of BAZ1B, which are genes whose expression tightly follows BAZ1B levels and whose enhancers are bound by BAZ1B and clearly differentially modified (Fig. 3G, fig. S4H, and text S1B). Together, this first dosage-faithful analysis of BAZ1B occupancy in a diverse cohort of human NCSCs establishes its pervasive and mostly distal targeting of the NC-specific epigenome, with a preferential activator role on the critical transcriptional circuits that define NC fate and function.
Intersection with paleogenomic datasets uncovers a key evolutionary role for BAZ1B
Mild NC deficits have been put forth as a unifying explanatory framework for the defining features of the so-called domestication syndrome, with BAZ1B listed among the putative underlying genes because of its previously reported role in the NC of model organisms (5, 10, 11). The recent observation that its expression is affected by domestication-related mobile element insertion methylation in gray wolves (12) further supported its role in domestication, offering an intriguing parallel to the paleogenomic results that had detected BAZ1B within the regions of the modern genome reflective of selective sweeps and found it enriched for putatively regulatory mutations in AMHs (15).
Having defined the molecular circuits through which BAZ1B regulates NC, and since NC changes have been implicated in the domestication syndrome (5), since craniofacial differences correlate with self-domestication (1), and since 7q11.23 dosage-related craniofacial differences in humans relate to the H. sapiens versus Neanderthal comparison (fig. S1A), we set out to test the role of BAZ1B dosage in the differences between modern and archaic humans. For this, we carried out a systematic integrative analysis of the overlaps between our empirically defined BAZ1B dosage–sensitive genes (blue Venn in Fig. 4B) and a combination of uniquely informative datasets highlighting differences between modern humans and archaics (Neanderthals/Denisovans) (represented in Fig. 4A by skulls illustrating the more “gracile” and “juvenile” profile in AMH relative to Neanderthals visible in the overall shape of the neurocranium, reduced prognathism, brow ridges, and nasal projections) (1, 13–15). Specifically, as shown in Fig. 4B, these datasets include (i) genes associated with signals of positive selection in the modern branch compared to archaic lineages (purple Venn) (13, 14); (ii) genes harboring (nearly) fixed mutations in moderns versus archaics (pink Venn); and (iii) genes associated with signals of positive selection in the four paradigmatic domesticated species dog, cat, cattle, and horse (1) (orange Venn), to reveal statistically significant overlaps between them and genes associated with signals of positive selection in the modern branch compared to archaic lineages. In turn, the list of genes harboring (nearly) fixed mutations in moderns versus archaics contains three classes: (i) genes harboring high-frequency changes (15), (ii) genes harboring high-frequency missense mutations (red barplot), and (iii) genes enriched for high-frequency mutations in regulatory regions (green barplot) [data based on (15)] (Fig. 4C). As shown in the barplots, the obviously very limited number of high-quality coverage archaic genomes available results in a much higher number of nearly fixed changes identified in archaics (left/negative side of the plot) versus modern humans (right side) (Fig. 4C), setting a comparatively much higher threshold for the identification of nearly fixed modern changes.
Fig. 4. Exploration of paleogenomic datasets supports a key evolutionary role for BAZ1B and validates the self-domestication hypothesis.
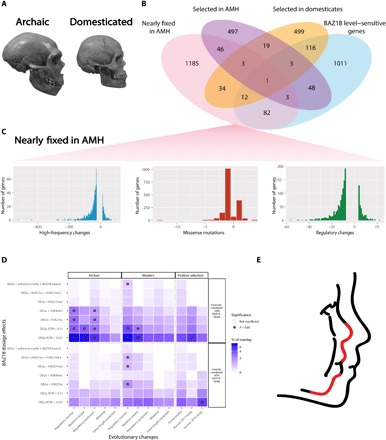
(A) Archaic (Neanderthal) and modern skulls, illustrating the target domesticated phenotype that was captured by our analysis. Skull images were derived from work under a CC BY-SA 2.0 license (https://creativecommons.org/licenses/by-sa/2.0/deed.en) by hairymuseummatt. (B) Overlap between BAZ1B level–sensitive genes and datasets, which bring out differences between AMHs and archaics, as well as genes under positive selection in modern humans and domesticates. (C) Barplots showing the occurrence of high-frequency changes, missense mutations, and mutations in regulatory regions in genes from the AMH (nearly) fixed mutation dataset (pink Venn in B). (D) Heat map representing the amount of overlaps for each list selected from (B). Gene overlaps and detailed list descriptions are reported in table S2. (E) Rendering of a typical WBS face (left) against the background of a typical modern face (right). Red segments indicate areas of the lower face where the two faces most sharply depart (nose, philtrum, and lower front of the mandible). The lower midface region is most often associated with mutations in genes figuring prominently in our intersections, as discussed in the text and table S3.
These analyses are visualized in Fig. 4D (and detailed in tables S2 and S3) through a matrix that intersects all BAZ1B dosage–dependent genes (partitioned in the two categories of directly and inversely correlated and ordered across the full range of biological significance and regulatory proximity, from simply DEGs to bona fide direct targets) with the evolutionary changes underlying domestication and self-domestication, yielding the following key insights (color coded for degree of overlap and marked for significance in hypergeometric tests). First, the most significant pattern was obtains at the intersection with the top 10% genes showing an excess of (nearly) fixed mutations in the regulatory regions of modern humans compared to archaics, across both directly and inversely BAZ1B level–dependent genes (table S2). This same category of nearly fixed modern regulatory changes is also the only one that returns a statistically significant overlap with the most stringent core of BAZ1B dosage–dependent targets (i.e., DEGs whose enhancers are both directly bound by BAZ1B and differentially marked upon its decrease), demonstrating that BAZ1B directly controls, in an exquisitely dosage-dependent manner, this coherent and particularly relevant set of genes that underwent regulatory changes in human evolution. Second, the overall strongest overlaps map to the class of genes that are inversely correlated to BAZ1B levels, which we found to be strongly and specifically enriched for head morphogenesis and NC categories (Fig. 2C), thereby confirming craniofacial morphogenesis as the key domain of functionally relevant overlap between BAZ1B dosage and (self-) domestication changes relevant to the evolution of AMHs. Third, despite the spuriously inflated number of apparently fixed mutations in archaics (15), the overall extent of overlap between genes affected by BAZ1B dosage and our modern and archaic sets does not reveal significantly more hits for archaics. Globally, we found consistently more overlapping genes between the BAZ1B targets and the modern human data and even no statistically significant overlap for any list of the archaic-specific mutations when crossed against genes directly correlated to BAZ1B level. We find this noteworthy, given the evidence that the Neanderthal face also displays derived characteristics (42) that could be the result of modifications of genes that could overlap with those highlighted in this work. Last, the (lower) midface emerges as a particularly salient area of functionally relevant overlap (as illustrated in Fig. 4E and detailed in table S3), given the specific genes that our analysis unearthed: (i) COL11A1, one of the few craniofacial genes highlighted across domestication studies (dog, house sparrow, and pig breeds), which lies in a region of the human genome that resisted archaic introgression (13) and is associated with Marshall syndrome; (ii) XYLT1, one of the five genes (along with ACAN, SOX9, COL2A1, and NFIX) that affect lower and midfacial protrusion, are among the top differentially methylated genes compared to archaics and were also highlighted in a recent study on regulatory changes that shaped the modern human facial and vocal anatomy (tables S1 and S3) (43); and (iii) NFIB, which belongs to the same gene family as NFIX and shares some of its functions. In sum, the direct and dosage-sensitive control by BAZ1B of genes that underwent regulatory changes in human evolution and whose altered expression underlies neurocristopathic facial dysmorphisms is consistent with the hypothesis of mild neurocristopathy as the mechanistic core selected in the self-domestication of the modern human face.
DISCUSSION
As recently reconstructed (3), the idea of human self-domestication dates back, at least in terms of scientific record, to Johann Friedrich Blumenbach at the onset of the 19th century. Following on his seminal account of domestication systematized in Variations of Animals and Plants under Domestication (44), Charles Darwin also considered the analogy between modern humans and domesticated species in The Descent of Man (45), yet his emphasis on controlled breeding as a key aspect of domestication led him to frame domestication and self-domestication as distinct phenomena and thereby leave Blumenbach’s intuition largely undeveloped (46). Since then, the possibility that the anatomical and cognitive-behavioral hallmarks of AMHs could result from an evolutionary process bearing such significant similarities to the domestication of animals as to share the same underlying cause has been refined into the full-fledged self-domestication hypothesis (1, 2). As recently argued (1, 3), convergent lines of evidence also indicate that self-domestication is temporally aligned with the emergence of AMH, although the process may have acquired further momentum with the gradual expansion of our species (1, 3). However, despite spurring considerable interest, the self-domestication hypothesis has thus far failed to marshal conclusive evidence largely because of two factors: (i) the lack of a coherent explanation, even at a theoretical level, of what developmental and genetic mechanisms could underlie domestication in general and (ii) the absence of suitable experimental systems in which those mechanisms could be specifically tested in the case of human self-domestication. The first problem was tackled by the recent proposition of mild NC deficits as a central and unifying functional layer underlying domestication (5). This constituted a major conceptual advance, particularly because it generated the testable hypothesis of an altered NC gene expression program in domesticated species relative to their wild-type ancestors. For humans, given the obvious lack of gene expression data from archaic hominins, we reasoned that this hypothesis could be verified by examining the genetic changes between archaic and modern humans in light of the gene regulatory networks directly inferred from human neurocristopathies. We thus set out to test whether specific human neurodevelopmental disorders, carefully selected on the basis of both craniofacial and cognitive-behavioral traits relevant to domestication, could illuminate the regulatory circuits shaping the modern human face and hence be harnessed for an empirical validation of the self-domestication hypothesis. Specifically, we reasoned that WBS and 7dupASD, through their uniquely informative set of symmetrically opposite phenotypes at the level of face morphology (fig. S1A) and sociality, constituted a paradigmatic test case to probe the heuristic potential of neurodevelopmental disease modeling for the experimental understanding of human evolution. The following key insights confirm the validity of this approach.
First, we identified the 7q11.23 region BAZ1B gene as a master regulator of the modern human face on the basis of a molecular and functional dissection in the thus far largest cohort of WBS patient– and 7dupASD patient–specific NCSCs and across an exhaustive range of BAZ1B dosages. Notably, our cohort also included NCSCs from a patient with rare WBS featuring a much milder WBS gestalt and harboring an atypical, BAZ1B-sparing deletion that served as a particularly informative control, as confirmed by the clustering of atypical NCSC lines with controls when probed for BAZ1B occupancy. In particular, exploiting the fine-grained resolution of BAZ1B dosages recapitulated in our cohort, we could couple classical pairwise comparisons with a more sophisticated regression analysis on BAZ1B levels, thereby revealing major BAZ1B dosage–dependent transcriptional alterations pivoting around clusters of pathways that are crucial for NC development and maintenance, as well as for its downstream skeletal and cardiac outputs.
Second, we repurposed the versatility of CRISPR-Cas9 to generate an allelic series of endogenously tagged BAZ1B across 7q11.23 dosages (including the BAZ1B-sparing atypical patient as uniquely relevant control) to define its dosage-dependent genome-wide occupancy. Taking advantage of previous extensive work on the NCSC chromatin landscape (41, 47–49), we were able to define a pivotal role for BAZ1B in NCSC enhancer regulation, consistent with its preferential binding of distal regulatory regions, and to partition its dosage-dependent regulation into bona fide direct and indirect targets. The overall balance between the numbers of genes up- or down-regulated upon BAZ1B KD—together with the greater overlap, sheer size, and significance of enrichments in chromatin remodeling categories over other domains of transcription regulation—further corroborates the inclusion of BAZ1B among the factors acting upstream of enhancer and promoter modulations to enable or reinforce rather than specify their net outcome. Last, this molecular readout was translated to the functional level with the definition of an impairment in both NCSC migration and outgrowth from EBs upon decrease in BAZ1B, providing the first validation of BAZ1B involvement in key functions of the developing human NC.
Third, our investigation provides the first experimental evidence for the neurocristopathic hypothesis that had been put forth to explain the domestication syndrome and had pointed to BAZ1B as one of the candidates underlying this syndrome (5). Among the key NC hubs affected by BAZ1B dosage, we uncovered three additional critical genes—EDN3, MAGOH, and ZEB2—that had also been predicted in the same model because they are associated with behavioral changes found in domesticates, thereby defining a regulatory hierarchy for this coherent set of genes underlying domestication.
Last, the empirical determination of BAZ1B dosage–sensitive genes in NC models from AMHs with accentuated domestication-relevant traits allowed us to expose, in a functionally relevant manner, the genetic differences between modern versus archaic. This brought to the fore the significant convergence between BAZ1B-dependent circuits and genes harboring regulatory changes in the human lineage, reinforcing the notion that regulatory regions contain some of the most significant changes relevant for the modern lineage. This is also reinforced by the recent identification of AMH-specific hypermethylation in the regulatory region of BAZ1B itself (43).
Last, it is noteworthy that genes implicated in NC development also play significant roles in the establishment of brain circuits that are critical for cognitive processes like language or theory of mind prominently affected in 7q11.23 syndromes. Among the genes downstream of BAZ1B that we uncovered in this study, FOXP2, ROBO1, and ROBO2 have long been implicated in brain wiring processes critical for vocal learning in several species (50, 51), including humans, and will warrant further mechanistic dissection in light of the distinctive linguistic profile of WBS individuals. In conclusion, our findings establish the heuristic power of neurodevelopmental disease modeling for the study of human evolution.
MATERIALS AND METHODS
Human samples
Ethics approvals were reported in the study that established the original iPSC cohort (16) and also apply to the additional samples included in this study (7dupASD3 and CTL4R).
Fibroblast reprogramming and iPSC culture
WBS1, WBS2, WBS3, WBS4, 7dupASD1, atWBS1, and CTL2 fibroblasts were reprogrammed using the mRNA Reprogramming Kit (Stemgent), while the 7dupASD2 and CTL1R lines were reprogrammed with the microRNA Booster Kit (Stemgent). The CTL3 line was reprogrammed by transfection with the STEMCCA polycistronic lentiviral vector followed by Cre-mediated excision of the integrated polycistron. 7dupASD3 and CTL4R fibroblasts were reprogrammed using the Simplicon RNA Reprogramming Kit (Millipore).
Before differentiation, iPSC lines were cultured on Matrigel hESC-qualified Matrix (BD Biosciences)–coated plates, diluted 1:40 in Dulbecco’s minimum essential medium/F-12, and grown in mTeSR 1 medium (STEMCELL Technologies). They were passaged upon treatment with Accutase (Sigma-Aldrich) and then plated in mTeSR 1 medium supplemented with 5 μM Y-27632 (Sigma-Aldrich).
Differentiation
Differentiation into NCSCs was performed as previously described (52), with the exception of NCSCs used in the experiment reported in Fig. 1 (D and E) (19).
Flow cytometry
NCSCs were detached using Accutase and counted, and 1 × 106 cells per experimental condition were fixed in 4% paraformaldehyde and then blocked in 10% bovine serum albumin. Cells were incubated for 1 hour with primary antibodies conjugated to fluorophores (HNK1–fluorescein isothiocyanate and nerve growth factor receptor–Alex Fluor 647; BD Biosciences). Analyses were performed on a FACSCalibur instrument (BD Biosciences), and data were analyzed with FCS express software (Tree Star). Fluorescence-activated cell sorting characterization for 7dupASD3 and CTL4R lines is reported in fig. S1B; for all the other lines, see (16).
Lentiviral vector production and NCSC transfection
BAZ1B KD was performed using validated pLKO.1 TRC vector TRCN0000013338 (referred to as sh1) and TRCN0000013341 (referred to as sh2). A pLKO.1 TRC vector containing a scrambled short hairpin sequence was used as a negative control.
Second generation lentiviral vectors were produced through calcium phosphate transfection of human embryonic kidney 293T cells and ultracentrifugation (2 hours, 20°C, 20,000 rpm).
NCSCs (3 to 4 × 105) were infected upon splitting and then selected by adding puromycin (1 μg/ml) to the medium.
RNA extraction, retrotranscription, and real-time qPCR
RNA was extracted using the RNeasy Micro Plus Kit (QIAGEN) according to the manufacturer’s instructions. Retrotranscribed cDNA was obtained from 0.5 to 1 μg of total RNA using the SuperScript VILO retrotranscription kit (Thermo Fisher Scientific).
Real-time qPCR was performed on a 7500 Fast Real-Time PCR system (Applied Biosystems) using SYBR Green Master Mix (Applied Biosystems) as the detecting reagent. A total cDNA amount corresponding to 15 ng of starting RNA was used for each reaction. Each sample was analyzed in triplicate and normalized to GAPDH. Relative mRNA quantity was calculated by the comparative cycle threshold (Ct) method using the formula 2−∆Ct [BAZ1B, CCTCGCAGTAAGAAAGCAAAC (forward) and ACTCATCCAGCTCCTTTTGAC (reverse); GAPDH, GCACCGTCAAGGCTGAGAAC (forward) and AGGGATCTCGCTCCTGGAA (reverse); NR2F1, AGAAGCTCAAGGCGCTACAC (forward) and GGGTACTGGCTCCTCACGTA (reverse); NR2F2, GCAAGTGGAGAAGCTCAAGG (forward) and GCTTTCCACATGGGCTACAT (reverse); TFAP2A, GCCTCTCGCTCCTCAGCTCC (forward) and CGTTGGCAGCTTTACGTCTCCC (reverse); and SOX9, AGTACCCGCACTTGCACAAC (forward) and GTAATCCGGGTGGTCCTTCT (reverse)].
RNA-seq libraries preparation
Library preparation for RNA-seq was performed according to the TruSeq Total RNA sample preparation protocol (Illumina), starting from 250 ng to 1 μg of total RNA. cDNA library quality was assessed in an Agilent 2100 Bioanalyzer using the High Sensitivity DNA Kit. Libraries were sequenced with the Illumina HiSeq machine at a read length of 50–base pair (bp) paired end and a coverage of 35 million of reads per sample.
Protein extraction and Western blot
NCSCs were lysed in radioimmunoprecipitation assay buffer [10 mM tris (pH 8.0), 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, and 1 mM EDTA] supplemented with protease inhibitor cocktail (Sigma-Aldrich) and 0.5 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich) for 1 hour at 4°C.
Protein extracts (30 to 50 μg per sample) were supplemented with NuPAGE LDS sample buffer (Thermo Fisher Scientific) and 50 mM dithiothreitol (Thermo Fisher Scientific) and denatured at 95°C for 3 min. Then, extracts were run on a precast NuPAGE 4 to 12% bis-tris Gel (Thermo Fisher Scientific) in NuPAGE MOPS SDS Running Buffer (Thermo Fisher Scientific) and transferred to a 0.45-μm nitrocellulose membrane (GE Healthcare) for 1 hour at 100 V in a buffer containing 20% absolute ethanol and 10% 0.25 M tris base and 1.9 M glycine. The membranes were blocked in TBST [50 mM tris (pH 7.5), 150 mM NaCl, and 0.1% Tween 20] and 5% milk for 1 hour, incubated with primary antibodies overnight at 4°C and with secondary antibodies for 1 hour at room temperature. Primary [BAZ1B (Abcam) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Millipore)] and secondary antibodies were diluted in TBST and 5% milk. Blots were detected with the ECL Prime Western Blotting Detection Reagents (Sigma-Aldrich) and scanned using the ChemiDoc system (Bio-Rad).
Wound-healing assay
Cells (5 × 104 to 7 × 104) were plated in each of the two Matrigel-coated wells of silicone culture-inserts (Ibidi) attached to six-well culture plates. After 24 hours, the insert was removed, medium was changed to remove dead cells, and time lapse was performed for 24 hours at the rate of one image every 10 min at ×10 magnification; each condition was analyzed in duplicate. Images were acquired with the BX61 upright microscope equipped with a motorized stage from Olympus or the Nikon Eclipse Ti inverted microscope equipped with a motorized stage from Nikon and analyzed with ImageJ.
Endogenous BAZ1B tagging via CRISPR-Cas9
iPSCs were pretreated with 10 μM rho kinase inhibitor for 4 hours, and then 2 × 106 cells were electroporated using the Neon system with the Cas9/single-guide RNA ribonucleoprotein complex and the donor plasmid (synthesized by GeneArt). The donor plasmid contained three FLAG tags followed by a self-cleaving peptide (P2A) and a hygromycin resistance (HygroR). The 3xFLAG-P2A-HygroR cassette was flanked by BAZ1B-specific homology arms (5′ HA and 3′ HA) to promote homologous recombination and then subcloned into a bacterial backbone (Fig. 3A).
After 48 hours, iPSC medium was supplemented with hygromycin B (50 μg/μl), and selection medium was maintained for 15 days. Fifteen to 20 clones per iPSC line were then subjected to PCR to (i) evaluate the presence of the cassette and the insertion in the correct genomic locus and (ii) distinguish heterozygously tagged from homozygously tagged clones (fig. S3A). We could isolate a clone with a homozygous integration from the CTL, the atWBS, and the typical WBS but not from the 7dupASD line. In the 7dupASD clone, the FLAG tag was present in two of three copies, as shown by a digital PCR analysis (fig. S3B).
Digital PCR
DNA (60 ng) was amplified in a reaction volume containing the following reagents: QuantStudio 3D Digital PCR Master Mix v2 (Thermo Fisher Scientific), Custom TaqMan Copy Number Assays SM 20× FAM labeled (Thermo Fisher Scientific), and TaqMan Copy Number Reference Assay 20× (Thermo Fisher Scientific) VIC labeled (Thermo Fisher Scientific). The mix was loaded on a chip using the QuantStudio 3D Digital PCR Chip Loader. The chips were then loaded on the ProFlex PCR System (Thermo Fisher Scientific), and data were analyzed using the “QuantStudio 3D AnalysisSuite Cloud Software.” The entire process was performed by the qPCR Service at Cogentech, Milano [Custom (FLAG) TaqMan Copy Number Assays: forward primer, TGGACAGTCCAGAGGACGAA; reverse primer, CACCCTTGTCGTCATCGTCTT; and probe, FAMACAGAAGAAGGACTACAAAGACG and TaqMan Copy Number Reference Assay: TERT (VIC) (catalog number 4403316)].
ChIP coupled with sequencing
Approximately 2 × 105 cells were used (~100 μg of chromatin) for histone mark IP, and 1 mg of chromatin was used for BAZ1B-FLAG IP. Cells were fixed with phosphate-buffered saline, containing 1% formaldehyde (Sigma-Aldrich), for 10 min to cross-link proteins and DNA, when the reaction was then stopped by adding 125 mM glycine for 5 min. Cells were lysed with SDS buffer containing 100 mM NaCl, 50 mM tris-HCl (pH 8.0), 5 mM EDTA (pH 8.0), and 10% SDS, at which point chromatin pellets were resuspended in IP buffer containing 1 volume of SDS buffer and 0.5 volume of Triton dilution buffer [100 mM tris-HCl (pH 8.5), 5 mM EDTA (pH 8.0), and 5% Triton X-100]. Chromatin was then sonicated using the S220 Focused-ultrasonicator (Covaris) to generate <300 bp DNA fragments (for histone mark IPs) or the Branson Digital Sonifier to generate 500 to 800 bp DNA fragments (for BAZ1B-FLAG IP).
Sonicated chromatin was incubated overnight at 4°C with primary antibodies [H3K27ac (Abcam), H3K4me1 (Abcam), H3K4me3 (Abcam), H3K27me3 (Cell Signaling Technology), and FLAG (Sigma-Aldrich)] and then for 3 hours with Dynabeads Protein G (Thermo Fisher Scientific). Beads were washed three times with low-salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl (pH 8.0), and 150 mM NaCl] and once with high-salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl (pH 8.0), and 500 mM NaCl]. Immunocomplexes were eluted in decross-linking buffer (1% SDS and 100 mM NaHCO3) at 65°C for 2 hours. DNA was purified using QIAquick PCR columns (QIAGEN) and quantified with a Qubit dsDNA HS assay kit (Thermo Fisher Scientific). DNA libraries were prepared by the sequencing facility at European Institute of Oncology according to the protocol described by Blecher-Gonen and colleagues (53), and DNA was sequenced on the Illumina HiSeq 2000 platform. For the FLAG ChIP, samples were run in duplicate.
RNA-seq analysis
RNA-seq data were quantified using Salmon 0.91 to calculate read counts and transcripts per million in a transcript- and gene-wise fashion, using the quasi-mapping offline algorithm (54) on the GRCh38 (National Center for Biotechnology Information) database. edgeR was used for differential gene expression analysis (DEA), using generalized linear regression methods, to identify pattern of differential expression following two different schemes:
1) A factorial analysis based on the definition of one group of scrambled and one group of KD samples to identify genes dysregulated similarly across short hairpins characterized by different efficiencies.
2) A numerical analysis in which log-normalized [Trimmed Mean of M-values (TMM)] BAZ1B levels, as quantified by RNA-seq, was used as independent variable.
All analyses were performed dropping individual variations (~individual+KD or ~individual+BAZ1B) to account for the genetic background of each individual. In particular, this design is expected to permit the identification of genes, which change expression level upon KD even in situations in which genotype-specific makeups would lead BAZ1B-dependent genes to have unique expression levels in scramble lines. In the factorial analysis, DEGs were identified and characterized by filtering for fold change (FC) > 1.25 and FDR < 0.05 unless explicitly indicated.
To our knowledge, performing a regression analysis at a gene-specific level has never been performed. We were able to do this because of the availability of a large set of samples (11 individuals) and because of the two short hairpins robustly respectively reducing BAZ1B expression levels, respectively by ~40 and ~70% in all individuals lines. To validate the quality of our numerical differential expression analysis, we took advantage of HipSci data (55, 56) and iPSCpoweR tools (29). We took 50 of 105 possible combinations of 13 random individual RNA-seq data from the healthy HipSci cohort, representing both sexes and having at least two technical replicates per individual. Unfortunately, HipSci does not contain at least 13 individuals with three clones per individual. Thus, we performed four alternative DEAs with edgeR (table S4) on the 50 different random combinations of 13 individuals identified (200 DEAs in total, on 22 samples, two clones per individual), using the same model matrix used for the regression analysis (~individual+BAZ1B) and using BAZ1B levels of scramble and sh2 lines. All analyses identified very low number of spurious DEGs (fig. S2E). Thus, we used the “Edg2” pipeline (table S4) because it does not discard genes with higher variability (Edg2 and Edg4 versus Edg1 and Edg3), and it is based on a better suited algorithm (Edg2 versus Edg4). With our model matrix, filtering by P < 0.01 (and FDR < 0.25), using Edg2 on a random HipSci data, we obtained an average of 93.32 DEGs (on average) with a median equal to 43 (table S5). GO enrichments were performed using topGO R package version 2.28.0.
Master regulatory analysis was performed via hypergeometric test by measuring gene set enrichments in lists of transcription factor targets provided by the TFBS tools database (57). Both GO and transcription factor enrichment analyses were performed considering background genes expressed in at least two samples in our NCSC cohort.
ChIP-seq analysis
ChIP-seq experiments were analyzed both qualitatively and quantitatively. Reads were trimmed with the FASTX-Toolkit (-Q33 -t 20 -l 22), aligned with Bowtie 1.0 (-v 2 -m 1) on the Human hg38 reference genome, and peaks were called using MACS 2.1.1. H3K4me1, H3K27ac, H3K4me3, and H3K27me3 peaks were called with --broad using default parameters and q < 0.05.
Qualitative analysis, including intersection and comparison of bed files, was performed using BedTools version 2.23.
To define enhancer regions, we intersected those marked by H3K4me1 and H3K27ac in at least two samples, discarded regions with H3K4me3 in at least two samples, and discarded regions overlapping with TSS. Motif enrichment was performed by using HOMER v4.10.
Quantification of reads per region was performed with DeepTools 3.0.2. Differential mark deposition was conducted by means of edgeR 3.24.1 inside R 3.3.3. To define mark deposition following BAZ1B levels, we used the same design as for RNA-seq data (~individual+BAZ1B).
To identify BAZ1B bound regions and to avoid losing identification of lowly covered regions, we resorted to (i) aggregation of all sample aligned reads and (ii) peak calling with MACS2 using –extsize 800 and q < 0.25. BAZ1B binding coverage was calculated with DeepTools, with the same parameters used for histone marks, on the identified peak regions. Differentially bound regions were identified with edgeR.
Assembly of archaic and modern human lists
The archaic/modern lists were generated from the material presented in (15). We used high-coverage genotypes for three archaic individuals: one Denisovan (58), one Neanderthal from the Denisova cave in Altai mountains (59), and another Neanderthal from Vindija cave, Croatia (60). The data are publicly available at http://cdna.eva.mpg.de/neandertal/Vindija/VCF/, with the human genome version hg19 as reference. High-frequency (HF) differences were defined as positions where more than 90% of present-day humans carry a derived allele, while at least the Denisovan and one Neanderthal carry the ancestral allele. High-frequency changes in archaics were defined as occurring at less than 1% in present-day humans, while at least two archaic individuals carry the derived allele. The HF lists used here were examined as presented in (15), with the exception of the HF lists in regulatory regions, which were extracted from the same dataset but not presented as such in the original paper.
Supplementary Material
Acknowledgments
We thank the IFOM imaging unit and the IEO genomic unit for the help with, respectively, wound-healing assays and sequencing, and G. Scita and his group for suggestions on migration experiments. Funding: This work was funded by the Telethon Foundation (grant number GGP14265 to G.T. and G.M.), the EPIGEN Flagship Project of the Italian National Research Council (to G.T.), the European Research Council (consolidator grant number 616441-DISEASEAVATARS to G.T.), the Horizon 2020 Innovative Training Network EpiSyStem (to G.T.), Ricerca Corrente granted by the Italian Ministry of Health (to G.T. and G.M.) Giovani Ricercatori granted by the Italian Ministry of Health (to G.T.), Daunia Plast (to G.M.), Fondazione Umberto Veronesi (to P.-L.G.), the Foundation IEO-CCM (fellowship to A.V.), the Spanish Ministry of Economy and Competitiveness/FEDER funds (grant FFI2016-78034-C2-1-P to C.B.), the Generalitat de Catalunya (grant 2017-SGR-341 to C.B. and doctoral fellowships to T.O. and S.S.), the MEXT/JSPS Grant-in-Aid for Scientific Research on Innovative Areas 4903 [Evolinguistics: JP17H06379 (C.B.; PI: K. Okanoya)], a Marie Curie International Reintegration Grant from the European Union (PIRG-GA-2009-256413 to C.B.), the European Social Fund (grant BES-2017-080366 to A.A.), and the Portuguese Foundation for Science and Technology (PhD grant number SFRH/BD/131640/2017 to P.T.M.). M.Z., A.V., A.S., and S.T. conducted this study as fulfillment of their Ph.D. within the European School of Molecular Medicine (SEMM), Milan, Italy. Author contributions: M.Z. carried out all the experiments including the generation and characterization of both BAZ1B-interfered lines and BAZ1B-tagged lines, the preparation of RNA-seq libraries, ChIPs, and wound-healing assays. A.V. performed RNA-seq and ChIP-seq bioinformatic analyses with the supervision of P.-L.G. A.A., P.T.M., S.S., T.O., and C.B. performed analyses in Fig. 4. M.L. and A.R.-I. characterized the NC induction phenotype reported in Fig. 1 (D and E). N.M. reprogrammed 7dupASD3 and CTL4R fibroblasts and generated the corresponding NC lines. A.S. helped with cell culture. M.Z. and S.T. designed the tag strategy. M.M. provided theoretical support in IP experiments whose results reinforced the rationale for BAZ1B endogenous tagging. G.M. provided 7DupASD3 and CTL4R fibroblast samples through the Genomic and Genetic Disorders Biobank, Telethon Network of Genetic Biobanks, and EuroBioBank Network for biobanking biospecimens (Telethon Foundation, grant number GTB12001G). M.Z., C.B., and G.T. wrote the manuscript with contributions from A.V. and A.S. G.T. conceived, designed, and supervised the study. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The cell lines can be provided by G.T. pending scientific review and a completed material transfer agreement. Requests for the cell lines should be submitted to giuseppe.testa@ieo.it. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaaw7908/DC1
Fig. S1. BAZ1B KD validation in iPSC-derived NCSCs and evaluation of its impact on NCSC migration.
Fig. S2. BAZ1B KD affects the transcriptome of iPSC-derived NCSCs.
Fig. S3. Generation of BAZ1B-FLAG iPSC lines and differentiation to NCSCs.
Fig. S4. BAZ1B KD induces a significant chromatin remodeling at distal regions.
Text S1A. Detailed description of HOMER motif enrichments performed on BAZ1B ChIP-seq data.
Text S1B. List of key direct targets of BAZ1B involved in neural- and NC-related development and relevant associated literature.
Table S1. Genes relevant for NC and NC-derived features whose expression follows BAZ1B levels.
Table S2A. Significant genes in human evolution.
Table S2B. Regulatory excess in archaic humans, overlap with BAZ1B targets.
Table S2C. Mutation excess in archaic humans, overlap with BAZ1B targets.
Table S2D. Regulatory changes (exclusive) in archaic humans, overlap with BAZ1B targets.
Table S2E. Missense mutations in archaic humans, overlap with BAZ1B targets.
Table S2F. Mutation excess in archaic humans corrected for length, overlap with BAZ1B targets.
Table S2G. Regulatory excess in modern humans, overlap with BAZ1B targets.
Table S2H. Mutation excess in modern humans, overlap with BAZ1B targets.
Table S2I. Regulatory changes (exclusive) in modern humans, overlap with BAZ1B targets.
Table S2J. Missense mutations in modern humans, overlap with BAZ1B targets.
Table S2K. Mutation excess in modern humans corrected for length, overlap with BAZ1B targets.
Table S2L. Genes under positive selection in domesticated animals, overlap with BAZ1B targets.
Table S2M. Genes under positive selection from Peyrégne et al. (13) in modern humans, overlap with BAZ1B targets.
Table S2N. Genes under positive selection from Racimo (14) in modern humans, overlap with BAZ1B targets.
Table S3. Crucial genes identified in the overlap between BAZ1B datasets and archaic versus modern human datasets reported in this study.
Table S4. Alternative differential expression analysis functions tested with iPSCpower to assess the efficacy of our design matrix (~individual+BAZ1B). R code provided.
Table S5. Number of genes differentially expressed following BAZ1B data in our numerical analysis compared to an analysis conducted on randomized HipSci data, using Edg2 function (see table S4).
REFERENCES AND NOTES
- 1.Theofanopoulou C., Gastaldon S., O’Rourke T., Samuels B. D., Messner A., Martins P. T., Delogu F., Alamri S., Boeckx C., Self-domestication in Homo sapiens: Insights from comparative genomics. PLOS ONE 12, e0185306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hare B., Survival of the friendliest: Homo sapiens evolved via selection for prosociality. Annu. Rev. Psychol. 68, 155–186 (2017). [DOI] [PubMed] [Google Scholar]
- 3.R. W. Wrangham, The Goodness Paradox: How Evolution Made Us Both More and Less Violent (Profile Books Ltd., 2019). [Google Scholar]
- 4.Sánchez-Villagra M. R., van Schaik C. P., Evaluating the self-domestication hypothesis of human evolution. Evol. Anthropol. 28, 133–143 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Wilkins A. S., Wrangham R. W., Fitch W. T., The “domestication syndrome” in mammals: A unified explanation based on neural crest cell behavior and genetics. Genetics 197, 795–808 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pober B. R., Williams-Beuren syndrome. N. Engl. J. Med. 362, 239–252 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Sanders S. J., Ercan-Sencicek A. G., Hus V., Luo R., Murtha M. T., Moreno-de-Luca D., Chu S. H., Moreau M. P., Gupta A. R., Thomson S. A., Mason C. E., Bilguvar K., Celestino-Soper P. B. S., Choi M., Crawford E. L., Davis L., Davis Wright N. R., Dhodapkar R. M., DiCola M., DiLullo N. M., Fernandez T. V., Fielding-Singh V., Fishman D. O., Frahm S., Garagaloyan R., Goh G. S., Kammela S., Klei L., Lowe J. K., Lund S. C., McGrew A. D., Meyer K. A., Moffat W. J., Murdoch J. D., O'Roak B. J., Ober G. T., Pottenger R. S., Raubeson M. J., Song Y., Wang Q., Yaspan B. L., Yu T. W., Yurkiewicz I. R., Beaudet A. L., Cantor R. M., Curland M., Grice D. E., Günel M., Lifton R. P., Mane S. M., Martin D. M., Shaw C. A., Sheldon M., Tischfield J. A., Walsh C. A., Morrow E. M., Ledbetter D. H., Fombonne E., Lord C., Martin C. L., Brooks A. I., Sutcliffe J. S., Cook E. H. Jr., Geschwind D., Roeder K., Devlin B., State M. W., Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.vonHoldt B. M., Shuldiner E., Koch I. J., Kartzinel R. Y., Hogan A., Brubaker L., Wanser S., Stahler D., Wynne C. D. L., Ostrander E. A., Sinsheimer J. S., Udell M. A. R., Structural variants in genes associated with human Williams-Beuren syndrome underlie stereotypical hypersociability in domestic dogs. Sci. Adv. 3, e1700398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kukekova A. V., Johnson J. L., Xiang X., Feng S., Liu S., Rando H. M., Kharlamova A. V., Herbeck Y., Serdyukova N. A., Xiong Z., Beklemischeva V., Koepfli K. P., Gulevich R. G., Vladimirova A. V., Hekman J. P., Perelman P. L., Graphodatsky A. S., O’Brien S. J., Wang X., Clark A. G., Acland G. M., Trut L. N., Zhang G., Red fox genome assembly identifies genomic regions associated with tame and aggressive behaviours. Nat. Ecol. Evol. 2, 1479–1491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnett C., Yazgan O., Kuo H. C., Malakar S., Thomas T., Fitzgerald A., Harbour W., Henry J. J., Krebs J. E., Williams syndrome transcription factor is critical for neural crest cell function in Xenopus laevis. Mech. Dev. 129, 324–338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashe A., Morgan D. K., Whitelaw N. C., Bruxner T. J., Vickaryous N. K., Cox L. L., Butterfield N. C., Wicking C., Blewitt M. E., Wilkins S. J., Anderson G. J., Cox T. C., Whitelaw E., A genome-wide screen for modifiers of transgene variegation identifies genes with critical roles in development. Genome Biol. 9, R182 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.vonHoldt B. M., Ji S. S., Aardema M. L., Stahler D. R., Udell M. A. R., Sinsheimer J. S., Activity of genes with functions in Human Williams-Beuren syndrome is impacted by mobile element insertions in the gray wolf genome. Genome Biol. Evol. 10, 1546–1553 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peyrégne S., Boyle M. J., Dannemann M., Prüfer K., Detecting ancient positive selection in humans using extended lineage sorting. Genome Res. 2017, 1563–1572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Racimo F., Testing for ancient selection using cross-population allele frequency differentiation. Genetics 202, 733–750 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuhlwilm M., Boeckx C., A catalog of single nucleotide changes distinguishing modern humans from archaic hominins. Sci. Rep. 9, 8463 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adamo A., Atashpaz S., Germain P. L., Zanella M., D'Agostino G., Albertin V., Chenoweth J., Micale L., Fusco C., Unger C., Augello B., Palumbo O., Hamilton B., Carella M., Donti E., Pruneri G., Selicorni A., Biamino E., Prontera P., McKay R., Merla G., Testa G., 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat. Genet. 47, 132–141 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Fusco C., Micale L., Augello B., Teresa Pellico M., Menghini D., Alfieri P., Cristina Digilio M., Mandriani B., Carella M., Palumbo O., Vicari S., Merla G., Smaller and larger deletions of the Williams Beuren syndrome region implicate genes involved in mild facial phenotype, epilepsy and autistic traits. Eur. J. Hum. Genet. 22, 64–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng J., Zhang X. T., Liu X. L., Fan L., Li C., Sun Y., Liang X. H., Wang J. B., Mei Q. B., Zhang F., Zhang T., WSTF promotes proliferation and invasion of lung cancer cells by inducing EMT via PI3K/Akt and IL-6/STAT3 signaling pathways. Cell. Signal. 28, 1673–1682 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Bajpai R., Chen D. A., Rada-Iglesias A., Zhang J., Xiong Y., Helms J., Chang C. P., Zhao Y., Swigut T., Wysocka J., CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnett C., Krebs J. E., WSTF does it all: A multifunctional protein in transcription, repair, and replication. Biochem. Cell Biol. 89, 12–23 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poot R. A., Bozhenok L., van den Berg D. L. C., Steffensen S., Ferreira F., Grimaldi M., Gilbert N., Ferreira J., Varga-Weisz P. D., The Williams syndrome transcription factor interacts with PCNA to target chromatin remodelling by ISWI to replication foci. Nat. Cell Biol. 6, 1236–1244 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Culver-Cochran A. E., Chadwick B. P., Loss of WSTF results in spontaneous fluctuations of heterochromatin formation and resolution, combined with substantial changes to gene expression. BMC Genomics 14, 740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilderman A., VanOudenhove J., Kron J., Noonan J. P., Cotney J., High-resolution epigenomic atlas of human embryonic craniofacial development. Cell Rep. 23, 1581–1597 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spokony R. F., Aoki Y., Saint-Germain N., Magner-Fink E., Saint-Jeannet J.-P., The transcription factor Sox9 is required for cranial neural crest development in Xenopus. Development 129, 421–432 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Santagati F., Rijli F. M., Cranial neural crest and the building of the vertebrate head. Nat. Rev. Neurosci. 4, 806–818 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Bhatt S., Diaz R., Trainor P. A., Signals and switches in mammalian neural crest cell differentiation. Cold Spring Harb. Perspect. Biol. 5, a008326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ko S. O., Chung I. H., Xu X., Oka S., Zhao H., Cho E. S., Deng C., Chai Y., Smad4 is required to regulate the fate of cranial neural crest cells. Dev. Biol. 312, 435–447 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mishina Y., Snider T. N., Neural crest cell signaling pathways critical to cranial bone development and pathology. Exp. Cell Res. 325, 138–147 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Germain P.-L., Testa G., Taming human genetic variability: Transcriptomic meta-analysis guides the experimental design and interpretation of iPSC-based disease modeling. Stem Cell Rep. 8, 1784–1796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schanze I., Bunt J., Lim J. W. C., Schanze D., Dean R. J., Alders M., Blanchet P., Attié-Bitach T., Berland S., Boogert S., Boppudi S., Bridges C. J., Cho M. T., Dobyns W. B., Donnai D., Douglas J., Earl D. L., Edwards T. J., Faivre L., Fregeau B., Genevieve D., Gérard M., Gatinois V., Holder-Espinasse M., Huth S. F., Izumi K., Kerr B., Lacaze E., Lakeman P., Mahida S., Mirzaa G. M., Morgan S. M., Nowak C., Peeters H., Petit F., Pilz D. T., Puechberty J., Reinstein E., Rivière J. B., Santani A. B., Schneider A., Sherr E. H., Smith-Hicks C., Wieland I., Zackai E., Zhao X., Gronostajski R. M., Zenker M., Richards L. J., NFIB haploinsufficiency is associated with intellectual disability and macrocephaly. Am. J. Hum. Genet. 103, 752–768 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabriele M., Vulto-van Silfhout A. T., Germain P. L., Vitriolo A., Kumar R., Douglas E., Haan E., Kosaki K., Takenouchi T., Rauch A., Steindl K., Frengen E., Misceo D., Pedurupillay C. R. J., Stromme P., Rosenfeld J. A., Shao Y., Craigen W. J., Schaaf C. P., Rodriguez-Buritica D., Farach L., Friedman J., Thulin P., McLean S. D., Nugent K. M., Morton J., Nicholl J., Andrieux J., Stray-Pedersen A., Chambon P., Patrier S., Lynch S. A., Kjaergaard S., Tørring P. M., Brasch-Andersen C., Ronan A., van Haeringen A., Anderson P. J., Powis Z., Brunner H. G., Pfundt R., Schuurs-Hoeijmakers J. H. M., van Bon B. W. M., Lelieveld S., Gilissen C., Nillesen W. M., Vissers L. E. L. M., Gecz J., Koolen D. A., Testa G., de Vries B. B. A., YY1 haploinsufficiency causes an intellectual disability syndrome featuring transcriptional and chromatin dysfunction. Am. J. Hum. Genet. 100, 907–925 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gabriele M., Lopez Tobon A., D’Agostino G., Testa G., The chromatin basis of neurodevelopmental disorders: Rethinking dysfunction along the molecular and temporal axes. Prog. Neuropsychopharmacol. Biol. Psychiatry 84, 306–327 (2018). [DOI] [PubMed] [Google Scholar]
- 33.McMahon A. P., Champion J. E., McMahon J. A., Sukhatme V. P., Developmental expression of the putative transcription factor Egr-1 suggests that Egr-1 and c-fos are coregulated in some tissues. Development 108, 281–287 (1990). [DOI] [PubMed] [Google Scholar]
- 34.Dalcq J., Pasque V., Ghaye A., Larbuisson A., Motte P., Martial J. A., Muller M., RUNX3, EGR1 and SOX9B form a regulatory cascade required to modulate BMP-signaling during cranial cartilage development in zebrafish. PLOS ONE 7, e50140 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Werner A., Iwasaki S., McGourty C. A., Medina-Ruiz S., Teerikorpi N., Fedrigo I., Ingolia N. T., Rape M., Cell-fate determination by ubiquitin-dependent regulation of translation. Nature 525, 523–527 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanford L. P., Ormsby I., Gittenberger-de Groot A. C., Sariola H., Friedman R., Boivin G. P., Cardell E. L., Doetschman T., TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development 124, 2659–2670 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boileau C., Guo D. C., Hanna N., Regalado E. S., Detaint D., Gong L., Varret M., Prakash S. K., Li A. H., d’indy H., Braverman A. C., Grandchamp B., Kwartler C. S., Gouya L., Santos-Cortez R. L., Abifadel M., Leal S. M., Muti C., Shendure J., Gross M. S., Rieder M. J., Vahanian A., Nickerson D. A., Michel J. B.; National, Lung Heart Project Blood Institute (NHLBI) Go Exome Sequencing, Jondeau G., Milewicz D. M., TGFβ2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 44, 916–921 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wapinski O. L., Vierbuchen T., Qu K., Lee Q. Y., Chanda S., Fuentes D. R., Giresi P. G., Ng Y. H., Marro S., Neff N. F., Drechsel D., Martynoga B., Castro D. S., Webb A. E., Südhof T. C., Brunet A., Guillemot F., Chang H. Y., Wernig M., Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155, 621–635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong S. J., Choi H. J., Hong S., Huh Y., Chae H., Kim K. S., Transcription factor GATA-3 regulates the transcriptional activity of dopamine beta-hydroxylase by interacting with Sp1 and AP4. Neurochem. Res. 33, 1821–1831 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Crozé N., Maczkowiak F., Monsoro-Burq A. H., Reiterative AP2a activity controls sequential steps in the neural crest gene regulatory network. Proc. Natl. Acad. Sci. U.S.A. 108, 155–160 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rada-Iglesias A., Prescott S. L., Wysocka J., Human genetic variation within neural crest enhancers: molecular and phenotypic implications. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20120360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lacruz R. S., Stringer C. B., Kimbel W. H., Wood B., Harvati K., O’Higgins P., Bromage T. G., Arsuaga J.-L., The evolutionary history of the human face. Nat. Ecol. Evol. 3, 726–736 (2019). [DOI] [PubMed] [Google Scholar]
- 43.D. Gokhman, L. Agranat-Tamir, G. Housman, M. Nissim-Rafinia, M. Nieves-Colón, H. Gu, Recent regulatory changes shaped human facial and vocal anatomy. bioRxiv 106955 [Preprint]. 10.1101/106955. [DOI]
- 44.C. Darwin, The Variation of Animals and Plants under Domestication (J. Murray, 1868). [PMC free article] [PubMed] [Google Scholar]
- 45.C. Darwin, The Descent of Man: And Selection in Relation to Sex (J. Murray, 1871). [Google Scholar]
- 46.Brüne M., On human self-domestication, psychiatry, and eugenics. Philos. Ethics Humanit. Med. 2, 21 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Claes P., Roosenboom J., White J. D., Swigut T., Sero D., Li J., Lee M. K., Zaidi A., Mattern B. C., Liebowitz C., Pearson L., González T., Leslie E. J., Carlson J. C., Orlova E., Suetens P., Vandermeulen D., Feingold E., Marazita M. L., Shaffer J. R., Wysocka J., Shriver M. D., Weinberg S. M., Genome-wide mapping of global-to-local genetic effects on human facial shape. Nat. Genet. 50, 414–423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rada-Iglesias A., Bajpai R., Prescott S., Brugmann S. A., Swigut T., Wysocka J., Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell 11, 633–648 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buecker C., Wysocka J., Enhancers as information integration hubs in development: Lessons from genomics. Trends Genet. 28, 276–284 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vernes S. C., Oliver P. L., Spiteri E., Lockstone H. E., Puliyadi R., Taylor J. M., Ho J., Mombereau C., Brewer A., Lowy E., Nicod J., Groszer M., Baban D., Sahgal N., Cazier J. B., Ragoussis J., Davies K. E., Geschwind D. H., Fisher S. E., Foxp2 regulates gene networks implicated in neurite outgrowth in the developing brain. PLOS Genet. 7, e1002145 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang R., Chen C. C., Hara E., Rivas M. V., Roulhac P. L., Howard J. T., Chakraborty M., Audet J. N., Jarvis E. D., Convergent differential regulation of SLIT-ROBO axon guidance genes in the brains of vocal learners. J. Comp. Neurol. 523, 892–906 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Menendez L., Kulik M. J., Page A. T., Park S. S., Lauderdale J. D., Cunningham M. L., Dalton S., Directed differentiation of human pluripotent cells to neural crest stem cells. Nat. Protoc. 8, 203–212 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Blecher-Gonen R., Barnett-Itzhaki Z., Jaitin D., Amann-Zalcenstein D., Lara-Astiaso D., Amit I., High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat. Protoc. 8, 539–554 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Germain P.-L., Vitriolo A., Adamo A., Laise P., Das V., Testa G., RNAontheBENCH: Computational and empirical resources for benchmarking RNAseq quantification and differential expression methods. Nucleic Acids Res. 44, 5054–5067 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Streeter I., Harrison P. W., Faulconbridge A.; The HipSci Consortium, Flicek P., Parkinson H., Clarke L., The human-induced pluripotent stem cell initiative-data resources for cellular genetics. Nucleic Acids Res. 45, D691–D697 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kilpinen H., Goncalves A., Leha A., Afzal V., Alasoo K., Ashford S., Bala S., Bensaddek D., Casale F. P., Culley O. J., Danecek P., Faulconbridge A., Harrison P. W., Kathuria A., McCarthy D., McCarthy S. A., Meleckyte R., Memari Y., Moens N., Soares F., Mann A., Streeter I., Agu C. A., Alderton A., Nelson R., Harper S., Patel M., White A., Patel S. R., Clarke L., Halai R., Kirton C. M., Kolb-Kokocinski A., Beales P., Birney E., Danovi D., Lamond A. I., Ouwehand W. H., Vallier L., Watt F. M., Durbin R., Stegle O., Gaffney D. J., Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 546, 370–375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan G., Lenhard B., TFBSTools: An R/Bioconductor package for transcription factor binding site analysis. Bioinformatics 32, 1555–1556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meyer M., Kircher M., Gansauge M. T., Li H., Racimo F., Mallick S., Schraiber J. G., Jay F., Prufer K., de Filippo C., Sudmant P. H., Alkan C., Fu Q., Do R., Rohland N., Tandon A., Siebauer M., Green R. E., Bryc K., Briggs A. W., Stenzel U., Dabney J., Shendure J., Kitzman J., Hammer M. F., Shunkov M. V., Derevianko A. P., Patterson N., Andres A. M., Eichler E. E., Slatkin M., Reich D., Kelso J., Paabo S., A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prüfer K., Racimo F., Patterson N., Jay F., Sankararaman S., Sawyer S., Heinze A., Renaud G., Sudmant P. H., de Filippo C., Li H., Mallick S., Dannemann M., Fu Q., Kircher M., Kuhlwilm M., Lachmann M., Meyer M., Ongyerth M., Siebauer M., Theunert C., Tandon A., Moorjani P., Pickrell J., Mullikin J. C., Vohr S. H., Green R. E., Hellmann I., Johnson P. L., Blanche H., Cann H., Kitzman J. O., Shendure J., Eichler E. E., Lein E. S., Bakken T. E., Golovanova L. V., Doronichev V. B., Shunkov M. V., Derevianko A. P., Viola B., Slatkin M., Reich D., Kelso J., Pääbo S., The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prüfer K., de Filippo C., Grote S., Mafessoni F., Korlević P., Hajdinjak M., Vernot B., Skov L., Hsieh P., Peyrégne S., Reher D., Hopfe C., Nagel S., Maricic T., Fu Q., Theunert C., Rogers R., Skoglund P., Chintalapati M., Dannemann M., Nelson B. J., Key F. M., Rudan P., Kućan Ž., Gušić I., Golovanova L. V., Doronichev V. B., Patterson N., Reich D., Eichler E. E., Slatkin M., Schierup M. H., Andrés A. M., Kelso J., Meyer M., Pääbo S., A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 358, 655–658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barembaum M., Moreno T. A., LaBonne C., Sechrist J., Bronner-Fraser M., Noelin-1 is a secreted glycoprotein involved in generation of the neural crest. Nat. Cell Biol. 2, 219–225 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Huang D., Wang Y., Xu L., Chen L., Cheng M., Shi W., Xiong H., Zalli D., Luo S., GLI2 promotes cell proliferation and migration through transcriptional activation of ARHGEF16 in human glioma cells. J. Exp. Clin. Cancer Res. 37, 247 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Südhof T. C., Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen G., Sima J., Jin M., Wang K. Y., Xue X. J., Zheng W., Ding Y. Q., Yuan X. B., Semaphorin-3A guides radial migration of cortical neurons during development. Nat. Neurosci. 11, 36–44 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Betters E., Liu Y., Kjaeldgaard A., Sundström E., García-Castro M. I., Analysis of early human neural crest development. Dev. Biol. 344, 578–592 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu Q., Tang W., Luo Z., Li Y., Shu Y., Yue Z., Xiao B., Feng L., DISC1 regulates the proliferation and migration of mouse neural stem/progenitor cells through Pax5, Sox2, Dll1 and Neurog2. Front. Cell. Neurosci. 11, 261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Horowitz M. C., Xi Y., Pflugh D. L., Hesslein D. G. T., Schatz D. G., Lorenzo J. A., Bothwell A. L. M., Pax5-deficient mice exhibit early onset osteopenia with increased osteoclast progenitors. J. Immunol. 173, 6583–6591 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Hsu H., Lacey D. L., Dunstan C. R., Solovyev I., Colombero A., Timms E., Tan H. L., Elliott G., Kelley M. J., Sarosi I., Wang L., Xia X. Z., Elliott R., Chiu L., Black T., Scully S., Capparelli C., Morony S., Shimamoto G., Bass M. B., Boyle W. J., Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci. U.S.A. 96, 3540–3545 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McGourty C. A., Akopian D., Walsh C., Gorur A., Werner A., Schekman R., Bautista D., Rape M., Regulation of the CUL3 ubiquitin ligase by a calcium-dependent co-adaptor. Cell 167, 525–538.e14 (2016). [DOI] [PubMed] [Google Scholar]
- 70.Oka K., Honda M. J., Tsuruga E., Hatakeyama Y., Isokawa K., Sawa Y., Roles of collagen and periostin expression by cranial neural crest cells during soft palate development. J. Histochem. Cytochem. 60, 57–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rios H., Koushik S. V., Wang H., Wang J., Zhou H. M., Lindsley A., Rogers R., Chen Z., Maeda M., Kruzynska-Frejtag A., Feng J. Q., Conway S. J., Periostin null mice exhibit dwarfism, incisor enamel defects, and an early-onset periodontal disease-like phenotype. Mol. Cell. Biol. 25, 11131–11144 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Golding J. P., Trainor P., Krumlauf R., Gassmann M., Defects in pathfinding by cranial neural crest cells in mice lacking the neuregulin receptor ErbB4. Nat. Cell Biol. 2, 103–109 (2000). [DOI] [PubMed] [Google Scholar]
- 73.Burden S., Yarden Y., Neuregulins and their receptors: A versatile signaling module in organogenesis and oncogenesis. Neuron 18, 847–855 (1997). [DOI] [PubMed] [Google Scholar]
- 74.Andreoletti G., Seaby E. G., Dewing J. M., O'Kelly I., Lachlan K., Gilbert R. D., Ennis S., AMMECR1: Single point mutation causes developmental delAay, midface hypoplasia and elliptocytosis. J. Med. Genet. 54, 269–277 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tumiene B., Čiuladaitė Ž., Preikšaitienė E., Mameniškienė R., Utkus A., Kučinskas V., Phenotype comparison confirms ZMYND11 as a critical gene for 10p15.3 microdeletion syndrome. J. Appl. Genet. 58, 467–474 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Cobben J. M., Weiss M. M., van Dijk F. S., de Reuver R., de Kruiff C., Pondaag W., Hennekam R. C., Yntema H. G., A de novo mutation in ZMYND11, a candidate gene for 10p15.3 deletion syndrome, is associated with syndromic intellectual disability. Eur. J. Med. Genet. 57, 636–638 (2014). [DOI] [PubMed] [Google Scholar]
- 77.Oram K. F., Carver E. A., Gridley T., Slug expression during organogenesis in mice. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 271, 189–191 (2003). [DOI] [PubMed] [Google Scholar]
- 78.Wu W. S., Heinrichs S., Xu D., Garrison S. P., Zambetti G. P., Adams J. M., Look A. T., Slug antagonizes p53-mediated z progenitors by repressing puma. Cell 123, 641–653 (2005). [DOI] [PubMed] [Google Scholar]
- 79.Garbes L., Kim K., Rieß A., Hoyer-Kuhn H., Beleggia F., Bevot A., Kim M. J., Huh Y. H., Kweon H. S., Savarirayan R., Amor D., Kakadia P. M., Lindig T., Kagan K. O., Becker J., Boyadjiev S. A., Wollnik B., Semler O., Bohlander S. K., Kim J., Netzer C., Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am. J. Hum. Genet. 96, 432–439 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Griffith A. J., Sprunger L. K., Sirko-Osadsa D. A., Tiller G. E., Meisler M. H., Warman M. L., Marshall syndrome associated with a splicing defect at the COL11A1 locus. Am. J. Hum. Genet. 62, 816–823 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roessler E., Ma Y., Ouspenskaia M. V., Lacbawan F., Bendavid C., Dubourg C., Beachy P. A., Muenke M., Truncating loss-of-function mutations of DISP1 contribute to holoprosencephaly-like microform features in humans. Hum. Genet. 125, 393–400 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Punetha J., Kesari A., Hoffman E. P., Gos M., Kamińska A., Kostera-Pruszczyk A., Hausmanowa-Petrusewicz I., Hu Y., Zou Y., Bönnemann C. G., JȨdrzejowska M., NovelCol12A1variant expands the clinical picture of congenital myopathies with extracellular matrix defects. Muscle Nerve 55, 277–281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.T. O’Rourke, C. Boeckx, Converging roles of glutamate receptors in domestication and prosociality. bioRxiv 439869 [Preprint]. 10.1101/439869. [DOI]
- 84.Srivastava S., Engels H., Schanze I., Cremer K., Wieland T., Menzel M., Schubach M., Biskup S., Kreiß M., Endele S., Strom T. M., Wieczorek D., Zenker M., Gupta S., Cohen J., Zink A. M., Naidu S. B., Loss-of-function variants in HIVEP2 are a cause of intellectual disability. Eur. J. Hum. Genet. 24, 556–561 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lei H., Yan Z., Sun X., Zhang Y., Wang J., Ma C., Xu Q., Wang R., Jarvis E. D., Sun Z., Axon guidance pathways served as common targets for human speech/language evolution and related disorders. Brain Lang. 174, 1–8 (2017). [DOI] [PubMed] [Google Scholar]
- 86.Lausch E., Hermanns P., Farin H. F., Alanay Y., Unger S., Nikkel S., Steinwender C., Scherer G., Spranger J., Zabel B., Kispert A., Superti-Furga A., TBX15 mutations cause craniofacial dysmorphism, hypoplasia of scapula and pelvis, and short stature in Cousin syndrome. Am. J. Hum. Genet. 83, 649–655 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee M. K., Shaffer J. R., Leslie E. J., Orlova E., Carlson J. C., Feingold E., Marazita M. L., Weinberg S. M., Genome-wide association study of facial morphology reveals novel associations with FREM1 and PARK2. PLOS ONE 12, e0176566 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Iwata J., Parada C., Chai Y., The mechanism of TGF-β signaling during palate development. Oral Dis. 17, 733–744 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y., Su J., Yu J., Bu X., Ren T., Liu X., Yao L., An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. J. Bone Miner. Res. 26, 604–617 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaaw7908/DC1
Fig. S1. BAZ1B KD validation in iPSC-derived NCSCs and evaluation of its impact on NCSC migration.
Fig. S2. BAZ1B KD affects the transcriptome of iPSC-derived NCSCs.
Fig. S3. Generation of BAZ1B-FLAG iPSC lines and differentiation to NCSCs.
Fig. S4. BAZ1B KD induces a significant chromatin remodeling at distal regions.
Text S1A. Detailed description of HOMER motif enrichments performed on BAZ1B ChIP-seq data.
Text S1B. List of key direct targets of BAZ1B involved in neural- and NC-related development and relevant associated literature.
Table S1. Genes relevant for NC and NC-derived features whose expression follows BAZ1B levels.
Table S2A. Significant genes in human evolution.
Table S2B. Regulatory excess in archaic humans, overlap with BAZ1B targets.
Table S2C. Mutation excess in archaic humans, overlap with BAZ1B targets.
Table S2D. Regulatory changes (exclusive) in archaic humans, overlap with BAZ1B targets.
Table S2E. Missense mutations in archaic humans, overlap with BAZ1B targets.
Table S2F. Mutation excess in archaic humans corrected for length, overlap with BAZ1B targets.
Table S2G. Regulatory excess in modern humans, overlap with BAZ1B targets.
Table S2H. Mutation excess in modern humans, overlap with BAZ1B targets.
Table S2I. Regulatory changes (exclusive) in modern humans, overlap with BAZ1B targets.
Table S2J. Missense mutations in modern humans, overlap with BAZ1B targets.
Table S2K. Mutation excess in modern humans corrected for length, overlap with BAZ1B targets.
Table S2L. Genes under positive selection in domesticated animals, overlap with BAZ1B targets.
Table S2M. Genes under positive selection from Peyrégne et al. (13) in modern humans, overlap with BAZ1B targets.
Table S2N. Genes under positive selection from Racimo (14) in modern humans, overlap with BAZ1B targets.
Table S3. Crucial genes identified in the overlap between BAZ1B datasets and archaic versus modern human datasets reported in this study.
Table S4. Alternative differential expression analysis functions tested with iPSCpower to assess the efficacy of our design matrix (~individual+BAZ1B). R code provided.
Table S5. Number of genes differentially expressed following BAZ1B data in our numerical analysis compared to an analysis conducted on randomized HipSci data, using Edg2 function (see table S4).