

Abstract
By reducing amino acid alphabet, the protein complexity can be significantly simplified, which could improve computational efficiency, decrease information redundancy and reduce chance of overfitting. Although some reduced alphabets have been proposed, different classification rules could produce distinctive results for protein sequence analysis. Thus, it is urgent to construct a systematical frame for reduced alphabets. In this work, we constructed a comprehensive web server called RAACBook for protein sequence analysis and machine learning application by integrating reduction alphabets. The web server contains three parts: (i) 74 types of reduced amino acid alphabet were manually extracted to generate 673 reduced amino acid clusters (RAACs) for dealing with unique protein problems. It is easy for users to select desired RAACs from a multilayer browser tool. (ii) An online tool was developed to analyze primary sequence of protein. The tool could produce K-tuple reduced amino acid composition by defining three correlation parameters (K-tuple, g-gap, λ-correlation). The results are visualized as sequence alignment, mergence of RAA composition, feature distribution and logo of reduced sequence. (iii) The machine learning server is provided to train the model of protein classification based on K-tuple RAAC. The optimal model could be selected according to the evaluation indexes (ROC, AUC, MCC, etc.). In conclusion, RAACBook presents a powerful and user-friendly service in protein sequence analysis and computational proteomics. RAACBook can be freely available at http://bioinfor.imu.edu.cn/raacbook.
Database URL: http://bioinfor.imu.edu.cn/raacbook
Introduction
With the development of various biotechnologies, the number of protein sequences is growing at a rapid pace. However, the three-dimensional structures and function of most proteins are still not determined. For example, in August 2019, there are 154 939 structures, 560 537 reviewed proteins and 167 761 270 unreviewed protein sequences in Protein Data Bank (PDB) (1, 2), the Swiss-Prot and TrEMBL (3), respectively. Obviously, the gaps between structure data, function data and protein sequences are increasing fast. Although X-ray crystallography is a powerful tool in determining these structures, it is time-consuming and expensive, and not all proteins can be successfully crystallized. Membrane proteins are difficult to crystallize, and most of them will not dissolve in normal solvents. Therefore, so far few membrane protein structures have been determined. NMR is indeed a very powerful tool in determining the 3D structures of membrane proteins (4–21), but it is also time-consuming and costly. Thus, it is urgent to design efficient computational methods based on sequence information for rapidly and accurately identifying biological features in primary protein sequences.
Subsequently, experimental interest in reduced alphabet was firstly proposed in the 1960s (22). Alphabet reduction techniques play high-potential roles for sequence alignment and topological estimation (23), which have been widely used in almost all of protein classification (24–32). Meanwhile, a series of 3D protein structures have been developed by means of structural bioinformatics tools (33–45). Facing the explosive growth of biological sequences discovered in the postgenomic age, to timely use them for drug development, a lot of important sequence-based information, such as PTM (post-translational modification) sites in proteins (46–87), protein–drug interaction in cellular networking (88), protein–protein interactions (89), DNA-methylation sites (90), recombination spots (91) and sigma-54 promoters (92), have been deducted by various sequential bioinformatics tools such as the PseAAC approach and PseKNC approach (93). Recently, success of AlphaFold on creating 3D protein models proved that the sequence-dependent inference has incredible potential in computational proteomics (94). Actually, rapid development in sequential bioinformatics and structural bioinformatics has driven the medicinal chemistry undergoing an unprecedented revolution (54), in which the computational biology has played increasingly important roles in stimulating the development of finding novel drugs (95, 96).
By clustering around 20 amino acids to smaller alphabet based on some similar rules, the protein complexity will be dramatically reduced, and some functional conserved regions will be more clearly displayed (97). For example, Figure 1A shows a schematic view of a protein 5TCD, which is ectonucleotide pyrophosphatase. Its decreased levels may be involved in colon cancer. By utilizing the analysis of amino acid reduction, we can clearly find the correlation between the primary sequence and its 3D structure (Figure 1B). The unique sequence bias of the three-dimensional structure can be visualized in a one-dimensional interface, which shows that reduced amino acid clusters (RAACs) have sufficient capability to identify the consensus domain in sequence alignment (98). Recent work demonstrated that the specific codes endow sequence motifs with unique structures or functions, and the differential combination and arrangement of the motifs with specific codes determine the protein isoforms that possesses multiple functions (99).
Figure 1.
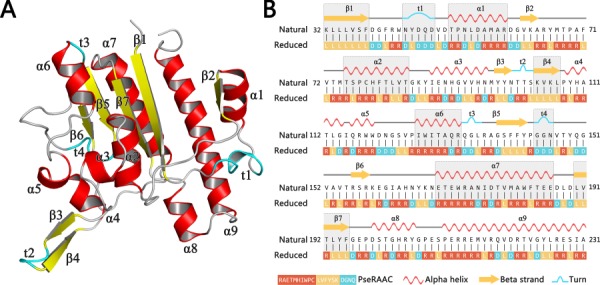
A schematic view of a protein 5TCD in PDB with secondary structures. Subfigure (A) shows the three-dimensional structure of this protein. All secondary structural elements are indicated as different labels. Subfigure (B) shows its corresponding chain view, where the gray background represents the portion of the reduced amino acid sequence that matches the protein secondary structural elements.
With the explosive growth of biological sequences in the postgenomic era, one of the most important but also most difficult problems in computational biology is how to express a biological sequence with a discrete model or a vector, yet still keep considerable sequence-order information or key pattern characteristic. This is because all the existing machine-learning algorithms (such as ‘Optimization’ algorithm (100), ‘Covariance Discriminant’ or ‘CD’ algorithm (101, 102), ‘Nearest Neighbor’ or ‘NN’ algorithm (103) and ‘Support Vector Machine’ or ‘SVM’ algorithm (103, 104)) can only handle vectors as elaborated in a comprehensive review (105).
However, a vector defined in a discrete model may completely lose all the sequence-pattern information. To avoid completely losing the sequence-pattern information for proteins, the pseudo amino acid composition (93) or PseAAC (106) was proposed. Ever since the concept of Chou’s PseAAC was proposed, it has been extensively used in nearly all the areas of computational proteomics (107–116). Because it has been widely and increasingly used, four powerful open-access softwares, called ‘PseAAC’ (117), ‘PseAAC-Builder’ (118), ‘propy’ (119) and ‘PseAAC-General’ (120), were established: the former three are for generating various modes of Chou’s special PseAAC (121), the fourth one for those of Chou’s general PseAAC (122), including not only all the special modes of feature vectors for proteins but also the higher level feature vectors such as ‘Functional Domain’ mode, ‘Gene Ontology’ mode and ‘Sequential Evolution’ or ‘PSSM’ mode (122). Encouraged by the successes of using PseAAC to deal with protein/peptide sequences, the concept of PseKNC (Pseudo K-tuple Nucleotide Composition) (105) was developed for generating various feature vectors for DNA/RNA sequences (123–125) that have proved very useful as well. Particularly, recently a very powerful web server called ‘Pse-in-One’ (126) and its updated version ‘Pse-in-One 2.0’ (100) have been established that can be used to generate any desired feature vectors for protein/peptide and DNA/RNA sequences according to the need of users’ studies.
Since the reduced amino acids perform powerful ability, we firstly developed the flexible web server for generating pseudo K-tuple reduced amino acid composition (27). During the last 2 years, amounts of users’ feedback show that this online server must be updated by providing additional online services. Therefore, we update the online server, called RAACBook, which not only contained reduced amino acid analysis but also newly added the visualization report module, the comprehensive RAAC repository and the machine learning online tool. It performed more robust and powerful for simplifying protein complexity, providing feature files for prediction, training classification models and showing clearer conservative regions.
Materials and Methods
Data collection and curation
The framework for the development of the RAACBook is described in Figure 2. The reduced amino acid alphabets of RAACBook were derived from over 1000 PubMed’s original literature, which were filtered by keywords as follows: ‘amino acid alphabet’, ‘reduced amino acid’, ‘amino acid cluster’, ‘amino acid group’, ‘simplified amino acid’ etc. Till 14 August 2019, 74 types of reduced amino acid alphabets were manually curated in RAACBook, which can generate 673 reduced amino acid descriptors for analyzing protein sequence (Supplementary Table 1). There are more than 40 clustering algorithms involved in this online repository, including BLOSUM matrix, maximum information gain, data mining and physico-chemical properties. In addition, the deep learning method has been also applied to the amino acid reduction (26). For better selecting desired alphabets, we developed a multilayer browser tool, which supports filtering of different keywords. The returned entries will link to the annotation information of RAAC, including description, reference and visualized clusters, which can be conveniently selected to reduced analysis.
Figure 2.
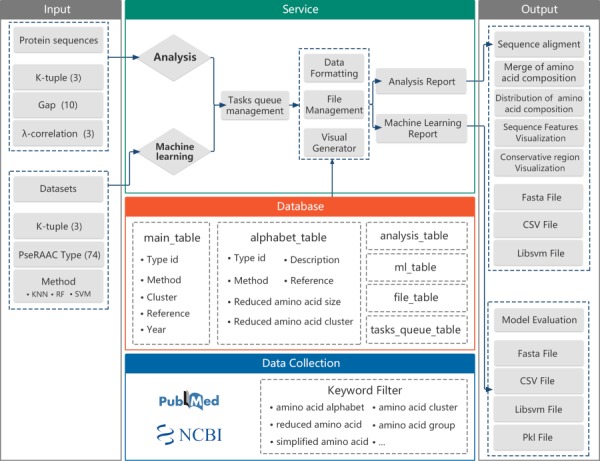
The framework of the RAACBook. Block diagrams showing the modules and functions of RAACBook. Input data are on the left, output data presented on the right. The data of the manually curated database is collected in PubMed by a keyword filter. Users can provide protein sequences in the webpage to generate reduced sequence vector files and visualizations. The users can also upload the protein sequence datasets as input to obtain the corresponding classifier model and evaluation.
Implementation
The current version of the RAACBook database is constructed on MariaDB. The system is operated on Linux servers with 28 physical cores, which uses Apache as a proxy. The analysis engine and the visualization module are developed by Python.
After users upload protein sequences to the data management and set essential parameters, each job from the client can be submitted into the task queue with a unique Job ID. Job ID and parameters will be stored for the user to call again. As the function of the task queue management, if there are not enough computing resources available, the job is put on waiting schedule. For the user, job status is refreshed in the analysis report page. When the job is completed, the analysis report could be generated online.
As pointed out in (127) and demonstrated in a series of recent publications (67, 71, 72, 82, 128–144), user-friendly and publicly accessible web servers represent the future direction for developing practically more useful prediction methods and computational tools. Actually, many practically useful web servers have significantly increased the impacts of bioinformatics on medical science (54), driving medicinal chemistry into an unprecedented revolution (116).
Results
Overview of the RAACBook web service
To reduce complexity and understand topological estimation of protein sequences, natural protein sequences with the 20-letter amino acid alphabet can be commonly compressed to simplified alphabet based on some amino acid similar standards (98). RAACBook is an online repository of reduced amino acid alphabets. The current version contains RAAC database, reduction analysis, visualization and machine learning of protein classification (Figure 2). Firstly, The RAAC database provides a comprehensive resource of reduced amino acid alphabets. A multilayer browser is applied for filtering the desired RAAC from the database. Secondly, the analysis server can produce K-tuple reduced amino acid composition by defining three correlation parameters. The result provides reduced fasta files, csv and libsvm vector files for downloading. For different protein studies, we visualized alignment of sequences, mergence of amino acids, distribution of reduced amino acid composition, heat map of sequence features and logo of reduced sequence. Thirdly, the machine learning server enables users to build interesting classifier models, using different machine learning algorithms based on reduced sequence features, which can generate corresponding performance evaluation and downloadable file for each model.
Reduction analysis of primary protein sequences
The major challenge in sequence-dependent inference is to extract the most efficient features and withdraw the information buried in primary protein sequences. To solve that, we developed the reduction analysis and the workflow is as below.
Data input and RAAC-type selection
Users only need to upload the primary sequence with fasta format as input with three parameters and RAAC Type (Figure 3A, Step 1). The Types can be filtered in a two-dimensional selection box with the different alphabet types and cluster sizes, which are recorded in the RAACBook database (Figure 3A, Step 2).
Figure 3.

RAACBook analysis and machine learning workflow. Subfigure (A) The workflow shows the reduction analysis of natural amino acid sequence. Settings pane: After uploading primary sequences in fasta format (Step 1) and the alphabet types of interest were used as input (Step 2). If only these parameters are submitted, the server can generate reduced sequence files (Step 3). If the aim is to produce sequence feature files for machine learning, users need to select three parameters (Step 4) and submit (Step 5). Analysis panel: there are three files for download (Step 6). The reduced amino acid sequences are visualized by clicking ‘Visualization’ button (Step 7). Sequences visualization: three charts were exhibited: alignment between natural and reduced amino acid sequences (Step 8), mergence of natural amino acid composition (Step 9), and distribution of amino acid composition (Step 10). Features visualization (Steps 11–13): according to different reduced alphabets and parameters, service will generate the K-tuple reduced amino acid composition heat map of multiple sequences and the distribution of single reduced sequence peptides. Reduced logo visualization (Steps 14–15): the figure represents each amino acid information of each position in protein sequence based on the reduced alphabet. Subfigure (B) Machine learning workflow shows the acquisition of the classifier model by uploading datasets and setting parameters. Settings panel (Steps 1–3): K-tuple, the alphabet type and the machine learning algorithm are selected (Step 2), after uploading fasta files containing positive and negative datasets (Step 1). Subsequently, the machine learning service was executed (Step 3). Model evaluation: the chart of Sp, Sn, Acc, Mcc and the diagram of the ROC curve are generated (Steps 4 and 5), and the classifier model and vector files can be downloaded (Steps 6–8).
Parameter selection
(i) K-tuple. The K-tuple value represents the number of peptide. For example, K = 1 means a monopeptide or amino acid, K = 2 represents a dipeptide, K = 3 represents a tripeptide, and so on. In a typical K-tuple analysis, one usually slides the window of width K amino acids along the protein by one residue at a time. For a protein with N amino acids, with k = 2, the dipeptide frequency be counted as follows, R1R2, R2R3, R3R4, etc. (Supplementary Figure 1A) (126,145). (ii) g-gap. The value of gap represents the inter-gap number between two nearest amino acid or K-tuple peptides along the protein. That is, the gap between each K-tuple peptide is represented. With k = 2, g = 1, the aim is to count the dipeptide frequency along the protein by skipping one residue in every slide as follows, R1R2, R3R4, R5R6, etc. (Supplementary Figure 1B) (126). (iii) λ-correlation. The λ-correlation of parameters, also called parallel correlation, which represents the gap number of each two adjacent amino acids in the K-tuple peptide interval. It is an integer greater than 2 and less than L-K, which reflects the protein sequence correlation between the nearest residue when K-tuple is determined (Supplementary Figure 1C). Taking K = 3, λ = 1, g = 2 as an example, the intra-gap number of within tripeptide interval is 1, and the number of skipping residue of tripeptide in each slide is 2. In the calculation process, the combination is R1R3R5, R4R6R8, R7R9R11 and so on (Supplementary Figure 1D).
The biological meaning of three parameters: it is well-known that the protein with specific domain codes endows sequence motifs with unique consensus, and the differential combination and arrangement determine the protein function. Here the K-tuple parameter was applied to calculate the total composition of n-peptide in the whole protein sequence. For example, when K = 3, the reduced tripeptide composition of protein sequence will be counted for 5TCD protein in Figure 1. It reflects the global composition of the secondary structure in 5TCD protein. The g-gap was introduced to compute the interspersed frequency of specific n-peptide with position bias. For example, ‘RR’ or ‘RRR’ can represent feature preference of alpha helix, which is usually scattered throughout the protein sequence. The λ-correlation was defined to extract feature information of function domain with low internal conservatism. Taking CXXC domains as example (Supplementary Figure 2), ‘X’ can be any amino acids, but the two external C residue is extremely conserved (146). This domain is ubiquitous in TET, DNMT and MBD protein. When K = 2, λ = 2, the feature preference of CXXC domain will be precisely captured by our feature extraction. Therefore, these three quantities will help researchers to obtain features with effective biological significance in meaningfully characterizing proteins.
Analysis report
The server will finally generate an analysis report of reduced amino acid, which consisted of parameter information, reduced feature and sequence files and visualization. The parameter information includes RAAC, K-tuple, g-gap, λ-correlation and job id. The download supports fasta, csv and libsvm vector files, and they are packaged as downloadable zip file (Figure 3A, Steps 6 and 7). Filtering to the desired feature vectors and sequence files is necessary for the prediction of protein three-dimensional structures or the construction of protein classification models. Therefore, the sequence reduction analysis includes the visualization to meet different studies, which consists of three parts: the sequence visualization, the feature visualization and the reduced logo visualization.
Sequence visualization includes three presentation methods (Figure 3A, Steps 8–10): firstly, in the alignment of the natural and reduced sequences, the colors used by the reduced amino acids are only a subset of those used by the natural ones. With such a color reduction in visualization, the primary structures of the proteins show clearer physical and chemical characteristics than the natural amino acid sequences. Also, the relevant protein complexity will be minimally degraded with nonessential information being suppressed, in some cases leading to more clearly display functionally conserved regions (147). Secondly, the matching relates the natural amino acids with the reduced ones. The lines represent association, and the width of a line is drawn in proportion to the frequency of involved amino acids in the reduction process. In general, some amino acids are more likely to reduce to certain amino acids, which show the preference in protein sequences. It plays a major role in structural biology, synthetic biology and new drug development. Thirdly, the frequency map of the natural and reduced amino acids is drawn.
Feature visualization (Figure 3A, Steps 11–13): according to different reduced alphabets and parameters, the server will generate the K-tuple reduced amino acid composition heat map of multiple sequences and the distribution of single reduced sequence peptides. Generally, the tool can provide the reference and helps users to find suitable vector files for their protein research.
Reduced logo visualization (Figure 3A, Steps 14 and 15): each logo is the representation of a multiple-protein sequence alignment, including natural sequence alignment and reduced sequence alignment. Each position of the sequence is a stack. Each stack is a collection of each amino acid frequency at this position (148). After the reduction, visualization provides a richer and clearer way to depict the sequences, such as binding sites and functional conserved regions.
Machine learning of protein classification
As demonstrated by a series of recent publications (57, 60, 76–78, 82, 84, 88, 90–92, 128–132, 149–160) and summarized in two comprehensive review papers (122, 161), to develop a really useful predictor for a biological system, one needs to follow Chou’s five-step rule to go through the following five steps: (i) the valid benchmark datasets are firstly submitted as inputs for training the classifier model (Figure 3B, Step 1); (ii) the samples with an effective formulation can truly reflect their intrinsic correlation with the target to be predicted; (iii) the support vector machine (SVM), K-nearest neighbor (KNN) and random forest (RF) algorithm are introduced to operate the classifier; (iv) the 5-fold cross-validation is the default test for using to evaluate the anticipated accuracy of the classifier; and (v) establishment of a user-friendly web server for a classifier that is accessible to the public user. A new sequence-analyzing method or statistical predictor by observing the guidelines of Chou’s five-step rule has the following notable merits: (i) crystal clear in logic development, (ii) completely transparent in operation, (iii) easily to repeat the reported results by other investigators, (iv) with high potential in stimulating other sequence-analyzing methods and (v) very convenient to be used by the majority of experimental scientists.
The result of the machine learning server includes parameter information and model evaluation. The machine learning method, job ID, K-tuple and alphabet type are listed at the top of the report. The details of prediction for every alphabet are shown in the model evaluation section, including specificity (Sp), sensitivity (Sn), accuracy (Acc) and Mathew correlation coefficient (MCC) (Figure 3B, Step 5). The results of different alphabets are displayed in the receiver operating characteristic (ROC) curve, and the AUC value is given for reflecting the overall prediction ability (Figure 3B, Step 4). Finally, researchers can use the above indexes to select the appropriate classifier model or feature file to apply to their study. At the bottom of the report, users can conveniently download the fasta, csv, libsvm files and classifier models (Figure 3B, Steps 6–8).
Applications
The reduced amino acid alphabets combined with machine leanings have been shown to have the ability for functional annotation of protein, such as iDPF-PseRAAAC (28), iHSP-PseRAAAC (149), Antimicrobial Peptide Scanner (26), Bastion6 (25) and iDNA-Prot|dis (162). A recent example of a collaborative focus within the RAACBook is the identification of secretory protein using RAAC, implemented as the ISP-PseRAAC. This is an online implementation of the SVM method, which can use protein sequences as input and provides prediction scores of secreted proteins. The valid benchmark datasets contain secretory proteins and non-secretory proteins of the malaria parasite. We use the SVM method to train the model by extracting three kinds of sequence feature including amino acid composition, dipeptide composition and tripeptide composition based on reduced amino acid sequence. Finally, based on dipeptide compositions of alphabet type 11 with cluster size 10, the prediction result of leave-one-out cross validation achieves 91.67% accuracy with 0.84 Mathew’s correlation coefficient, which demonstrates that the reduced alphabet has sufficient discriminatory power to predict protein function.
New Features
The first version of RAACBook, PseKRAAC, was released as described by Zuo (27). The innovative features of the current version are as follows: (i) we increased the types of reduced amino acids alphabet from 16 to 74 and built a database for updating systematically the latest reduction type. These types contain nearly 700 clusters from literatures, each of which has a detailed method, description, reference, etc. (ii) The current web server rebuilt the user interface and background services, providing more friendly user interaction and a stronger system. Detailed tutorial and help are also supported throughout the use of the web server. (iii) The reduced analysis script was rewritten to improve efficiency, and analysis results were shown as a report, including a variety of downloadable files and images. In particular, we have added visualizations of reduced amino acid, sequence features and conservative region. (iv) We developed a machine learning tool to train online the classifier model and generated the evaluation report for users.
Conclusion
The major challenge in protein sequence research, however, remains, for extracting precise information. The RAACs performed sufficient ability for decreasing protein complexity and withdrawing the conservative feature hidden in the noise signals that affect protein sequence researches. As new literature on amino acid reductions is published, we will update timely the database and server workflows to support the latest reduced alphabets and complicated studies. In short, RAACBook is a flexible and comprehensive web online platform where the hidden value of a large number of protein sequences can be explored by a wide range of users. With continuous user feedback and further enhancement, RAACBook has the potential to become an integral part of routine data of protein analysis for computational and experimental biologists.
Supplementary Material
Funding
National Nature Scientific Foundation of China (61561036, 61702290); Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (NJYT-18-B01); Fund for Excellent Young Scholars of Inner Mongolia (2017JQ04). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Conflict of interest. None declared.
References
- 1. Berman H.M., Westbrook J., Feng Z. et al. (2000) The protein data bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burley S.K., Berman H.M., Christie C. et al. (2018) RCSB Protein Data Bank: sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci., 27, 316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bairoch A. and Apweiler R. (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res., 28, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chou J.J., Matsuo H., Duan H. et al. (1998) Solution structure of the RAIDD CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell, 94, 171–180. [DOI] [PubMed] [Google Scholar]
- 5. Oxenoid K., Dong Y., Cao C. et al. (2016) Architecture of the mitochondrial calcium uniporter. Nature, 533, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dev J., Park D., Fu Q. et al. (2016) Structural basis for membrane anchoring of HIV-1 envelope spike. Science, 353, 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schnell J.R. and Chou J.J. (2008) Structure and mechanism of the M2 proton channel of influenza A virus. Nature, 451, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berardi M.J., Shih W.M., Harrison S.C. et al. (2011) Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature, 476, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chou J.J., Li S., Klee C.B. et al. (2001) Solution structure of Ca(2+)-calmodulin reveals flexible hand-like properties of its domains. Nat. Struct. Biol., 8, 990–997. [DOI] [PubMed] [Google Scholar]
- 10. OuYang B., Xie S., Berardi M.J. et al. (2013) Unusual architecture of the p7 channel from hepatitis C virus. Nature, 498, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J., Pielak R.M., McClintock M.A. et al. (2009) Solution structure and functional analysis of the influenza B proton channel. Nat. Struct. Mol. Biol., 16, 1267–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fu Q., Fu T.M., Cruz A.C. et al. (2016) Structural basis and functional role of intramembrane trimerization of the Fas/CD95 death receptor. Mol. Cell, 61, 602–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chou J.J., Li H., Salvesen G.S. et al. (1999) Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell, 96, 615–624. [DOI] [PubMed] [Google Scholar]
- 14. Oxenoid K. and Chou J.J. (2005) The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc. Natl. Acad. Sci. USA, 102, 10870–10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Call M.E., Schnell J.R., Xu C. et al. (2006) The structure of the zetazeta transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell, 127, 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Call M.E., Wucherpfennig K.W. and Chou J.J. (2010) The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat. Immunol., 11, 1023–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gagnon E., Xu C., Yang W. et al. (2010) Response multilayered control of T cell receptor phosphorylation. Cell, 142, 669–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bruschweiler S., Yang Q., Run C. et al. (2015) Substrate-modulated ADP/ATP-transporter dynamics revealed by NMR relaxation dispersion. Nat. Struct. Mol. Biol., 22, 636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cao C., Wang S., Cui T. et al. (2017) Ion and inhibitor binding of the double-ring ion selectivity filter of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. USA, 114, E2846–E2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Piai A., Dev J., Fu Q. et al. (2017) Stability and water accessibility of the trimeric membrane anchors of the HIV-1 envelope spikes. J. Am. Chem. Soc., 139, 18432–18435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pan L., Fu T.M., Zhao W. et al. (2019) Higher-order clustering of the transmembrane anchor of DR5 drives signaling. Cell, 176, e1414, 1477–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan H.S. (1999) Folding alphabets. Nat. Struct. Biol., 6, 994–996. [DOI] [PubMed] [Google Scholar]
- 23. Stephenson J.D. and Freeland S.J. (2013) Unearthing the root of amino acid similarity. J. Mol. Evol., 77, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Z.R., Lin H.H., Han L.Y. et al. (2006) PROFEAT: a web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res., 34, W32–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang J., Yang B., Leier A. et al. (2018) Bastion6: a bioinformatics approach for accurate prediction of type VI secreted effectors. Bioinformatics, 34, 2546–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Veltri D., Kamath U. and Shehu A. (2018) Deep learning improves antimicrobial peptide recognition. Bioinformatics, 34, 2740–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zuo Y., Li Y., Chen Y. et al. (2017) PseKRAAC: a flexible web server for generating pseudo K-tuple reduced amino acids composition. Bioinformatics, 33, 122–124. [DOI] [PubMed] [Google Scholar]
- 28. Zuo Y., Lv Y., Wei Z. et al. (2015) iDPF-PseRAAAC: a web-server for identifying the defensin peptide family and subfamily using pseudo reduced amino acid alphabet composition. PloS One, 10, e0145541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pan Y., Wang S., Zhang Q. et al. (2019) Analysis and prediction of animal toxins by various Chou's pseudo components and reduced amino acid compositions. J. Theor. Biol., 462, 221–229. [DOI] [PubMed] [Google Scholar]
- 30. Zuo Y.C., Chang Y., Huang S.H. et al. (2019) iDEF-PseRAAC: identifying the defensin peptide by using reduced amino acid composition descriptor. Evol Bioinform, 15, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zuo Y.C. and Li Q.Z. (2009) Using reduced amino acid composition to predict defensin family and subfamily: integrating similarity measure and structural alphabet. Peptides, 30, 1788–1793. [DOI] [PubMed] [Google Scholar]
- 32. Zuo Y.C. and Li Q.Z. (2010) Using K-minimum increment of diversity to predict secretory proteins of malaria parasite based on groupings of amino acids. Amino Acids, 38, 859–867. [DOI] [PubMed] [Google Scholar]
- 33. Chou K.C., Tomasselli A.G. and Heinrikson R.L. (2000) Prediction of the tertiary structure of a caspase-9/inhibitor complex. FEBS Lett., 470, 249–256. [DOI] [PubMed] [Google Scholar]
- 34. Chou K.C., Jones D. and Heinrikson R.L. (1997) Prediction of the tertiary structure and substrate binding site of caspase-8. FEBS Lett., 419, 49–54. [DOI] [PubMed] [Google Scholar]
- 35. Chou K.C. (2004) Insights from modelling the 3D structure of the extracellular domain of alpha7 nicotinic acetylcholine receptor. Biochem Biophys. Res. Commun., 319, 433–438. [DOI] [PubMed] [Google Scholar]
- 36. Chou K.C. (2005) Coupling interaction between thromboxane A2 receptor and alpha-13 subunit of guanine nucleotide-binding protein. J. Proteome Res., 4, 1681–1686. [DOI] [PubMed] [Google Scholar]
- 37. Chou K.C. and Howe W.J. (2002) Prediction of the tertiary structure of the beta-secretase zymogen. Biochem. Biophys. Res. Commun., 292, 702–708. [DOI] [PubMed] [Google Scholar]
- 38. Chou K.C. (2004) Insights from modeling the tertiary structure of human BACE2. J. Proteome Res., 3, 1069–1072. [DOI] [PubMed] [Google Scholar]
- 39. Chou K.C. (2004) Insights from modeling three-dimensional structures of the human potassium and sodium channels. J. Proteome Res., 3, 856–861. [DOI] [PubMed] [Google Scholar]
- 40. Chou K.C. (2005) Modeling the tertiary structure of human cathepsin-E. Biochem. Biophys. Res. Commun., 331, 56–60. [DOI] [PubMed] [Google Scholar]
- 41. Chou K.C. (2005) Insights from modeling the 3D structure of DNA-CBF3b complex. J. Proteome Res., 4, 1657–1660. [DOI] [PubMed] [Google Scholar]
- 42. Wang S.Q., Du Q.S. and Chou K.C. (2007) Study of drug resistance of chicken influenza A virus (H5N1) from homology-modeled 3D structures of neuraminidases. Biochem. Biophys. Res. Commun., 354, 634–640. [DOI] [PubMed] [Google Scholar]
- 43. Wang S.Q., Du Q.S., Huang R.B. et al. (2009) Insights from investigating the interaction of oseltamivir (Tamiflu) with neuraminidase of the 2009 H1N1 swine flu virus. Biochem. Biophys. Res. Commun., 386, 432–436. [DOI] [PubMed] [Google Scholar]
- 44. Li X.B., Wang S.Q., Xu W.R. et al. (2011) Novel inhibitor design for hemagglutinin against H1N1 influenza virus by core hopping method. PLoS One, 6, e28111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ma Y., Wang S.Q., Xu W.R. et al. (2012) Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PLoS One, 7, e38546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xie H.L., Fu L. and Nie X.D. (2013) Using ensemble SVM to identify human GPCRs N-linked glycosylation sites based on the general form of Chou's PseAAC. Protein Eng. Des. Sel., 26, 735–742. [DOI] [PubMed] [Google Scholar]
- 47. Xu Y., Ding J., Wu L.Y. et al. (2013) iSNO-PseAAC: predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition. PLoS One, 8, e55844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jia C., Lin X. and Wang Z. (2014) Prediction of protein S-nitrosylation sites based on adapted normal distribution bi-profile Bayes and Chou’s pseudo amino acid composition. Int. J. Mol. Sci., 15, 10410–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qiu W.R., Xiao X., Lin W.Z. et al. (2014) iMethyl-PseAAC: identification of protein methylation sites via a pseudo amino acid composition approach. Biomed. Res. Int., 2014, 947416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu Y., Wen X., Shao X.J. et al. (2014) iHyd-PseAAC: predicting hydroxyproline and hydroxylysine in proteins by incorporating dipeptide position-specific propensity into pseudo amino acid composition. Int. J. Mol. Sci., 15, 7594–7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu Y., Wen X., Wen L.S. et al. (2014) iNitro-Tyr: prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. PLoS One, 9, e105018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang J., Zhao X., Sun P. et al. (2014) PSNO: predicting cysteine S-nitrosylation sites by incorporating various sequence-derived features into the general form of Chou’s PseAAC. Int. J. Mol. Sci., 15, 11204–11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen W., Feng P., Ding H. et al. (2015) iRNA-methyl: identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem., 490, 26–33. [DOI] [PubMed] [Google Scholar]
- 54. Chou K.C. (2015) Impacts of bioinformatics to medicinal chemistry. Med. Chem., 11, 218–234. [DOI] [PubMed] [Google Scholar]
- 55. Qiu W.R., Xiao X., Lin W.Z. et al. (2015) iUbiq-Lys: prediction of lysine ubiquitination sites in proteins by extracting sequence evolution information via a gray system model. J. Biomol. Struct. Dyn., 33, 1731–1742. [DOI] [PubMed] [Google Scholar]
- 56. Chen W., Tang H., Ye J. et al. (2016) iRNA-PseU: Identifying RNA pseudouridine sites. Mol. Ther. Nucleic Acids, 5, e332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jia J., Liu Z., Xiao X. et al. (2016) iSuc-PseOpt: identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal. Biochem., 497, 48–56. [DOI] [PubMed] [Google Scholar]
- 58. Jia J., Liu Z., Xiao X. et al. (2016) pSuc-Lys: predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J. Theor. Biol., 394, 223–230. [DOI] [PubMed] [Google Scholar]
- 59. Jia J., Liu Z., Xiao X. et al. (2016) iCar-PseCp: identify carbonylation sites in proteins by Monte Carlo sampling and incorporating sequence coupled effects into general PseAAC. Oncotarget, 7, 34558–34570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jia J., Zhang L., Liu Z. et al. (2016) pSumo-CD: predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics, 32, 3133–3141. [DOI] [PubMed] [Google Scholar]
- 61. Ju Z., Cao J.Z. and Gu H. (2016) Predicting lysine phosphoglycerylation with fuzzy SVM by incorporating k-spaced amino acid pairs into Chous general PseAAC. J. Theor. Biol., 397, 145–150. [DOI] [PubMed] [Google Scholar]
- 62. Liu Z., Xiao X., Yu D.J. et al. (2016) pRNAm-PC: predicting N(6)-methyladenosine sites in RNA sequences via physical-chemical properties. Anal. Biochem., 497, 60–67. [DOI] [PubMed] [Google Scholar]
- 63. Qiu W.R., Sun B.Q., Xiao X. et al. (2016) iHyd-PseCp: identify hydroxyproline and hydroxylysine in proteins by incorporating sequence-coupled effects into general PseAAC. Oncotarget, 7, 44310–44321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Qiu W.R., Sun B.Q., Xiao X. et al. (2016) iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics, 32, 3116–3123. [DOI] [PubMed] [Google Scholar]
- 65. Qiu W.R., Xiao X., Xu Z.C. et al. (2016) iPhos-PseEn: identifying phosphorylation sites in proteins by fusing different pseudo components into an ensemble classifier. Oncotarget, 7, 51270–51283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu Y. and Chou K.C. (2016) Recent progress in predicting posttranslational modification sites in proteins. Curr. Top. Med. Chem., 16, 591–603. [DOI] [PubMed] [Google Scholar]
- 67. Feng P., Ding H., Yang H. et al. (2017) iRNA-PseColl: identifying the occurrence sites of different RNA modifications by incorporating collective effects of nucleotides into PseKNC. Mol. Ther. Nucleic Acids, 7, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ju Z. and He J.J. (2017) Prediction of lysine crotonylation sites by incorporating the composition of k-spaced amino acid pairs into Chou’s general PseAAC. J. Mol. Graph. Model., 77, 200–204. [DOI] [PubMed] [Google Scholar]
- 69. Liu L.M., Xu Y. and Chou K.C. (2017) iPGK-PseAAC: identify lysine phosphoglycerylation sites in proteins by incorporating four different tiers of amino acid pairwise coupling information into the general PseAAC. Med Chem, 13, 552–559. [DOI] [PubMed] [Google Scholar]
- 70. Qiu W.R., Jiang S.Y., Sun B.Q. et al. (2017) iRNA-2methyl: identify RNA 2'-O-methylation sites by incorporating sequence-coupled effects into general PseKNC and ensemble classifier. Med. Chem., 13, 734–743. [DOI] [PubMed] [Google Scholar]
- 71. Qiu W.R., Jiang S.Y., Xu Z.C. et al. (2017) iRNAm5C-PseDNC: identifying RNA 5-methylcytosine sites by incorporating physical-chemical properties into pseudo dinucleotide composition. Oncotarget, 8, 41178–41188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Qiu W.R., Sun B.Q., Xiao X. et al. (2017) iPhos-PseEvo: identifying human phosphorylated proteins by incorporating evolutionary information into general PseAAC via Grey system theory. Mol. Inform., 36, 1–9. [DOI] [PubMed] [Google Scholar]
- 73. Xu Y., Wang Z., Li C. et al. (2017) iPreny-PseAAC: identify C-terminal cysteine prenylation sites in proteins by incorporating two tiers of sequence couplings into PseAAC. Med. Chem., 13, 544–551. [DOI] [PubMed] [Google Scholar]
- 74. Akbar S. and Hayat M. (2018) iMethyl-STTNC: identification of N(6)-methyladenosine sites by extending the idea of SAAC into Chou’s PseAAC to formulate RNA sequences. J. Theor. Biol., 455, 205–211. [DOI] [PubMed] [Google Scholar]
- 75. Chandra A., Sharma A., Dehzangi A. et al. (2018) PhoglyStruct: prediction of phosphoglycerylated lysine residues using structural properties of amino acids. Sci. Rep., 8, 17923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen W., Ding H., Zhou X. et al. (2018) iRNA(m6A)-PseDNC: identifying N(6)-methyladenosine sites using pseudo dinucleotide composition. Anal. Biochem., 561–562, 59–65. [DOI] [PubMed] [Google Scholar]
- 77. Chen W., Feng P., Yang H. et al. (2018) iRNA-3typeA: identifying three types of modification at RNA's adenosine sites. Mol. Ther. Nucleic Acids, 11, 468–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ghauri A.W., Khan Y.D., Rasool N. et al. (2018) pNitro-Tyr-PseAAC: predict nitrotyrosine sites in proteins by incorporating five features into Chou’s general PseAAC. Curr. Pharm. Des., 24, 4034–4043. [DOI] [PubMed] [Google Scholar]
- 79. Ju Z. and Wang S.Y. (2018) Prediction of citrullination sites by incorporating k-spaced amino acid pairs into Chou’s general pseudo amino acid composition. Gene, 664, 78–83. [DOI] [PubMed] [Google Scholar]
- 80. Khan Y.D., Rasool N., Hussain W. et al. (2018) iPhosT-PseAAC: identify phosphothreonine sites by incorporating sequence statistical moments into PseAAC. Anal. Biochem., 550, 109–116. [DOI] [PubMed] [Google Scholar]
- 81. Khan Y.D., Rasool N., Hussain W. et al. (2018) iPhosY-PseAAC: identify phosphotyrosine sites by incorporating sequence statistical moments into PseAAC. Mol. Biol. Rep., 45, 2501–2509. [DOI] [PubMed] [Google Scholar]
- 82. Qiu W.R., Sun B.Q., Xiao X. et al. (2018) iKcr-PseEns: identify lysine crotonylation sites in histone proteins with pseudo components and ensemble classifier. Genomics, 110, 239–246. [DOI] [PubMed] [Google Scholar]
- 83. Sabooh M.F., Iqbal N., Khan M. et al. (2018) Identifying 5-methylcytosine sites in RNA sequence using composite encoding feature into Chou’s PseKNC. J. Theor. Biol., 452, 1–9. [DOI] [PubMed] [Google Scholar]
- 84. Hussain W., Khan Y.D., Rasool N. et al. (2019) SPalmitoylC-PseAAC: a sequence-based model developed via Chou’s 5-steps rule and general PseAAC for identifying S-palmitoylation sites in proteins. Anal. Biochem., 568, 14–23. [DOI] [PubMed] [Google Scholar]
- 85. Li F., Zhang Y., Purcell A.W. et al. (2019) Positive-unlabelled learning of glycosylation sites in the human proteome. BMC Bioinformatics, 20, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wang L., Zhang R. and Mu Y. (2019) Fu-SulfPred: identification of protein S-sulfenylation sites by fusing forests via Chou’s general PseAAC. J. Theor. Biol., 461, 51–58. [DOI] [PubMed] [Google Scholar]
- 87. Kumar V.S. and Vellaichamy A. (2019) Sequence and structure-based characterization of ubiquitination sites in human and yeast proteins using Chou’s sample formulation. Proteins, 87, 646–657. [DOI] [PubMed] [Google Scholar]
- 88. Xiao X., Min J.L., Lin W.Z. et al. (2015) iDrug-Target: predicting the interactions between drug compounds and target proteins in cellular networking via benchmark dataset optimization approach. J. Biomol. Struct. Dyn., 33, 2221–2233. [DOI] [PubMed] [Google Scholar]
- 89. Jia J., Liu Z., Xiao X. et al. (2015) iPPI-Esml: an ensemble classifier for identifying the interactions of proteins by incorporating their physicochemical properties and wavelet transforms into PseAAC. J. Theor. Biol., 377, 47–56. [DOI] [PubMed] [Google Scholar]
- 90. Liu Z., Xiao X., Qiu W.R. et al. (2015) iDNA-methyl: identifying DNA methylation sites via pseudo trinucleotide composition. Anal. Biochem., 474, 69–77. [DOI] [PubMed] [Google Scholar]
- 91. Chen W., Feng P.M., Lin H. et al. (2013) iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res., 41, e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lin H., Deng E.Z., Ding H. et al. (2014) iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res., 42, 12961–12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chou K.C. (2001) Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins, 43, 246–255. [DOI] [PubMed] [Google Scholar]
- 94. Silver D., Hubert T., Schrittwieser J. et al. (2018) A general reinforcement learning algorithm that masters chess, shogi, and go through self-play. Science, 362, 1140–1144. [DOI] [PubMed] [Google Scholar]
- 95. Long C.S., Li W., Liang P.F. et al. (2019) Transcriptome comparisons of multi-species identify differential genome activation of mammals embryogenesis. IEEE Access, 7, 7794–7802. [Google Scholar]
- 96. Hu B., Zheng L., Long C., et al. (2019) EmExplorer: a database for exploring time activation of gene expression in mammalian embryos. Open Biol., 9, 190054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Riddle D.S., Santiago J.V., Bray-Hall S.T. et al. (1997) Functional rapidly folding proteins from simplified amino acid sequences. Nat. Struct. Biol., 4, 805–809. [DOI] [PubMed] [Google Scholar]
- 98. Solis A.D. (2015) Amino acid alphabet reduction preserves fold information contained in contact interactions in proteins. Proteins, 83, 2198–2216. [DOI] [PubMed] [Google Scholar]
- 99. Liu D., Li G. and Zuo Y. (2018) Function determinants of TET proteins: the arrangements of sequence motifs with specific codes. Brief. Bioinform.. doi: 10.1093/bib/bby1053. [DOI] [PubMed] [Google Scholar]
- 100. Zhang C.T. and Chou K.C. (1992) An optimization approach to predicting protein structural class from amino acid composition. Protein Sci., 1, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chou K.C. and Elrod D.W. (2002) Bioinformatical analysis of G-protein-coupled receptors. J. Proteome Res., 1, 429–433. [DOI] [PubMed] [Google Scholar]
- 102. Chou K.C. and Cai Y.D. (2003) Prediction and classification of protein subcellular location-sequence-order effect and pseudo amino acid composition. J. Cell Biochem., 90, 1250–1260. [DOI] [PubMed] [Google Scholar]
- 103. Hu L., Huang T., Shi X. et al. (2011) Predicting functions of proteins in mouse based on weighted protein-protein interaction network and protein hybrid properties. PLoS One, 6, e14556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Cai Y.D., Feng K.Y., Lu W.C. et al. (2006) Using LogitBoost classifier to predict protein structural classes. J. Theor. Biol., 238, 172–176. [DOI] [PubMed] [Google Scholar]
- 105. Chen W., Lei T.Y., Jin D.C. et al. (2014) PseKNC: a flexible web server for generating pseudo K-tuple nucleotide composition. Anal. Biochem., 456, 53–60. [DOI] [PubMed] [Google Scholar]
- 106. Chou K.C. (2005) Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics, 21, 10–19. [DOI] [PubMed] [Google Scholar]
- 107. Dehzangi A., Heffernan R., Sharma A. et al. (2015) Gram-positive and gram-negative protein subcellular localization by incorporating evolutionary-based descriptors into Chous general PseAAC. J. Theor. Biol., 364, 284–294. [DOI] [PubMed] [Google Scholar]
- 108. Behbahani M., Mohabatkar H. and Nosrati M. (2016) Analysis and comparison of lignin peroxidases between fungi and bacteria using three different modes of Chou’s general pseudo amino acid composition. J. Theor. Biol., 411, 1–5. [DOI] [PubMed] [Google Scholar]
- 109. Kabir M. and Hayat M. (2016) iRSpot-GAEnsC: identifing recombination spots via ensemble classifier and extending the concept of Chou’s PseAAC to formulate DNA samples. Mol. Genet. Genomics, 291, 285–296. [DOI] [PubMed] [Google Scholar]
- 110. Meher P.K., Sahu T.K., Saini V. et al. (2017) Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci Rep, 7, 42362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yu B., Li S., Qiu W.Y. et al. (2017) Accurate prediction of subcellular location of apoptosis proteins combining Chou’s PseAAC and PsePSSM based on wavelet denoising. Oncotarget, 8, 107640-107665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ahmad J. and Hayat M. (2019) MFSC: multi-voting based feature selection for classification of Golgi proteins by adopting the general form of Chou’s PseAAC components. J. Theor. Biol., 463, 99–109. [DOI] [PubMed] [Google Scholar]
- 113. Contreras-Torres E. (2018) Predicting structural classes of proteins by incorporating their global and local physicochemical and conformational properties into general Chou’s PseAAC. J. Theor. Biol., 454, 139–145. [DOI] [PubMed] [Google Scholar]
- 114. Zhang S. and Liang Y. (2018) Predicting apoptosis protein subcellular localization by integrating auto-cross correlation and PSSM into Chou’s PseAAC. J. Theor. Biol., 457, 163–169. [DOI] [PubMed] [Google Scholar]
- 115. Tahir M., Hayat M. and Khan S.A. (2019) iNuc-ext-PseTNC: an efficient ensemble model for identification of nucleosome positioning by extending the concept of Chou’s PseAAC to pseudo-tri-nucleotide composition. Mol. Genet. Genomics, 294, 199–210. [DOI] [PubMed] [Google Scholar]
- 116. Chou K.C. (2017) An unprecedented revolution in medicinal chemistry driven by the progress of biological science. Curr. Top. Med. Chem, 17, 2337–2358. [DOI] [PubMed] [Google Scholar]
- 117. Shen H.B. and Chou K.C. (2008) PseAAC: a flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem., 373, 386–388. [DOI] [PubMed] [Google Scholar]
- 118. Du P., Wang X., Xu C. et al. (2012) PseAAC-Builder: a cross-platform stand-alone program for generating various special Chou’s pseudo-amino acid compositions. Anal. Biochem., 425, 117–119. [DOI] [PubMed] [Google Scholar]
- 119. Cao D.S., Xu Q.S. and Liang Y.Z. (2013) propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics, 29, 960–962. [DOI] [PubMed] [Google Scholar]
- 120. Du P., Gu S. and Jiao Y. (2014) PseAAC-General: fast building various modes of general form of Chou’s pseudo-amino acid composition for large-scale protein datasets. Int. J. Mol. Sci., 15, 3495–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Chou K.-C. (2009) Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr. Proteomics, 6, 262–274. [Google Scholar]
- 122. Chou K.C. (2011) Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol., 273, 236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Chen W., Lin H. and Chou K.C. (2015) Pseudo nucleotide composition or PseKNC: an effective formulation for analyzing genomic sequences. Mol. Biosyst., 11, 2620–2634. [DOI] [PubMed] [Google Scholar]
- 124. Liu B., Yang F., Huang D.S. et al. (2018) iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics, 34, 33–40. [DOI] [PubMed] [Google Scholar]
- 125. Tahir M., Tayara H. and Chong K.T. (2019) iRNA-PseKNC(2methyl): identify RNA 2'-O-methylation sites by convolution neural network and Chou’s pseudo components. J. Theor. Biol., 465, 1–6. [DOI] [PubMed] [Google Scholar]
- 126. Liu B., Liu F., Wang X. et al. (2015) Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res., 43, W65–W71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Chou K.-C. and Shen H.-B. (2009) Recent advances in developing web-servers for predicting protein attributes. Natural Science, 1, 63. [Google Scholar]
- 128. Chen W., Feng P., Yang H. et al. (2017) iRNA-AI: identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget, 8, 4208–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Cheng X., Xiao X. and Chou K.C. (2018) pLoc_bal-mPlant: predict subcellular localization of plant proteins by general PseAAC and balancing training dataset. Curr. Pharm. Des., 24, 4013–4022. [DOI] [PubMed] [Google Scholar]
- 130. Chou K.C., Cheng X. and Xiao X. (2019) pLoc_bal-mEuk: predict subcellular localization of eukaryotic proteins by general PseAAC and quasi-balancing training dataset. Med. Chem., 15, 472–485. [DOI] [PubMed] [Google Scholar]
- 131. Xiao X., Cheng X., Chen G. et al. (2019) pLoc_bal-mGpos: predict subcellular localization of gram-positive bacterial proteins by quasi-balancing training dataset and PseAAC. Genomics, 111, 886–892. [DOI] [PubMed] [Google Scholar]
- 132. Xiao X., Cheng X., Chen G. et al. (2019) pLoc_bal-mVirus: predict subcellular localization of multi-label virus proteins by Chou’s general PseAAC and IHTS treatment to balance training dataset. Med. Chem., 15, 496–509. [DOI] [PubMed] [Google Scholar]
- 133. Cheng X., Xiao X. and Chou K.-C. (2017) pLoc-mPlant: predict subcellular localization of multi-location plant proteins by incorporating the optimal GO information into general PseAAC. Mol. Biosyst., 13, 1722–1727. [DOI] [PubMed] [Google Scholar]
- 134. Cheng X., Xiao X. and Chou K.C. (2017) pLoc-mVirus: predict subcellular localization of multi-location virus proteins via incorporating the optimal GO information into general PseAAC. Gene, 628, 315–321. [DOI] [PubMed] [Google Scholar]
- 135. Cheng X., Xiao X. and Chou K.C. (2018) pLoc-mEuk: predict subcellular localization of multi-label eukaryotic proteins by extracting the key GO information into general PseAAC. Genomics, 110, 50–58. [DOI] [PubMed] [Google Scholar]
- 136. Cheng X., Xiao X. and Chou K.C. (2018) pLoc-mGneg: predict subcellular localization of gram-negative bacterial proteins by deep gene ontology learning via general PseAAC. Genomics, 110, 231–239. [DOI] [PubMed] [Google Scholar]
- 137. Cheng X., Zhao S.-G., Lin W.-Z. et al. (2017) pLoc-mAnimal: predict subcellular localization of animal proteins with both single and multiple sites. Bioinformatics, 33, 3524–3531. [DOI] [PubMed] [Google Scholar]
- 138. Xiao X., Cheng X., Su S. et al. (2017) pLoc-mGpos: incorporate key gene ontology information into general PseAAC for predicting subcellular localization of gram-positive bacterial proteins. Natural Science, 9, 330. [Google Scholar]
- 139. Cheng X., Xiao X. and Chou K.C. (2018) pLoc-mHum: predict subcellular localization of multi-location human proteins via general PseAAC to winnow out the crucial GO information. Bioinformatics, 34, 1448–1456. [DOI] [PubMed] [Google Scholar]
- 140. Cheng X., Xiao X. and Chou K.C. (2018) pLoc_bal-mGneg: predict subcellular localization of gram-negative bacterial proteins by quasi-balancing training dataset and general PseAAC. J Theor Biol, 458, 92–102. [DOI] [PubMed] [Google Scholar]
- 141. Chou K.C., Cheng X. and Xiao X. (2018) pLoc_bal-mHum: predict subcellular localization of human proteins by PseAAC and quasi-balancing training dataset. Genomics. doi: 10.1016/j.ygeno.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 142. Cheng X., Lin W.Z., Xiao X. et al. (2019) pLoc_bal-mAnimal: predict subcellular localization of animal proteins by balancing training dataset and PseAAC. Bioinformatics, 35, 398–406. [DOI] [PubMed] [Google Scholar]
- 143. Zuo Y.C., Peng Y., Liu L. et al. (2014) Predicting peroxidase subcellular location by hybridizing different descriptors of Chou’s pseudo amino acid patterns. Anal. Biochem., 458, 14–19. [DOI] [PubMed] [Google Scholar]
- 144. Zuo Y.C., Su W.X., Zhang S.H. et al. (2015) Discrimination of membrane transporter protein types using K-nearest neighbor method derived from the similarity distance of total diversity measure. Mol. Biosyst., 11, 950–957. [DOI] [PubMed] [Google Scholar]
- 145. Liu B., Liu F., Fang L. et al. (2015) repDNA: a Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics, 31, 1307–1309. [DOI] [PubMed] [Google Scholar]
- 146. Hashimoto H., Vertino P.M. and Cheng X. (2010) Molecular coupling of DNA methylation and histone methylation. Epigenomics, 2, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Melo F. and Marti-Renom M.A. (2006) Accuracy of sequence alignment and fold assessment using reduced amino acid alphabets. Proteins, 63, 986–995. [DOI] [PubMed] [Google Scholar]
- 148. Crooks G.E., Hon G., Chandonia J.M. et al. (2004) WebLogo: a sequence logo generator. Genome Res., 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Feng P.M., Chen W., Lin H. et al. (2013) iHSP-PseRAAAC: identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem., 442, 118–125. [DOI] [PubMed] [Google Scholar]
- 150. Chen W., Feng P.M., Deng E.Z. et al. (2014) iTIS-PseTNC: a sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal. Biochem., 462, 76–83. [DOI] [PubMed] [Google Scholar]
- 151. Ding H., Deng E.Z., Yuan L.F. et al. (2014) iCTX-type: a sequence-based predictor for identifying the types of conotoxins in targeting ion channels. Biomed. Res. Int., 2014, 286419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Liu B., Fang L., Wang S. et al. (2015) Identification of microRNA precursor with the degenerate K-tuple or Kmer strategy. J. Theor. Biol., 385, 153–159. [DOI] [PubMed] [Google Scholar]
- 153. Liu B., Fang L., Long R. et al. (2016) iEnhancer-2L: a two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition. Bioinformatics, 32, 362–369. [DOI] [PubMed] [Google Scholar]
- 154. Feng P., Yang H., Ding H. et al. (2019) iDNA6mA-PseKNC: identifying DNA N(6)-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics, 111, 96–102. [DOI] [PubMed] [Google Scholar]
- 155. Hussain W., Khan Y.D., Rasool N. et al. (2019) SPrenylC-PseAAC: a sequence-based model developed via Chou’s 5-steps rule and general PseAAC for identifying S-prenylation sites in proteins. J. Theor. Biol., 468, 1–11. [DOI] [PubMed] [Google Scholar]
- 156. Jia J., Li X., Qiu W. et al. (2019) iPPI-PseAAC (CGR): identify protein-protein interactions by incorporating chaos game representation into PseAAC. J. Theor. Biol., 460, 195–203. [DOI] [PubMed] [Google Scholar]
- 157. Khan Y.D., Jamil M., Hussain W. et al. (2019) pSSbond-PseAAC: prediction of disulfide bonding sites by integration of PseAAC and statistical moments. J. Theor. Biol., 463, 47–55. [DOI] [PubMed] [Google Scholar]
- 158. Lu Y., Wang S., Wang J. et al. (2019) An epidemic avian influenza prediction model based on Google trends. Lett. Org. Chem., 16, 303–310. [Google Scholar]
- 159. Khan Y.D., Batool A., Rasool N. et al. (2019) Prediction of nitrosocysteine sites using position and composition variant features. Lett. Org. Chem., 16, 283–293. [Google Scholar]
- 160. Li J.X., Wang S.Q., Du Q.S. et al. (2018) Simulated protein thermal detection (SPTD) for enzyme Thermostability study and an application example for Pullulanase from Bacillus deramificans. Curr. Pharm. Des., 24, 4023–4033. [DOI] [PubMed] [Google Scholar]
- 161. Chou K.C. (2019) Advance in predicting subcellular localization of multi-label proteins and its implication for developing multi-target drugs. Curr. Med. Chem. doi: 10.2174/0929867326666190507082559. [DOI] [PubMed] [Google Scholar]
- 162. Liu B., Xu J., Lan X. et al. (2014) iDNA-Prot|dis: identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition. PLoS One, 9, e106691. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.