

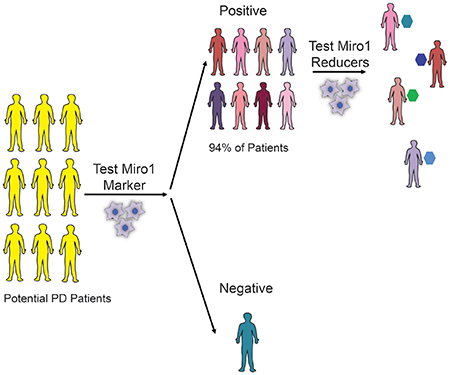
SUMMARY
The identification of molecular targets and pharmacodynamic markers for Parkinson’s disease (PD) will empower more effective clinical management and experimental therapies. Miro1 is localized on the mitochondrial surface and mediates mitochondrial motility. Miro1 is removed from depolarized mitochondria to facilitate their clearance via mitophagy. Here we explore the clinical utility of Miro1 for detecting PD and for gauging potential treatments. We measure the Miro1 response to mitochondrial depolarization using biochemical assays in skin fibroblasts from a broad spectrum of PD patients, and discover that more than 94% of the patients’ fibroblast cell lines fail to remove Miro1 following depolarization. We identify a small molecule that can repair this defect of Miro1 in PD fibroblasts. Treating patients-derived neurons and fly models with this compound rescues the locomotor deficits and dopaminergic neurodegeneration. Our results indicate that tracking this Miro1 marker and engaging in Miro1-based therapies could open new avenues to personalized medicine.
Graphical Abstract
eTOC SUMMARY
Hsieh et al. discover a molecular failure in removing Miro1 from depolarized mitochondria in a large population of Parkinson’s patients, and a small molecule that can eliminate this defect of Miro1. This compound rescues neuron loss in Parkinson’s iPSC neuron and fly models.
INTRODUCTION
Parkinson’s disease (PD) is a debilitating movement disorder, characterized by a selective loss of dopaminergic neurons in the substantia nigra. Currently, there is no effective treatment or reliable pharmacodynamic biomarker to monitor the response to treatment and efficacy of drug targeting (Bartels, 2016). The challenges in PD clinical practice limit moving experimental therapies from bench to bedside. Miro is an outer mitochondrial membrane (OMM) protein and anchors mitochondria to motors and microtubules. Miro is removed from the surface of depolarized mitochondria to stop their motility and to facilitate their following clearance via mitophagy. Miro removal from damaged mitochondria is mediated by at least two pathways— LRRK2 and the PINK1-Parkin axis (Hsieh et al., 2016; Wang et al., 2011) (Figure 1A). Mutations in LRRK2, PINK1, or Parkin cause familial PD (Bonifati, 2002; Kitada et al., 1998; Valente et al., 2004; Zimprich et al., 2004), while the majority of PD cases are sporadic without an inheritance pattern. The link between Miro and multiple PD-causing genes (Hsieh et al., 2016) prompted us to test the utility of Miro for detecting the disease and for gauging potential treatments.
Figure 1. Miro1 Response to CCCP in Fibroblasts.

(A) Schematic representation of our readouts. (B, C) Examples of the readouts using Healthy-1, PD-2, and risk-2. (D) Heat maps show the relative mitochondrial protein levels. The intensity of each band in the mitochondrial fraction is normalized to that of the mitochondrial loading control VDAC from the same blot and expressed as a fraction of the mean of Healthy-1 with DMSO treatment; this control was included in every experiment. Mean values are imported into heat maps. n=3-35. (E-F) ELISA of Miro1. n=4 with duplicates each time. Comparison within the same subject. Throughout the paper, *: P<0.05; **: P<0.01; ***: P<0.001. See also Figure S1–S3, Table S1–S2.
RESULTS AND DISCUSSION
Miro1 Is Resistant to Removal from Depolarized Mitochondria in Skin Fibroblasts from a Large Population of PD Patients.
Skin fibroblasts can be easily obtained from patients by a minimally-invasive, painless procedure (Le et al., 2017). We aimed to determine the frequency of the Miro1 phenotype in skin fibroblasts (Hsieh et al., 2016) from a large cohort of both sporadic and familial PD patients. We fractioned mitochondria after CCCP treatment that depolarizes the mitochondrial membrane potential (ΔΨm) (Hsieh et al., 2016). In wild-type controls at 6 hrs following treatment, both Miro1 and Mitofusin2 were removed from damaged mitochondria as detected by Western blotting (Figure 1A, B) (Hsieh et al., 2016). The OMM protein Mitofusin2 is a target of the PINK1-Parkin pathway, but not of LRRK2, for depolarization-triggered degradation (Hsieh et al., 2016; Tanaka et al., 2010). We included Mitofusin2 in our readout to compare its phenotypic frequency in PD with Miro1’s. We screened a total of 71 PD and 3 at-risk fibroblast lines comprising the entire PD fibroblast collection at the National Institute of Neurological Disorders and Stroke (NINDS) human and cell repository and the first released PD-control cohort from the Parkinson’s Progression Markers Initiative (PPMI). All patients were diagnosed with PD and without the presence of signs for other neurological disorders. At-risk subjects are younger asymptomatic family members of the probands and harbor the same genetic mutations (in LRRK2 or SNCA). Twenty-eight patients have a positive family history. We included 22 controls that consisted of 12 age/gender/race-matched healthy subjects recruited from the same cohorts and 10 subjects with other neurological disorders including Huntington’s (HD) or Alzheimer’s disease (AD) (Table S1A). We conducted our assay in a blinded manner. Notably, we discovered a unifying impairment in removing Miro1 from the mitochondrial fractions at 6 hrs after CCCP treatment in 69 PD and risk lines (93.2%). By contrast, Miro1 was efficiently removed following depolarization in every single control subject (0%) (Table S1A). The phenotype was more strikingly demonstrated when we imported the mean band intensities into a heat map where a lack of color change after treatment reflects the failure to remove Miro1 (Figure 1D). The lack of color change of Miro1 occurred broadly in PD patients. A smaller fraction of PD cell lines also failed to remove Mitofusin2 after CCCP treatment (Figure 1D). Basal protein levels of Miro1 and Mitofusin2 were largely comparable among all lines (Figure 1D, Table S1A; P>0.0906). This phenotype in Miro1 removal was significantly associated with PD (P<0.00001; Table S2A). The ratio of Miro1 intensity (with CCCP/with DMSO) was also significantly correlated with PD (P<0.0001; Figure S1A), but not with age (at sampling and onset) or gender (Figure S1B–D). There was no significant correlation between the Miro1 ratio and the disease progression (years with PD) or the clinical manifestations in patients with the Unified Parkinson’s Disease Rating Scale (UPDRS), Hoehn and Yahr Scale, or Mini-Mental Status Examination (Figure S1E–H). We confirmed that the cell passaging numbers within the range of 5-19 had no influence on the phenotype (Table S2B). Taken together, these observations show that the failure to remove Miro1 from damaged mitochondria is a common cellular defect in a large population of PD patients.
The LRRK2 and PINK-Parkin Pathways Are Broadly Affected in PD Fibroblasts.
We have previously identified two parallel molecular pathways, both of which are essential for removing Miro from the OMM of depolarized mitochondria–LRRK2 and the PINK1-Parkin axis (Hsieh et al., 2016; Wang et al., 2011). To investigate the mechanisms underlying Miro1 accumulation on damaged mitochondria in PD fibroblasts, we tested the hypothesis that the buildup was due to the impairments of the LRRK2 or PINK1-Parkin pathway. We have established that in wild-type control fibroblasts mitochondrial depolarization by CCCP treatment for only 1 hr triggers the recruitment of cytosolic LRRK2 and Parkin to mitochondria and Miro1, prior to Miro1 removal at 6 hrs (Figure 1A, C) (Hsieh et al., 2016). Antibodies against LRRK2, Parkin, and Miro1 have been validated in human cells lacking the corresponding genes (Hsieh et al., 2016). We used this readout to screen all 96 fibroblast lines, and found a variety of phenotypes in patients’ cells. Some cell lines were impaired only in recruiting LRRK2 to depolarized mitochondria, some impaired only in recruiting Parkin, and some cell lines were defective in recruiting both proteins. Because the failure to relocate LRRK2 to damaged mitochondria disrupts the following removal of Miro1 but not of Mitofusin2 (Hsieh et al., 2016), this result may explain the lower frequency of the Mitofusin2 phenotype than Miro1 in those patients (Figure 1D). We discovered that in 7 PD cell lines the recruitment of both LRRK2 and Parkin to damaged mitochondria appeared as normal when compared to the controls, while Miro1 removal was still compromised (Figure 1D, Table S1A), suggesting that additional mechanisms might be at play in those cells. Basal levels of LRRK2 and Parkin were comparable in all lines (P>0.8684). Collectively, our results provide evidence that the LRRK2 and PINK1-Parkin pathways are largely affected in PD patients’ fibroblasts, leading to a convergent downstream failure to remove Miro1 from damaged mitochondria.
Confirmation of the High Frequency and Specificity of the Miro1 Phenotype for PD Fibroblasts.
We established an enzyme-linked immunosorbent assay (ELISA) to detect Miro1 response to CCCP (Figure S2) which is useful for clinical laboratory use. We examined 14 PD/risk and 15 control cell lines used in Figure 1D. For each individual line, the result of Miro1 response to mitochondrial depolarization (Figure S2A) was consistent with that from using mitochondrial fractionation and Western blotting (Figure 1D, Table S1A). We used this ELISA to validate additional independent cohorts. We included 40 healthy controls and 12 PD patients from the Stanford Alzheimer’s Disease Research Center (ADRC) and the Coriell Institute (Table S1B). We discovered that Miro1 was efficiently degraded upon depolarization in all control lines, but in none of the PD patients (Figure 1E, S2A–B); this finding verified the reliability of the high frequency of Miro1 accumulation on depolarized mitochondria in the PD population (Figure 1, Table S2A). We also obtained cell lines from patients with movement disorders that exhibit clinical presentations similar to PD, including 4 of sporadic Dementia with Lewy Bodies (DLB; Stanford), 3 of Frontotemporal Degeneration (FTD; Coriell and NINDS), 2 of sporadic Corticobasal Degeneration (CBD; Mayo Clinic), and 3 of sporadic Progressive Supranuclear Palsy (PSP; Mayo Clinic) (Table S1B). Miro1 was effectively degraded after CCCP treatment in all lines (Figure 1F, S3A–B, Table S2A), demonstrating the specificity of Miro1 accumulation for PD. The establishment of the ELISA to detect Miro1 could facilitate clinical applications of our discovery.
A Small Molecule Predicted to Bind to Miro1 Eliminates the Miro1 Defect in PD Fibroblasts.
Now that we have discovered a Miro1 molecular phenotype in skin cells from a large number of PD patients, we next explored the possibility to use this defect as a pharmacodynamic marker for gauging Miro1-based therapies. In future clinical trials, we could first determine whether a potential PD patient has the Miro1 phenotype using his/her cultured fibroblasts in a dish, then use those fibroblasts to test multiple potential treatments/drugs to identify the best treatment that ameliorates the Miro1 defect, and lastly administer the individual-tailored treatment to each patient. To prove our concept, we sought for a therapeutic strategy that can eliminate this marker in PD fibroblasts and then tested the neuroprotective effect of this strategy in PD models. We have previously shown that genetically knocking down Miro1 by RNAi in both human neuron and fly PD models rescues Miro1 accumulation, slower mitophagy, and dopaminergic neurodegeneration (Hsieh et al., 2016; Shaltouki et al., 2018). We searched for pharmacological approaches to reduce Miro1, which are more applicable for clinical treatments and could remove this Miro1 phenotype from PD fibroblasts. We assessed the binding ability of 6,835,320 commercialized small molecules to one of the two atypical Rho GTPase domains of Miro1 (amino acids 411-592) (Klosowiak et al., 2016) (Figure S3C) by a customized virtual screen using the artificial intelligence (AI) technology (Wallach et al., 2015). Small molecules were filtered for oral availability, toxicophores, and blood-brain barrier permeability. We conducted a preliminary functional screen by feeding fruit flies with the top 11 compounds at 250 μM for 7 days. We identified 4 hits (compound 3-6 in the order of AI ranking) that partially but significantly reduced fly Miro (DMiro) protein levels (Figure S3D). We picked compound 3 for further validation (Figure 2A, S3C). This compound did not significantly affect another OMM GTPase Mitofusin (Marf) (Figure 2B). We applied compound 3 to fibroblasts from one sporadic PD patient (PD-2) and tested the Miro1 response to CCCP treatment for 6 hrs using Western blotting. Treating PD fibroblasts with this small molecule at 10 μM for 30 hrs robustly promoted Miro1 degradation following CCCP treatment (Figure 2C). We named compound 3 Miro1 Reducer. We found that Miro2 was also subject to depolarization-triggered degradation in healthy controls, although to a lesser extent than Miro1 (Figure 2C), consistent with previous reports showing that Parkin prefers Miro1 over Miro2 for ubiquitination and proteasome degradation (Klosowiak et al., 2016). Miro1 Reducer had no influence on Miro2 in either healthy or PD cells (Figure 2C). It is worthwhile to note that CCCP treatment for only 6 hrs in healthy human fibroblasts triggers degradation of the OMM proteins Miro and Mitofusin but not of other OMM, inner mitochondrial membrane (IMM), and matrix proteins, because mitophagy has not yet started at this early time point (Figure 2C, S4A) (Hsieh et al., 2016). To measure mitophagy, we detected multiple mitochondrial matrix and membrane proteins at a later time point (14 hrs after CCCP treatment) in those cells using both Western blotting and immunocytochemistry (Hsieh et al., 2016), and confirmed that Miro1 Reducer treatment rescued the delayed mitophagy phenotype in PD fibroblasts (Figure S4A–B). Miro1 protein levels responded to Miro1 Reducer in a dose-dependent manner (IC50: 7.8 μM; Figure 2D). High concentrations (>25 μM) of Miro1 Reducer combined with CCCP treatment for 6 hrs impacted multiple mitochondrial matrix and cytosolic proteins (Figure 2D), indicating cytotoxicity. The IMM dynamin GTPase OPA1 was not reduced by Miro1 Reducer treatment (Figure 2D, right). Mitochondrial depolarization promotes Miro degradation through proteasomes in a wild-type background (Figure 2E) (Chan et al., 2011; Wang et al., 2011). Miro1 Reducer facilitated proteasome Miro1 degradation following CCCP treatment in PD-2 fibroblasts, because the reduction of Miro1 protein triggered by Miro1 Reducer was blocked by the proteasome inhibitor MG132 (Figure 2E). A brief CCCP treatment for only 1 hr in healthy fibroblasts boosted ubiquitination of immunoprecipitated (IPed) Miro1, prior to Miro1 degradation at 6 hrs (Hsieh et al., 2016); this depolarization-elicited increase of ubiquitination did not occur in PD-2 fibroblasts, regardless of the presence of Miro1 Reducer (Figure S4C). It is likely that Miro1 Reducer alters E3 ligase kinetics, functions as a PROTAC, or even recruits proteasomes to Miro1 directly. Miro1 Reducer did not affect the GTPase activity of Miro1 IPed from PD-2 fibroblasts under either basal or depolarized condition (Figure S4D), nor did it change Miro1 mRNA expression examined by quantitative PCR (qPCR) (Figure S4E). We next generated a drug-resistant form by mutating K427, N428, S432, Q446, K454, K528, and D530 to Alanine in the Myc-Miro1 construct. These residues showed high probability of binding to Miro1 Reducer in our structural model (Figure S3C). This mutant Myc-Miro1 (named Myc-Miro1-7A) was degraded after CCCP treatment in healthy fibroblasts, but it failed to respond to Miro1 Reducer when expressed in PD-2 (Figure 2F). These results suggest a possible mechanism of action of Miro1 Reducer in PD cells: once bound to Miro1, it promotes proteasome degradation of Miro1 upon mitochondrial depolarization.
Figure 2. Miro1 Reducer Eliminates the Miro1 Defect in PD Fibroblasts.

(A) Chemical properties of Miro1 Reducer. (B) Wild-type flies (w1118) were fed with DMSO alone (0 μM Miro1 Reducer) or Miro1 Reducer in DMSO at 250 μM for 7 days, and lysed and blotted as indicated. The band intensities are normalized to those of Tubulin from the same blots. n=4. (C-E) Fibroblasts were treated, lysed, and blotted as indicated. (C) Band intensities are normalized to those of GAPDH from the same blots and compared to “Healthy-1, no treatment” except otherwise indicated. n=4. (D) Band intensities are normalized to those of ATP5β from the same blots and expressed as a percentage of the mean of “0 μM Miro1 Reducer with CCCP”. Mean±S.E.M is shown. n=3. (E) Band intensities are normalized to those of GAPDH from the same blots and compared to “Healthy-1, no CCCP, no MG132” except otherwise indicated. n=4. (F) Healthy-1 and PD-2 fibroblasts were transfected as indicated, and immunostained with anti-Myc (Miro1, green) and stained with Dapi (blue). The Miro1 intensity is normalized to that of mito-dsRed from the same cell and quantified across 37-62 cells from 5 fields each transfection, 3 independent transfections. Scale bars: 50 μm. For (C-F), both CCCP and Miro1 Reducer were dissolved in DMSO. Cells were pretreated with Miro1 Reducer 24 hrs before the application of CCCP for another 6 hrs. The same volume of DMSO was applied at the same time in negative controls. Cell line information is in Table S1. See also Figure S3–S4.
We found that the therapeutic dose of Miro1 Reducer (10 μM for 30 hrs) did not affect baseline mitochondrial reactive oxygen species levels stained by MitoSox (Figure S4F), general mitochondrial morphology and the ΔΨm detected by TMRM staining (Figure S4G), or peroxisome movement or distribution (Figure S4H) in either Healthy-1 or PD-2 fibroblasts. We administered Miro1 Reducer to additional 4 control and 4 sporadic PD fibroblast cell lines and observed the same beneficial change of Miro1 degradation after depolarization in PD cells (Figure 3A). Collectively, Miro1 Reducer appears to be reliable and selective to abolish the Miro1 marker in PD fibroblasts.
Figure 3. Miro1 Reducer Rescues Parkinson’s Neurodegeneration.

(A) Fibroblasts were treated, lysed, and blotted as indicated. Band intensities are normalized to those of ATP5β from the same blots and compared to “Healthy-2, no treatment”. n=4. (B-C) iPSC-derived neurons were treated as indicated and immunostained with anti-TH (B) or stained with PI and Dapi (C). Both Antimycin A and Miro1 Reducer were dissolved in ethanol. Neurons were pretreated with Miro1 Reducer 24 hrs before the application of Antimycin A for another 6 hrs. The same volume of ethanol was applied at the same time in negative controls. (B) The density of TH-positive neurons is calculated in each condition under 20×, from 10 fields each experiment, 3-4 experiments. The neuron densities without Antimycin A are not significantly different among all conditions (p=0.0682). (C) The percentage of Pi-positive cells out of total cells (Dapi-positive) is calculated. n=20 fields each experiment, 4 experiments. For (B-C), comparisons with “Wild-type-I, no treatment” except otherwise indicated. (D) Mitochondrial movement (mito-dsRed) in representative axons pretreated with 5 μM Miro1 Reducer for 24 hrs, and then after treatment with 100 μM Antimycin A. The first frame of each live-imaging series is shown above a kymograph generated from the movie. Graphs of Mean±S.E.M show the percent of time each mitochondrion is in motion (red and blue, compared to “Wild-type-I, before treatment”), and the mitochondrial intensity normalized to that of the same axonal region at “0 min” and expressed as a fraction of the mean of “Wild-type-I, 0 min”. n=33-77 mitochondria from 5-8 axons from 5-8 separate transfections. Scale bars: (B, C) 100 μm; (D) 10 μm. See also Figure S3–S4.
Miro1 Reducer Rescues Parkinson’s Phenotypes in Human Neuron and in vivo PD Models.
Now we have discovered an organic compound that can eliminate the Miro1 marker in skin cells from PD patients. We determined the extent to which it alleviated PD-relevant phenotypes in PD models. We tested Miro1 Reducer in two independent models: the human neuron and fly models. We have previously shown that induced pluripotent stem cell (iPSC)-derived dopaminergic neurons from PD patients are more vulnerable to stress than those from healthy controls (Hsieh et al., 2016; Shaltouki et al., 2018). When live neurons are challenged by Antimycin A, the complex III inhibitor, for 6 hrs, significantly more patients-derived dopaminergic neurons die compared to those from healthy controls. We examined Miro1 Reducer using iPSCs from one familial patient with the A53T mutation in SNCA (two independent clones: PD-I; PD-II) and one sporadic patient (PD-III), and from three healthy control subjects and one isogenic wild-type control for PD-I (see Method) (Figure S4I). We differentiated iPSCs to neurons expressing tyrosine hydroxylase (TH), the rate-limiting enzyme for dopamine synthesis as previously described (Hsieh et al., 2016; Shaltouki et al., 2018). All iPSCs and their derived neurons have been fully validated in our previous publications (Figure S4I). We applied 10 μM Antimycin A for 6 hrs to induce mitochondrial stress (Hsieh et al., 2016). We identified dopaminergic neurons by TH-immunostaining (Shaltouki et al., 2018) and cell death by propidium iodide (PI)-staining (Tsai et al., 2018). Antimycin A treatment for 6 hrs caused acute neuronal cell death leading to the loss of TH and increase of PI signals in PD patients-derived neurons, compared to healthy controls (Shaltouki et al., 2018) (Figure 3B–C). Notably, Miro1 Reducer treatment at 5 μM for 30 hrs completely rescued the stress-induced neurodegeneration of dopaminergic neurons derived from both PD patients (Figure 3B–C). We found that Miro1 Reducer at 5 μM for 24 hrs (therapeutic dose) did not affect the basal motility of polarized, healthy mitochondria, but rescued the delayed arrest and clearance of depolarized mitochondria in iPSC-derived neurons from the PD patient (Shaltouki et al., 2018) (Figure 3D). Therefore, Miro1 Reducer promotes damaged mitochondrial clearance and protects PD neurons against mitochondrial stress.
In order to cross-validate the neuroprotective effect of Miro1 Reducer in vivo, we fed the compound to three well-established fly PD models: (1) Actin-GAL4>UAS-LRRK2G2019S (ubiquitous expression of human pathogenic LRRK2); (2) PINK1 null; and (3) Elav-GAL4>UAS-SNCAA53T (neuronal expression of human pathogenic alpha-synuclein). Those flies show robust age-dependent locomotor deficits and dopaminergic neurodegeneration (Hsieh et al., 2016; Shaltouki et al., 2018; Tsai et al., 2018). We administered 2.5 μM Miro1 Reducer (therapeutic dose) to flies from adulthood (Figure S4J), and examined the lifespan, dopaminergic neuron number, and adult climbing ability. Miro1 Reducer treatment had no long-term toxicity in either wild-type or PD flies, and extended the shortened lifespan of SNCAA53T flies (Figure 4A–C). It significantly rescued the age-dependent dopaminergic neuronal loss in all three PD models (Figure 4D–F), and locomotor decline in two models (LRRK2 and PINK1) (Figure 4G–I). PINK1 null flies display a wide array of additional phenotypes during development or at eclosure, including lowered ATP levels, thoracic indentation, abnormal wing posture, and defective flying ability (Tsai et al., 2018). We found that feeding PINK1 null flies from adult onset with 2.5 μM Miro1 Reducer did not improve those non-progressive phenotypes (Figure S4K–N). Our results demonstrate that Miro1 Reducer antagonizes age-dependent Parkinson’s relevant phenotypes in vivo.
Figure 4. Miro1 Reducer Rescues PD-Relevant Phenotypes in Flies.

For all panels, when Miro1 Reducer was not added, the same volume of the solvent, DMSO, was supplied in fly food. Drug administration (2.5 μM) was started from day 2. (A-C) Survival curves of LRRK2G2019S (A), PINK1B9 (imprecise excision, null) and PINK1RV (the precise excision control for PINK1B9) (B), and SNCAA53T flies (C). n=58-150. (D) Dopaminergic (DA) neurons in the PPM1/2 clusters of 22-day old flies were immunostained with anti-TH. (D-F) Quantification of the DA neuron number in the PPL1 or PPM1/2 cluster (total number divided by 2 for each brain). (D) LRRK2 G2019S. n=6-8 brains. (E) PINK1B9 (“−”) and PINK1RV (“+”). n=9-16. (F) SNCAA53T. n=9-11. (G-I) Climbing ability (Average PI±S.E.M) of adult flies. n=24-47 flies. See also Figure S3–S4.
In this study, we have explored the mitochondrial protein Miro1 both as a common molecular signature for detecting PD and as a drug target for treating the disease. Biopsied fibroblasts have not been tested extensively for PD biomarker research. Our work is the largest study to date that reveals a distinct molecular defect using PD fibroblasts. Notably, the impairment in Miro1 clearance occurs in some PD patients without a known mutation or with normal functions of LRRK2 and Parkin (Figure 1), indicating that varying upstream defects may converge on mitochondrial quality control in PD mechanism. Our results indicate that Miro1 may play a more vital role in mediating mitophagy than Miro2. Using the AI technology that tackles complex drug discovery optimization, we have searched for small molecules from a vast chemical space in silico that are predicted to bind to Miro1. Treating fibroblasts from PD patients with the selected compound eliminates the Miro1 molecular pathology, and treating PD flies and patients-derived neurons with the compound rescues their locomotor deficits and dopaminergic neurodegeneration. These results demonstrate the clinical utility of our Miro1 marker in skin cells for monitoring Miro1-based target engagements and the therapeutic promise of Miro1 reducers. Both our genetic and pharmacological approaches (therapeutic dose) in human cell and fly PD models only cause a nominal reduction of basal Miro1 levels without significantly disrupting the motility of polarized mitochondria (Figure 2C, 3A, 3D, S4I, S4J) (Hsieh et al., 2016; Shaltouki et al., 2018). This small reduction of basal Miro1 levels can facilitate Miro1 removal from depolarized mitochondria (Hsieh et al., 2016; Shaltouki et al., 2018) (Figure 2C, 3A, S4I). Therefore, to minimize cytotoxicity, an ideal therapeutic dose for a lead compound should cause significant Miro1 reduction following mitochondrial depolarization but minimum Miro1 degradation at baseline. Our findings will aid in guiding precision health strategies and stratifying participants for clinical trials, by first identifying patients with the Miro1 phenotype, and then testing the responses of the Miro1 marker to multiple Miro1 reducers using their own skin cells to determine personalized prognosis.
Limitations of Study:
Compared with PD, the sample sizes for non-PD were much smaller. It will be valuable to test our Miro1 marker in larger cohorts across diverse neurodegenerative diseases.
STAR METHOD TEXT
LEAD CONTACT AND MATERIALS AVAILABILITY
One plasmid was generated in this study and is available upon request. Please contact Xinnan Wang (xinnanw@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fly Stocks
The following fly stocks were used: w1118, Actin-GAL4, UAS-hLRRK2G2019S-Flag (Lin et al., 2010), Elav-GAL4, UAS-SNCAA53T (Trinh et al., 2008), PINK1RV (Park et al., 2006), and PINK1B9 (Park et al., 2006). The age of flies for each experiment was stated in figures. Male flies of PINK1 null (PINK1B9) and their control (PINK1RV) were used because male PINK1 null flies are infertile and PINK1 is on the X chromosome. For the rest of the genotypes, females were used (Sofola et al., 2010). For males, 1-146 flies were used for each experiment, and for non-life-span experiments the average age was 20 days old. For females, 42-150 flies were used for each experiment, and for non-life-span experiments the average age was 29 days old. All fly stocks were maintained at 25°C with a 12:12 hr light: dark cycle and constant humidity (65%) on standard sugar-yeast (SY) medium (15 g/l agar, 50 g/l sugar, 100 g/l autolyzed yeast, 6 g/l nipagin, and 3 ml/l propionic acid) (Castillo-Quan et al., 2016). For all experiments, flies were raised at standard density on standard SY medium in 200 ml bottles unless otherwise stated. All fly lines were backcrossed 6 generations into a w1118 background to ensure a homogeneous genetic background across all lines.
Human Subjects
Written informed consent was obtained from all participants and the study was approved by the institutional review boards at Mayo Clinic and Stanford University. Skin biopsies were performed and fibroblasts were dissociated as described in (Wren et al., 2015). Briefly, dermal skin biopsy specimens were harvested in a routine fashion from the medical aspect of non-dominant forearm. Skin biopsy tissue was dissected into small pieces, and fibroblasts were amplified and maintained in DMEM (Invitrogen) containing 10% fetal bovine serum (Gemini Bio-Products), supplemented with 1% non-essential amino acids (Invitrogen), 1% Penicillin-Streptomycin (Invitrogen), and 1% Amphotecerin B (Gemini Bio-Products).
METHOD DETAILS
Fly Behavior and Biochemical Assays
Newly eclosed flies were allowed to mature and mate for 24-48 hrs, and then females and males were separated and collected. Miro1 Reducer was dissolved in DMSO at 100 mM and supplemented into SY medium. For lifespan, mated males and females were separated, and split into vials (15 flies each) containing SY medium with or without compound. Flies were transferred into fresh vials and the numbers of dead and escaped flies were scored 3 times a week. The final n was the total number of dead flies. Data were presented as cumulative survival curves, and survival rates were compared using Log-Rank Test. For Figure 4A, P=0.2, Actin-Gal4>UAS-LRRK2G2019S 0 μM v.s. UAS-LRRK2G2019S 0 μM; P=0.2, Actin-Gal4>UAS-LRRK2G2019S 0 μM v.s. 2.5 μM; P=0.6, UAS-LRRK2G2019S 0 μM v.s. 2.5 μM. n=132-150 flies. For Figure 4B, P=8.5×10−7, PINK1B9 0 μM v.s. PINK1RV 0 μM; P=0.36, PINK1B9 0 μM v.s. 2.5 μM; P=0.15, PINK1rv 0 μM v.s. 2.5 μM. n=58-146 flies. For Figure 4C, P=3.19×10−11, Elav-Gal4>UAS-SNCAA53T 0 μM v.s. UAS-SNCAA53T 0 μM; P=0.00019, Elav-Gal4>UAS-SNCAA53T0 μM v.s. 2.5 μM; P=0.44, UAS-SNCAA53T 0 μM v.s. 2.5 μM. n=120-143 flies. Fly climbing (negative geotaxis) ability was evaluated as previously described (Kinghorn et al., 2015; Rival et al., 2004; Sofola et al., 2010). Briefly, adult flies were gently tapped to the base of a modified 25 ml climbing tube and their climbing progress was recorded after 45 sec. Three populations of flies were assessed, and for each population, flies were examined 3 times per experiment. The recorded values were used to calculate Average Performance Index (PI). For Figure 4G, data of mutant LRRK2 flies were presented as mean PI±S.E.M at the indicated days and compared using Ordinary Two-Way ANOVA Dunnett’s Test. n=3 independent tests, 44-47 flies per genotype/condition. P<0.0001, UAS-LRRK2G2019S 0 μM v.s. Actin-GAL4>UAS-LRRK2G2019S 0 μM; P<0.0001, Actin-GAL4>UAS-LRRK2 0 μM v.s. 2.5 μM. For Figure 4H, mean PI±S.E.M of PINK1B9 (“−”) and PINK1RV (“+”) flies was shown. n=3 independent tests, 24-37 flies per genotype/condition. For Figure 4I, mean PI±S.E.M of mutant SNCA flies was shown. n=3 independent tests, 42-46 flies per genotype/condition. For Figure 4H and 4I, One-Way ANOVA Fisher’s LSD Test was used. To assess flying ability, flies were tapped one at a time out of a vial held upside down 1 foot above a benchtop. If the fly flew away, it was recorded as “1”, and if it was unable to fly, it was recorded as “0”. Wing posture was assessed by viewing each fly through a vial under a dissecting microscope. Thoracic indentation was observed by placing flies on a CO2 pad and viewing their thoraxes under a dissecting microscope. A normal wing posture or thorax (Course et al., 2018) was scored as “1”, otherwise “0”. For Figure S4L–N, Chi-Square Test was used because the data were categorical. ATP concentrations were determined using the Roche ATP Bioluminescence Assay Kit HS II (Sigma-Aldrich, Cat:11699709001). Briefly, 1 whole fly was homogenized in 150 μl ice-cold lysis buffer using a Kontes pellet pestle. Lysate was then boiled for 5 min and centrifuged at 20,000 g at 4°C for 1 min. Cleared lysate was diluted 1:200 in dilution buffer and loaded with 10 μl luciferase. Luminescence was immediately measured using a Glomax Multi Jr. Reader (Promega). Total protein amount was measured using the bicinchoninic acid protein (BCA) assay (Thermo Scientific). The ATP level in each sample was normalized to the total protein amount. For all experiments, 1-150 flies were used.
Cell Culture and Biochemistry
Adult flies were lysed as previously described (Shaltouki et al., 2018) and immunoblotted by the following antibodies: guinea pig anti-DMiro (GP5) (Tsai et al., 2014) at 1:20,000, mouse anti-tubulin (T6199, Sigma) at 1:3,000, mouse anti-β-actin (ab8224, Abcam) at 1:5,000, mouse anti-VDAC (ab14734, Abcam) at 1:3,000, and rabbit anti-marf (Ziviani et al., 2010) at 1:1,000. HEK cell culture and transfection were described in (Hsieh et al., 2016). Fibroblasts were cultured in high-glucose DMEM (SH30243.01, Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (F0926, Sigma-Aldrich, and 900-108, Gemini Bio Products) and maintained in a 37°C, 5% CO2 incubator with humidified atmosphere. The media were refreshed every 3-4 days and split every 7-8 days. CCCP (C2759, Sigma-Aldrich) was prepared at 40 mM in DMSO fresh every time and applied at 40 μM in fresh culture medium (1:1000 dilution). IP was performed as described in (Hsieh et al., 2016). For transfection in fibroblasts, medium was replaced with Opti-MEM (Gibco) prior to transfection. 0.5 μg of DNA or 2 μl of Lipofectamine 2000 was diluted in Opti-MEM at room temperature (22°C) to a final volume of 50 μl in two separate tubes, and then contents of the two tubes were gently mixed, incubated for 25 min at room temperature, and subsequently added onto fibroblasts. After transfection for 6 hrs, Opti-MEM containing DNA-Lipofectamine complexes was replaced with regular culture medium. After transfection for 18 hrs, fibroblasts were live imaged, or treated with Miro1 reducer and/or CCCP. Mitochondria were isolated from cultured human fibroblasts as described previously (Wang and Schwarz, 2009b) with minor modifications. Briefly, CCCP in DMSO or the same volume of DMSO treated fibroblasts were lifted by a cell scraper, and mechanically homogenized with a Dounce homogenizer in 750 μl isolation buffer (200 mM sucrose, 10 mM TRIS/MOPS, pH 7.4). After centrifugation at 500 g for 10 min, crude supernatant was spun at 10,000 g for 10 min to pellet intact mitochondria. Mitochondrial pellet was washed twice with isolation buffer. After this step, supernatant was referred to “cytosolic fraction (Cyto)”, and pellet was resuspended in 50 μl lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, and 1% Triton X-100–T8787, Sigma-Aldrich) with 0.25 mM phenylmethanesulfonylfluoride (P7626, Sigma-Aldrich) and protease inhibitors (Roche) referred to “mitochondrial fraction (Mito)”. Samples were mixed 1:1 with 2× laemmli buffer (4% SDS, 20% Glycerol, 120 mM Tris-HCl, 0.02% bromophenol blue, 700 mM 2-mercaptoethanol) and boiled for 5 min prior to being loaded (Mito:Cyto=25:1) into an SDS-PAGE. 10% polyacrylamide gels (acrylamide:bis-acrylamide=29:1) and Tris-Glycine-SDS buffer (24.8 mM Tris, 192 mM glycine, 0.1% SDS) were used for electrophoresis. After electrophoresis, nitrocellulose membranes (1620115, Bio-Rad) were used in wet transfer with Tris-Glycine buffer (24.8 mM Tris, 192 mM glycine) at 360 mA for 2 hrs on ice. Transferred membranes were first blocked overnight in phosphate-buffered saline (PBS) containing 5% fat-free milk and 0.1% tween-20 at 4°C, and then incubated with the following primary antibodies: mouse anti-Miro1 (WH0055288M1, Sigma-Aldrich) at 1:1,000, rabbit anti-Miro1 (HPA010687, Sigma-Aldrich) at 1:1,000, rabbit anti-Miro2 (HPA012624, Sigma-Aldrich) at 1:800, rabbit anti-VDAC (4661S, Cell Signaling Technology) at 1:1,000, mouse anti-Mitofusin2 (H00009927-M01, Abnova) at 1:1,000, mouse anti-Parkin (sc32282, Santa Cruz Biotechnology) at 1:500, rabbit anti-LRRK2 (NB300-268, Novus Biologicals) at 1:500, rabbit anti-OPA1 (ab42364, Abcam) at 1:750, mouse anti-β-actin (A00702, Genscript) at 1:1,000, mouse anti-ubiquitin (A-104, Boston Biochem) at 1:500 or rabbit anti-GAPDH (5174S, Cell Signaling Technology) at 1:3,000, at 4°C overnight in blocking buffer. HRP-conjugated goat anti-mouse or rabbit IgG (Jackson ImmunoResearch Laboratories) were used at 1:5-10,000. West Dura ECL Reagents (34075, GE Healthcare) were used for ECL immunoblotting. Membranes were exposed to UltraCruz autoradiography films (Santa Cruz Biotechnology) and developed on a Konica Minolta SRX-101A developer. For fluorescent Western, blots were probed with Cy5-conjugated goat anti-mouse IgG (PA45009, GE Healthcare) at 1:5,000, and scanned at 635nm with a Molecular Dynamics Storm 860 Imager (Amersham BioSciences, Piscataway, NJ) in a linear range for fluorescent detection. Representative raw blots are in supplementary tables. Experiments were repeated for more than 3 times.
Statistics of Fibroblast Western Blotting Data
All experiments were performed in a blinded format, and the identities of the lines were unblinded either by us (NINDS lines) or by the PPMI researchers (PPMI lines). Films were scanned or digital blots were exported as 16-bit tiff format. The intensities of protein bands were measured by ImageJ (ver. 1.48V, NIH). The intensity of each band in the mitochondrial fraction was normalized to that of the mitochondrial loading control VDAC from the same blot, and expressed as a fraction of the mean of Healthy-1 with DMSO treatment (Hsieh et al., 2016); this control was included in every independent experiment. Values of Mean±S.E.M of Miro1 were reported in Table S1A. Values of Mean of Miro1, Mitofusin2, LRRK2, and Parkin were imported into heat maps in Figure 1D. The band intensities of VDAC were not significantly different among all fibroblast lines and conditions (p=0.8490, One-Way ANOVA Post-Hoc Tukey Test with adjustment). n=3-35 independent experiments. Mann-Whitney U Test was performed for comparing normalized Miro1 band intensities within the same subject (DMSO v.s. CCCP), and the P values were reported in Table S1A. The numbers of subjects with a P value >0.05 and <0.05 were counted respectively and used in Fisher Exact Test in Table S2A. Linear Regression Analysis was used to determine the correlation with the ratio of Miro1 intensity (mean intensity at CCCP/DMSO). One-Way ANOVA Post-Hoc Tukey Test with adjustment was performed for band intensities at baseline (P>0.3509 for all markers in “Cyto”+”Mito”). Statistical analyses were two-sided and performed using the Prism software (ver. 5.01, GraphPad).
PPMI is an international, multi-center and progressing study designed to identify PD biomarkers by the MJFF (http://www.ppmi-info.org/study-design/). The study design, subject recruitment criteria, site selection, and study assessment have been detailed in (Parkinson Progression Marker, 2011).
ELISA
All experiments were performed as blinded tests. 40 μM CCCP in DMSO or the same volume of DMSO alone was applied to fibroblasts for 6 hrs, and then cells were lysed in lysis buffer (100 mM Tris, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 0.5% Sodium deoxycholate) with protease inhibitor cocktail (539134, Calbiochem). Cell debris was removed by centrifugation at 17,000 g for 10 min at 4°C. Microplates (MaxiSorp, NUNC) were coated with mouse anti-Miro1 (clone 4H4, WH0055288M1, Sigma-Aldrich) at 1:1,000, chicken anti-β-actin (LS-C82919, LifeSpan BioSciences) at 1:750, mouse anti-β-actin (A00702, Genscript) at 1:1000, or mouse anti-ATP5β (ab14730, Abcam) at 1:1000 in 0.1 M sodium carbonate-bicarbonate buffer (3:7, pH=9.6) overnight at room temperature with cover to avoid evaporation. After plates were washed in wash buffer (0.05% Tween 20 in PBS, pH 7.3), nonspecific binding sites were blocked in PBS with 2% BSA (BP-1600-100, Fisher scientific) for 1 hr. Next, 50 μl of cell lysate prepared from above, purified full-length Miro1 protein (0-900 ng/ml, ab163047, Abcam), or serial dilutions of cell lysates of fibroblasts (Healthy-1) or HEK cells (1/16×-2×) were added and incubated at room temperature for 2 hrs. After washes, plates were incubated with biotinylated rabbit anti-Miro1 (ARP44818_P050, Aviva Systems Biology) at 1:1000, or biotinylated rabbit anti-β-actin (#5057S, Cell Signaling Technology) at 1:500, in 100 μl diluent (1% BSA in PBS, pH=7.3) for 2 hrs. Next, plates were washed and incubated with horseradish peroxidase-conjugated streptavidin (21130, Thermo Scientific) at 1:2000 in 100 μl diluent for 20 min. Plates were washed again, and 100 μl of the tetramethylbenzidine liquid substrate (SB01, Life Technologies) was added and incubated for another 20 min. The colorimetric reactions were stopped by 50 μl 1 M H2SO4 and absorbance was read at 450 nm by a microplate reader (FlexStation 3, Molecular devices). An experiment for generating the standard curve was included in each plate and representative standard plots were shown in figures. Each data point was from 4 independent experiments with 2 technical repeats each time. Mann-Whitney U test was performed for comparing Miro1 signals within the same subject (DMSO v.s. CCCP). Basal Miro1 signals were not significantly different among all lines (P>0.1177, One-Way ANOVA Post-Hoc Tukey Test with adjustment). The distribution of data points was expressed as box-whisker plots (Extreme, Quartile, Median).
AI
We employed the AtomNet neural network, the first deep neural network for structure-based drug design (Wallach et al., 2015). A single global AtomNet model was trained to predict binding affinity using Ki and IC50 values of several million small molecules and protein structures of several thousand different protein families that Atomwise has curated from both public databases and proprietary sources. Next, this model was used prospectively, even against novel binding sites with no known ligands, while the use of a single global model helped prevent overfitting. Training proceeded as the followings. First, we defined the binding site on a given protein structure using a flooding algorithm (Hendlich et al., 1997) based on an initial seed. The initial starting point of the flooding algorithm may be determined from a variety of methods, including a bound ligand annotated in the PDB database, crucial residues as revealed by mutagenesis studies, or identification of catalytic motifs previously reported. Second, we shifted the coordinates of the co-complex to a three-dimensional Cartesian system with an origin at the center-of-mass of the binding site. We then performed data augmentation by randomly rotating and translating the protein around the center-of-mass of the binding site. This prevents the neural network from learning a preferred protein orientation; as gravity rarely makes a significant contribution to binding, any such uncovered signal would be an inadvertent artifact from our data curation process. Third, for a given ligand, we sampled multiple poses within the binding site cavity. Each of these poses represents a putative co-complex and therefore, unlike previous structure-based predictive methods such as docking, our method does not require experimental co-complexes for either training or prediction. Each generated co-complex was then rasterized into a fixed-size regular three-dimensional grid, where the values at each grid point represent the structural features that are present at each grid point. Similar to a photo pixel containing three separate channels representing the presence of red, green, and blue colors, our grid points represent the presence of different atom types. These grids serve as the input to a convolutional neural network, defining the network’s receptive field. We used a network architecture of a 30×30×30 grid with a 1Å spacing for the input layer, followed by five convolutional layers of 32×33, 64×33, 64×33, 64×33, 64×23 (number of filters×filer-dimension), and a fully-connected layer with 256 ReLU hidden units. The scores for each pose in the ensemble were then combined through a weighted Boltzmann averaging to produce a final score. These scores were compared against the experimentally-measured Ki or IC50 of the protein and ligand pair, and the weights of the neural network were adjusted to reduce the error between the predicted and experimentally-measured affinity using a mean-square-error loss function. Training was done using the ADAM adaptive learning method, the backpropagation algorithm, and mini-batches with 64 examples per gradient step.
We used the 2.25Å structure of the C-terminal GTPase domain of human Miro1 with a bound GDP (PDB ID: 5KSO, chain A, position 411-592) (Klosowiak et al., 2016). We identified the predicted GDP binding sites V426, K427, N428, C429, G430, K431, S432, G433, V434, L443, Q446, K447, K454, D475, K528, S529, D530, L531, F557, T558, C559, N560 of Miro1 protein as the binding sites to small molecules, which were used for virtual screening by AtomNet. The crystalized GDP, phosphate ions, and all water molecules were removed from the sites before the AtomNet virtual screen. We screened the Mcule small-molecule library version v20180722, containing 6,835,320 small organic molecules for drug discovery purchasable from the chemical vendor Mcule (https://mcule.com/). For each small molecule, we generated a set of 64 poses within the binding site. Each of these poses was scored by the trained model, and the molecules were ranked by their scores. We then applied the filters for oral availability, toxicophores, and blood-brain barrier permeability using Lipinski’s rule of five (Lipinski et al., 2001), Eli Lilly’s MedChem Rules (Bruns and Watson, 2012), and Lipinski’s rule for CNS drugs (Pajouhesh and Lenz, 2005), respectively. These filters removed compounds with undesirable substructures and molecular properties including molecular weight, logP, and number of hydrogen-bond donors and acceptors. Our method does not introduce a manual bias caused by visual inspections of compounds, and yields an actual assessment of the ability of the software to identify hits with minimal operator intuition. After we identified Miro1 Reducer, we performed a visual inspection and revealed 7 amino acids residues (K427, N428, S432, Q446, K528, D530, K454) which make frequent molecular interactions, especially via hydrogen-bonds, with the poses of Miro1 Reducer. For Figure 2A, MW: molecular weight (Dalton); nof RotB: number of rotatable bonds; nof HBD: number of hydrogen-bond donors; nof HBA: number of hydrogen-bond acceptors; molLogP: calculated logP; molPSA: polar surface area (square Ångström).
Neuronal Derivation from iPSCs and Transfection
All iPSC lines in this study are summarized below and have been fully characterized by the previous studies (Hsieh et al., 2016; Shaltouki et al., 2018), the NINDS human and cell repository, and Stanford Stem Cell Core.
| iPSC line | Source | Etiology | PD mutation | Age | Sex |
|---|---|---|---|---|---|
| PD-I | NINDS (ND50050); Episomal reprogramming | Genetic | SNCA (A53T) | 51 | Female |
| PD-II | In house, Shaltouki et al., 2018; Sendai Virus reprogramming | Genetic | SNCA (A53T) | 51 | Female |
| PD-III | NINDS (ND39896) | Sporadic | Unknown | 77 | Male |
| Wild-type-I | NINDS (ND50085) | Isogenic control for PD-I | Wild-type | 51 | Female |
| Wild-type-II | Drs. Pera and Palmer, Hsieh et al., 2016 | Healthy Control | Wild-type | 62 | Male |
| Wild-type-III | NINDS (ND41864) | Healthy Control | Wild-type | 64 | Male |
| Wild-type-IV | Stanford Stem Cell Core, Shaltouki et al., 2018 | Healthy Control | Wild-type | 42 | Female |
iPSCs were derived to midbrain dopaminergic neurons as previously described with minor modifications (Byers et al., 2011; Hsieh et al., 2016; Kriks et al., 2011; Nguyen et al., 2011; Wichterle et al., 2002). Briefly, neurons were generated using an adaptation of the dual-smad inhibition method with the use of smad inhibitors dorsormorphin (Sigma) and SB431542 (Tokris), and the addition of GSK3β inhibitor CHIR99021 (Stemgent). SHH was replaced with the smoothened agonist SAG. To gain a higher purity of neural precursor cells, 12 days after neural induction, rosette-forming neuroectodermal cells were manually lifted and detached en bloc, and then cultured in suspension in a low-attachment dish (430589, Corning Inc.) with N2 medium with 20 ng/ml BDNF, 200 μM Ascorbic Acid, 500 nM SAG, and 100 ng/ml FGF8a. On day 17, neurons were transferred onto poly-ornithine and laminin-coated glass coverslips in a 24-well plate. On day 18, medium was switched to N2 medium supplemented with 20 ng/ml BDNF, 200 μM Ascorbic Acid, 20 ng/ml GDNF, 1 ng/ml TGFβ3, and 500 μM Dibutyryl-cAMP for maturation of dopaminergic neurons. Neurons were used at day 21-23 after neuronal induction, when about 80-90% of total cells expressed the neuronal marker TUJ-1, and 12% of total cells expressed TH and markers consistent with ventral midbrain neuronal subtypes (Sanders et al., 2014; Shaltouki et al., 2018). Antimycin A (A8674, Sigma-Aldrich) was applied to neurons at 10 μM.
For transfection, on day 20 after neuronal induction, culture medium was replaced with Opti-MEM (Gibco) prior to transfection. 0.5 μg EGFP and 0.5 μg mito-dsRed, or 5 μl Lipofectamine 2000 was diluted in Opti-MEM at room temperature (22°C) to a final volume of 50 μl in two separate tubes, and then contents of the two tubes were gently mixed, incubated for 20 min at room temperature, and subsequently added onto neurons. After transfection for 6 hrs, Opti-MEM containing DNA-Lipofectamine complexes was replaced with regular N2 medium. After transfection for 3 days, neurons were imaged.
Live Image Acquisition and Quantification
We transfected neurons with mito-dsRed and EGFP at day 20 after neuronal induction, and live imaged mito-dsRed from EGFP-positive axons 3 days later. As described previously (Wang and Schwarz, 2009a, b; Wang et al., 2011), neurons on glass coverslips were placed in a 35-mm petridish containing the Hibernate E low-fluorescence medium (BrainBits) on a heated stage of 37°C, and imaged with a 63×/N.A.0.9 water-immersion objective with excitation at 561 nm or 488 nm. Axons were identified by morphology, and axons longer than 50 μm were selected for recording. Time-lapse movies were obtained continually with 5 sec intervals before and after Antimycin A (100 μM, Sigma-Aldrich) was added. Movie length ranged from 120 to 300 min. For quantification, kymographs were generated from time-lapse movies by ImageJ, representing a 100-sec period either right before, or following different time points after addition of Antimycin A. Each kymograph was then imported into a macro written in Labview (NI, TX), and individual mito-dsRed puncta were traced using a mouse-driven cursor at the center of the mito-dsRed object. Using Matlab (The MathWorks, MA), we determined the following parameters: 1) the instantaneous velocity of each mitochondrion, 2) the average velocity of those mitochondria that are in motion, 3) the percent of time each mitochondrion is in motion, 4) stop frequency, and 5) turn back frequency. The intensity of mitochondria was measured using ImageJ. n=5-8 axons from 5-8 separate transfections.
Immunocytochemistry and Confocal Microscopy
PI (P4170, Sigma) at 2.5 μg/ml or 4’, 6-Diamidino-2-phenylindole (Dapi-D9542, Sigma) at 0.5 μg/ml was applied in culture media at room temperature in the dark for 10 min, MitoSox (M36008, Invitrogen) at 2 μM was applied in culture media at 37°C for 10 min, or TMRM (T668, Molecular Probes) at 25 nM was applied in culture media at 37°C for 30 min, and cells were washed with HBSS and imaged immediately in a 35-mm petridish containing the Hibernate E low-fluorescence medium with a 63×/N.A.0.9 water-immersion objective. For immunostaining, cells were fixed in 2-4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for 15 min, then washed three times in PBS, and blocked in PBS with 0.1% Saponin (Sigma-Aldrich) for 60 min. Cells were then immunostained with rabbit anti-TH (AB-152, Millipore) at 1:500 overnight, or mouse anti-Myc (sc-40, SantaCruz) at 1:1,000, rabbit anti-Tom20 (13929S, Cell Signaling Technology) at 1:1000, or mouse anti-ATP5β (ab14730, Abcam) at 1:500 overnight followed with Dapi at 0.5 μg/ml for 10 min. Adult fly brains were dissected in PBST (0.3% Triton X-100 in PBS), and incubated with fixative solution (4% formaldehyde in PBST) for 15 min. Fixed samples were immunostained with rabbit anti-TH (AB-152, EMD Millipore) at 1:200. Samples were imaged at room temperature with a 20×/N.A.0.60 or a 63×/N.A.1.30 oil Plan-Apochromat objective on a Leica SPE laser scanning confocal microscope, with identical imaging parameters among different genotypes in a blinded fashion. Images were processed with ImageJ (Ver. 1.48, NIH) using only linear adjustments of contrast and color. n=3-11 independent experiments.
GTPase Assay
IPed Miro1 was eluted from protein-A beads by incubation with 60 μl 0.2 M glycine (pH 2.5) for 10 min, followed by centrifugation at 3000 g for 2 min. Supernatants (eluates) were collected and neutralized by adding an equal volume of Tris pH 8.0. Eluates were processed for colorimetric analysis of GTPase activity at room temperature by using a GTPase Activity Kit according to the manufacturer’s instructions (602-0120, Novus Biologicals). 100 μl eluates were mixed with 100 μl substrate/buffer mix (20 μl 0.5 M Tris Buffer, 5 μl 0.1 M MgCl2, 10 μl 10 mM GTP, and 65 μl ddH2O). 200 μl of inorganic phosphate group (Pi) standard (0-50 μM) was prepared in water. Next, 50 μl PiColorLock™ mix was added into either Pi standards or samples.
After 2 min, stabilizer was added and mixed thoroughly. 30 min later, absorbance was read at 650 nm by a microplate reader (FlexStation 3, Molecular devices). The assay was validated using purified Miro1 protein (ab163047, Abcam). The concentrations of purified Miro1 protein ranging from 0-900 ng/ml showed a linear dependency on the absorbances at 650 nm which reflect released Pi (R2=0.9711, P=0.0021). Omitting GTP or Miro1 eliminated the signals (Pi<3 μM). Experiments were repeated twice.
qPCR
Total RNA was extracted from at least 106 cells per sample using TRIzol® (GIBCO) according to the manufacturer’s instructions. Concentrations of total RNA were measured using a Nano Drop. 1 μg of total RNA was then subjected to DNA digestion using DNase I (Ambion), immediately followed by reverse transcription using the iScript Reverse Transcription Supermix (1708841, BIO-RAD). qPCR was performed using the StepOnePlus™ instrument (Thermo Fisher Scientific) and SYBR® Green Supermix (172-5270, BIO-RAD) by following the manufacturer’s instructions. Human GAPDH was amplified as internal standards. Expression levels were analyzed by the StepOne™ Software (Version 2.2.2). The relative expression level of Miro1 was divided by that of GAPDH from the same experiment. Each sample was analyzed in duplicate from 4 independent biological repeats. The following primers were used:
GAPDH forward: 5’- ACCACAGTCCATGCCATCAC-3’
GAPDH reverse: 5’- TCCACCACCCTGTTGCTGT-3’
Miro1 forward: 5’- GGGAGGAACCTCTTCTGGA-3 ‘
Miro1 reverse: 5’- ATGAAGAAAGACGTGCGGAT-3 ‘
QUANTIFICATION AND STATISTICAL ANALYSIS
Throughout the paper, the distribution of data points was expressed as box-whisker or dot-plot, except otherwise stated. One-Way ANOVA Post-Hoc Tukey Test was performed for comparing multiple groups, except otherwise stated. Mann-Whitney U Test was performed for comparing two groups. Statistical analyses were performed using the Prism software (ver. 5.01, GraphPad). For all experiments, between 3 and 150 animals or independent experiments were used. The number of animals and experimental replications (n) can be found in Figure Legends and Method Details. No statistical methods were used to predetermine sample sizes, but the number of flies, experiments, and biological replicates were chosen based on the nature of the experiments (it is usually difficult to assess an outcome that follows a normal distribution in our experiments), degree of variations, and published papers describing similar experiments. We did not exclude any data. * P<0.05, ** P<0.01, *** P<0.001 for all figures.
DATA AND CODE AVAILABILITY
Representative raw Western blots for Figure 1 in the paper are available at Mendeley Data DOI: 10.17632/z2g8wbvhgx.2
Supplementary Material
Table S1. Demographic Information and Miro1 Values. Related to Figure 1. (A) Demographic information and clinical scores are from the online databases of the consortia. Not all information is available for all subjects. Particular analyses use data from the subjects with the available information. P values are from comparing the normalized Miro1 intensities within the same subject (CCCP v.s. DMSO). (B) Demographic Information of Additional Cohorts for ELISA.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-Miro1 | Sigma | HPA010687, RRID:AB_1079813 |
| mouse anti-Miro1 | Sigma | WH0055288M1, RRID:AB_1843347 |
| rabbit anti-Miro2 | Sigma | HPA012624, RRID:AB_1856272 |
| rabbit anti-VDAC | Cell Signaling Technology | 4661S, RRID:AB_10557420 |
| mouse anti-Mitofusin2 | Abnova | H00009927-M01, RRID:AB_714775 |
| mouse anti-Parkin | Santa Cruz Biotechnology | sc32282, RRID:AB_628104 |
| rabbit anti-LRRK2 | Novus Biologicals | NB300-268, RRID:AB_10126471 |
| rabbit anti-OPA1 | Abcam | ab42364, RRID:AB_944549 |
| mouse anti-β-actin | Genscript | A00702, RRID:AB_914102 |
| mouse anti-β-actin | Abcam | Ab8224, RRID: AB_449644 |
| rabbit anti-GAPDH | Cell Signaling Technology | 5174S, RRID:AB_10622025 |
| chicken anti-β-actin | LifeSpan BioSciences | LS-C82919, RRID:AB_1648164 |
| rabbit anti-TH | EMD Millipore Corporation | AB152, RRID:AB_390204 |
| mouse anti-ATP5β | Abcam | ab14730, RRID:AB_301438 |
| Rabbit anti-Tom20 | Cell Signaling Technology | 13929S, RRID:AB_2631994 |
| biotinylated rabbit anti-Miro1 | Aviva Systems Biology | ARP44818_P050, RRID:AB_2047790 |
| biotinylated rabbit anti-β-actin | Cell Signaling Technology | 5057, RRID:AB_10694076 |
| guinea pig anti-DMiro | Tsai et al., 2014 | GP5 |
| rabbit anti-marf | Ziviani et al., 2010 | |
| mouse anti-tubulin | Sigma | T6199, RRID:AB_477583 |
| mouse anti-VDAC | Abcam | ab14734, RRID:AB_443084 |
| mouse anti-Myc | Santa Cruz | sc-40, RRID:AB_627268 |
| mouse anti-ubiquitin | Boston Biochem | A-104, RRID:AB_2687460 |
| HRP-conjugated goat anti-mouse IgG | Jackson Laboratories | 115-035-166, RRID:AB_2338511 |
| HRP-conjugated goat anti-rabbit IgG | Jackson Laboratories | 111-035-144, RRID:AB_2307391 |
| Cy5-conjugated goat anti-mouse IgG | GE Healthcare | PA45009, RRID:AB_772199 |
| Alexa Fluor 555-conjugated donkey anti-rabbit IgG | Thermo Fisher | A-31572, RRID:AB_162543 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| TMRM | Molecular Probes | T668 |
| PI | Sigma | P4170 |
| MitoSox | Invitrogen | M36008 |
| MG132 | Sigma | M7449 |
| DAPI | Sigma | D9542 |
| Antimycin A | Sigma | A8674 |
| CCCP | Sigma | C2759 |
| Recombinant Human Miro1 | Abcam | ab163047 |
| 5-bromo-7-methyl-1H,1’H,2H,2’H-[3,3’-biindolylidene]-2,2’-dione | Mcule | P-6825976 |
| ethyl 6-(2-chloro-6-fluorophenyl)-4-methyl-2-sulfanyl-1,6-dihydropyrimidine-5-carboxylate | Mcule | P-7500881 |
| 3-(3-fluorophenyl)-1-(4-{[6-(1H-imidazol-1-yl)pyrimidin-4-yl]amino}phenyl)urea | Mcule | P-3876569 |
| 3-(2,3-dimethylphenyl)-1-(4-{[6-(1H-imidazol-1-yl)pyrimidin-4-yl]amino}phenyl)urea | Mcule | P-4007459 |
| 3-(2-chlorophenyl)-1-(4-{[4-(dimethylamino)-6-methylpyrimidin-2-yl]amino}phenyl)urea | Mcule | P-4123009 |
| 5-chloro-N4-[(3R,4S)-4-[(3,5-dimethyl-1,2-oxazol-4-yl)methoxy]oxan-3-yl]-6-methylpyrimidine-2,4-diamine | Mcule | P-427170230 |
| 2-amino-6-(2,2-dimethyloxan-4-yl)-4-[2-(ethylamino)pyrimidin-5-yl]-5-propylpyridine-3-carbonitrile | Mcule | P-9111737 |
| 4-(4-chloro-1-methyl-1H-indazol-3-yl)-N-methyl-6-oxo-1H,4H,5H,6H,7H-pyrazolo[3,4-b]pyridine-3-carboxamide | Mcule | P-7687498 |
| ethyl 2-{[5-(2-amino-3-cyano-5,6,7,8-tetrahydroquinolin-4-yl)pyrimidin-2-yl]amino}acetate | Mcule | P-9346473 |
| 2-amino-6-(4-hydroxyphenyl)-5-methyl-4-[2-(methylsulfanyl)pyrimidin-5-yl]pyridine-3-carbonitrile | Mcule | P-8406267 |
| 2-amino-6-(3-fluoro-4-methoxyphenyl)-4-(5-hydroxy-2-methoxyphenyl)pyridine-3-carbonitrile | Mcule | P-8629452 |
| Critical Commercial Assays | ||
| ATP Determination Kit | Sigma | 11699709001 |
| GTPase Activity Kit | Novus Biologicals | 602-0120 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human iPSCs (see method) | NINDS, Shaltouki et al., 2018 | Healthy controls and PD patients |
| Human fibroblasts (see method) | NINDS, PPMI, Mayo Clinic, Stanford ADRC, Coriell | Healthy controls and PD patients |
| HEK293T cells | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| PINK1RV | Park et al., 2006 | N/A |
| PINK1B9 | Park et al., 2006 | N/A |
| UAS-hLRRK2G2019S-Flag | Lin et al., 2010 | N/A |
| UAS-SNCA-A53T | Trinh et al., 2008 | N/A |
| Elav-GAL4 | Lab stock | N/A |
| Actin-GAL4 | Lab stock | N/A |
| W1118 | Lab stock | N/A |
| Oligonucleotides | ||
| See Methods | ||
| Recombinant DNA | ||
| pRK5-Myc-Miro1 | Wang et al., 2011 | N/A |
| pRK5-Myc-Miro1-7A | This paper, custom-made by Synbio Technologies | N/A |
| pcDNA3-HA-Miro1 | Hsieh et al., 2016 | N/A |
| pLevP1-PLK-mito-dsRed | Hsieh et al., 2016 | N/A |
| pCMV-GFP | Hsieh et al., 2016 | N/A |
| pEGFP-peroxisomes-0 | Addgene | #54501 |
| Software and Algorithms | ||
| Prism | ver. 5.01, GraphPad | |
| Other | ||
HIGHLIGHTS.
Miro1 accumulates on depolarized mitochondria in a large cohort of PD fibroblasts
A small molecule can promote Miro1 degradation in PD fibroblasts
Treating PD models with this compound rescues dopaminergic neurodegeneration
CONTEXT AND SIGNIFICANCE.
There are quite a few un-met needs in the Parkinson’s disease (PD) field which limit efforts in finding a cure. In particular, a more effective and reliable molecular target for PD is urgently needed. Here researchers at Stanford University and their collaborators explore the mitochondrial protein Miro1 both as a common molecular signature for detecting PD and as a drug target for treating the disease. The researchers discover a unifying defect of Miro1 in skin cells of a large cohort of PD patients, and a small molecule that can repair this Miro1 defect and rescue Parkinson’s phenotypes. These results demonstrate the clinical utility of Miro1 marker in skin cells for monitoring Miro1-based target engagements and the therapeutic promise of compounds targeting Miro1.
ACKNOWLEDGEMENTS
We thank M. Kim, Drs. W. Weis, S. Pokutta, V. Bharat for technical support, the Neuroregeneration Lab at Mayo Clinic, NINDS, Michael J. Fox Foundation (MJFF), Coriell Institute, and Stanford ADRC Neuropathology and Biospecimens Core (P50 AG047366) for cell lines, Drs. A. Whitworth and K. Zinsmaier for antibodies, and Dr. S. Omlid for sourcing compounds. This work is supported by Klingenstein Fund (X.W.), CIRM (X.W.), NINDS (X.W. RO1NS089583), Archer Fund (X.W.), Stanford Parkinson’s Disease Seed Grant (X.W.), Stanford SPARK (X.W.), Belgian American Education Foundation (R.V.), Sol Goldman Charitable Trust (Z.K.W.), Donald G. and Jodi P. Heeringa Family (Z.K.W.), Haworth Family Professorship in Neurodegenerative Diseases Fund (Z.K.W.), Albertson Parkinson’s Research Foundation (Z.K.W.), and Mayo Clinic Center for Regenerative Medicine (M.D.D., Z.K.W., G.B.). PPMI is funded by MJFF and funding partners, including Abbvie, Avid, Biogen, Bristol-Myers Squibb, COVANCE, GE Healthcare, Genentech, GlaxoSmithKline, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Piramal, Roche, Servier, and UCB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
NO COMPETING FINANCIAL INTERESTS
REFERENCES
- Bartels T (2016). Conformation-Specific Detection of alpha-Synuclein: The Search for a Biomarker in Parkinson Disease. JAMA Neurol. [DOI] [PubMed] [Google Scholar]
- Bonifati V (2002). Deciphering Parkinson’s disease--PARK8. The Lancet Neurology 1, 83. [DOI] [PubMed] [Google Scholar]
- Bruns RF, and Watson IA. (2012). Rules for identifying potentially reactive or promiscuous compounds. J Med Chem 55, 9763–9772. [DOI] [PubMed] [Google Scholar]
- Byers B, Cord B, Nguyen HN, Schule B, Fenno L, Lee PC, Deisseroth K, Langston JW, Pera RR, and Palmer TD (2011). SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PloS one 6, e26159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Quan JI, Li L, Kinghorn KJ, Ivanov DK, Tain LS, Slack C, Kerr F, Nespital T, Thornton J, Hardy J, et al. (2016). Lithium Promotes Longevity through GSK3/NRF2-Dependent Hormesis. Cell Rep 15, 638–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, and Chan DC (2011). Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Human molecular genetics 20, 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Course MM, Scott AI, Schoor C, Hsieh CH, Papakyrikos AM, Winter D, Cowan TM, and Wang X (2018). Phosphorylation of MCAD selectively rescues PINK1 deficiencies in behavior and metabolism. Molecular biology of the cell 29, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendlich M, Rippmann F, and Barnickel G (1997). LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model 15, 359–363, 389. [DOI] [PubMed] [Google Scholar]
- Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, Schule B, Krainc D, Palmer TD, and Wang X (2016). Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell stem cell 19, 709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinghorn KJ, Castillo-Quan JI, Bartolome F, Angelova PR, Li L, Pope S, Cocheme HM, Khan S, Asghari S, Bhatia KP, et al. (2015). Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain : a journal of neurology 138, 1801–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, and Shimizu N (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. [DOI] [PubMed] [Google Scholar]
- Klosowiak JL, Park S, Smith KP, French ME, Focia PJ, Freymann DM, and Rice SE (2016). Structural insights into Parkin substrate lysine targeting from minimal Miro substrates. Scientific reports 6, 33019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le W, Dong J, Li S, and Korczyn AD (2017). Can Biomarkers Help the Early Diagnosis of Parkinson’s Disease? Neurosci Bull 33, 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Tsai PI, Wu RM, and Chien CT (2010). LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ss. The Journal of neuroscience : the official journal of the Society for Neuroscience 30, 13138–13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, and Feeney PJ (2001). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schule B, Dolmetsch RE, Langston W, et al. (2011). LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell stem cell 8, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajouhesh H, and Lenz GR (2005). Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161. [DOI] [PubMed] [Google Scholar]
- Parkinson Progression Marker I (2011). The Parkinson Progression Marker Initiative (PPMI). Progress in neurobiology 95, 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rival T, Soustelle L, Strambi C, Besson MT, Iche M, and Birman S (2004). Decreasing glutamate buffering capacity triggers oxidative stress and neuropil degeneration in the Drosophila brain. Current biology : CB 14, 599–605. [DOI] [PubMed] [Google Scholar]
- Sanders LH, Laganiere J, Cooper O, Mak SK, Vu BJ, Huang YA, Paschon DE, Vangipuram M, Sundararajan R, Urnov FD, et al. (2014). LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiology of disease 62, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaltouki A, Hsieh CH, Kim MJ, and Wang X (2018). Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol 136, 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofola O, Kerr F, Rogers I, Killick R, Augustin H, Gandy C, Allen MJ, Hardy J, Lovestone S, and Partridge L (2010). Inhibition of GSK-3 ameliorates Abeta pathology in an adult-onset Drosophila model of Alzheimer’s disease. PLoS genetics 6, e1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, and Youle RJ (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. The Journal of cell biology 191, 1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh K, Moore K, Wes PD, Muchowski PJ, Dey J, Andrews L, and Pallanck LJ (2008). Induction of the phase II detoxification pathway suppresses neuron loss in Drosophila models of Parkinson’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PI, Course MM, Lovas JR, Hsieh CH, Babic M, Zinsmaier KE, and Wang X (2014). PINK1-mediated phosphorylation of Miro inhibits synaptic growth and protects dopaminergic neurons in Drosophila. Scientific reports 4, 6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PI, Lin CH, Hsieh CH, Papakyrikos AM, Kim MJ, Napolioni V, Schoor C, Couthouis J, Wu RM, Wszolek ZK, et al. (2018). PINK1 Phosphorylates MIC60/Mitofilin to Control Structural Plasticity of Mitochondrial Crista Junctions. Molecular cell 69, 744–756 e746. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. [DOI] [PubMed] [Google Scholar]
- Wallach I, Dzamba M, and Heifets A (2015). Atomnet: A deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv arXiv.org > cs > arXiv:1510.02855.
- Wang X, and Schwarz TL (2009a). Imaging axonal transport of mitochondria. Methods in enzymology 457, 319–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, and Schwarz TL (2009b). The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, and Schwarz TL (2011). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, and Jessell TM (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397. [DOI] [PubMed] [Google Scholar]
- Wren MC, Zhao J, Liu CC, Murray ME, Atagi Y, Davis MD, Fu Y, Okano HJ, Ogaki K, Strongosky AJ, et al. (2015). Frontotemporal dementia-associated N279K tau mutant disrupts subcellular vesicle trafficking and induces cellular stress in iPSC-derived neural stem cells. Molecular neurodegeneration 10, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. [DOI] [PubMed] [Google Scholar]
- Ziviani E, Tao RN, and Whitworth AJ (2010). Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proceedings of the National Academy of Sciences of the United States of America 107, 5018–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Demographic Information and Miro1 Values. Related to Figure 1. (A) Demographic information and clinical scores are from the online databases of the consortia. Not all information is available for all subjects. Particular analyses use data from the subjects with the available information. P values are from comparing the normalized Miro1 intensities within the same subject (CCCP v.s. DMSO). (B) Demographic Information of Additional Cohorts for ELISA.
Data Availability Statement
Representative raw Western blots for Figure 1 in the paper are available at Mendeley Data DOI: 10.17632/z2g8wbvhgx.2