

Summary
Neuroferritinopathy (NF) is a movement disorder caused by alterations in the L-ferritin gene that generate cytosolic free iron. NF is a unique pathophysiological model for determining the direct consequences of cell iron dysregulation. We established lines of induced pluripotent stem cells from fibroblasts from two NF patients and one isogenic control obtained by CRISPR/Cas9 technology. NF fibroblasts, neural progenitors, and neurons exhibited the presence of increased cytosolic iron, which was also detectable as: ferritin aggregates, alterations in the iron parameters, oxidative damage, and the onset of a senescence phenotype, particularly severe in the neurons. In this spontaneous senescence model, NF cells had impaired survival and died by ferroptosis. Thus, non-ferritin-bound iron is sufficient per se to cause both cell senescence and ferroptotic cell death in human fibroblasts and neurons. These results provide strong evidence supporting the primary role of iron in neuronal aging and degeneration.
Keywords: iron, induced pluripotent stem cells, senescence, ferroptosis, neurodegeneration, aging
Graphical Abstract
Highlights
-
•
NF is a unique pathophysiological model for analyzing cellular iron dysregulation
-
•
NF fibroblast/iPSC-derived NPCs and neurons show a phenotype of iron mobilization
-
•
Free iron is sufficient per se to cause both cell senescence and ferroptosis
-
•
Iron has a primary role in neuronal aging and degeneration
In this article Sonia Levi and colleagues show that non-ferritin-bound iron is able to cause cell senescence and ferroptotic cell death in human fibroblasts and neurons, underlining the primary role of iron in accelerating the processes of aging and neurodegeneration. These findings provide implications for studies investigating the role of iron in a more general context of neurodegenerative diseases.
Introduction
Neuroferritinopathy (NF) (OMIM, no. 606159) is a rare dominantly inherited late-onset monogenic disorder with 100% penetrance caused by mutations in the L-ferritin gene (FTL1) (Curtis et al., 2001). NF belongs to a group of monogenic extrapyramidal disorders called neurodegeneration with brain iron accumulation (NBIA) and is characterized by the focal accumulation of iron in the basal ganglia (Levi and Finazzi, 2014). The main common symptoms of NBIA include movement disorders, spasticity, and cognitive impairment; the age of onset varies among these diseases (Di Meo and Tiranti, 2018), averaging around 40 years. At present, 11 different causative mutations have been described worldwide, all of which are located in the last part of exon 4 in the FTL1 sequence (Levi and Rovida, 2015). These mutations affect both length and sequence of the C terminus peptide, disturbing the amino acid contacts involved in the shaping of the hydrophobic channels along the 4-fold axis of the molecule (Levi and Rovida, 2015). In humans, cytosolic ferritin is a heteropolymeric protein with a spherical shape obtained by the assembly of 24 structurally similar subunits of two different types, namely H and L, and encoded by two genes, FTH1 and FTL1, with iron stored in the internal cavity as ferrihydrite (Arosio and Levi, 2010). The overexpression of disease-associated gene variants in vitro (Cozzi et al., 2010) and in vivo (Maccarinelli et al., 2015, Vidal et al., 2008) revealed that the NF causative mutations act in a negative-dominant manner to impair the iron-storage function of ferritin, resulting in increased level of intracellular free iron (Cozzi et al., 2010, Luscieti et al., 2010). Emerging evidence supports the key role of iron in aging (Zecca et al., 2004) and neurodegeneration processes (Rouault, 2013), mainly because iron accumulates in the brain during aging (Ward et al., 2014) and its surplus renders cells more susceptible to oxidative stress (Koskenkorva-Frank et al., 2013). Thus, NF cellular models represent valuable tools for investigations of the controversial role of this metal in the cellular processes occurring during aging and neurodegeneration. However, the precise role of iron in the development of these two cellular processes is not completely elucidated, and its role in the neuronal compartment is particularly obscure due to the lack of faithful experimental models recapitulating spontaneous occurrence of these alterations.
Cellular senescence is normally induced in vitro by several stressful events (radiation, oxidants, and oncogenes) and in vivo by ablation of anti-senescent genes, such as p66 (Berry et al., 2008) and nuclear receptor co-activator 4 (NCOA4) (Bellelli et al., 2014). Ferroptosis is prevalently studied in cancer cell lines (Dixon et al., 2012), where it is revealed only after ferroptosis-inducing reagents (Xu et al., 2019).
The scarcity of human primary neuronal models to study the action of iron in aging and neurodegeneration stimulated us to develop a model characterized by the presence of excess free iron. We applied cellular reprogramming techniques (Orellana et al., 2016) to fibroblasts, obtaining induced pluripotent stem cell (iPSC)-derived neuronal precursor cells (NPCs) and neurons derived from two patients affected by NF, one isogenic control and three healthy subjects. A significant increase of cytosolic free iron content, alteration of iron homeostasis, DNA/protein/lipid oxidative damage, a clear senescence phenotype, and spontaneous death by ferroptosis were observed in NF fibroblasts, iPSC-derived NPCs, and neurons compared with controls.
These results, when interpreted in view of the pathogenetic mechanism of NF, confirm the detrimental effect of free iron in neuronal cells. In fact, in conditions such as NF in which iron is not safely removed from cytosol due to alterations of ferritin structure/function, it triggers a cascade of damaging events leading to senescence and ferroptosis, thereby accelerating the aging process.
Results
Development and Characterization of NF Fibroblasts and iPSC-Derived Neuronal Models
Fibroblasts were obtained from skin biopsies of two NF affected patients: one with heterozygous FTL1 469_484dup (Storti et al., 2013), and the other with heterozygous FTL1 351delG_InsTTT (hereafter referred to as NF1 and NF2, respectively) (Figure S1). Control fibroblasts from three healthy adult subjects were purchased from ATTC (hereafter referred to as Ctr1, Ctr2, and Ctr3). To develop a neuronal model we established multiple iPSC lines by reprogramming fibroblasts from all subjects as previously described (Orellana et al., 2016). Isogenic control cells were obtained by CRISPR/Cas9 technology on clone no. 7 of NF1-iPSC. We used one clone from each healthy subject (Ctr1 no. 203, Ctr2 no. 37, and Ctr3 no. 151), and three from each patient and isogenic control (NF1 no. 1, no. 7, and no. 8; NF2 no. 8, no. 11, and no. 12; and R-NF1 no. 38, no. 40, and no. 41). Characterization of the obtained clones of iPSCs, embryoid bodies (EBs), derived NPCs, and neurons are shown and described in Supplemental Information (Figures S2 and S3). Electrophysiological recordings in iPSC-derived neurons are reported in Figure S4. All clones were subjected to the same reported analysis, an example of which is provided in each panel.
NF Fibroblast/iPSC-Derived NPCs and Neurons Showed Cellular Iron Mobilization and Ferritin/Iron Aggregates
NF mutations caused structural modification in the ferritin E-helix, which is involved in the formation of the hydrophobic pores of the molecule (Figure S5A) (Cozzi et al., 2010, Levi and Rovida, 2015). In-silico-generated structural models of the ferritins containing the mutated subunits (Figures S5B, S5C, S1B, and S1C) showed the 4-fold channel dramatically altered, with a larger pore that likely impairs the retention of iron into the cavity. In the NF2 variant (Figure S1C), this effect is directly related to the premature stop site caused by the mutation, resulting in a shortened sequence and a smaller subunit lacking the E-helix and a part of the D-helix. In the NF1 variant (Figure S1B), this effect can be attributed to conformational changes that expose the E-helix to proteolytic cleavage, as confirmed experimentally by proteinase K digestion (Figure S1D).
To investigate the ferritin functionality, we treated the fibroblasts with 2 μCi 55Fe-ammonium citrate (FeAC) for 18 h. The incorporation of iron into ferritin was reduced to approximately 20% in the NF1 and NF2 fibroblasts (Figure S5D) and to 50% in the iPSC-derived neurons (Figure S5H) compared with controls, probably leaving a high amount of intracellular free iron. To confirm this effect, we explored the activity of the IRE/IRP machinery, which post-transcriptionally controls the expression of the major iron proteins (ferritin, transferrin receptor 1, and ferroportin) as a function of cytosolic iron concentration (Muckenthaler et al., 2008).
ELISA assay, specific for cytosolic ferritin-L (FtL) and ferritin-H (FtH), performed in fibroblasts, revealed similar contents of FtL and of FtH among the three controls; in patients the concentration of FtL was found to be similar to controls, while FtH was 2-fold higher (Table S1). Immunoblotting revealed that the expression of transferrin receptor 1 was decreased ∼2.5-fold in the NF fibroblasts and NPCs (Figures S5E and S5F) and was undetectable in neurons (Figure S5I); accordingly, ferroportin appeared unchanged in fibroblasts (not shown) and NPCs (Figure S5G), and showed a tendency to increase in neurons (Figure S5L). These data suggest that the reduced efficiency of iron incorporation of mutant ferritin results in an increased free cellular iron availability.
To stress this phenotype, the NF fibroblasts were treated with 0.1 mM ferric ammonium citrate (FAC) for 14 days and subjected to immunostaining using an antibody specific for H-ferritin. The higher number of ferritin-positive granules observed in the NF1 and NF2 fibroblasts in basal conditions (Figure 1A, upper panel) was further increased after treatment with iron (Figure 1A, lower panel). Analysis at ultrastructural level by electron microscopy (EM) and electron spectroscopy imaging (ESI) in patient cells showed that the granules were localized in the cytosol, mainly associated with lipid droplets and surrounded by bilayer membranous formations (Figure 1B), similarly to the ones detected in the NF disease mouse model (Maccarinelli et al., 2015). ESI defined iron as a component of these aggregates (Figure 1B), the number of which was ∼3-fold greater in NF than in control cells.
Figure 1.

Representative Images of NF Fibroblasts and iPSC-Derived Neurons Showed Formation of Ferritin/Iron Aggregates
(A) Control and variant fibroblasts untreated (UT) or treated (Fe) with 100 μM FeAC for 14 days and stained with an anti-human H-ferritin antibody (Hoechst staining to detect the nuclei). The arrows show the ferritin aggregates. Scale bars, 20 μm.
(B) Ultrastructural analysis of fibroblasts examined under an electron microscope (EM). The arrows indicate the aggregates. Cells untreated (UT) or treated (Fe) as described in (A) were subjected to an ESI analysis. The images showing the ultrastructural organization observed at 250 eV with a superimposed iron map represented by pseudo-colors. The iron granules were counted and are represented as a ratio to the total number in the counted fields (means ± SD of three independent experiments). The data were analyzed by unpaired, two-tailed t test, ∗∗∗p < 0.001.
(C) Immunofluorescence of β-III-tubulin (Tuj1) and H-ferritin (FtH) in neurons obtained from a control subject and patient NF1 (Hoechst staining for the nuclei), and co-treatment with the iron chelator (5 μM DFO) and antioxidant (100 μM NAC). Digital higher-magnification of images (dashed rectangles) is shown. Scale bars, 20 μm.
(D) Control and NF1 Perls staining under basal conditions and after treatment with 5 μM DFO and 100 μM NAC. The arrows show the presence of iron aggregates. Scale bars, 20 μm.
A similar phenotype was present in the NF iPSC-derived neurons. Immunofluorescence analysis on NF iPSC-derived neurons showed increased FtH, visualized as granular formations localized to the cytosolic part of the soma and along the axon (Figure 1C). These formations were also detectable by Perls staining (a specific dye for ferric iron deposits), suggesting the presence of a large amount of iron (Figure 1D). A 3-week treatment with the iron chelator deferoxamine (DFO) (5 μM) and the antioxidant n-acetyl-cysteine (NAC) (100 μM) reverted this phenotype (Figures 1C and 1D).
Cellular Iron Mobilization-Induced Oxidative Damage and Cellular Death in the NF Fibroblast/iPSC-Derived NPCs and Neurons
The free redox-active form of iron promotes the formation of free oxygen/nitrogen radicals, and thus increased cellular iron availability may affect cell resistance to oxidative stress. We searched for any sign of an altered cellular oxidative status under basal conditions and compared NF fibroblasts, iPSC-derived NPCs, and neurons with controls. For each cell type, we set up appropriate methods to reveal different types of oxidative damage products. The ROS levels were assayed by the specific fluorescent probe 2′,7′-dichlorofluorescein (DCF). Carbonylated proteins were assessed using an Oxy-Blot analysis, while lipid peroxidation was assessed using an Oxy-MDA assay (Cozzi et al., 2010) and C11-Bodipy (581/591) staining. A low but significant increase in the ROS levels was revealed in the NF fibroblasts (Figure 2A), which was corroborated by an enhancement of oxidized proteins in NF fibroblasts, iPSC-derived NPCs, and neurons (Figures 2B, 2D, and 2F). Lipid peroxidation was significantly increased only in the NF1, but not NF2, fibroblasts (Figure 2C); however, the phenotype was more severe in the NPCs and neurons, and the cells from both patients showed a statistically significant difference from the controls (Figures 2E and 2G). Furthermore, the reduced form of glutathione was significantly decreased in the NF iPSC-derived neurons (Figure 2H).
Figure 2.

Cellular Iron Mobilization-Induced Oxidative Damage in NF Fibroblast/iPSC-Derived NPCs and Neurons
(A) Fibroblasts were incubated with the ROS-sensitive 2′,7′-dichlorofluorescein. The results are presented as the mean of three independent experiments in octuplicate.
(B and C) Quantification by densitometry of Oxyblot (B) and lipid-malondialdehyde (MDA) peroxidation (C) of fibroblasts.
(D and E) Quantification of Oxyblot (D) and lipid-MDA peroxidation (E) of NPCs.
(F) Oxyblot of neurons.
(G) BODIPY of neurons.
(H) Glutathione (GSH)-sensitive ThiolTracker Violet fluorescence signal detected on Tuj1-positive human neurons.
The data are presented as the means ± SD (B–F) or ± SEM (G and H) of at least three independent experiments. All the data were analyzed by unpaired, two-tailed t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To investigate whether this altered oxidative status was sufficient to promote cellular death, we performed DAPI and trypan blue staining and LDH determination. No significant differences were detected between controls and NF fibroblasts (not shown), indicating that the NF fibroblasts did not die by apoptosis or necrosis. However, the MTT assay revealed a significantly decreased number of viable cells in the NF fibroblasts (NF1 ∼27%, NF2 ∼22%), which was more consistent in the iPSC-derived NPCs (NF1 ∼46%, NF2 ∼52%) and neurons (NF1 ∼52%, NF2 ∼51%) compared with the controls. To confirm the specific involvement of free iron in eliciting cell death, neurons were grown for 3 weeks in the presence of the iron chelator deferiprone (5 μM) (Pinto et al., 2018). The addition of deferiprone prevented the death of the NF-derived neurons (∼90% NF1 and ∼94% NF2 viable cells), demonstrating that iron is the leading cause of neuronal death.
Iron-Induced Senescence in NF Fibroblast/iPSC-Derived NPCs and Neurons
Patient fibroblasts showed a disorganized layer that did not reach cellular confluence in the plate/flask (Figure 3A, bright field), suggesting the presence of a senescence phenotype in these cells. We stained the cells to detect SA-β-galactosidase (SA-β-Gal) activity at pH 6. In control cells this activity was almost undetectable, while NF fibroblasts showed positive staining that boosted in intensity as the cell passages increased (not shown); in particular, at the 13th passage the SA-β-Gal-positive cells constituted ∼90% of all NF1 and 60% of all NF2 fibroblasts versus 10% of all controls (Figure 3A).
Figure 3.

Iron-Induced Senescence in NF Fibroblasts
(A and B) (A) Cells morphology (upper panel) and SA-β-Gal activity (lower panel) in bright field. Scale bars, 20 μm. (B) SA-β-Gal activity of NF1 cells treated with iron (Fe) and NAC. Scale bars, 20 μm. Positive stained cells in (A) and (B) were counted and plotted as a percentage of the total cells. The results are presented as the mean ± SD of five fields in three independent experiments.
(C) Ultrastructural analysis of fibroblasts examined by EM evidencing vacuoles (white arrows) and mitochondria (black arrows).
(D) Immunoblotting of LC3I and II expression in soluble fibroblast homogenates. Results are presented as the mean ± SD of three independent experiments.
(E) Levels of IL-1β and IL-6 determined by ELISAs on cell media. The results are presented as the mean ± SD of three independent experiments in octuplicate. The data were analyzed by unpaired, two-tailed t test, except for (B) in which one-way ANOVA were used, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To investigate whether the increased cytosolic free iron availability was the primary cause of the cellular senescence, we treated fibroblasts at an intermediate passage (i.e., the fifth passage) with 500 μM FAC for 24 h and analyzed their morphology and SA-β-Gal activity. The NF1 fibroblasts showed an evident change in morphology and a ∼30% increase of SA-β-Gal activity compared with the untreated cells, while control cells were almost unaffected (Figure 3B). When the iron treatment was performed in the presence of 500 μM NAC for 24 h, the cellular morphology and SA-β-Gal activity of the NF1 fibroblasts were similar to those of the untreated cells (Figure 3B), implying that the antioxidant treatment was able to counteract the effect of iron supplementation.
Ultrastructural analysis of NF fibroblasts showed vacuolar formations, similar to those already detected in senescent cells (Malatesta et al., 2011), which were almost undetectable in the control cells (Figure 3C, upper panel). EM analysis did not show alterations in the mitochondria ultrastructure in the NFs fibroblasts (Figure 3C, lower panel), suggesting that mitochondria function was not impaired in this cellular model. However, we cannot exclude mitochondrial impairment in neurons differentiated for longer time.
The NF fibroblasts showed a ∼3- to 4-fold increase in the LC3II protein compared with the controls (Figure 3D). In addition, the content of interleukin-1β (IL-1β) was significantly increased in both NF1 and NF2. The amount of IL-6 appeared extremely high only in NF1 (Figure 3E), while IL-8 and tumor necrosis factor alpha were unchanged. All these interleukins were undetectable in growth media from the iPSC-derived NPCs and neurons (not shown).
Interestingly, analysis of SA-β-Gal activity revealed a high number of positive cells in NF1 and NF2 iPSC-derived NPCs and neurons, reaching ∼70% and 20% in NF1 and NF2 NPCs, respectively (Figure 4A), and above 70% in neurons of both patients (Figure 4B). Moreover, the analysis of p62 expression (Masaldan et al., 2018) confirmed the increase of this peptide in neurons from both patients (Figure 4C), further proving the cellular senescence status.
Figure 4.

Iron-Induced Senescence in NF iPSC-Derived NPCs and Neurons
(A and B) (A) SA-β-Gal activity in NPCs. Scale bars, 20 μm. (B) SA-β-Gal activity in neurons. Scale bars, 20 μm. Positive stained cells in (A) and (B) were counted and plotted as a percentage of the total cells. The results are presented as the mean ± SD of five fields in three independent experiments.
(C) Immunoblotting of p62 expression in soluble neuron homogenates. Load represents Coomassie blue staining. Results are presented as the mean ± SD of three independent experiments. The data were analyzed by unpaired, two-tailed t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Iron-Dependent Senescence Phenotype Involves a NCOA4 Decrease and p53 Activation
NCOA4 is a mediator of ferritinophagy (Mancias et al., 2014) and is involved in senescence (Bellelli et al., 2014). We analyzed its expression in NF fibroblasts and NPCs to investigate the origin of the senescence phenotype. In the NF fibroblasts, the level of the NCOA4 protein was substantially undetectable (Figure 5A), although the level of NCOA4-mRNA was not significantly changed compared with the controls (Figure 5D). A similar result was obtained in the control cells exposed to 100 μM FeAC (Figure 5B), suggesting that the addition of iron to cells mimics the effect of the increased free cytosolic iron caused by ferritin dysfunction and is sufficient to stimulate NCOA4 degradation. The addition of the protease inhibitor leupeptin (50 μM) to inhibit the lysosomal enzymatic activity resulted in an increased amount of NCOA4 in the NF fibroblasts (Figure 5C).
Figure 5.

Iron-Dependent Senescence Phenotype Involves an NCOA4 Decrease and p53 Activation
(A–C) Immunoblotting of NCOA4 expression in soluble fibroblast homogenates, untreated (A), treated with iron (B), or leupeptin (C). The results are presented as the mean ± SD of three independent experiments.
(D) NCOA4 mRNA levels of fibroblasts determined by qRT-PCR and normalized to the mRNA expression of GAPDH. The data are presented as the mean ± SD of three independent experiments.
(E) Immunoblotting of NCOA4 expression in soluble NPC homogenates, ∗ unspecific band. The results are presented as the mean ± SD of three independent experiments.
(F) Immunofluorescence of p53 and NCOA4 in NPCs. The results are presented as the mean ± SD of three independent experiments. Scale bars, 20 μm.
(G and H) Immunoblotting of NCOA4 expression (G) and H2A.X (H) in neuron soluble homogenates, ∗ unspecific band.
(I) qRT-PCR analysis of SLC7A11 expression. The data are presented as the mean ± SD values of three independent experiments.
The data were analyzed by unpaired, two-tailed t test, ∗∗p < 0.01, ∗∗∗p < 0.001.
The iPSC-derived NPCs and neurons from the controls showed a low level of NCOA4 expression, which was further reduced in the patients' cells (Figures 5E–5G). One of the main roles of NCOA4 is to maintain DNA stability (Bellelli et al., 2014); thus, we investigated DNA damage by testing the presence of correlated post-translational histone modifications. Immunoblotting analysis showed a large increase in Ser139 phosphorylation on γ-H2A.X histone in the NF neurons compared with a negligible level in the control cells (Figure 5H), suggesting that the telomere uncapping starts to support the initiation of the dsDNA break response (d'Adda di Fagagna et al., 2003).
In addition, the immunofluorescence staining revealed that a massive decrease in NCOA4 was concurrent with the translocation of p53 to the nucleus in the NF-derived NPCs (Figure 5F). To determine whether p53 also acquires its transcriptional activity in iPSC-derived neurons, we evaluated the transcription of one of its target genes, i.e., SLC7A11, a key component of the cystine/glutamate antiporter. In the patient neurons, the amount of SLC7A11 mRNA was increased by ∼6-fold (Figure 5I). Overall, these data suggest that the NF fibroblasts, iPSC-derived NPCs, and neurons exhibited a consistent senescent phenotype in multiple aspects, and that this cellular status was associated with the activation of the p53-signalling pathway.
Iron Triggers Cell Death via Ferroptosis in iPSC-Derived Neurons
p53 has been described as an activator of ferroptosis, thus the p53-nuclear translocation is suggestive of a possible pathway of cellular death via ferroptosis (Jiang et al., 2015). iPSC-derived NPCs were incubated with 80 μM erastin for 18 h in the presence or not of 20 μM ferrostatin-1. Cell viability was then evaluated by an MTT assay (Figure 6A). The data suggested that erastin reduced the cell viability to 50% in the isogenic control and ∼25% in the NFs (Figure 6A). Ferrostatin-1 rescued the viability of the cells (Figure 6A), suggesting that ferroptosis is crucially involved in the cell death program. To confirm the hypothesis that cytosolic iron mobilization triggers ferroptosis, iPSC-derived NPCs and neurons were grown in the presence of ferrostatin-1 (2.6 μM for five passages and 1 μM for 3 weeks, respectively). Then, we performed the MTT analysis in the absence of erastin. A reduction in cell viability was observed in the NF cells compared with their isogenic counterparts and was comparable with the reduction observed in the erastin-treated isogenic control (Figures 6A and 6B). The addition of ferrostatin-1 did not affect the isogenic control cells but was able to significantly prevent cell death in the NF NPCs and neurons, indicating that cytosolic iron mobilization is sufficient to cause cell death via ferroptosis (Figures 6B and 6C).
Figure 6.

Iron Triggers Cell Death via Ferroptosis in iPSC-Derived NPCs and Neurons
(A) MTT assay of NPCs treated with/without 80 μM of erastin and 20 μM of ferrostatin-1 for 18 h.
(B) NPCs re-plated for five passages in the presence of 2.6 μM ferrostatin-1.
(C) Neurons treated with 1 μM ferrostatin-1 during differentiation.
(D) Scheme representing the percentage of senescent and ferroptotic cells in neurons.
All results are presented as a percentage of cell viability compared with that in the isogenic controls and as the mean ± SD of three independent experiments. The data were analyzed by unpaired, two-tailed t test, ∗∗p < 0.01, ∗∗∗p < 0.001.
Furthermore, from the above experiments that were performed in parallel starting from the same cell number we calculated the percentage of neurons that died by ferroptosis and those in senescence (Figure 6D). We found that the percentage of senescent cells was higher than the β-galactosidase-negative cells (73% versus 27% in NF1 and 67% versus 33% in NF2), while in ferroptotic cells we reported a lower percentage compared with the viable cells (43% versus 57% in NF1 and 42% versus 58% in NF2). Thus, the number of ferroptotic cells is higher than non-senescent (negative) cells (Figure 6D), indicating that the difference may be attributed to a portion of senescent cells dying by ferroptosis.
Discussion
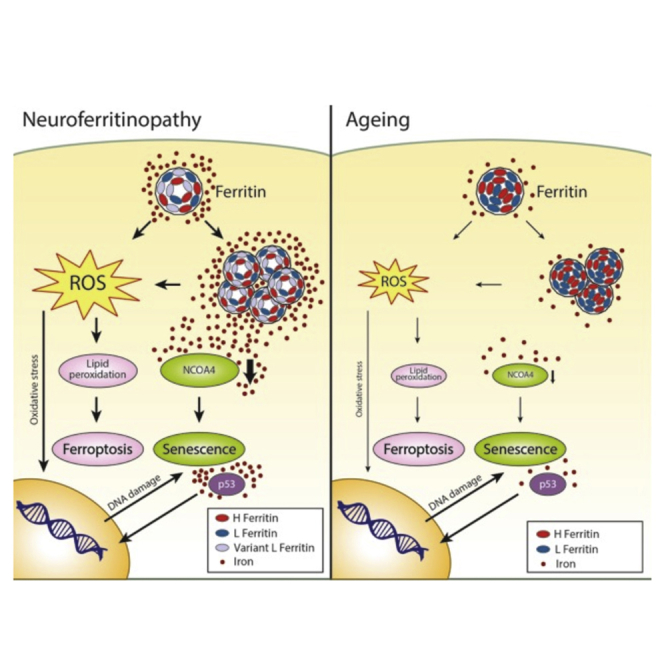
Using cell-reprogramming technology, we clarified the pathogenesis of NF in an in vitro human neuronal model and outlined the molecular mechanism by which iron plays a primary role in triggering the cascade of events that leads to neuronal death. The defective ferritin shell promotes redox-active iron excess in the cytosol, thereby inducing (1) the upregulation of iron-dependent ferritin translation, which generates the continuous overproduction of defective peptides and facilitates ferritin aggregation; (2) a decrease in NCOA4 that reduces ferritinophagy favors ferritin/iron aggregation, and implements DNA instability; and (3) an increase in ROS, causing oxidative injury followed by DNA damage and protein and lipid oxidation. As a consequence, the cells either activate pathways leading to senescence (possibly as a defensive mechanism) or, alternatively, have an increased tendency to die by ferroptosis (Figure 7).
Figure 7.

Scheme of the Proposed Molecular Mechanism Stimulated by Iron in NF and Aging
The insertion of the variant L-ferritin peptide into the protein shell causes the enhancement of redox-active iron in the cytosol, inducing iron-dependent ferritin translation that generates the overproduction of protein, facilitating its aggregation. Furthermore, an iron-dependent decrease in NCOA4 occurs that abolishes the physiological degradation of ferritin by the lysosome, further increasing ferritin/iron aggregation. The augmentation of ROS promotes cell oxidative injury with consequent DNA damage and protein and lipid oxidation. DNA instability is also enhanced by the diminished level of NCOA4. The cells seem to respond by triggering a p53-dependent pathway that further promotes senescence. However, the endless ROS-induced lipid peroxidation leads to death by ferroptosis. Although over a much longer time course, this mechanism might also occur physiologically during aging due to the ferritin/iron accumulation mainly in non-dividing cells.
The appearance of an evident senescence phenotype in the fibroblasts demonstrated that free iron alone may induce a cell-cycle arrest to counteract iron imbalance. This phenotype is even more pronounced in NF neurons, indicating that even quiescent cells can activate cell-cycle-associated events in response to DNA damage (Kruman et al., 2004). Thus, these results provide strong evidence supporting the direct involvement of iron in the aging process, especially in the neuronal compartment, as demonstrated by iron accumulation in the brain during both physiological and pathological aging (Ward et al., 2014). The precise mechanism of regulation of iron uptake into the brain is currently not fully defined; however, growing evidence suggests that, during aging, the blood-brain barrier becomes more permeable to iron, thus favoring its deposition (Marques et al., 2013). It is conceivable that, in neuronal cells, when accumulated iron exceeds the ability of ferritin to incorporate it, an iron-dependent detrimental process is triggered, leading to cell aging (Figure 7). Although with a premature onset and higher severity, a similar chain of events is initiated in the disease state as shown here in NF.
The molecular mechanism responsible for triggering senescence in these cellular models appeared to be mediated by the iron-dependent reduction of the ferritin-cargo NCOA4, which, in addition to failing to maintain ferritinophagy (Mancias et al., 2015), induced ferritin accumulation and facilitated DNA instability. In fact, NCOA4 also acts as a regulator of DNA stability as demonstrated in Ncoa4−/− MEFs in which Ncoa4 ablation caused aberrant DNA replication origin activation, generating replication stress and premature senescence (Bellelli et al., 2014). In addition, the iron-dependent development of ROS exacerbates DNA instability as confirmed by the increased Ser139 phosphorylation on the H2A.X histone (Burma et al., 2001). Cells appeared to respond to the insult by activating the p53-dependent anti-cellular stress pathway, as demonstrated by the p53-nuclear translocation and the increase in its transcriptional activity in neurons. This latter result seems contradictory because, recently, the tumor-suppression activity of p53 has been reported to be involved in the regulation of cystine metabolism, and p53 exerts its effect by repressing the expression of SLC7A11 and inducing ferroptosis (Jiang et al., 2015). However, previous studies investigating mouse models have suggested that iron excess might negatively regulate p53 signaling by both destabilizing the p53 protein and interfering with its DNA binding activity (Shen et al., 2014). Thus, we cannot exclude the possibility that p53 might progressively lose its ability to protect cells. Indeed, p53 accumulation has been detected in autoptic NF brain samples as a component of the protein aggregates that are pathognomonic of the disease (Curtis et al., 2001).
Remarkably, in our cellular models, ferroptosis can also occur in senescent cells. In fact, in the NF-derived NPCs and neurons, the development of senescence occurred in parallel with consistent cellular death, which was significantly reduced by ferrostatin-1, a widely used inhibitor of ferroptosis. Moreover, we can formulate some hypotheses about the fate of senescent cells among non-dividing cells, such as neurons. By simultaneously measuring senescence and ferroptosis in parallel experiments, we noticed that the percentage of senescent iPSC-derived neurons is higher than that in neurons that did not die by ferroptosis. This finding implies that at least some neurons showed a senescence phenotype before ferroptotic death. This intriguing result might be explained by considering that senescence and ferroptosis are not induced by other activating agents but instead develop consecutively over a relatively long period. Moreover, this study is the first work reporting a simultaneous analysis of senescence and ferroptosis in human neurons, highlighting a peculiar behavior of neuronal cells. It is well known that neuronal cells are more sensitive to iron toxicity than MEFs or cancer cell lines generally used in these experiments (Stockwell et al., 2017). Thus, the development of ferroptosis soon after senescence can be a peculiarity of this cell type. Alternatively, the continuous cellular deterioration due to the excess of cytoplasmic free iron counteracts the ability of neurons to maintain the senescence status, leading to premature death. Although the present in vitro experimental model does not include all cellular types that are involved in iron management in the nervous system, such as the glial components, we believe that it represents an innovative and valid approach to study the direct involvement of iron in neurodegenerative processes. In fact, even in this simplified model we have been able to detect iron accumulation in human NF neurons, similarly to that occurring in patients, and thus we show that iron triggers neuronal death via ferroptosis.
In summary, we demonstrated that non-ferritin-bound iron is able to cause both cell senescence and ferroptotic cell death in human fibroblasts and neurons, indicating a primary role of iron in accelerating the processes of aging and neurodegeneration. These data have also important implications for studies investigating the role of iron in a more general context of neurodegenerative diseases. The pathogenetic mechanism that we described in the setting of NF might be common in various neurodegenerative disorders, such as Alzheimer (Liu et al., 2018), Parkinson (McLeary et al., 2019), and Huntington diseases (Mi et al., 2019), amyotrophic lateral sclerosis (Yu et al., 2019), and frontotemporal lobar dementia (Zhang et al., 2017), in which dysfunction of the lysosome-autophagy pathway can lead to ferritin engulfment with consequent impairment of iron homeostasis (Biasiotto et al., 2016). Accordingly, growing evidence suggests that iron imbalance is a hallmark of neurodegenerative diseases, suggesting that iron chelators may be therapeutic options for preventing these diseases (Ward et al., 2014). Our data support this idea and suggest considering the monitoring of iron deposition in the brain during aging and carefully evaluating iron-supplemented diets in elderly people.
Experimental Procedures
The primary fibroblast cell lines were generated from skin biopsies from two patients, obtained with informed consent from the patients, and three healthy adult subjects purchased from ATCC.
Cases
The case of the patient carrying the FTL1 469_484dup mutation (named NF1) has been described previously (Storti et al., 2013).
The second patient carrying the heterozygous nucleotide insertion 351delG_InsTTT in FTL1 (named NF2) is a 58-year-old woman who presented with mild tremor in both hands at the age of 52 years. The patient did not exhibit alterations in muscular tone or an unsteady gait with broadened and multidirectional retropulsion after the pushing test without spontaneous rest. No optical alterations were observed. The neuropsychological evaluation showed moderate cognitive deterioration. These symptoms appeared within a few weeks with progressive worsening. No familiarity was present. The laboratory examinations showed normal vitamin B12 values and hepatic and renal function parameters. A low level of serum ferritin (1.1 ng/mL, normal range 15–150 ng/mL) and normal saturated transferrin and sideremia values were obtained. Brain MRI showed hyperintensity with surrounding hypointensity on T2∗-weighted imaging involving the pallidum, substantia nigra, and red nucleus. The clinical symptoms, radiological evidence, and low level of serum ferritin suggested an analysis of the FTL1 gene; the DNA sequence identified the abovementioned mutation associated with NF.
iPSC Generation
The fibroblasts were grown in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 100 mg/mL streptomycin, 100 U/mL penicillin and 2 mM L-glutamine (Sigma-Aldrich) and reprogrammed into iPSCs as in (Orellana et al., 2016) using CytoTune-iPS 2.0 Sendai reprogramming kit (Life Technologies) according to the manufacturer's instructions. Colonies started to appear 30 days later, and on approximately day 40, the cells were selected according to their morphology and transferred to a new feeder layer in DMEM/F12 (Sigma-Aldrich), 20% knockout serum replacement (Life Technologies), 1% penicillin/streptomycin (P/S), 2 mM L-glutamine, 1% non-essential amino acids MEM NEAA (Life Technologies), 1 mM NaPyr (Sigma-Aldrich), 0.1 mM β-mercaptoethanol (Life Technologies), and 10 ng/mL fibroblast growth factor 2 (FGF2) (Life Technologies). Subsequently, the iPSC clones were passed into feeder-free conditions on Matrigel hESC-qualified coated plates (Corning) while gradually increasing the percentage of Essential 8 medium (Life Technologies). The iPSCs were passaged every 5–7 days with ReLeSR (STEMCELL Technologies) on Matrigel-coated wells. The fibroblasts and iPSCs were periodically tested for mycoplasma contamination by PCR. To detect the presence of FTL1 mutations in the iPSCs clones, the genomic DNA was amplified by PCR, and the purified PCR products were analyzed by Sanger sequencing (GATC Biotech). The DNA sequences of the primers are listed in Table S2 (primers designated P1Fw and Re were used for the sequencing of the NF1 and R-NF1 clones, and P2 Fw and Re were used for the sequencing of the NF2 clones). In vitro three germ layer differentiation and karyotype analysis were performed as in Orellana et al. (2016).
Generation of R-NF1 Clones
We performed a gene knockin of the FTL1 WT allele in the NF1 iPSCs by combining CRISPR/Cas9 with a single-stranded oligodeoxynucleotide donor template and an sgRNA directed specifically to the 469_484dup mutation as listed in Table S2. The gene correction was corroborated by a sequence analysis of the iPSC clones. The potential off-target sites were selected according to http://crispr.mit.edu. We analyzed the seven most likely off-target hits present in the gene coding regions as described previously (Rubio et al., 2016). These sites were amplified by genomic PCR using the following primers: DPF1 Fw, DPF1 Re, ACADS Fw, ACADS Re, TUBB4 Fw, TUBB4 Re, SUZ12 Fw, SUZ12 Re, GPR150 Fw, GPR150 Re, FTL 2 Fw, FTL 2 Re, METTL25 Fw, and METTL25 Re (Table S2). Then, we performed Sanger sequencing.
Generation of Human NPCs and Neurons
The NPCs and EBs were formed by the dissociation of the iPSC colonies following the procedure described in (Orellana et al., 2016). To obtain the rosettes, the EBs were plated onto Matrigel growth factor reduced (Corning)-coated dishes in DMEM/F12 (Sigma-Aldrich) plus N2 (1:200), 1% NEAA (Life Technologies), and 1% P/S (Marchetto et al., 2010). After 10 days, the rosettes were dissociated with accutase and plated again onto Matrigel-coated dishes with the NPC medium DMEM/F12, N2 (1:200), B27 (1:100, Life Technologies), 1% P/S, and FGF2 (20 ng/mL). Homogeneous NPC populations were achieved after 3–5 passages with accutase under the same conditions. The neurons were obtained as described previously (Orellana et al., 2016). The NPCs were transduced with a lentivirus-expressing Ngn2 cDNA under the control of the tetracycline-responsive promoter TetO-Ngn2-t2a-Puro (kindly provided by T.C. Sudhof) and a lentivirus-expressing rtTA. The lentiviruses were produced as described previously (Indrigo et al., 2010). The NPCs were seeded on Matrigel-coated wells and differentiated in medium containing Neurobasal (Life Technologies), BDNF (10 ng/mL, Peprotech), NT-3 (10 ng/mL, Peprotech), B27, P/S, glutamine, and doxycycline (2 μg/mL, Sigma-Aldrich). NT-3 was present in the medium only during the first week of differentiation, and doxycycline was present only for 3 weeks. Half of the medium was changed every 2–3 days.
Electrophysiology
Whole-cell patch-clamp recordings were performed after 45 days of differentiation. Cells were continuously perfused with standard artificial cerebrospinal fluid containing the following (in mM): 125 NaCl, 2.5 KCl, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, and 11 glucose bubbled with 95% O2 and 5% CO2 at pH 7.3 at room temperature. Glass pipettes were filled with a solution containing the following (in mM): 124 KH2PO4, 2 MgCl2, 10 NaCl, 10 HEPES, 0.5 EGTA, 2 Na2-ATP, and 0.02 Na-GTP (pH 7.2, adjusted with KOH; tip resistance, 5–6 MΩ). Action potential firing was evoked in current clamp mode by current injection steps (10–50 pA, 500 ms). Traces were recorded using a digital amplifier (Multiclamp 700B, Molecular Devices) interfaced with a PC through an analog-to-digital board (Digidata 1440A, Molecular Devices). Signals were acquired and analyzed using pClamp software (Molecular Devices). All reagents were supplied by Sigma-Aldrich.
Iron Parameter Analysis and Immunofluorescence
Cellular 55Fe incorporation, EM, ESI, ferritin quantification, iron-protein expression, analysis of cellular oxidative status, and immunofluorescence staining were described previously (Cozzi et al., 2010, Maccarinelli et al., 2015, Orellana et al., 2016). The used antibodies were listed in Table S3.
Cell Viability Assay
In total, 2 × 104 cells grown at 37°C in the appropriate medium were treated with or without deferiprone, erastin, or ferrostatin at the concentrations and times indicated in the figure legends and finally incubated with 10 μL MTT solution (5 mg/mL in phosphate-buffered saline) for 2 h at 37°C. The color absorbance was read at 570 nm following the manufacturer's instructions (Sigma-Aldrich) (Cozzi et al., 2010).
SA-β-Gal Activity Detection
The cells were analyzed according to the protocol “Senescence Cells Histochemical Staining” (Sigma). This assay is based on a histochemical stain for β-galactosidase activity at pH 6. In brief, the cells were fixed with a solution containing 20% formaldehyde and 2% glutaraldehyde, stained with a solution of X-gal, and incubated at 37°C for 18 h. The cells were also treated with 0.5 mM FAC, 0.5 mM NAC, and 0.1 mM DFO for 18 h, and β-galactosidase activity was analyzed as described above. The cells were observed under bright light; the blue-stained cells and total number of cells were counted, and the percentage of cells expressing β-galactosidase was calculated.
qRT-PCR Analysis
RNA was extracted using an RNeasy kit (QIAGEN) and then retrotranscribed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). For the real-time qPCR analysis, HOT FIREPol EvaGreen qPCR Mix Plus (Solis Biodyne) was used, and the expression levels were normalized to GAPDH expression. The primers specific for SLC7A11 and NCOA4 are listed in Table S2.
Computational Analysis
The secondary and tertiary structure information of wild-type ferritin was obtained from X-ray coordinates of the human ferritin L chain (PDB: 2FFX). For the mutants, the secondary structure prediction was performed with the online tool “Psi-pred” (http://bioinf.cs.ucl.ac.uk/psipred/), while the 3D structure was modeled with a threading approach using the iTasser server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). The electrostatic potential was calculated with Delphi software package using the default parameters (Li et al., 2012). The surface representation of the electrostatic potential was obtained with PyMol Molecular Graphics System (Schrödinger), which was also used for all molecular visualizations.
Statistical Analyses
Statistical methods were not employed to predetermine sample size in the in vitro experiments. All the experiments were performed at least in triplicate; data were analyzed using GraphPad Prism. In general, for normally distributed data two-tailed unpaired Student's t test and one-way ANOVA followed by Bonferroni post-test was used. Data are reported as means ± SD or ± SEM. A p value < 0.05 was considered statistically significant.
Author Contributions
A.C., D.I.O., A.R., and C.C. developed the neuronal models and maintained iPSCs. A.C. and D.I.O. performed in vitro analysis. P.S. performed biochemical analysis. M.R. performed electrophysiological recordings. S.G. performed the CRISPR/Cas9 gene correction. E.R. performed molecular modeling. G.D.L. measured interleukin levels. M.F. provided the genetic analysis of iPSCs. G.L.F. and C.F. provided fibroblasts from patients. S.T. analyzed electrophysiological data and wrote the manuscript. V.B. supervised iPSC and gene editing work and collaborate to write the manuscript. S.L. conceived the study and wrote the manuscript. All authors have given their approval of the final version of the manuscript.
Acknowledgments
A part of this work was carried out at ALEMBIC, which is an advanced microscopy laboratory established by the San Raffaele Scientific Institute and the Vita-Salute San Raffaele University. The authors are grateful to Filippo Santorelli and Alessandra Tessa, Molecular Medicine Lab, IRCCS Stella Maris, for caring for the patients and providing the skin biopsies, and to Eufemia Putortì for the graphical assistance in drawing the scheme showed in Figure 7. Deferiprone was kindly provided by Dr. Fernando Tricta, ApoPharma, Toronto, Canada. The financial support from Telethon-Italia (grant nos. GGP10099, GGP11088, and GGP16234 to S.L.), AISNAF (to S.L.), and the European Research Council (AdERC no. 340527 to V.B.) is gratefully acknowledged.
Published: October 3, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.09.002.
Supplemental Information
References
- Arosio P., Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta. 2010;1800:783–792. doi: 10.1016/j.bbagen.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Bellelli R., Castellone M.D., Guida T., Limongello R., Dathan N.A., Merolla F., Cirafici A.M., Affuso A., Masai H., Costanzo V. NCOA4 transcriptional coactivator inhibits activation of DNA replication origins. Mol. Cell. 2014;55:123–137. doi: 10.1016/j.molcel.2014.04.031. [DOI] [PubMed] [Google Scholar]
- Berry A., Greco A., Giorgio M., Pelicci P.G., de Kloet R., Alleva E., Minghetti L., Cirulli F. Deletion of the lifespan determinant p66(Shc) improves performance in a spatial memory task, decreases levels of oxidative stress markers in the hippocampus and increases levels of the neurotrophin BDNF in adult mice. Exp. Gerontol. 2008;43:200–208. doi: 10.1016/j.exger.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Biasiotto G., Di Lorenzo D., Archetti S., Zanella I. Iron and neurodegeneration: is ferritinophagy the link? Mol. Neurobiol. 2016;53:5542–5574. doi: 10.1007/s12035-015-9473-y. [DOI] [PubMed] [Google Scholar]
- Burma S., Chen B.P., Murphy M., Kurimasa A., Chen D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- Cozzi A., Rovelli E., Frizzale G., Campanella A., Amendola M., Arosio P., Levi S. Oxidative stress and cell death in cells expressing L-ferritin variants causing neuroferritinopathy. Neurobiol. Dis. 2010;37:77–85. doi: 10.1016/j.nbd.2009.09.009. [DOI] [PubMed] [Google Scholar]
- Curtis A.R., Fey C., Morris C.M., Bindoff L.A., Ince P.G., Chinnery P.F., Coulthard A., Jackson M.J., Jackson A.P., McHale D.P. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat. Genet. 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna F., Reaper P.M., Clay-Farrace L., Fiegle R.H., Carr P., Von Zglinicki T., Saretzki G., Carter N.P., Jackson S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- Di Meo I., Tiranti V. Classification and molecular pathogenesis of NBIA syndromes. Eur. J. Paediatr. Neurol. 2018;22:272–284. doi: 10.1016/j.ejpn.2018.01.008. [DOI] [PubMed] [Google Scholar]
- Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indrigo M., Papale A., Orellana D., Brambilla R. Lentiviral vectors to study the differential function of ERK1 and ERK2 MAP kinases. Methods Mol. Biol. 2010;661:205–220. doi: 10.1007/978-1-60761-795-2_12. [DOI] [PubMed] [Google Scholar]
- Jiang L., Kon N., Li T., Wang S.J., Su T., Hibshoosh H., Baer R., Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskenkorva-Frank T.S., Weiss G., Koppenol W.H., Burckhardt S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013;65:1174–1194. doi: 10.1016/j.freeradbiomed.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Kruman I.I., Wersto R.P., Cardozo-Pelaez F., Smilenov L., Chan S.L., Chrest F.J., Emokpae R., Jr., Gorospe M., Mattson M.P. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 2004;41:549–561. doi: 10.1016/s0896-6273(04)00017-0. [DOI] [PubMed] [Google Scholar]
- Levi S., Finazzi D. Neurodegeneration with brain iron accumulation: update on pathogenic mechanisms. Front. Pharmacol. 2014;5:99. doi: 10.3389/fphar.2014.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi S., Rovida E. Neuroferritinopathy: from ferritin structure modification to pathogenetic mechanism. Neurobiol. Dis. 2015;81:134–143. doi: 10.1016/j.nbd.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Li C., Sarkar S., Zhang J., Witham S., Zhang Z., Wang L., Smith N., Petukh M., Alexov E. DelPhi: a comprehensive suite for DelPhi software and associated resources. BMC Biophys. 2012;5:9. doi: 10.1186/2046-1682-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.L., Fan Y.G., Yang Z.S., Wang Z.Y., Guo C. Iron and Alzheimer's disease: from pathogenesis to therapeutic implications. Front. Neurosci. 2018;12:632. doi: 10.3389/fnins.2018.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscieti S., Santambrogio P., Langlois d'Estaintot B., Granier T., Cozzi A., Poli M., Gallois B., Finazzi D., Cattaneo A., Levi S. Mutant ferritin L-chains that cause neurodegeneration act in a dominant-negative manner to reduce ferritin iron incorporation. J. Biol. Chem. 2010;285:11948–11957. doi: 10.1074/jbc.M109.096404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarinelli F., Pagani A., Cozzi A., Codazzi F., Di Giacomo G., Capoccia S., Rapino S., Finazzi D., Politi L.S., Cirulli F. A novel neuroferritinopathy mouse model (FTL 498InsTC) shows progressive brain iron dysregulation, morphological signs of early neurodegeneration and motor coordination deficits. Neurobiol. Dis. 2015;81:119–133. doi: 10.1016/j.nbd.2014.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malatesta M., Giagnacovo M., Renna L.V., Cardani R., Meola G., Pellicciari C. Cultured myoblasts from patients affected by myotonic dystrophy type 2 exhibit senescence-related features: ultrastructural evidence. Eur. J. Histochem. 2011;55:e26. doi: 10.4081/ejh.2011.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias J.D., Pontano Vaites L., Nissim S., Biancu R.D.E., Kim A.J., Wang X., Liu Y., Goessling W., Kimmelman A.C., Harpe R.J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife. 2015;4:e10308. doi: 10.7554/eLife.10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias J.D., Wang X., Gygi S.P., Harper J.W., Kimmelman A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto M.C., Carromeu C., Acab A., Yu D., Yeo G.W., Mu Y., Chen G., Gage F.H., Muotri A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques F., Sousa J.C., Sousa N., Palha J.A. Blood-brain-barriers in aging and in Alzheimer's disease. Mol. Neurodegener. 2013;8:38. doi: 10.1186/1750-1326-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaldan S., Clatworthy S.A.S., Gamell C., Meggyesy P.M., Rigopoulos A.T., Haupt S., Haupt Y., Denoyer D., Adlard P.A., Bush A.I. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018;14:100–115. doi: 10.1016/j.redox.2017.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeary F.A., Rcom-H'cheo-Gauthier A.N., Goulding M., Radford R.A.W., Okita Y., Faller P., Chung R.S., Pountney D.L. Switching on endogenous binding proteins in Parkinsons's disease cell. Cells. 2019;8:179. doi: 10.3390/cells8020179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi Y., Gao X., Xu H., Cui Y., Zhang Y., Gou X. The emerging roles of ferroptosis in Huntington's disease. Neuromolecular. Med. 2019;21:110–111. doi: 10.1007/s12017-018-8518-6. [DOI] [PubMed] [Google Scholar]
- Muckenthaler M.U., Galy B., Hentze M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [DOI] [PubMed] [Google Scholar]
- Orellana D.I., Santambrogio P., Rubio A., Yekhlef L., Cancellieri C., Dusi S., Giannelli S.G., Venco P., Mazzara P.G., Cozzi A. Coenzyme A corrects pathological defects in human neurons of PANK2-associated neurodegeneration. EMBO Mol. Med. 2016;8:1197–1211. doi: 10.15252/emmm.201606391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto V.M., Balocco M., Quintino S., Bacigalupo L., Gianesin B., Rizzi M., Malago R., De Franceschi L., Forni G.L. Daily alternating deferasirox and deferiprone therapy successfully controls iron accumulation in untreatable transfusion-dependent thalassemia patients. Am. J. Hematol. 2018;93:E79–E82. doi: 10.1002/ajh.25222. [DOI] [PubMed] [Google Scholar]
- Rouault T.A. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat. Rev. Neurosci. 2013;14:551–564. doi: 10.1038/nrn3453. [DOI] [PubMed] [Google Scholar]
- Rubio A., Luoni M., Giannelli S.G., Radice I., Iannielli A., Cancellieri C., Di Berardino C., Regalia G., Lazzari G., Menegon A. Rapid and efficient CRISPR/Cas9 gene inactivation in human neurons during human pluripotent stem cell differentiation and direct reprogramming. Sci. Rep. 2016;6:37540. doi: 10.1038/srep37540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Zhang S., Huang X., Chen K., Shen J., Wang Z. Involvement of p53 mutation and mismatch repair proteins dysregulation in NNK-induced malignant transformation of human bronchial epithelial cells. Biomed. Res. Int. 2014;2014:920275. doi: 10.1155/2014/920275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell B.R., Friedmann Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J., Fulda S., Gascon S., Hatzios S.K., Kagan V.E. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storti E., Cortese F., Di Fabio R., Fiorillo C., Pierallini A., Tessa A., Valleriani A., Pierelli F., Santorelli F.M., Casali C. De novo FTL mutation: a clinical, neuroimaging, and molecular study. Mov. Disord. 2013;28:252–253. doi: 10.1002/mds.25275. [DOI] [PubMed] [Google Scholar]
- Vidal R., Miravalle L., Gao X., Barbeito A.G., Baraibar M.A., Hekmatyar S.K., Widel M., Bansal N., Delisle M.B., Ghetti B. Expression of a mutant form of the ferritin light chain gene induces neurodegeneration and iron overload in transgenic mice. J. Neurosci. 2008;28:60–67. doi: 10.1523/JNEUROSCI.3962-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward R.J., Zucca F.A., Duyn J.H., Crichton R.R., Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014;13:1045–1060. doi: 10.1016/S1474-4422(14)70117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T., Ding W., Ji X., Ao X., Liu Y., Yu W., Wang J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell Mol. Med. 2019;23:4900–4912. doi: 10.1111/jcmm.14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Wang N., Qi F., Wang X., Zhu Q., Lu Y., Zhang H., Che F., Li W. Serum ferritin is a candidate biomarker of disease aggravation in amyotrophic lateral sclerosis. Biomed. Rep. 2019;9:333–338. doi: 10.3892/br.2018.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L., Youdim M.B., Riederer P., Connor J.R., Crichton R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Schmid B., Nikolaisen N.K., Rasmussen M.A., Aldana B.I., Agger M., Calloe K., Stummann T.C., Larsen H.M., Nielsen T.T. Patient iPSC-derived neurons for disease modeling of frontotemporal dementia with mutation in CHMP2B. Stem Cell Reports. 2017;14:648–658. doi: 10.1016/j.stemcr.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.