

Abstract
Background:
Wnt signaling is a cellular pathway involved in embryogenesis, development, and neoplasia. Wnt-pathway activation may accelerate prostate cancer androgen-independent growth and mediate antiandrogen resistance. Since 10–20% of advanced prostate cancers harbor Wnt-activating mutations, we aimed to characterize the clinical features and response to novel antiandrogens in such patients.
Objective:
To determine whether men with metastatic castration-resistant prostate cancer (mCRPC) who harbor Wnt-pathway mutations have poorer responses to first-line novel hormonal therapies: abiraterone/enzalutamide.
Design, setting, and participants:
Patients with mCRPC who received first-line abiraterone or enzalutamide were retrospectively evaluated. Using tumor DNA analyses, we queried for activating mutations in CTNNB1 or inactivating mutations in APC or RNF43, all of which are predicted to stimulate Wnt signaling. Presence or absence of at least one Wnt-activating alteration was correlated with clinical-pathologic characteristics and treatment outcomes.
Outcome measurements and statistical analysis:
Time to prostate-specific antigen (PSA) progression, overall survival (OS), and PSA response were measured. Cox regression models were used to test associations between Wnt status and clinical-pathologic outcomes; Kaplan-Meier and log-rank analyses were used to compare time-to-event data.
Results and limitations:
Of 137 patients evaluated, 11% (n = 15) had tumor DNA analysis showing at least one Wnt-stimulating alteration. Patients with Wnt-activating mutations had numerically fewer T3/T4 tumors than Wnt wild-type patients (31% vs 51%), but were otherwise generally balanced. Median time to PSA progression on first-line abiraterone/enzalutamide was shorter in Wnt-activated patients (6.5 vs 9.6 mo, hazard ratio [HR] 2.34, p = 0.003), as was OS (23.6 vs 27.7 mo, HR 2.28, p = 0.01). PSA responses were numerically worse in Wnt-activated patients (53% vs 75%, p = 0.12). Presence of Wnt-activating alterations (adjusted HR [aHR] 2.33, p = 0.007) and use of previous chemotherapy (aHR 1.83, p = 0.003) were both independently associated with increased hazard of progression.
Conclusions:
Patients with somatic Wnt-pathway activating mutations have worse outcomes to first-line abiraterone/enzalutamide than Wnt wild-type patients. Our data suggest that additional genomically informed therapies are needed for this relevant subset of mCRPC patients.
Patient summary:
In this report, we retrospectively examined outcomes of metastatic prostate cancer patients with or without Wnt-pathway mutations who received abiraterone or enzalutamide for the first time, in order to examine whether these mutations affect the prognosis. Our study suggested that patients who have Wnt-pathway activating mutations derived less benefit from abiraterone and enzalutamide when compared with patients without these mutations. We conclude that Wnt-pathway mutations might decrease the effectiveness of abiraterone and enzalutamide, and we propose that the Wnt pathway might be a good therapeutic target for these patients, in order to potentially reverse or prolong resistance to abiraterone and enzalutamide in men with Wnt mutations.
Keywords: Wnt, Prostate, Cancer, Antiandrogens, Abiraterone, Enzalutamide
Patients with metastatic castration-resistant prostate cancer who harbor Wnt-pathway–activating mutations have inferior outcomes with first-line abiraterone and enzalutamide when compared with patients without these abnormalities. Prospective validation of this finding is needed.
1. Introduction
Prostate cancer has an intrinsic dependency on androgens and androgen receptor (AR) signaling, with androgen deprivation therapy (ADT) being the backbone of systemic treatment of metastatic disease [1]. Unfortunately, almost all patients invariably develop hormonal resistance over time, progressing to a state called metastatic castration-resistant prostate cancer (mCRPC) [2]. The most common mechanisms of escape include amplification and/or mutation of the AR [3], expression of constitutively active AR splice variants [4], increased production of intracrine androgens, and changes in the activity or expression levels of AR coactivators and corepressors [3]. In this context, novel antiandrogens such as abiraterone [5,6] and enzalutamide [7,8] provide clinical and survival benefits, suggesting that mCRPC often remains addicted to androgens and AR signaling [2]. However, in addition to these AR-dependent mechanisms of escape, additional AR-independent mechanisms are involved in the emergence of castration resistance, as well as innate or acquired antiandrogen resistance; one of these relevant players is the Wnt signaling pathway [9,10].
Wnt signaling pathways are a group of three major signal transduction pathways that are physiologically responsible for many functions including cell growth, organ formation, stem cell renewal, cell-cycle progression, and survival [11,12]. The “canonical” Wnt pathway primarily comprises the cytoplasmatic protein β-catenin that is negatively regulated by the tumor suppressor protein APC. The “noncanonical” Wnt pathways include the complexes of planar cell polarity and Wnt calcium (Wnt/Ca2+), which are responsible for morphogenetic cell movements [11,13]. Mutated Wnt-pathway genes cause multiple growth-related pathologies [14] and are implicated in the development, progression, and mechanisms of resistance of multiple nonprostatic cancers, including colorectal [15,16], breast [17], lung [18], and hematopoietic malignancies [19].
Somatic mutations in genes that regulate the Wnt signaling pathway, including activating mutations in CTNNB1 and RSPO2, or inactivating mutations in APC, RNF43 and ZNRF3, are present in approximately 10–20% of patients with mCRPC [20,21]. Preliminary clinical data have suggested an association between Wnt-pathway activation and higher Gleason grades [22], higher prostate-specific antigen (PSA) levels [22], earlier-onset prostate cancer (diagnosed at <50 yr of age) [20], and higher hazard of recurrence after radical prostatectomy [23]. Preclinical data have also demonstrated that the AR may have an intrinsic interaction with canonical and noncanonical Wnt signaling [24–26]. Hormonal modulations such as ADT, and even abiraterone and enzalutamide, may activate the canonical Wnt/β-catenin signaling pathway, leading to androgen-independent prostate cancer growth [24,25]. In addition, activation of the noncanonical Wnt signaling pathway has been implicated in resistance to AR antagonism, especially to enzalutamide [26,27].
In the present study, we hypothesized that mCRPC patients who harbor somatic Wnt-pathway activating mutations would have poorer responses to first-line novel hormonal therapies such as abiraterone and enzalutamide, due to the AR-independent mechanism of continued growth that this pathway may confer to these tumors. To resolve this biological question, we utilized clinical-grade tumor DNA sequencing results to interrogate for Wnt-activating alterations and to correlate these with sensitivity or resistance to abiraterone or enzalutamide treatment in the first-line mCRPC setting.
2. Patients and methods
Men who received first-line abiraterone or enzalutamide therapy for mCRPC at the Johns Hopkins Hospital, over a 9-yr period (from August 2009 to November 2018), formed the study population. Genomic analyses were performed at each treating physician’s discretion, and there was no systematic reason why some patients underwent or did not undergo genetic testing. Therefore, this is a genomic-centric analysis that is not capable of capturing information about all treated patients (ie, no clinical data were entered on patients without available genomic results). We utilized data from clinical-grade next-generation DNA sequencing assays (Foundation Medicine, or Personal Genome Diagnostics [PGDx]) to interrogate for somatic mutations resulting in activation of the Wnt pathway. Our genes of interest were APC, CTNNB1, RNF43, RSPO2, and ZNRF3. In the case of inactivating mutations in APC or RNF43, pathogenic alterations were defined as those that were predicted to result in a truncated protein (nonsense mutations, frameshift insertions or deletions, splice site mutations at conserved splice donor or acceptor sites, or genomic/exonic deletions). In the case of activating mutations in CTNNB1, only missense mutations at the six hotspot residues (codons 32, 33, 34, 37, 41, and 45) that are known to increase stabilization of the protein were defined as pathogenic. In the case of RSPO2, only activating gene fusions were classified as pathogenic. Of note, only Foundation Medicine interrogates RSPO2 fusions, so this gene was only partially characterized in our cohort. APC, CTNNB1, and RNF43 are interrogated by both Foundation Medicine and PGDx, while ZNRF3 is not interrogated by either assay.
Tumor DNA analysis was performed using archival primary cancer specimens (n = 70), metastatic biopsies (n = 52), or circulating tumor DNA samples (n = 24). Men without available somatic DNA analysis were excluded from the study. All genomic analyses were performed retrospectively, and information about Wnt-pathway status was not used to influence the selection of therapy. The Johns Hopkins University Institutional Review Board and the Human Research Ethics Committee approved the conduct of this retrospective study.
Demographic, pathologic, and clinical characteristics of all patients were collected. We recorded the presence/absence of pathogenic or likely pathogenic somatic alterations predicted to result in Wnt-pathway stimulation (namely, activating mutations in CTNNB1 or RSPO2, or inactivating mutations in APC or RNF43), and classified patients into mutation-positive (Wnt-activated) and -negative (Wnt wild-type) groups. The primary endpoint was to estimate time to PSA progression on first-line abiraterone/enzalutamide treatment, defined as the time from starting the antiandrogen therapy to the second consecutive elevation in PSA above baseline or nadir. Secondary endpoints were overall survival (OS), defined as the time from starting abiraterone/enzalutamide therapy to death from any cause and PSA50 response, defined as a decline of ≥50% in PSA from baseline confirmed on a subsequent measurement at least 4 wk apart. In order for patients to be evaluable for PSA response, PSA data for a minimum of 12 wk were required following the initiation of treatment (all patients met this criterion). Fisher’s exact test was used to test for the association between PSA50 response and Wnt-pathway status. Kaplan-Meier curves were used to estimate PSA progression-free survival and OS probabilities, and univariable Cox proportional-hazards model was used to compare differences in time to PSA progression and OS between the mutation-positive and mutationnegative groups. Multivariable Cox proportional-hazards models were used to estimate hazard ratios (HRs) and corresponding 95% confidence intervals (CIs), and to test for the association between Wnt-pathway status or types (APC/RNF43 vs CTNNB1) and PSA progression or OS, after adjusting for the clinical-pathologic variables (age at diagnosis, time to castration resistance, Gleason grade group, PSA baseline level, and previous chemotherapy). Separate multivariable Cox proportional-hazards models were used to estimate the association between Wnt-pathway status and PSA progression or OS, adjusting for concurrent inactivating alterations in TP53, RB1, and PTEN. All statistical tests were two sided, and statistical significance was set at p ≤ 0.05. Since this study was hypothesis generating, we did not perform corrections for multiple comparisons.
3. Results
3.1. Baseline clinical and genomic characteristics
We identified 137 mCRPC patients with available somatic DNA sequencing results who had received first-line abiraterone or enzalutamide treatment at our institution. Of these, 11% (15 patients) harbored at least one Wnt-pathway stimulating alteration, including activating mutations in CTNNB1 (n = 8), and inactivating mutations in APC (n = 6) and RNF43 (n = 3). No RSPO2 fusions were identified. Two patients had more than one Wnt-pathway alteration in their tumor (Table 1). Concurrent deleterious TP53 mutations were seen in 33% (5/15) and 40% (49/122) of patients with and without Wnt-activating mutations, respectively. Of the 15 patients with Wnt-pathway alterations, 53% (8/15) had somatic DNA analysis from metastatic tissue, 33% (5/15) from the primary tumor, and 20% (3/15) from circulating tumor DNA (Table 2); one patient had analyses performed from both primary tumor and circulating tumor DNA.
Table 1 –
Wnt pathway alterations in our patient cohort
| Patient ID | Wnt mutation | Mutant allelic fraction (%) | Function | Other somatic mutations | Source of somatic DNA |
|---|---|---|---|---|---|
| Patient 27 | CTNNB1 (G34E) | 3 | Activating | AKT1 (D323N), MLH1 (E230V), ROS1 (A1381T) | Metastasis |
| Patient 46 | APC (Q215fs*) | 15 | Inactivating | PTEN (loss exons 3–9), RB1 (V654fs*14), TP53 (S215I) | Metastasis |
| Patient 48 | APC (H668Tfs*6) | 63 | Inactivating | BRCA2 (N8991fs*5), PDGFRA (A384S), SMO (R726Q), SRC (R208H), TSC2 (A678T) | Metastasis |
| Patient 136 | CTNNB1 (D32G) | 34 | Activating | TP53 (E271V), DDR2 (D621Y), FANCA (L278I), KIT (P467Q), BRCA2 (P3189H), TSC1 (Q898H) | Metastasis |
| Patient 157 | APC (D1425fs*) | 31 | Inactivating | ARID1A (E1780fs*) | Prostate |
| Patient 180 | APC (R1450X*) | 26 | Inactivating | AR (T878A), AKT1 (E17K), NF1 (Q1993X*) | Plasma |
| Patient 197 | CTNNB1 (T41A) | 7 | Activating | BRCA1 (Q1111fs*), NF1 | Prostate |
| RNF43 (G659fs*) | 41 | Inactivating | (K1387fs*), PTEN (T319fs*), CIC (P1116fs*), CSF1R (E317fs*), JAK1 (P430fs*), TMPRSS2 (-ERG fusion) | ||
| Patient 263 | CTNNB1 (D32H) | 37 | Activating | ARID2 (S1500X*), NTRK1 (A5T), SF3B1 (E622D), STAG2 (S1075X*), TP53 (H115fs*34) | Metastasis |
| Patient 266 | CTNNB1 (S37F) | 12 | Activating | ASXL1 (P808Lfs*10), POLH | Metastasis |
| APC (Q1621K) | 48 | Inactivating | (R371H), RUNX1 (D160Y), BRCA2 (C1913G), BLM (P126Q), ), MET (Q826K), NOTCH2 (C1060S), ERCC4 (D762V) | ||
| Patient 268 | CTNNB1 (S33F) | 8 | Activating | CDKN2A (S12L) | Plasma |
| Patient 271 | RNF43 (truncation exon 8) | Cannot be determined | Inactivating | PTEN (H123fs*2), TMPRSS2 (- ERG) | Prostate |
| Patient 276 | CTNNB1 (S37F) | 13 | Activating | ATM (D2708N), ), FLT3 (C368F), TP53 (A76Vfs*55), TMPRSS2 (- ERG) | Metastasis |
| Patient 277 | CTNNB1 (S45Y) | 43 | Activating | CDKN1B (S175X*) | Prostate |
| Patient 283 | APC (D1532fs*33) | 24 | Inactivating | AR (H875Y), BRCA2 (loss exons 19-20), ASXL1 (deletion exon 2), BCORL1 (c.3608-lG>C), CDKN2A (c.151-1G>C), VHL (L188V) | Metastasis |
| Patient 294 | RNF43 (G659Vfs*41) | 45 | Inactivating | ASXL1 (Q976X*), TP53 (C238G, M237I) | Prostate |
Table 2 –
Baseline clinical and pathologic characteristics, by Wnt status
| Wnt mutation (N = 15) | No Wnt mutation (N = 122) | |
|---|---|---|
| Age at diagnosis, median (Q1, Q3) | 60 (58–66) | 62 (56–68) |
| Age at ADT starting, median (Q1, Q3) | 64 (59–66) | 65 (58–69) |
| Race, N (%) | ||
| White | 13 (87) | 98 (81) |
| Black | 2 (13) | 17 (14) |
| Other | 0 (0) | 6 (5) |
| Baseline PSA (ng/ml) prior to starting abiraterone or enzalutamide, median (Q1, Q3) | 21.8 (6.3–75.8) | 14.5 (5.6–46.2) |
| Time from diagnosis to first-line abi/enza, mo (Q1, Q3) | 56.3 (22.6–79.7) | 41.9 (19.4–83.4) |
| Time to castration-resistance, mo (Q1, Q3) | 14.6 (8.3–33.4) | 17.3 (11.9–28.5) |
| Gleason grade group, N (%) | ||
| Grade 1 | 0 (0) | 2 (1.7) |
| Grade 2 | 1 (6.7) | 11 (9.6) |
| Grade 3 | 3 (20) | 13 (11) |
| Grade 4 | 2 (13) | 20 (17) |
| Grade 5 | 9 (60) | 69 (60) |
| Radical prostatectomy, N (%) | 9 (60) | 46 (38) |
| Pathologic/clinical T stage, N (%) | ||
| T1/T2 | 9 (69) | 52 (49) |
| T3/T4 | 4 (31) | 55 (51) |
| Pathologic pN+, N (%) | 3 (33) | 8 (18) |
| Metastasis at diagnosis, N (%) | 4 (27) | 50 (42) |
| Ductal or intraductal histology, N (%) | 3 (20) | 15 (14) |
| Perineural invasion, N (%) | 8 (57) | 63 (61) |
| Lymph vascular invasion, N (%) | 1 (7.1) | 13 (13) |
| Previous taxane chemotherapy, N (%) | 11 (73) | 62 (51) |
| Taxane chemotherapy for mHSPC, N (%) | 2 (13) | 16 (13) |
| Concurrent p53 mutation, N (%) | 5 (33) | 49 (40) |
| Sites of metastatic disease, N (%) | ||
| Bone | 12 (80) | 109 (89) |
| Lymph node | 9 (60) | 83 (68) |
| Visceral | 7 (47) | 38 (31) |
| Source of somatic DNA, N (%) a | ||
| Prostate | 5 (33) | 65 (53) |
| Metastasis | 8 (53) | 44 (36) |
| Plasma tumor DNA | 3 (20) | 21 (17) |
abi = abiraterone; ADT = androgen deprivation therapy; enza = enzalutamide; mHSPC = metastatic hormone-sensitive prostate cancer; PSA = prostate-specific antigen.
The sum of percentages exceeds 100% because some patients had more than one test.
The timing of collection of tumor DNA samples for genomic analysis was also evaluated. Of the 16 tumor DNA samples from 15 patients with Wnt-pathway alterations, 63% (10/16) were collected during castration-resistant disease and 38% (six/16) during hormone-sensitive disease. In patients without Wnt-pathway alterations, 53% (69/130) and 47% (61/130) of somatic DNA samples were collected during the castration-resistant and hormone-sensitive disease states, respectively (Supplementary Table 1). All somatic DNA analyses were performed from samples obtained prior to initiation of first-line abiraterone or enzalutamide.
The median follow-up for survivors only was 26.5 mo. Overall, 53% of patients had received prior taxane chemotherapy in our cohort, with 73% of Wnt-activated versus 51% of Wnt wild-type patients having received previous chemotherapy. Patients with Wnt-activating mutations had numerically fewer T3/T4 tumors (31% vs 51%) than Wnt wild-type patients. Ductal and intraductal histologies were seen at similar frequencies in Wnt-activated and wild-type groups, respectively (20% vs 14%). All demographic, clinical, and pathologic characteristics of our patients are detailed in Table 2. Groups generally appeared balanced with respect to common prognostic variables.
3.2. Efficacy of first-line abiraterone or enzalutamide
The median time to PSA progression in our overall cohort was 8.3 mo (range, 0.8–62.0 mo). Patients with Wnt-activating alterations had a median time to PSA progression of 6.5 versus 9.6 mo in patients without Wnt mutations (HR 2.34, 95% CI 1.32–4.17, p = 0.003; Fig. 1A). Multivariable analysis showed that the presence of a Wnt-activating mutation was independently associated with increased hazard of PSA progression (adjusted HR [aHR] 2.33, 95% CI 1.25–4.32, p = 0.007). Previous taxane chemotherapy was also independently associated with higher hazard of PSA progression (aHR 1.83, 95% CI 1.21–2.76, p = 0.004; Table 3). The multivariable results for PSA progression are depicted in Table 3.
Fig. 1 –

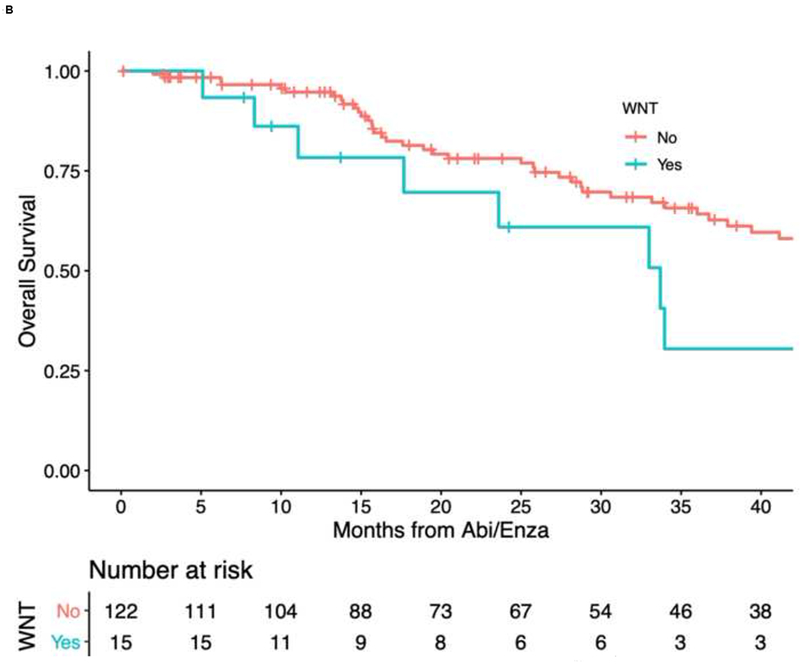
Kaplan-Meier curves of (A) time to PSA progression and (B) overall survival, by Wnt status.
Abi = abiraterone; Enza = enzalutamide; PSA = prostate-specific antigen.
Table 3 –
Multivariable analyses of time to PSA progression and overall survival
| Time to PSA progression |
Overall survival |
|||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p value | HR | 95% CI | p value | |
| Model 1 | ||||||
| Any Wnt-activating mutation | 2.33 | 1.25–4.32 | 0.007 | 2.14 | 1.03–4.43 | 0.041 |
| Age at diagnosis | 1.03 | 1.01–1.06 | 0.015 | 1.05 | 1.01–1.09 | 0.01 |
| Time to castration resistance | 0.56 | 0.41–0.75 | 0.0001 | 0.65 | 0.42–1.0 | 0.050 |
| Gleason score | 1.07 | 0.66–1.74 | 0.8 | 1.56 | 0.76–3.19 | 0.2 |
| Baseline PSA | 0.99 | 0.87–1.12 | 0.8 | 1.17 | 0.99–1.39 | 0.061 |
| Previous chemotherapy | 1.83 | 1.21–2.76 | 0.004 | 2.49 | 1.33–4.65 | 0.004 |
| Model 2 | ||||||
| APC/RNF43 (ref. wild type) | 2.58 | 1.14–5.83 | 0.023 | 2.39 | 0.83–6.90 | 0.10 |
| CTNNB1 (ref. wild type) | 2.12 | 0.93–4.81 | 0.072 | 1.98 | 0.79–4.96 | 0.14 |
| Age at diagnosis | 1.03 | 1.01–1.06 | 0.015 | 1.05 | 1.01–1.09 | 0.01 |
| Time to castration resistance | 0.56 | 0.41–0.76 | 0.0002 | 0.64 | 0.41–1.00 | 0.050 |
| Gleason score | 1.08 | 0.67–1.74 | 0.8 | 1.57 | 0.77–3.21 | 0.2 |
| Baseline PSA | 0.99 | 0.88–1.12 | 0.9 | 1.18 | 1.34–4.69 | 0.062 |
| Previous chemotherapy | 1.84 | 1.22–2.78 | 0.004 | 2.51 | 1.34–4.69 | 0.004 |
CI = confidence interval; HR = hazard ratio; PSA = prostate-specific antigen; ref. = reference.
Model 1 evaluated the effect of any Wnt-activating mutation versus wild type, and model 2 evaluated the effect of various types of Wnt-activating mutations.
Exploratory analysis of the different types of Wnt-pathway mutations (inactivating APC or RNF43 mutations vs activating CTNNB1 mutations) showed that APC/RNF43 mutations were independently associated with higher hazard of PSA progression than Wnt wild type (aHR 2.58, 95% CI 1.14–5.83, p = 0.023). Despite a strong trend in the same direction, CTNNB1 mutations showed no statistically significant association with higher hazard of PSA progression (aHR 2.12, 95% CI 0.93–4.81, p = 0.072). Finally, after adjusting for concurrent inactivating alterations in TP53, RB1, and PTEN, presence of Wnt-activating mutations remained independently prognostic (aHR 2.38, 95% CI 1.33–4.24, p = 0.003; Supplementary Table 2).
With respect to PSA >50% responses to first-line abiraterone/enzalutamide treatment, Wnt-activated patients had numerically lower PSA responses than patients without Wnt mutations (53% vs 75%), but no statistical difference was observed between the two groups (p = 0.12). The PSA response waterfall plot is demonstrated in Figure 2.
Fig. 2 –
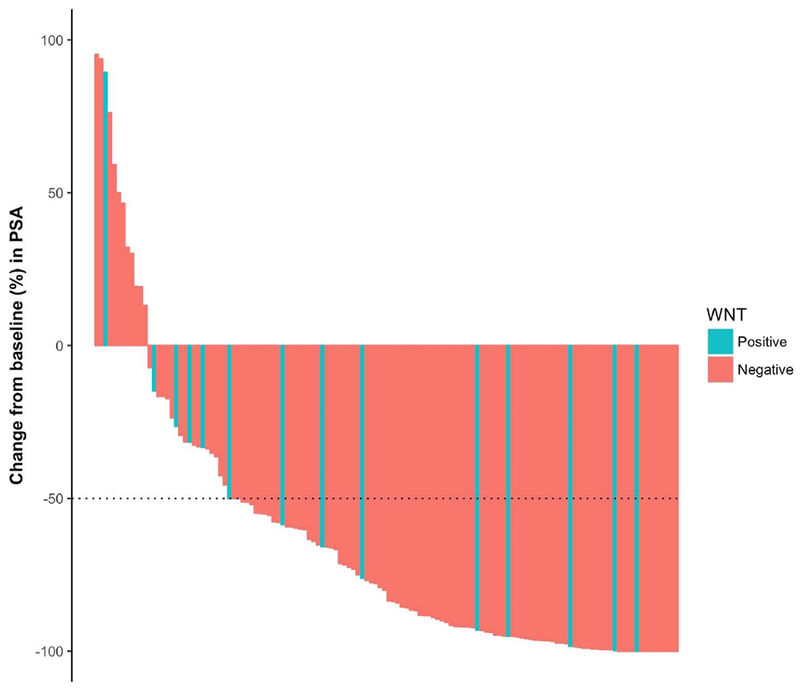
PSA waterfall plot (best PSA response), by Wnt status.
PSA = prostate-specific antigen.
3.3. Overall survival
The median OS of the whole cohort was 26.5 mo (range, 1.0–98.4 mo). Median survival time among patients with Wnt-activating mutations was shorter than those without Wnt mutations (median OS 23.6 vs 27.7 mo, HR 2.28, 95% CI 1.15–4.53, p = 0.01; Fig. 1B). Multivariable analysis confirmed that patients with Wnt-activating mutations had higher hazard of death than those without Wnt mutations (aHR 2.14, 95% CI 1.03–4.43, p = 0.041). Other factors were also independently associated with higher hazard of death in multivariable analysis, such as age at diagnosis and previous chemotherapy use (Table 2). Finally, after adjusting for concurrent inactivating alterations in TP53, RB1, and PTEN, presence of Wnt-activating mutations remained independently prognostic of OS (aHR 2.27, 95% CI 1.13–4.56, p = 0.021; Supplementary Table 3).
4. Discussion
The findings of our hypothesis-generating study support the rationale that tumors harboring Wnt signaling pathway activating mutations may be more resistant to antiandrogen therapies, such as abiraterone and enzalutamide. Our study showed that Wnt-activated tumors are more resistant to antiandrogen therapies in the first-line mCRPC context, translating into more rapid PSA progression in these patients. Moreover, patients with activating Wnt-pathway mutations have a trend for lower PSA responses, compared with patients without these abnormalities. Finally, our data suggest that OS may also be impaired in men harboring Wnt-activated tumors compared with those with Wnt wild-type tumors.
The rationale underlying this acquired resistance is that Wnt-activating mutations confer to these tumors an earlier androgen-independent pathway of growth [24,25] and resistance to AR antagonism [26]. Previous studies have analyzed the prognostic and predictive significance of Wnt alterations in mCRPC, and have produced conflicting results. Geng et al [28] designed a study that correlated germline single nucleotide polymorphisms (SNPs) in Wnt-pathway genes with clinical outcomes in 465 patients with metastatic prostate cancer treated with ADT. This study found some protective alleles that reduced APC gene expression. The median time to castration resistance was 17 mo in patients without any of these protective alleles versus 29 mo in patients with three to four protective APC alleles. Multivariable analysis showed that two SNPs in APC were independently associated with decreased hazard of progression and also lower mortality [28].
The hypothesis that somatically derived Wnt-pathway mutations would negatively influence novel hormonal therapy sensitivity was also recently tested in a single-center prospective study designed to identify predictive biomarkers of primary resistance to abiraterone using molecular analyses of tumor biopsies [29]. In that study of 82 mCRPC patients receiving first-line abiraterone, those with activating mutations in the canonical Wnt/β-catenin signaling pathway (eg, stabilizing CTNNB1 alterations) had a higher chance of primary resistance to therapy (56% vs 17%, p = 0.001), suggesting that constitutively active Wnt/γ-catenin signaling might be responsible for this intrinsic antiandrogen resistance.
Another study also tried to evaluate sensitivity and resistance to first-line abiraterone and enzalutamide in 202 patients with mCRPC (7.9% of whom had Wnt-activating mutations), using whole-exome and deep targeted sequencing of plasma cell-free DNA [30]. Although this study showed greater hazard to disease progression on these drugs in patients who harbored different genomic abnormalities such as BRCA2/ATM, TP53, RB1, PI3K, and AR gain, no statistically significant differences were seen in patients with Wnt signaling pathway mutations in this study [30]. However, this study was limited by the fact that approximately one-third of patients did not have enough cell-free tumor DNA content to permit somatic genomic analyses.
Despite the clinical successes of abiraterone and enzalutamide as single-agent therapies for mCRPC, it is clear that additional non–AR-targeted therapeutic advances are needed. Further, now that antiandrogen therapies have been moved to earlier disease stages such as nonmetastatic CRPC (eg, apalutamide [31] and enzalutamide [32]) and hormone-sensitive metastatic disease [33,34] (eg, abiraterone), medical oncologists will possibly face the earlier development of different mechanisms of antiandrogen resistance. It is now clear that several of these mechanisms are acquired more frequently after the widespread and earlier use of abiraterone and enzalutamide [35,36], and knowledge of these potential resistance mechanisms (especially the AR-independent mechanisms) will be crucial to develop new strategies to improve outcomes in mCRPC moving forward.
Although our study offers some new insights into one important and emerging pathway of primary and acquired antiandrogen resistance, it also raises some clinical questions inherent to the limitations of its design. First, since Wnt-pathway mutations are generally not inherited in prostate cancer, at what stage of the disease are they somatically acquired? Second, is the development of Wnt-pathway mutations related to treatment selective pressures, and if yes, how could we avoid or delay these pressures? Third, once we detect a Wnt-pathway mutation, how should we change our practice, if at all? Fourth, given the possibility of emerging drugs that specifically target activated Wnt signaling components [37], the question that arises is whether canonical or noncanonical Wnt signals are more critical in prostate cancer progression. Fifth, what is the inter-relationship between Wnt-pathway signaling mutations and other treatment-dependent mechanisms of antiandrogen resistance, such as AR overexpression and mutations, AR splice variants [4,35], and treatment-emergent small-cell neuroendrocrine prostate cancer [36]?
Our study has several limitations that should be considered when interpreting the results. First, this was a single-center retrospective analysis. While several provocative associations have been demonstrated between Wnt status and novel antiandrogen resistance, causal relationships are difficult to assess and the results may have been influenced by other clinical factors. For example, almost three-fourths of patients with Wnt-activating alterations (73%) had received previous chemotherapy, compared with only half (50%) of patients without these alterations. This difference may have impacted our results, since antiandrogens have lower efficacy after chemotherapy. Second, other inherent limitations of this type of study include the introduction of information bias. In addition, because this is a single-institution study, selection bias should be considered while interpreting our results. To this end, we tried to mitigate selection bias by including consecutive patients who received first-line abiraterone or enzalutamide and had next-generation DNA sequencing data available. In addition, we were unable to compare the clinical outcomes to abiraterone/enzalutamide between patients with and without available genomic information, because we do not routinely collect clinical data on patients who have not undfergone genetic analyses and this information is not readily available. Therefore, it is possible that outcomes in genomically selected patients might not be reflective of the broader mCRPC population at our institution. Finally, our assessment of somatic mutational status relied on multiple clinical-grade assays performed on primary or metastatic tumors; the use of primary tumor materials may have partially underestimated (or misclassified) Wnt-pathway mutations in these patients. In addition, not all relevant Wnt-pathway genes were interrogated by the commercial next-generation sequencing platforms used here; RSPO2 fusions were evaluated only by the Foundation Medicine assay and not by the PGDx assay, and the ZNRF3 gene was not evaluated in either assay. Therefore, defects in the full complement of genes potentially involved in Wnt-pathway signaling are likely underreported in our study, although probably only slightly.
5. Conclusions
In summary, our preliminary data suggest that mCRPC patients with Wnt-pathway activating somatic mutations have faster PSA progression on first-line abiraterone and enzalutamide, as well as worse OS. The negative prognostic impact of Wnt alterations remained after multivariable adjustments and after accounting for concurrent alterations in other key tumor suppressor genes (TP53, RB1, and PTEN). Our data support the rationale for testing Wnt-pathway inhibitors (eg, porcupine inhibitors or other agents), either alone or in combination with antiandrogen therapies, in clinical trials for Wnt-activated mCRPC patients.
Supplementary Material
Acknowledgments
Financial disclosures: Emmanuel S. Antonarakis certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Emmanuel S. Antonarakis is a paid consultant/advisor to Janssen, Astellas, Sanofi, Dendreon, AstraZeneca, Clovis, and Merck; has received research funding to his institution from Janssen, Johnson & Johnson, Sanofi, Dendreon, Bristol Myers-Squibb, AstraZeneca, Clovis, and Merck; and is the coinventor of a biomarker technology that has been licensed to Qiagen. The remaining authors report no relevant conflicts of interest.
Funding/Support and role of the sponsor: This work was partially supported by National Institutes of Health Grant P30 CA006973 (Emmanuel S. Antonarakis) and Department of Defense grant W81XWH-16-PCRP-CCRSA (Emmanuel S. Antonarakis).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Mohler JL, Lee RJ, Antonarakis ES, Higano CS, Richey S. NCCN clinical practice guidelines in oncology—prostate cancer. Prostate Cancer 2018;151. [Google Scholar]
- [2].Armstrong AJ, Antonarakis ES, Taplin M-E, et al. Naming disease states for clinical utility in prostate cancer: a rose by any other name might not smell as sweet. Ann Oncol 2018;29:23–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wang Q, Li W, Zhang Y, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009;138:245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011;364:1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013;368:138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014;371:424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012;367:1187–97. [DOI] [PubMed] [Google Scholar]
- [9].Yokoyama NN, Shao S, Hoang BH, Mercola D, Zi X. Wnt signaling in castration-resistant prostate cancer: implications for therapy. Am J Clin Exp Urol 2014;2:27–44. [PMC free article] [PubMed] [Google Scholar]
- [10].Murillo-Garzón V, Kypta R. Wnt signalling in prostate cancer. Nat Rev Urol 2017;14:683–96. [DOI] [PubMed] [Google Scholar]
- [11].Croce JC, McClay DR. Evolution of the Wnt pathways. Methods Mol Biol 2008;469:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Acebron SP, Niehrs C. β-Catenin-independent roles of Wnt/LRP6 signaling. Trends Cell Biol 2016;26:956–67. [DOI] [PubMed] [Google Scholar]
- [13].Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017;169:985–99. [DOI] [PubMed] [Google Scholar]
- [14].Clevers H Wnt/β-catenin signaling in development and disease. Cell 2006;127:469–80. [DOI] [PubMed] [Google Scholar]
- [15].Nishisho I, Nakamura Y, Miyoshi Y, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991;253:665–9. [DOI] [PubMed] [Google Scholar]
- [16].Tenbaum SP, Ordóñez-Morán P, Puig I, et al. β-Catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med 2012;18:892–901. [DOI] [PubMed] [Google Scholar]
- [17].Pohl S-G, Brook N, Agostino M, Arfuso F, Kumar AP, Dharmarajan A. Wnt signaling in triplenegative breast cancer. Oncogenesis 2017;6:e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Teng Y, Wang X, Wang Y, Ma D. Wnt/beta-catenin signaling regulates cancer stem cells in lung cancer A549 cells. Biochem Biophys Res Commun 2010;392:373–9. [DOI] [PubMed] [Google Scholar]
- [19].Peterson LF, Turbiak AJ, Giannola DM, et al. Wnt-pathway directed compound targets blast crisis and chronic phase CML leukemia stem progenitors. Blood 2009;114:2168.19589924 [Google Scholar]
- [20].Robinson D, Van Allen EM, Wu Y-M, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Beltran H, Yelensky R, Frampton GM, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol 2013;63:920–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jung SJ, Oh S, Lee GT, et al. Clinical significance of Wnt/β-catenin signalling and androgen receptor expression in prostate cancer. World J Mens Health 2013;31:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Huang S-P, Ting W-C, Chen L-M, et al. Association analysis of Wnt pathway genes on rostatespecific antigen recurrence after radical prostatectomy. Ann Surg Oncol 2010;17:312–22. [DOI] [PubMed] [Google Scholar]
- [24].Lee E, Ha S, Logan SK. Divergent androgen receptor and beta-catenin signaling in prostate cancer cells. PLoS One 2015;10:e0141589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Terry S, Yang X, Chen M-W, Vacherot F, Buttyan R. Multifaceted interaction between the androgen and Wnt signaling pathways and the implication for prostate cancer. J Cell Biochem 2006;99:402–10. [DOI] [PubMed] [Google Scholar]
- [26].Miyamoto DT, Zheng Y, Wittner BS, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015;349:1351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen WS, Aggarwal R, Zhang L, et al. Genomic drivers of poor prognosis and enzalutamide resistance in metastatic castration-resistant prostate cancer. Eur Urol. In press. 10.1016/j.eururo.2019.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Geng J-H, Lin VC, Yu C-C, et al. Inherited variants in Wnt pathway genes influence outcomes of prostate cancer patients receiving androgen deprivation therapy. Int J Mol Sci 2016;17:1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang L, Dehm SM, Hillman DW, et al. A prospective genome-wide study of prostate cancer metastases reveals association of Wnt pathway activation and increased cell cycle proliferation with primary resistance to abiraterone acetate-prednisone. Ann Oncol 2018;29:352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Annala M, Vandekerkhove G, Khalaf D, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov 2018;8:444–57. [DOI] [PubMed] [Google Scholar]
- [31].Smith MR, Saad F, Chowdhury S, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med 2018;378:1408–18. [DOI] [PubMed] [Google Scholar]
- [32].Hussain M, Fizazi K, Saad F, et al. Enzalutamide in men with nonmetastatic, castration-resistant prostate cancer. N Engl J Med 2018;378:2465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fizazi K, Tran N, Fein L, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med 2017;377:352–60. [DOI] [PubMed] [Google Scholar]
- [34].James ND, de Bono JS, Spears MR, et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med 2017;377:338–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Scher HI, Lu D, Schreiber NA, et al. Association of AR-V7 on circulating tumor cells as a treatmentspecific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol 2016;2:1441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aggarwal R, Huang J, Alumkal JJ, et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J Clin Oncol 2018;36:2492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang Z, Cheng L, Li J, et al. Inhibition of the Wnt/β-catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res 2018;78:3147–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.