

Abstract
Carbohydrate intolerance is one of several syndromes and diseases which together are known as malabsorption syndromes. These include small intestinal bacterial overgrowth (SIBO), coeliac disease, intestinal lymphangiectasia, short bowel syndrome, tropical sprue and some inherited metabolic disorders such as galactosaemia and pyruvate kinase deficiency. Specifically, the malabsorption of sugars affects morbidity for millions of sufferers across the world. Disaccharidase measurement is used in the investigation of disorders of the gastrointestinal tract. Diagnosis is by endoscopic small bowel biopsy of the duodenum or jejunum with subsequent biochemical and histopathological analysis. The diagnosis of bowel disorders presents several challenges with numerous overlapping presentations and symptoms such as bloating, diarrhoea, constipation, flatulence, borborygmus, weight loss and severe discomfort.
Introduction
The incidence of carbohydrate malabsorption disorders is on the increase with increased public awareness and diets such as low FODMAP (Fermentable Oligosaccharides, Disaccharides, Monosaccharides and Polyols) gaining in popularity. Examples of low FODMAP foods include cucumber, pineapple, almond milk, eggs and dark chocolate.1 Other than lactase enzyme replacement (microbial beta galactosidases such as lactAid) which can be added to milk, no specific clinical intervention is required such as surgery or medication and the management of these disorders primarily involves the avoidance of the offending sugar.2 The first step in the clinical investigation is the dietary removal of the sugar causing an immediate cessation of the symptoms and re-introduction causing their sudden and unwelcome return. Children are more of a diagnostic challenge with clinicians less likely to opt for invasive testing than for adults. Subsequently more hospital visits are required with a longer time for diagnosis. This leads to increased days off school with socio-economic consequences for both child and parent.
Carbohydrates are one of three macronutrients in the human diet and along with proteins and fats are the primary source of energy. Although energy provision is their major function, carbohydrates also have an important role to play in maintaining the cell integrity as well as the formation of carbohydrate structures on the external surface of cells for cell signalling and trafficking purposes such as proteoglycans, glycoproteins and the glycolipids. As their name implies, all carbohydrates are made only of carbon, hydrogen and oxygen. Monosaccharides have a fixed 1:2:1 ratio of carbon (C), hydrogen (H) and oxygen (O) and are organic compounds organised in the forms of aldehydes or ketones with multiple hydroxyl groups off the carbon chain. Glucose is a polyhydroxyaldehyde. It was Emil Fischer, a carbohydrate chemist who demonstrated proof of the structure of glucose for which he received the Nobel Prize in 1902.
In normal physiology, carbohydrate metabolism begins with mastication of carbohydrate-rich foods such as pasta and rice and the digestive action of salivary α-amylase on starch and glycogen abundant in these foods to produce the glucose oligomers amylose and amylopectin. The monosaccharides glucose and fructose, found in fruit and honey, require no further digestion and are absorbed as they are. Two simple sugars (monosaccharides) together form a disaccharide. Dietary disaccharides include maltose (a product of the digestion of starch), sucrose (table sugar), and lactose (the sugar in milk). These disaccharides must be broken down by enzymes into two simple sugars so that they can be absorbed by the intestine. Amylose consists of linear helical chains of roughly 500–20,000 α-D-glucose monomers linked through α-1,4 glycosidic bonds (Figure 1a). Amylopectin, on the other hand, consists of much larger, branched polymers of glucose containing up to 2 million residues. It contains multiple amylose-like chains of up to 30 glucose residues linked through α-1,4 bonds, connected to each other through α-1,6 branch points (Figure 1b).3 Salivary (s) amylase, encoded by the gene AMY1 found on chromosome 1, is an endoenzyme which carries out multiple breaks on linear portions of both amylose and amylopectin by hydrolysing the α(1-4) linkage bonds.4 The main short chain products obtained are maltose and maltotriose. Interestingly, it is the absence of β-amylase in humans which prevents the digestion of cellulose – a carbohydrate of plant cell wall origin containing β-1,4 glycosidic bonds between glucose molecules. Cellulose is considered to be one of the most abundant organic materials on earth. The action of the s-amylase isoform is inhibited by the acidic (pH 1.5–3.5) stomach environment. The optimum pH for s-amylase is 6.7–7.0 therefore no further digestion of carbohydrate can occur. However, the pancreatic isoform of amylase, encoded by the gene AMY2, is able to continue this process once the pancreas releases sufficient bicarbonate to neutralise the acidic content of the lumen of the small intestine.4 This bicarbonate solution is released through the ampulla of Vater under the control of the sphincter of Oddi.
Figure 1.
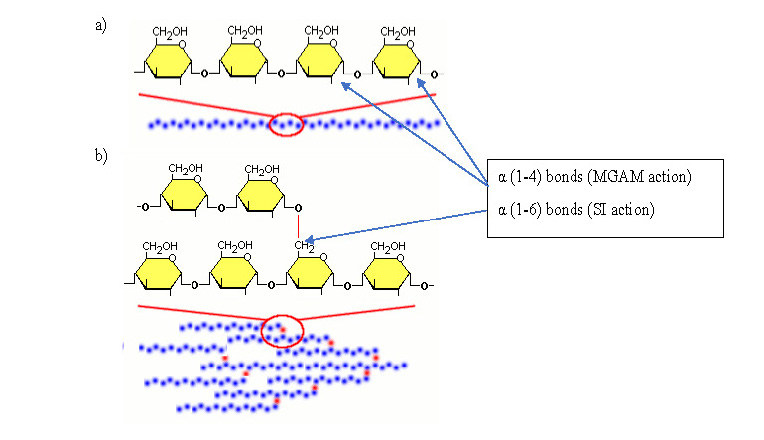
Structure of Amylose and Amylopectin. (A) α(1-4) glycosidic bonds linking glucose molecules into linear helical chains to form amylose. (B) α(1-6) glycosidic bonds linking amylose molecules into branched polymers chains to form amylopectin. (adapted with permission from ref. 3)
Two disaccharidases with exoglucosidase activity, maltose-glucoamylase (MGAM) and sucrase-isomaltase (SI) then take over the next phase of carbohydrate digestion which is the removal of the terminal α(1-4) linked (MGAM) glucose molecules and digestion of the α(1-6) branched (SI) glucose molecules from the glucose oligomers.5 Lactase-phlorozin hydrolase (LPH), with β-galactosidase activity, is the only enzyme to hydrolyse lactose (the sugar found in milk) in the intestinal lumen. The SI complex also assists maltose-glucoamylase in breaking down the α(1-4) bonds.6 SI is a dimeric intestinal enzyme synthesised as a single-chain precursor protein and it is the isomaltase subunit which anchors the protein at its terminus via a type II membrane anchor.7 Trehalase, a disaccharidase found in mushrooms, is a β-galactosidase which catalyses the hydrolysis of trehalose to two glucose molecules for absorption.8 The resultant product of these disaccharidases is either free glucose, galactose or fructose, which are then rapidly absorbed across the serosal surface of the epithelial cells by one of several luminal membrane-located glucose transporters e.g. GLUT-2. All three monosaccharides, glucose, galactose and fructose, can be transported from the intestinal mucosa into the portal circulation by GLUT-2, however, the sodium dependent glucose transporter, SGLT-1, is responsible only for the active transport of glucose or galactose with equimolar amounts of sodium against a concentration gradient into the cytoplasm of the enterocyte. GLUT-5 is responsible for the facilitated transport of fructose only.9
This final step in carbohydrate digestion takes place on specialised epithelial cells of the intestinal mucosa which together constitute the brush border. The three membrane-bound disaccharidases, SI, MGAM and LPH, are heavily N- and O-glycosylated which allows for their trafficking, sorting and locating to the apical membrane to be carried out. SI and MGAM share a high degree of homology. Correct positioning of these integral membrane proteins near the glucose transporters allows for efficient uptake of the resultant product, glucose. The brush border consists of individual microvilli approximately 0.1 μm in diameter and 1 μm in length and each cell may have up to 1000 microvilli. The numerous folds in these specialised cells increase their surface area by a factor of 600 allowing for the maximum absorptive capacity possible. At the brush border, several enzymes as described earlier have key roles in the breakdown of disaccharides into their final products (Table 1).
Table 1.
Brush border disaccharides.
| Enzyme | Substrate | Product |
|---|---|---|
| Lactase | Lactose | Glucose and Galactose |
| Sucrase | Sucrose | Glucose and Fructose |
| Maltase | Maltose | 2 x Glucose |
| Isomaltase | Maltose | 2 x Glucose |
| Trehalase | Trehalose | 2 x Glucose |
Carbohydrate Intolerance
Carbohydrate intolerance is a worldwide phenomenon affecting millions of people across the world. An estimated 50 million Americans alone are unable to adequately hydrolyse lactose.10 Specifically carbohydrate intolerance is the inability to digest certain carbohydrates due to a lack of one or more of these brush border disaccharide enzymes. It may be congenital, primary or secondary. The most common type of carbohydrate intolerance is lactose intolerance due to a primary insufficiency of the lactose digesting enzyme, lactase. Humans, being mammals, are genetically programmed to have reduced lactase activity once the weaning process begins. There is an obvious and essential requirement to break down the abundant lactose found in mammalian milk into glucose for the nourishment of newborn infants. The age of onset of primary hypolactasia varies between different ethnic groups but it is important to remember that hypolactasia does not cause discomfort unless lactose-containing food is consumed. In most infants, intestinal lactase activity is at its peak during the perinatal period after which two distinct groups emerge: ‘lactase-persistence’ and ‘lactase non-persistence’.11 Lactase non-persistence is the most common phenotype in humans, estimated at 65–70% of the population.12 Lactase persistence is thought to be related to the domestication of dairy cattle during the last 10,000 years as a result of strong positive selection and is inherited as a dominant trait. Adult expression of the gene encoding lactase (LCT) is located on chromosome 2 (2q21). Two single nucleotide polymorphisms (SNPs), C/T-13910 and G/A-22018, are strongly associated with the ability to digest lactase late into adulthood in the European population whereas the Chinese genotype is predominantly C/C-13910 with no SNP associated with lactase persistence in this population.13,14 However, the presence of a lactase-persistence polymorphism (T/T-13910) does not offer protection against other causes of lactase deficiency such as gastrointestinal infection, abdominal surgery or inflammatory bowel diseases such as Crohn’s disease or ulcerative colitis. Congenital lactase deficiency (CLD) may be fatal if not detected soon after birth. CLD is an autosomal recessive disorder resulting in severe gastrointestinal symptoms such as copious watery diarrhoea after lactose intake, either breast-feeding or formula solutions. This osmotic diarrhoea leads to severe dehydration and loss of weight. Although there have been worldwide case reports of this condition, CLD is enriched in the Finnish population with an incidence of 1:60,000.14
Congenital sucrase-isomaltase deficiency (CSID) is a rare autosomal recessive inborn error of metabolism resulting from mutation in the sucrase-isomaltase enzyme producing a complete absence of sucrase and most of the maltase activity. Currently 37 genetic alleles resulting in seven Sucrase-Isomaltase Deficiency (SID) phenotypes have been characterised.15 Infants with SI deficiency present with history of chronic diarrhoea and nutritional evidence of malabsorption.16 The osmotic diarrhoea is again caused by increased luminal disaccharides and presentation is usually seen at 2–16 weeks, normally at weaning from breast milk and replacement of fluid intake with dietary oligosaccharides and disaccharides in the form of sugary drinks. Once the diagnosis has been established immediate dietary cessation and/or replacement of the causative disaccharides restores normal gastrointestinal (GI) function. Most of the SI mutations result in lack of enzyme protein (i.e. null mutations). Other mutations produce unanchored or wrongly placed (secreted rather than retained) proteins, adding further evidence that this membrane-bound protein is required to be at the enterocyte cell surface to function normally.
Maltase deficiency per se can be more difficult to diagnose than either lactase or sucrase deficiency because both the sucrase-isomaltase and maltase-glucoamylase proteins have overlapping maltase activities. In the 1960s Dahlqvist identified four major fractions of the maltase activity in the small intestine that were essential to two different peptides, SI and MGAM.17 A recent article by Nichols et al. eloquently describes the advances modern molecular biology has had on Dahlqvist’s seminal work on the study of intestinal disaccharidases.18 They subtyped maltase deficiency into four classes based on a large UK study (n=30k) of disaccharidase activity using duodenal biopsies collected for clinical indication between 2006 and 2011. These four classes are: Congenital Sucrase-Isomaltase deficiency (CSID) (maltase 1b and/or 1a deficiency), Sucrase deficiency (maltose 1b), Pandisaccharidase deficiency (PDD) and a novel fourth classification based on the sum of maltase activities 1a, II and III, but without sucrase deficiency which they hypothesise is a new category called Primary Maltase Deficiency (PMD). They derive this maltase non-sucrase deficiency by the following equation:
This article challenged Dahlqvist’s hypothesis that congenital maltose intolerance does not exist due to the redundancy of multiple maltase activities from mucosal tissue. They postulated that a negative result from the above equation would identify primary maltose deficiency.
Trehalase is also found on the brush border and hydrolyses trehalose, an α(1,1) disaccharide of glucose found in many species of plants and animals. Mushrooms are the only source of dietary trehalose in man.19 Its main function in nature appears to be protection against desiccation as trehalose has high water retention capabilities.20 Trehalose is produced in response to long periods without water with subsequent reanimation once a water supply is resumed e.g. resurrection plants. This useful cell-membrane preservation function is being increasingly used in the commercial, pharmaceutical and medical industries to potentially treat diseases such as Huntington’s chorea and Alzheimer’s disease. Due to its water retention capabilities, trehalose is used as an additive to dried foods.21 Trehalose is not synthesised by mammals. Mutations in the trehalase gene (TREH) result in similar symptoms as described with other disaccharidase deficiency e.g. vomiting, abdominal discomfort etc after eating foods containing trehalose. Both autosomal recessive and autosomal dominate inheritance patterns have been described.
Carbohydrate Intolerance – Diagnosis
The main diagnostic platforms available for the determination of carbohydrate intolerance are hydrogen breath tests, small bowel disaccharidase biopsy testing, lactose tolerance tests and lactose intolerance genetic testing.
Lactose intolerance can usually be diagnosed with a careful clinical history supported by dietary challenge with patients usually presenting with a long-standing history of diarrhoea or flatulence after milk ingestion.22 Other symptoms such as rash, wheezing or other anaphylactic symptoms suggest a cow’s milk allergy as opposed to a specific lactose intolerance. This is especially true for infants, as milk allergy is rare in adults and may cause symptoms such as vomiting or esophageal reflux, neither of which are manifestations of lactose intolerance.23 True lactose intolerance is also indicated in the presence of a chronic and intermittent acidic diarrhoea. Most people with lactose intolerance can ingest up to 6–12 g of lactose (120–240 mL milk) without developing symptoms.24
Diagnosis – Breath Tests
Hydrogen breath tests (HBT) are a relatively cheap, reliable and non-invasive test for diagnosing carbohydrate intolerance or small intestinal bacterial overgrowth (SIBO). The rationale behind the test is that anaerobic bacteria colonise the small bowel to produce hydrogen via the fermentation of incompletely digested carbohydrates, due to a lack or absence of brush border disaccharidase enzymes.3 This carbohydrate malabsorption can produce significant amounts of hydrogen which is then absorbed through the epithelial walls of the small intestine where it is transported via the blood to the lungs where it is then released with expired air. Partially digested sugars in the form of disaccharides and oligosaccharides pass through the small intestine to the large intestine which already contains large concentrations of bacteria. This new source of fuel allows even more bacteria to be produced overwhelming the ability of the ileocaecal valve to prevent colonisation of the small intestine. This significantly increased bacteria population can now out-compete the host for other essential nutrients in the form of vitamins and minerals such as iron, zinc, tryptophan and folic acid, exacerbating the symptoms of carbohydrate intolerance.3 Up to 30% of the population are colonised by Methanobrevibacter smithii which converts four atoms of hydrogen into one molecule of methane. This population may not produce much hydrogen in the breath despite having SIBO or carbohydrate intolerance. It is therefore essential to screen for both hydrogen and methane producers when testing for these conditions.
Malabsorption is usually diagnosed by ingestion of the offending sugar e.g. lactose, and measuring the concentration of breath hydrogen and methane over time. There does not appear to be a standardised approach to either patient preparation or laboratory investigation for this test. However, most laboratories advocate not to test if the patient is on antibiotics, or had a colonoscopy or barium enema within 14 days of the test. A consensus statement of working specialists in America in 2017 recommended that antibiotics should be avoided for four weeks prior to the breath test. Additionally, patients should stick to a low fibre diet the day before the test and fast overnight for a minimum of 12 hours.25 During the test smoking, exercising and sleeping are not permitted. It is also important to establish whether the patient is an insulin-dependent diabetic as they will need to take insulin prior to the sugar load. A baseline alveolar sample is collected into a bag and transferred to a specialised leak-proof syringe. A minimum of 20 mL of alveolar air is recommended. The patient is then given the relevant dissolved sugar solution e.g. lactose, sucrose, fructose or glucose (if collecting for SIBO) to consume. Timed collections at 30-minute intervals are required until 3 hours have elapsed. The level of hydrogen in alveolar air will rise significantly (usually within 2 hours) only if the sugar is not digested. A fasting breath hydrogen greater than 20 ppm suggests small bacterial overgrowth. A rise in breath hydrogen greater than 20 ppm over the baseline value suggests impaired absorption of the ingested sugar. A short interval between sugar ingestion and rise in hydrogen and/or methane is also used as a marker of SIBO. False-negatives range from 2.5–15%, however, many of these false-negative reports can be avoided by simultaneously measuring both hydrogen and methane. Modern gas chromatograph breath analysers have the ability to calculate the alveolar concentrations of components in breath samples which may have been imperfectly collected or accidentally contaminated with room air. The instrument applies a correction factor based on the degree of dilution of alveolar carbon dioxide. Both hydrogen and methane are carried by room air past a solid-state sensor within the analyser.
Diagnosis – Disaccharidase Testing
The gold standard for any suspected disaccharidase deficiency is endoscopic biopsy of the small intestine with subsequent biochemical disaccharidase analysis and routine histological testing.15 Although the levels of disaccharidase are up to 50% higher in the jejunum, most endoscopic samples are taken from the duodenum due to ease of access.26 Disaccharidase analysis measures the glucose released after incubating with homogenised tissue samples with the various disaccharides i.e. lactose, sucrose, maltose and isomaltose (also known as palatinose). Most laboratories use a variation of the original work by Dahlqvist i.e. biopsy samples are homogenised in a solution of either saline or potassium chloride, incubated at 37°C for a fixed time interval and expressed as units of activity per gram of protein. Due to the increasing requests for this test, some laboratories have moved onto automated rather than manual testing. To ascertain the integrity of the biopsy sample, alkaline phosphatase is used as a reference enzyme. Disaccharidase testing involves measuring the glucose product from all four reactions either using a glucose oxidase or hexokinase procedure. Hexokinase phosphorylates each glucose molecule using adenosine triphosphate (ATP) to create glucose-6-phosphate which is then oxidised to produce 6-phosphogluconate and NADH. The strong absorption of UV light at 340 nm by NADH allows for the determination of the concentration of NADH and therefore an indirect measure of the concentration of glucose present. Thus, for each mole of glucose, one mole of NADH is produced as follows:
Automated analysers can combine the disaccharidase-hexokinase method within one reaction cuvette, where the disaccharidase reaction occurs by pre-incubating each specimen in the presence of each substrate (lactose, sucrose, isomaltose and maltose) for a standard time interval. The reaction is then inhibited by tris buffer (pH 7.8) – a direct disaccharidase inhibitor – contained within the hexokinase buffer. The reaction then proceeds to produce NADH, following the addition of hexokinase and glucose-6-phosphate dehydrogenase enzymes. Several assumptions are made in this assay. Firstly, that all disaccharidase activity is inhibited by the tris buffer present at the end of the incubation period. Secondly, that for each mole of glucose present, one mole of NADH is produced via the hexokinase-G6PD reaction. Lastly, that the reaction goes to completion i.e. all glucose moles produced by the hexokinase-G6PD reaction are oxidised to NADH i.e. the activity of an enzyme is defined as the number of micromoles of substrate converted to product per minute. Disaccharidase activity is calculated by the following formula:
| ΔGlucose | Change in concentration of glucose (in μmol/L) between a water-blank and a reaction sample |
| n | Number of glucose molecules formed per molecule of substrate. For lactose and sucrose n=1; for maltose and isomaltose n=2. |
| t | Enzyme sample and substrate incubation time (of 15 min) |
In order to derive a specific disaccharidase activity, a two-point calibration curve is used to determine the activity of unknown samples. This calibration curve is derived by measuring the change in absorbance of a water-blank and a glucose calibrator of known concentration following fifteen minutes of incubation at 37°C in the presence of each substrate and a hexokinase-G6PD reaction. The water-blank is assigned a ‘zero-activity’ and the calibrator is assigned an activity based on the concentration of glucose present (as measured independently). Therefore, using the calibration curve, the measured change in absorbance at 340 nm can be directly related to the specific enzyme activity (U/L) for the sample. Disaccharidase results are normalised for protein concentration within the homogenate and expressed as U/g protein. Most laboratories determine their own reference interval but true reference intervals for normal brush border enzyme activities are obviously difficult to come by, given the invasive nature of the test and the skewed population which are subject to these tests, with probable GI tract malfunction. Historical reference intervals are given in Table 2.27 In 2016, Hackenmueller and Grenache analysed a large sample set of biopsy samples with the aim of validating these historical reference intervals. Using the Hoffman method to calculate a reference interval, they determined new cut-offs for lactase, maltase, isomaltase and sucrase to be 10, 100, 9 and 25 U/g protein respectively. They recommended increasing the isomaltase cut-off from 5 to 9 U/g protein and to decrease the lactase cut-off from 15 to 10 U/g protein. Applying these cut-offs to the data set, 61% had no enzyme deficiency, 35% were lactase deficient, 11% were maltase deficient, 13% were isomaltase deficient and 13% were sucrase deficient. Pandisaccharidase deficiency was present in 8%.28
Table 2.
Historical reference intervals for disaccharidase levels.
| Values for normal enzyme levels | U/g |
|---|---|
| Lactase | >15 |
| Maltase | >100 |
| Isomaltase | >5 |
| Sucrase | >25 |
Diagnosis – Lactose Tolerance Tests
Lactose tolerance tests require the patient to drink a liquid containing lactose (e.g. milk or weighed solutions of lactose dissolved in water) after a baseline blood glucose level is taken. A subsequent blood glucose measurement is taken after 2 hours to determine the difference between the pre and post results. The original paper by Newcomer in 1966 used two different doses of lactose on 18 healthy volunteers all of whom had proven normal lactase activity in the jejunum.27 All the tests were done after an overnight fast with blood collected at time 0, 15, 30, 60, 90 and 120 minutes after ingesting the lactose. A flat curve was defined as an increase in blood sugar of less than 20 mg per decilitre and five of the 18 normal subjects had no response which Newcomer pointed out would limit the usefulness of the lactose tolerance test to diagnose lactose deficiency. A paper by Hovde in 2009 compared lactose tolerance tests with breath hydrogen and methane tests.23 Sixty patients suspected of having lactose malabsorption (LM) were given 25 g of lactose each and a combined test of serum glucose and hydrogen and methane in expired air was performed. This study demonstrated poor agreement between commonly used diagnostic tests for lactose malabsorption including duodenal/jejunal biopsy and genetic testing.
Diagnosis – Lactose Intolerance Genetic Testing
The C/T-13910 genetic variant, previously described in this article, is used to genotype individuals suspected of having primary lactose intolerance and to differentiate those with lactase intolerance due to secondary causes. Although C/T-13910 is specific for a European population, several other polymorphisms more common in non-Europeans may assist in identifying lactase persistence, thus excluding primary lactose intolerance as a cause of the patient’s symptoms. Several chemical pathology laboratories in Australia now routinely offer genetic testing for lactose intolerance.
Discussion
As with any biochemical test, the quality of the test is only as good as the quality of the sample. There is little to be gained from attempting to quantify enzyme activities if the sample is taken from an incorrect location or stored inappropriately. Specimen treatment between biopsy sampling and analysis is critical. Even a brief exposure to formalin destroys enzyme activity and while the human olfactory system can detect formalin contamination, an increasing number of laboratories are moving to more sensitive detection systems to identify imperceptible contaminants which may affect the integrity of the result leading to an incorrect interpretation.
Standardisation of disaccharidase testing is essential. Different laboratories have different sample preparation procedures such as homogenisation with tissue grinders or sonication which may also affect the results. Inter-laboratory incongruity also exists for total protein and glucose methodologies, reporting units and interpretation of results. There is also a disparate range of reference intervals between laboratories most of which have been derived from historical data. A lack of both commercially available quality control material and external quality assurance schemes contributes to a lack of standardisation and harmonisation for these tests.
Most laboratories have experienced significant growth in these tests over the last 10 years. This has led to a move from manual assays to automated analysers. This should assist in standardisation of the technical parameters at least but will still require large, collaborative efforts from each laboratory.
Each test for carbohydrate malabsorption presents limitations and difficulties. The main problems with disaccharidase testing of biopsy samples are the invasive nature of the test which renders it unsuitable for children, irregular distribution of lactase in the intestine and surgical differences in location of the dissection as few samples are taken from the ligament of Trietz (the original biopsy location sited in Dahlqvist’s paper). Most biopsy samples received by routine chemical pathology laboratories are taken from the duodenum due to comparative ease of access. Additionally, coeliac disease has a confounding effect on disaccharidase tests as the resultant villous atrophy will cause a generalised reduction in all disaccharidase enzymes. Breath tests lack a standardised patient preparation protocol such as length of time since cessation of antibiotics, testing protocol such as duration of test, dose of sugar administered and are highly dependent on the gut microbiome i.e. hydrogen or methane producers. Serum glucose testing depends on glucose absorption and metabolism with variation between individuals. Genetic tests do not detect all genetic disorders related to carbohydrate insufficiency and also fail to pick up secondary disorders i.e. carbohydrate disorders are not caused by a single gene defect unlike diseases such as cystic fibrosis. They are multifactorial e.g. surgery, trauma, gut biome, diet, alcohol intake etc. However given all these deficiencies, the general consensus is that disaccharidase testing of biopsy samples with combined biochemical and histological investigation remain the test of choice.29
Conclusions
Carbohydrate intolerance may be primary or secondary in origin and there are several tests available to clinicians to elucidate the true cause of several overlapping symptoms. The most common cause of carbohydrate intolerance, lactose intolerance, is not a disease per se but a natural, evolutionary phenomenon which drives the decreased production of lactase during infancy. Molecular testing has greatly increased scientific knowledge of primary disaccharidase deficiency but more investigation into coding mutations and noncoding genetic variants is required.
The lack of clinical laboratory standardisation of disaccharidase testing urgently needs to be addressed with appropriate age-specific reference intervals. This may be the most difficult challenge of all.
The requirement for a specific, non-invasive, gold-standard approach to carbohydrate intolerance remains elusive.
Footnotes
Competing Interests: None declared.
References
- 1.High and low FODMAP foods. [Accessed 17 September 2019]. https://monashfodmap.com/about-fodmap-and-ibs/high-and-low-fodmap-foods/
- 2.Rosado JL, Solomons NW, Lisker R, Bourges H. Enzyme replacement therapy for primary adult lactase deficiency. Effective reduction of lactose malabsorption and milk intolerance by direct addition of beta-galactosidase to milk at mealtime. Gastroenterology. 1984;87:1072–82. [PubMed] [Google Scholar]
- 3.VIVO Pathophysiology. Dietary Polysaccharides. [Accessed 17 September 2019]. http://www.vivo.colostate.edu/hbooks/pathphys/digestion/basics/polysac.html.
- 4.Butterworth PJ, Warren FJ, Ellis PR. Human α-amylase and starch digestion: An interesting marriage. Starch. 2011;63:395–405. [Google Scholar]
- 5.Robayo-Torres CC, Quezada-Calvillo R, Nichols BL. Disaccharide digestion: clinical and molecular aspects. Clin Gastroenterol Hepatol. 2016;4:276–87. doi: 10.1016/j.cgh.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 6.Amiri M, Naim HY. Characterization of mucosal disaccharidases from human intestine. Nutrients. 2017;9:E1106. doi: 10.3390/nu9101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lentze MJ. Molecular and cellular aspects of hydrolysis and absorption. Am J Clin Nutr. 1995;61(Suppl):946S–51S. doi: 10.1093/ajcn/61.4.946S. [DOI] [PubMed] [Google Scholar]
- 8.Murray IA, Coupland K, Smith JA, Ansell ID, Long RG. Intestinal trehalase activity in a UK population: establishing a normal range and the effect of disease. Br J Nutr. 2000;83:241–5. doi: 10.1017/s0007114500000313. [DOI] [PubMed] [Google Scholar]
- 9.Mattar R, de Campos Mazo DF, Carrilho FJ. Lactose intolerance: diagnosis, genetic, and clinical factors. Clin Exp Gastroenterol. 2012;5:113–21. doi: 10.2147/CEG.S32368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Lacy Costello BP, Ledochowski M, Ratcliffe NM. The importance of methane breath testing: a review. J Breath Res. 2013;7:024001. doi: 10.1088/1752-7155/7/2/024001. [DOI] [PubMed] [Google Scholar]
- 11.Enattah NS, Sahi T, Savilahti E, Terwilliger JD, Peltonen L, Järvelä I. Identification of a variant associated with adult-type hypolactasia. Nat Genet. 2002;30:233–7. doi: 10.1038/ng826. [DOI] [PubMed] [Google Scholar]
- 12.Bayless TM, Brown E, Paige DM. Lactase non-persistence and lactose intolerance. Curr Gastroenterol Rep. 2017;19:23. doi: 10.1007/s11894-017-0558-9. [DOI] [PubMed] [Google Scholar]
- 13.Deng Y, Misselwitz B, Dai N, Fox M. Lactose Intolerance in Adults: Biological Mechanism and Dietary Management. Nutrients. 2015;7:8020–35. doi: 10.3390/nu7095380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuokkanen M, Kokkonen J, Enattah NS, Ylisaukko-Oja T, Komu H, Varilo T, et al. Mutations in the translated region of the lactase gene (LCT) underlie congenital lactase deficiency. Am J Hum Genet. 2006;78:339–44. doi: 10.1086/500053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen SA, Oloyede H. Cohen and Hannah Oloyede. Variable Use of Disaccharidase Assays When Evaluating Abdominal Pain. Gastroenterol Hepatol (N Y) 2018;14:26–32. [PMC free article] [PubMed] [Google Scholar]
- 16.Belmont JW, Reid B, Taylor W, Baker SS, Moore WH, Morriss MC, et al. Congenital sucrase-isomaltase deficiency presenting with failure to thrive, hypercalcemia, and nephrocalcinosis. BMC Pediatr. 2002;2:4. doi: 10.1186/1471-2431-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dahlqvist A. Specificity of the human intestinal disaccharidases and implications for hereditary disaccharide intolerance. J Clin Invest. 1962;41:463–70. doi: 10.1172/JCI104499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nichols BL, Baker SS, Quezada-Calvillo R. Metabolic aspects of maltase deficiencies. J Pediatr Gastroenterol Nutr. 2018;66(Suppl 3):S24–9. doi: 10.1097/MPG.0000000000001955. [DOI] [PubMed] [Google Scholar]
- 19.Madzarovovà-Nohejlova J. Trehalase deficiency in a family. Gastroenterology. 1973;65:130–3. [PubMed] [Google Scholar]
- 20.Luyckx J, Baudouin C. Trehalose: an intriguing disaccharide with potential for medical application in ophthalmology. Clin Ophthalmol. 2011;5:577–81. doi: 10.2147/OPTH.S18827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swallow DM, Poulter M, Hollox EJ. Intolerance to lactose and other dietary sugars. Drug Metab Dispos. 2001;29:513–6. [PubMed] [Google Scholar]
- 22.DiPalma JA, Narvaez RM. Prediction of lactose malabsorption in referral patients. Dig Dis Sci. 1988;33:303–7. doi: 10.1007/BF01535754. [DOI] [PubMed] [Google Scholar]
- 23.Hovde Ø, Farup PG. A comparison of diagnostic tests for lactose malabsorption – which one is the best? BMC Gastroenterol. 2009;9:82. doi: 10.1186/1471-230X-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rana SV, Bhasin DK, Katyal R, Singh K. Comparison of duodenal and jejunal disaccharidase levels in patients with non ulcer dyspepsia. Trop Gastroenterol. 2001;22:135–6. [PubMed] [Google Scholar]
- 25.Rezaie A, Buresi M, Lembo A, Lin H, McCallum R, Rao S, et al. Hydrogen and methane-based breath testing in Gastrointestinal disorders: The North American Consensus. Am J Gastroenterol. 2017;112:775–84. doi: 10.1038/ajg.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puertolas MV, Fifi AC. The role of disaccharidase deficiencies in functional abdominal pain disorders – a narrative review. Nutrients. 2018;10:E1835. doi: 10.3390/nu10121835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newcomer AD, McGill DB. Lactose tolerance tests in adults with normal lactase activity. Gastroenterology. 1966;50:340–6. [PubMed] [Google Scholar]
- 28.Hackenmueller SA, Grenache DG. Reference intervals for intestinal disaccharidase activities determined from a non-reference population. J Appl Lab Med. 2016;1:172–80. doi: 10.1373/jalm.2016.020388. [DOI] [PubMed] [Google Scholar]
- 29.Suligoj T, Ciclitira PJ, Bozic B. Diagnostic and research aspects of small intestinal disaccharidases in coeliac disease. J Immunol Res. 2017;2017 doi: 10.1155/2017/1042606. 1042606. [DOI] [PMC free article] [PubMed] [Google Scholar]