

Abstract
The mitochondria have emerged as a novel target for cancer chemotherapy primarily due to their central roles in energy metabolism and apoptosis regulation. Here, we report a new molecular approach to achieve high levels of tumor- and mitochondria-selective deliveries of the anticancer drug doxorubicin. This is achieved by molecular engineering, which functionalizes doxorubicin with a hydrophobic lipid tail conjugated by a solubility-promoting poly(ethylene glycol) polymer (amphiphilic doxorubicin or amph-DOX). In vivo, the amphiphile conjugated to doxorubicin exhibits a dual function: (i) it binds avidly to serum albumin and hijacks albumin’s circulating and transporting pathways, resulting in prolonged circulation in blood, increased accumulation in tumor, and reduced exposure to the heart; (ii) it also redirects doxorubicin to mitochondria by altering the drug molecule’s intracellular sorting and transportation routes. Efficient mitochondrial targeting with amph-DOX causes a significant increase of reactive oxygen species levels in tumor cells, resulting in markedly improved antitumor efficacy than the unmodified doxorubicin. Amphiphilic modification provides a simple strategy to simultaneously increase the efficacy and safety of doxorubicin in cancer chemotherapy.
Keywords: doxorubicin, mitochondria, amphiphiles, chemotherapy, drug delivery
Graphical Abstract
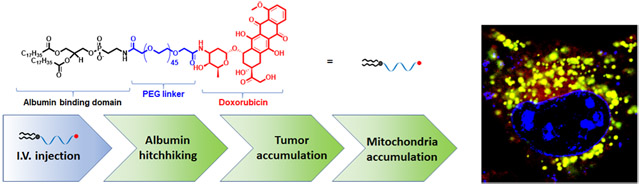
INTRODUCTION
Anthracyclines, especially doxorubicin (DOX), have broad-spectrum antineoplastic activities and have been extensively used in cancer chemotherapy for more than 40 years.1,2 However, intrinsic or acquired drug resistance greatly limited the success of DOX in the clinical management of cancers.2-5 Additionally, like many chemotherapeutic drugs, doxorubicin targets both proliferating cancer and normal cells, such treatment can lead to severe off-target toxicity and side effects,3-5 especially in patients with advanced disease requiring dose escalation.
An emerging strategy to simultaneously enhance efficacy and reduce toxicity is targeted delivery of doxorubicin to tumor mitochondria,6-17 the unique cellular organelles that play a central role in the regulation of fundamental tumor cellular functions, including cellular metabolism, adenosine triphosphate production, reactive oxygen species (ROS) generation, and apoptosis, among many others.7-9 Delivery of DOX to mitochondria may bypass the classical resistance pathways, while at the same time improving or maintaining its cytotoxic effects.9 Mitochondria-targeted anticancer therapeutics can eradicate resistant cancer cells through several possible mechanisms.9,14,15,18 For example, mitochondria-specific delivery of doxorubicin or similar anthracyclines has been shown to exert their cytotoxic effects by intercalating mitochondrial DNA9 or by oxidative damage of DNA, membrane-bound proteins and enzymes,14,15,18 resulting in a significantly enhanced cytotoxic effect in cancer cells.
Despite intensive research, to date, no mitochondria-targeting pharmaceutical formulations have been approved clinically. This is in part because in vivo, a successful mitochondriotropic delivery requires multilevels of targeting: it must achieve sufficient circulating time in blood for drug exposure and must achieve tumor tissue- and tumor cellspecific accumulation followed by mitochondria-specific accumulation.7-15 Although considerable research attempts have been made to incorporate multiple targeting ligands for mitochondria-targeted delivery,7-15 many of these strategies fail to overcome the multiple biological barriers in vivo. For example, delocalized lipophilic cations (DLCs) are compounds that efficiently accumulate within mitochondria, mainly in response to mitochondria membrane potential.19-25 However, the intrinsic toxicities associated with DLCs have hampered their clinical development.26,27 Further, such small molecular compounds fail to achieve the multilevels of targeting in vivo, in some cases, nonspecific accumulation in brain, heart, liver, and muscle was observed.19 Attempting to target mitochondria also includes the use of synthetic peptides and amino-acid-based transporters, which either derived from mitochondrial targeting sequence9,28-30 or comprised of altered lipophilicity and charge that exhibit strong affinity toward mitochondria.31-34 The major issues of these peptides are their considerable molecular sizes, poor water solubility, lack of membrane permeability, and low serum stability.8,23 Another strategy is to make use of emerging biopharmaceutical nanotechnologies, which have demonstrated to offer many advantages compared with traditional small molecular drugs alone. Drug carriers based on nanoparticles are modified with tumor- and/or mitochondria-specific ligands.13,35-38 However, multilevel drug targeting nanoparticles require complex designs to increase drug encapsulation efficiency, to evade host immune system, and to release drug upon intracellular exposure.39
Here, we show that a simple amphiphilic modification on doxorubicin (amph-DOX) can overcome multiple biological barriers and selectively target tumor mitochondria in vivo. This is achieved by molecular engineering, which functionalizes doxorubicin with a lipophilic diacyl lipid connected by a poly(ethylene glycol) linker (Figure 1A). This amphiphilic modification fulfills a two-fold purpose: first, amph-DOX reaches and penetrates solid tumor by “hitchhiking” on albumin protein.40-42 Albumin-binding increases the hydrodynamic size of doxorubicin and prolongs its circulating time in the blood.41 Albumin binding also increases DOX’s uptake in the tumor by the enhanced permeation and retention (EPR) effect and more importantly, by active metabolic uptake because tumors heavily use albumin as an energy and nutrient source.40,41 Second, amph-DOX accumulates in mitochondria following tumor cell uptake through a yet unknown mechanism. Compared with free DOX, iv injection of amph-DOX heavily accumulates in tumor but not in heart. Efficient mitochondria targeting with amph-DOX causes a significant increase in oxidative stress in tumor mitochondria, resulting in markedly improved antitumor efficacy. Thus, in vivo, amphiphilic functionalization improves the doxorubicin molecule’s physicochemical properties, which in turn redefines its bioavailability, organ and subcellular distributions. Amphiphilic modification represents a simple, effective, and nontoxic molecular approach for mitochondria-targeted delivery of doxorubicin in vivo.
Figure 1.

Amph-DOX binds to albumin in blood. (A) Molecular structure of amphiphilic doxorubicin (amph-DOX). (B–D) amph-DOX, but not free DOX binds to serum albumin in blood. Mouse blood samples were incubated with 0.5 μM DOX or amph-DOX for 4 h, after centrifugation, cells were analyzed by flow cytometry (B) and sera were analyzed by gel electrophoresis (D). DOX concentrations in serum were quantified by fluorescence spectroscopy (C).
EXPERIMENTAL SECTION
Materials and Chemicals.
All chemicals were purchased from Sigma-Aldrich and used without further purification unless noted otherwise. Doxorubicin hydrochloride was purchased from LC laboratories, 3-(N-succinimidyloxyglutaryl) aminopropyl, poly(ethylene glycol)-carbamyl distearoylphosphatidyl-ethanolamine (DSPE-PEG2000-NHS) were obtained from Biochempeg scientific Inc. Cholesterol poly(ethylene glycol) NHS and DSPE-PEG2000-hydrazide was purchased from Nanocs Inc.
Animals and Cells.
Animals were housed in the United States Department of Agriculture (USDA)-inspected Wayne State University animal facility under federal, state, local and NIH guidelines for animal care. Female C57BL/6 mice (5–8 weeks) were obtained from the Jackson Laboratory. B16F10, 4T1 cells were purchased from ATCC. The OVCAR-8 human ovarian carcinoma cell line and its doxorubicin resistant derivative NCI/ADR-RES cell line were obtained from NIH. Cells were cultured in complete medium (RPMI 1640, 10% fetal bovine serum (Greiner Bio-one), 100 U/mL penicillin G sodium, and 100 μg/mL streptomycin (Pen/Strep)).
Synthesis and Characterization of amph-DOX.
Doxorubicin hydrochloride (DOX, 5 mg) and 38 mg of DSPE-PEG2000-NHS (molar ratio of DSPE-PEG2000-NHS/DOX = 1.5:1) were dissolved in 500 μL and 4.5 mL of dimethyl sulfoxide (DMSO), respectively. These two solutions were mixed and activated with 3 μL of triethylamine (TEA). After stirred in the dark at 25 °C for 24 h, the solution was dried in a stream of air for 72 h. The remaining reaction residues were dissolved in 5 mL of phosphate-buffered saline (PBS) buffer (0.1 M, pH = 7.4) with sonication. Amph-DOX was purified by reverse-phase high-performance liquid chromatography (HPLC) with a C4 column (Thermo Scientific, 250 × 4.6 mm, 5 μm). Samples of 100 μL were injected and separations were performed at 25 °C using a flow rate of 1.0 mL/min by a liquid chromatography system (Agilent Technologies 1220 Infinity). DSPE-PEG2000-NHS and DOX were detected by measurement of the UV absorbance at 260 and 485 nm, respectively. DSPE-PEG2000-DOX was monitored by both wavelengths. A solvent gradient (Table S2) with methanol and triethylammonium acetate (TEAA) buffer (0.1 M pH = 7.4) was used for the separation. Amph-DOX was collected (typical retention time: 12–14 min). The solvent was air dried and the final product was dissolved in DMSO and concentration was determined by UV/vis spectrophotometry (Thermo Scientific). DSPE-PEG2000-DOX (amph-DOX) was confirmed by 1H NMR (Varian, 400 MHz) and mass spectrometry analysis.
Albumin-Binding Assay.
A gel electrophoresis mobility shift assay was used to detect albumin protein binding with amph-DOX. The solutions of free DOX and amph-DOX were incubated with freshly isolated mouse blood for 4 h at 37 °C. The resulting mixtures were separated into two equal volumes. Half of the sample was used for flow cytometry analysis and the other half for fluorescent spectroscopy and gel electrophoresis. Samples were loaded for electrophoresis run under 200 V for 30 min through 0.5% agarose gel. Images were recorded using a digital camera under UV illustration. For the Förster resonance energy transfer (FRET) assay, Alexa660 (ThermoFisher Scientific)-labeled bovine serum albumin (BSA-Alexa660) was incubated with 10 μM DOX or amph-DOX in PBS (pH 7.4) for 4 h at 37 °C, after that samples were analyzed by spectrofluorometer (JASCO FP-6500). DOX or amph-DOX was excited at 470 nm.
In Vitro Cell Viability Assay.
The antiproliferation activities of the free anticancer drug DOX and the amphiphilic drug amph-DOX against B16F10, 4T1, OVCAR-8, and NCI/ADR-RES cells were evaluated using the AlamarBlue assay method. B16F10, 4T1 cells (5 × 104 cells/well) and NCI/ADR-RES cells (1 × 105 cells/well) cultured with 100 μL of medium were seeded in 96-well plates, and incubated overnight to adhere. Cells were incubated with free DOX or amph-DOX at serial doxorubicin concentrations ranging from 0.05 to 10 μM for 24 or 48 h, following by the addition of 10 μL of alamarBlue reagent and incubated for another 1 h. Cells treated with complete medium were used as the controls. Finally, the absorbance was measured at 570 nm with 600 nm as a reference by a microplate reader (Thermo Scientific). The percentage of surviving cells was calculated as the absorbance ratio of treated to untreated cells. The half maximal inhibitory concentration (IC50) was determined from the dose–response curve. All of the experiments were carried out in triplicate.
In Vitro Uptake and Subcellular Distribution.
The cell uptakes of free DOX and amph-DOX were examined in B16F10 or NCI/ ADR-RES cells by flow cytometry. Cells were seeded to 48-well plate (1 × 106 cells/well) and incubated at 37 °C for overnight. The cell medium was removed and replaced with DOX and amph-DOX at a final concentration of 1.0 μM for different time periods. The cells were harvested and washed with 1× PBS buffer three times and analyzed by flow cytometry using an Attune acoustic focusing cytometer (Applied Biosystems). Each assay was performed in triplicate.
To determine the intracellular distribution of amph-DOX, cells (1 × 104 cells/well) were seeded on a coverslip in six-well plates and cultured at 37 °C for 24 h to achieve confluence. For tracking mitochondria by MitoTracker Green FM, cells were treated with free doxorubicin (DOX) or amph-DOX at the concentration of 1 μM (37 °C) for 4 h. After treatment, the cells were washed with PBS, fixed with 4% paraformaldehyde (PFA), stained with MitoTracker Green FM (500 nM) (Invitrogen) and 4′,6-diamidino-2-phenylindole (DAPI, 200 nM) (Invitrogen), and washed with PBS before imaging. For tracking mitochondria by CellLight Mitochondria-RFP BacMam 2.0 (Invitrogen), cells were transfected with 10 μL of CellLight reagent for 24 h. After that the cells were washed with PBS and incubated with free doxorubicin or amph-DOX (1 μM) at 37 °C for 4 h. Then, the cells were washed with PBS, fixed with 3% paraformaldehyde (PFA), stained with DAPI (200 nM) (Invitrogen), and washed with PBS. Images were captured by Zeiss confocal microscope (LSM 810) with a 63× oil-immersion objective. Images were obtained by the following excitation/emission settings: MitoTracker Green (excitation 488 nm, emission 515 nm bandpass filter), doxorubicin (excitation 488 nm, emission 560 nm bandpass filter), CellLight Mitochondria-RFP (excitation 561 nm, emission 585 nm bandpass filter).
Image Colocalization Analysis.
ImageJ (NIH) with Coloc 2 of Fiji’s plug-in was used for colocalization analysis. The colocalizations of mitochondria and DOX or amph-DOX were quantified based on the green/red signal intensities and those of nuclei and DOX or amph-DOX were based on blue/red signal intensities. Pearson’s (Ps) and Manders’ (M1/M2) coefficients were calculated from 1 individual field of view in each of the n = 3 independent experiments (total 12 fields).
Quantification of Free DOX and amph-DOX in the Intracellular Compartments.
Cells were plated at a concentration of 1 × 108 with 15 mL of media in 100 mm diameter tissue culture dishes and allowed to grow overnight. DOX and amph-DOX (10 μM) were added and incubated for different time periods. After internalization, the mitochondria and the nuclei were isolated using a mitochondria isolation kit (ThermoFisher Scientific) and a nuclei isolation kit (Sigma), respectively, following manufacturer’s instructions.
The amount of DOX or amph-DOX in each fraction was quantified by measuring fluorescence intensity from doxorubicin after solvent extraction. All of the experiments were carried out in triplicate.
DOX-Induced Reactive Oxygen Species (ROS) Measurement.
Cells (1 × 106) were precultured in 48-well plates for 12 h, cells were then incubated with DOX or amph-DOX at a final concentration of 1, 5, or 10.0 μM for 4 h. After treatment, cells were washed once with 1× PBS and incubated 30 min at 37 °C in PBS with a final concentration of H2DCFDA at 10 μM. No treatment group was used as a positive control for the quantifications of mitochondrial ROS production. Finally, the cells were washed and analyzed by flow cytometry.
For visualizing intracellular ROS, 1 × 104 cells were plated on coverslip in six-well plates and were treated with DOX or amph-DOX (10.0 μM final concentration) for 4 h. After treatment, cells were washed once with 1× PBS and incubated for 30 min at 37 °C in PBS with a final concentration of H2DCFDA at 10 μM. Finally, cells were washed with PBS, stained with DAPI (200 nM) (Invitrogen), MitoTracker Green (500 nM) (Invitrogen), and washed with PBS. Images were captured by Zeiss confocal microscope (LSM 810) with a 63× oil-immersion objective.
In Vivo Pharmacokinetics Evaluation.
To measure the pharmacokinetics, 1 × 106 B16F10 melanoma cells suspended in 100 μL of PBS buffer were inoculated in the flank region of 5 weeks old C57BL/6 mice. When the tumor volume reached ~50 mm3, mice were randomly assigned into three groups (n = 8 mice per group). Free DOX (5 mg/kg) or amph-DOX (5 mg/kg equivalent doxorubicin) was injected into the tumor bearing mice intravenously via the tail vein. Plasma was separated by centrifugation (15 000g for 10 min at 4 °C) after blood samples were collected at 30 and 60 min, 2, 4, 6, 12, and 24 h post drug administration (n = 4 at each time point). Sera were diluted three times in PBS, and drug concentrations in sera were calculated from standard curve by measuring the fluorescence intensity of DOX in each sample, correcting against sera from blood samples of nontreated animals. The fluorescence intensity was fitted into a calibration curve to determine the DOX concentration. Half-life (t1/2) was calculated from DOX concentrations in the area vs time curve and was fit by one-phase exponential decay (Graphpad prism).
In Vivo Biodistribution Study.
For in vivo biodistribution study, B16F10 tumor (volume ~500 mm3) bearing C57BL/6 mice (n = 8 mice per group) were injected intravenously with either free DOX (10 mg/kg) or amph-DOX (10 mg/kg equivalent doxorubicin). After drug administration for 2 or 24 h (n = 4 at each time point), mice were sacrificed, and the spleen, heart, brain, lung, kidney, tumor, and liver were collected. Tissue samples were flash frozen and stored at −80 °C until extraction. Tissue samples were weighed and homogenized by biomasher II tube (Kimble) and sonicated in 9 parts (v/w) of PBS. In a typical procedure, 200 μL of tissue homogenate was extracted with 50 μL of 10% Triton X-100 (v/v) and 750 μL of 0.75 N HCl in dichloromethane for 12 h at −20 °C in the dark. Fluorescence intensity was read, and background fluorescence was corrected by subtracting extracts from untreated animal samples. The concentrations were determined by comparing the fluorescence intensities to a calibration curve established by adding known amounts of doxorubicin to homogenates of untreated tissue samples.
Confocal Microscopy of Tumor Tissue.
Fresh tissue samples were washed with PBS and fixed in formaldehyde fixation buffer. After 48 h fixation, each tissue was merged in optimal cutting temperature compound, freeze at −80 °C in the dark, and cut into 10 μm thick tissue sections using a cryostat. The frozen tissue slides were incubated with 100 μL of diluted (1 μL MITO-ID Red in 10 mL 1× assay buffer, Enzo life sciences) reagent for 30 min and DAPI (200 nM) for additional 15 min. Finally, slides were washed three times by PBS and imaged. Images were captured by Zeiss confocal microscope (LSM 810) with a 63× oil-immersion objective.
Tumor Model.
B16F10 (5.0 × 105 cells) were subcutaneously inoculated into the right flank of 5–6 weeks old C57BL/6 mice. On day 5 (tumor volume ~50 mm3), mice were iv injected with 5 mg/kg of doxorubicin hydrochloride or amph-DOX on days 5, 8, and 11. Tumor length and width were measured with digital calipers, and the tumor volume was calculated using the following equation: tumor volume (V) = length × width2/2.
Statistical Analysis.
Comparisons of mean values of two groups were performed using unpaired Student’s t-tests. To analyze the statistical difference between groups, a one-way analysis of variance with the Bonferroni post-test was used. All of the values were expressed as means ± standard error of mean. GraphPad Prism software was used for all of the statistical analyses. ***P < 0.001, **P < 0.01, *P < 0.05. NS, not significant.
RESULTS AND DISCUSSION
Amph-DOX Binds to Serum Albumin in Blood.
Anticancer drug delivery based on endogenous serum proteins is an attractive “self-delivering” approach in targeting cancer cells in vivo.40,41 We recently developed an “albuminhitchhiking” molecular approach, which uniquely delivers subunit vaccines to lymph nodes after subcutaneous injection.42 In this approach, subunit vaccines are conjugated to diacyl lipid-poly(ethylene glycol), a structurally optimized albumin-binding domain, and following subcutaneous injection, accumulate in the draining lymph nodes by binding to and transporting with endogenous albumin.42 Diacyl lipidpolymer self-assembles into micelles in aqueous buffer.43 However, these micelles are kinetically unstable, especially in the presence of lipid-binding albumin.42-44 In addition to albumin binding, these amphiphilic molecules also exhibit intrinsic affinity toward plasma membrane, as demonstrated by the rapid uptake and confined intracellular membrane-domainselective accumulation.42,45,46 Thus, in the presence cells and serum, there exists a complicated three-way equilibrium: amph-DOX forms micelles, but amph-DOX can also insert its diacyl tails into cell membranes or bind to albumin protein. This three-way equilibrium is delicately controlled by (1) the concentrations of albumin; (2) the molecular weight (or length) of both lipid tails and PEG.42,43,47 We showed that the equilibrium shifts toward albumin binding when a long diacyl lipid (≥16 carbons) and a long poly(ethylene glycol) (≥36 ethylene glycol units) are used.42 To translate this albumin-hitchhiking vaccine approach to deliver anticancer drugs, we modified doxorubicin with a structure-optimized amphiphilic albumin-binding diacyl lipid linked by a poly(ethylene glycol) linker (Figures 1A and S1). We hypothesize that the amphiphilic functionalization alters doxorubicin’s physicochemical properties, which in turn redefines its bioavailability, organ and subcellular distributions, improves its therapeutic efficacy, and reduces DOX-associated toxicity.
The amph-DOX was synthesized and purified, as previously reported (Figure S1).48 Due to the molecular similarities between amph-DOX and DSPE-PEG2000-NHS, the complete separation of amph-DOX after reaction by a preparative HPLC was not practical (Figure S1). However, we found that DSPE-PEG2000-NHS or its hydrolyzed product did not affect the subsequent experiments. The self-assembled and albumin-binding properties were demonstrated by dynamic size scattering, transmission electron microscopy, and Forster resonance energy transfer (FRET) (Figure S2). To test whether amph-DOX can bind to albumin in blood, free DOX or amph-DOX was incubated with freshly isolated mouse blood for 4 h at 37 °C. The partition of DOX between serum albumin and blood cells was subsequently analyzed and quantified by fluorescence spectroscopy, gel electrophoresis, and flow cytometry. Upon incubation with freshly isolated blood, free DOX was detected in 9.8% of the blood cells, which was almost 3 times more than that of amph-DOX (3.6%) (Figure 1B). This observation suggests that free DOX interacts with erythrocytes, consistent with previous publications.49-52 In contrast, despite being in the possession of lipophilic diacyl lipid tail, amph-DOX had less association with the cells in the blood. Fluorescence measurements by spectroscopy indicated that around 92% of amph-DOX and 18% of free DOX remained in the blood serum (Figure 1C). Further, gel electrophoresis analysis (Figures 1D and S3) indicated that the vast majority of the amph-DOX in serum bound to serum albumin, showing a light-yellow fluorescent band co-migrated with albumin (Figure 1D, lane 5). This band was distinct from albumin as pure serum showed a major albumin band with green autofluorescence under ultraviolet light (254 nm) (Figure 1D, lane 3). In contrast, free DOX incubated with blood migrated as a single band toward the negative electrode (Figure 1D, lane 4), indicating a lack of interaction with albumin. These data strongly suggest that unlike unmodified DOX, which extensively interacts with erythrocytes,49-52 amph-DOX binds to albumin protein in whole blood and warrants further investigation of using this albumin-hitchhiking platform for targeted drug delivery.
Amph-DOX Selectively Accumulates in Mitochondria In Vitro.
To investigate the uptake and intracellular distribution of amph-DOX related to DOX parent compound in cancer, murine melanoma B16F10 cells were incubated with amph-DOX or DOX in the presence of bovine serum and analyzed by flow cytometry and confocal laser scanning microscopy (CLSM). The melanoma model was selected due to its intrinsic resistance to DOX.53 In vitro, amph-DOX showed a rapid and enhanced uptake, reaching high levels of DOX concentration 1 h after incubation in B16F10 cells (Figure 2A). In cells treated with free DOX, the drug concentration slowly increased over 12 h, reaching 30% of the level of that treated with amph-DOX (Figure 2A) when assayed by flow cytometry. The subcellular locations of amph-DOX in B16F10 cells were subsequently determined by confocal microscopy. As expected, free DOX exhibited strong nuclear accumulation following drug exposure, determined by using the intrinsic DOX fluorescence (Figure 2B). In contrast, amph-DOX fluorescence was mainly confined in the mitochondria (Figure 2B), demonstrated by analyzing the fluorescence colocalization with MitoTracker Green FM (Invitrogen), a mitochondria-specific dye (Figure 2B, upper two panels). The mitochondria-selective accumulation of amph-DOX was unexpected, as our previous amphiphilic oligonucleotides were mainly confined within the endolysosomal compartment.42,54 Analysis of LysoTracker Green (a lysosome-specific dye) colocalization by confocal laser scanning microscopy showed little overlap with amph-DOX (Figure S4), suggesting that amph-DOX does not accumulate within lysosomes. To verify the mitochondria accumulation, we used CellLight Mitochondria-RFP BacMam 2.0 (Invitrogen) to stain the mitochondrial matrix (Figure 2B lower two panels). CellLight Mitochondria-RFP is a highly selective transfection-based approach, which targets the red fluorescent protein to the mitochondria in live cells. Quantitative analysis using the ImageJ “Coloc 2” plug-in revealed significant spatial overlap between amph-DOX and both mitochondria dyes in B16F10 cells (Pearson coefficient, 0.57; Manders coefficient, 0.874/0.992; Table S1). For unmodified DOX, weak correlation of the red signals and the mitochondrial staining was demonstrated by low coefficient values (Pearson coefficient, 0.26; Manders coefficient, 0.196/0.039; Table S1).
Figure 2.

Amph-DOX selectively accumulates in mitochondria in vitro. (A) Kinetics of amph-DOX or DOX internalization showing amph-DOX is quickly internalized by B16F10 cells. The uptake is analyzed by measuring the mean fluorescence intensity by flow cytometry. (B) Confocal microscope characterization of B16F10 cells showing the cellular uptake and intracellular distribution of free doxorubicin or amph-DOX (concentration of 1 μM) at 4 h. B16F10 cells were treated with free DOX and amph-DOX (red) and stained for mitochondria (green) by MitoTracker Green (upper two panels) or Mitochondria-RFP (lower two panels). Cell nuclei were stained with DAPI (blue). Note that some cells were not transfected in the mitochondria-RFP-treated group. Scale bar = 10 μm. (C, D) Quantification of DOX or amph-DOX in (C) mitochondria and (D) nuclei of B16F10 cells. Cells (1 × 108) were incubated with 10 μM DOX or amph-DOX for 1, 4, 12, 24 h. Mitochondria and nuclei were isolated by isolating kits, and DOX fluorescence was quantified by fluorescence spectroscopy after solvent extraction.
Because DOX fluorescence is dramatically quenched upon DNA intercalation, the uptake quantification measured by flow cytometry (Figure 2A) might not reflect the DOX concentrations after being delivered to different subcellular locations. To verify the enhanced uptake and distribution results, we isolated the mitochondria and the nuclei from B16F10 cells, and the DOX concentrations were quantified by fluorescent spectroscopy after solvent extraction. Free DOX reached between 30 and 70% of the uptake from amph-DOX at different time points (Figure S5A). The uptake differences between flow cytometry and fluorescence spectroscopy are most likely due to the fluorescence quenching of DOX by different levels of DNA intercalation. Consistent with our confocal results, unmodified DOX accumulated primarily in the nuclei, accounting for 72% of the fluorescence within the cells in 24 h (Figures 2C,D and S5). In contrast, in cells treated with amph-DOX, approximately 40% of the intracellular DOX fluorescence was in isolated mitochondria (Figures 2C,D and S5). Though a fraction of the DOX might be lost during organelle isolation, these data clearly demonstrated the selective mitochondria accumulation in tumor cells after treatment with amph-DOX. Enhanced uptake and selective mitochondria accumulation of amph-DOX were not restricted to B16F10 cells, as similar intracellular distribution was observed in mouse breast tumor 4T1 cells (Figure S6).
Drug-resistant cancer cells are known to overexpress P-glycoprotein (P-gp), which acts as an efflux pump and reduces doxorubicin’s uptake and retention.3,55 To test whether amph-DOX can increase intracellular net drug uptake in DOX-resistant cells, human ovarian adenocarcinoma (NCI/ADR-RES) cells56 were used to incubate with amph-DOX or soluble DOX, and the DOX uptake, intracellular distribution were analyzed as before. These cells were established to resist doxorubicin treatment.56 Similar to previous observation, DOX uptake in NCI/ADR-RES cells remained low throughout the incubation, presumably due to the P-gp-mediated DOX efflux.57 In contrast, rapid DOX uptake and prolonged retention were observed in cells treated with amph-DOX (Figure 3A,B). Confocal microscopy analysis confirmed that in these DOX-resistant cells, amph-DOX selectively accumulated in mitochondria (Figure 3A). These results demonstrate that efficient uptake and mitochondria accumulation can be achieved in DOX-resistant cells, suggesting a plausible mechanism to overcome the drug-induced resistance.
Figure 3.

Amph-DOX enhances DOX uptake and accumulates in mitochondria in drug-resistant NCI/ADR-RES cells in vitro. (A) Confocal microscope characterization of NCI/ADR-RES cells showing the intracellular distribution of amph-DOX at 24 h (scale bar: 10 μm). (B) Kinetics of amph-DOX or DOX uptake in NCI/ADR-RES cells.
Structural Requirements of Amphiphiles in Mitochondrial Trafficking.
Targeting subcellular organelles via lipid modification on drugs have been extensively studied in the past.58 It is generally believed that the lipid structure governs the intracellular sorting mechanisms and thus determines where the lipid-modified molecules localize within the cell.58 However, no lipid has been shown to selectively accumulate in mitochondria. As amph-DOX exhibits an overall negative charge (Figure 1D, lane 2), it is unlikely that amph-DOX is concentrated in mitochondria in response to negative transmembrane potentials. To investigate the possible mechanisms for mitochondrial accumulation, we first set out to determine the uptake mechanisms of amph-DOX and compared that with DOX-encapsulated DSPE-PEG2000 micelles (micelle-DOX).59 Although micelle-DOX enhanced the levels of DOX uptake in B16F10 cells, it was primarily accumulated in the nuclei (Figure S8). In addition, amph-DOX employs multiple uptake mechanisms in typical cell culture conditions (Figure S9). To determine the role of albumin in the uptake and intracellular distribution, cell culture experiments were repeated in the absence or presence of FBS. In vitro, uptake of amph-DOX was inversely proportional to FBS content at first 2 h, reflecting the shift of equilibrium toward cellular membrane insertion at low albumin concentrations (Figure S10A,B). However, after longer time incubation, similar levels of uptake were observed for amph-DOX in the presence or absence of FBS in B16F10 cells (Figure S10B). At low albumin concentrations, amph-DOX equilibrate between albumin-binding state and membrane insertion state (Figure S10C,D), both of which showed significantly better cellular uptake than free DOX. It is worth to point out that these in vitro uptake assays may not accurately reflect the in vivo process as the blood albumin concentration is ~10 times higher than that in cell culture medium. Nevertheless, amph-DOX accumulated in the mitochondria in the absence of FBS (Figure S10E), suggesting albumin is not involved in the intracellular sorting and trafficking of amph-DOX, and that the intracellular release of amph-DOX from albumin/amph-DOX complex is highly possible (Figure S11). Finally, it appeared that intact amph-DOX conjugate traffics to mitochondria, as similar amphiphilic DOX linked via an acid labile hydrazone bond showed both mitochondrial and nuclear accumulation (Figure S12).
To investigate whether amphiphilic modification via diacyl lipid PEG can be a generalizable approach for mitochondria-specific targeting, we modified fluorescein with the same amphiphilic PEG and tested its intracellular uptake in B16F10 cells. Interestingly, no mitochondria accumulation of amph- Fluorescein was observed (Figure S13). To determine whether DSPE lipid is required in the mitochondria targeting, we conjugated DOX to cholesterol-PEG2000. Unlike DSPE lipid, which is negatively charged, cholesterol is neutral and is less hydrophobic. Similar to amph-DOX, cholesterol-PEG2000-DOX selectively accumulates in mitochondria (Figure S14). These data suggest that amphiphilic modification on DOX can alter its intracellular distribution, and that the mitochondria accumulation can tolerate the amphiphilic structure to a certain degree. This observation rules out the possibility that amph-DOX is sorted and transported by lipid-specific proteins, instead, it favors the notion that the unique chemical and biophysical properties of amphiphilic DOX conjugates have key roles in their intracellular trafficking and distribution. Although the detail structure–function relationship remains unclear (e.g., whether PEG plays a role), it appeared that the amphiphiles and DOX contributed jointly to the overall physicochemical characteristics, which govern the mitochondria targeting. Perhaps amphiphilic modification alters the overall hydrophilic/hydrophobic balance of DOX and subsequently affect its permeability, diffusion, and membrane partition. Together, these results clearly demonstrated that in vitro, amphiphilic modification on DOX enhanced the cellular uptake and selectively targeted DOX to mitochondria.
Amph-DOX Enhances Antiproliferation Efficacy by Increasing Reactive Oxygen Species Levels in Cancer Cells.
Targeting doxorubicin to mitochondria has recently been shown to enhance the cytotoxicity toward a number of tumor cells.9-14 To examine the impact of amphiphilic DOX modification on the antiproliferation efficacy, the viabilities of several cancer cells, including drug-resistant NCI/ADR-RES cells, were evaluated. Exposure of cells to amph-DOX caused a concentration-dependent toxicity, with an IC50 value of 0.2 μM in B16F10 cells, as compared with 2.0 μM in cells treated with free DOX (Figure 4A). Similarly, treatment with amph-DOX reduced the IC50 values in both OVCAR-8 cells (DOX sensitive, 0.1 μM as compared to 1.0 μM with free DOX) and the DOX-resistant NCI/ADR-RES cells (0.5 μM as compared to 1.8 μM with free DOX) (Figure 4B). It is worth to point out that DSPE-PEG2000-NHS or its hydrolyzed derivative exhibits negligible toxicity (Figure S15), suggesting amph-DOX exerts its cytotoxic effects via DOX instead of amphiphilic polymer. These results clearly demonstrated that amph-DOX was considerably more effective than free DOX in both drug sensitive and drug-resistant cell lines.
Figure 4.

Amph-DOX induces strong cytotoxicity in vitro by stimulating massive production of ROS in mitochondria. (A, B) In vitro cytotoxicities of free DOX and amph-DOX against B16F10 (A), OVCAR-8 (B), or NCI/ADR-RES (C) cells 24 h after exposure. Cells (1 × 106) were incubated with amph-DOX or free DOX with varying concentrations for 24 h. Cell viability was determined by alamarBlue viability assay. (D, E) Intracellular levels of ROS induced by DOX and amph-DOX. (D) Flow cytometer analyses of ROS production in 1 × 106 B16F10 cells treated with 1, 5, and 10 μM DOX or amph-DOX and (E) confocal microscopy images of B16F10 cells incubated with 10 μM DOX and amph-DOX (red) for 4 h, after which H2DCFDA (DCF, green) was added at a final concentration of 20 μM for 30 min. Scale bar = 10 μm.
The cytotoxic effect of doxorubicin is thought to be mediated primarily by nuclear DNA intercalation to disrupt topoisomerase-II-mediated DNA repair.1 However, oxidative damage of mitochondria functions has been observed in vitro following delivery of DOX to mitochondria.10 To gain insight into the source and potential mechanism of amph-DOX inducing ROS generation in cancer cells, we analyzed the production and spatial distribution of the intracellular ROS. B16F10 cells were continuously exposed to amph-DOX at different concentrations, and intracellular levels of ROS were measured after different times of drug exposure. By using the 2′,7′-dichlorodihydrofluorescein diacetate probe (H2DCFDA) that detects multiple ROS species within the cells, we observed significant increases in intracellular ROS levels in cells treated with amph-DOX compared with free DOX (Figure 4D,E). ROS production was dominantly amph-DOX in origin as demonstrated by colocalization of amph-DOX and dichlorofluorescein staining using confocal microscopy (Figure 4E). As amph-DOX accumulates in mitochondria (Figures 2B and 3A), our results suggest that mitochondria are the locations of amph-DOX-induced ROS response in cancer cells. Together, these data qualify amph-DOX as a promising drug for cancer chemotherapy, which significantly increases anticancer potency, through effective uptake of DOX to tumor cells and more importantly, through mitochondria-selective accumulation and ROS production.
Amphiphilic Conjugation Markedly Prolongs the Circulation Time, Enhances Tumor Accumulation, and Improves the Therapeutic Antitumor Efficacy of Doxorubicin.
Drugs associated with albumin are known to have long blood residence time.36,37,39 To test whether the albumin-binding amph-DOX has prolonged serum half-life, mice were injected intravenously with amph-DOX or free DOX. At various time points following injection, blood samples were collected from the tail for DOX measurements. In vivo, free DOX was rapidly cleared from the plasma, and its concentration was dropped below detectable level after 60 min (Figure 5A). In contrast, amph-DOX exhibited much higher serum concentrations after injection and had superior blood retention, with a half-life in blood increased to 3.0 h (Figure 5A). The area under the concentration–time curve (AUC) of amph-DOX was increased approximately 60 fold compared with that of free doxorubicin (Figure 5A).
Figure 5.

In vivo plasma pharmacokinetic analysis and biodistribution of amph-DOX in C57BL/6 mice bearing B16F10 tumor. (A) Plasma pharmacokinetic curves of amph-DOX and DOX. Doxorubicin concentrations in plasma as a function of time following a single dose of free doxorubicin (10 mg/kg) or amph-DOX (10 mg/kg equivalent doxorubicin). The values are the mean ± SEM (n = 4). (B, C) Tissue (tumor, liver, spleen, kidney, and heart) accumulation of doxorubicin at 2 h (B) and 24 h (C) following a single dose of free doxorubicin (10 mg/kg) or amph-DOX (10 mg/kg equivalent doxorubicin) (n = 4).
Albumin-bound DOX is also expected to accumulate in tumor via multiple mechanisms: (i) due to EPR effect, DOX–albumin complex accumulates in tumor instead of normal tissues; (ii) It is well known that tumor tissues utilize albumin as a source of amino acid and energy to fuel their growth.40,41 In contrast, the uptake of drug bound to albumin in normal tissues is expected to be low due to the FcRn-mediated albumin recycling pathway,60 (iii) albumin has an extraordinarily broad tissue penetration capability (by receptormediated transcytosis) in both normal and disease conditions.40,41 Compared with free DOX, iv injection of amph-DOX led to 14-fold increase in sc B16F10 tumor (mouse melanoma) 24 h postinjection (Figure 5C). Importantly, amph-DOX resulted in a significantly lower tissue accumulation of DOX compared to free DOX treatment in the heart (Figure 5B,C), where DOX can cause cardiotoxicity, suggesting that amph-DOX might lead to a reduction of the potential short-term and long-term side effects of the drug.
Next, the antitumor activities of amph-DOX were evaluated by therapeutical treatment of C57BL/6 mice bearing melanoma tumor. A total of 5 × 105 B16F10 cells were subcutaneously implanted into mice. Mice received three injections of 5 mg/kg of free DOX, equivalent amph-DOX, or saline on days 5, 8, and 11. As shown in Figure 6A, administration of free DOX only caused a transient regression of B16F10 tumor at the early stage of the treatment, and tumor quickly resumed growth. However, mice treated with the same doses of amph-DOX markedly delayed the growth of scimplanted B16F10 tumor (Figures 6A and S16). To examine whether amph-DOX accumulates in tumor mitochondria in vivo, tumors were isolated 24 and 48 h after injection, sectioned, and stained with MITO-ID, a mitochondria-selective dye suitable for fixed cells. Accumulation of amph-DOX was observed in tumor mitochondria 24 and 48 h postinjection (Figures 6B and S17), suggesting an improved EPR effect. In contrast, under the same conditions, soluble DOX fluorescence in the tumor section was undetectable. Treatment with amph-DOX also diminished doxorubicinrelated losses in total body weight in tumor-free mice (Figure 6C). Histopathological analysis of heart section (on day 15) of mice after three injections (on days 5, 8, and 11) of amph-DOX showed no sign of heart muscle damage and no acute cardiotoxicity, similar to those with no treatment control (Figure S17). However, DOX-treated animals showed noticeable, albeit mild damage to cardiac tissue, characterized by increased cytoplasmic vacuolization and distorted myocardial cell arrangement (Figure S18). Taken together, these data strongly suggest that amph-DOX is able to bind albumin protein in blood, prolong circulating time, accumulate in tumor mitochondria, and inhibit tumor growth. Though the long-term cardiotoxicity cannot be determined by our model, the reduced mouse cardiac tissue accumulation and no cardiomyocyte pathology also suggest a favorable cardiosafety profile in the preclinical model.
Figure 6.

In vivo antitumor activity of amph-DOX in B16F10 tumor. (A) C57BL/6 mice (n = 8 per group) were injected with free DOX, amph-DOX (3 × 5 mg/kg doxorubicin), or saline on days 5, 8, and 11 after tumor innoculation. Tumor volumes were measured on a daily basis during the experimental period. (B) Confocal laser scanning microscopy (CLSM) images of frozen sections of B16F10 tumor tissues. Tumor tissues were isolated at the end of tumor therapeutic period (day 16). Tumor sections were labeled and imaged. Images show mitochondria (green, stained with MITO-ID RED), nuclei (blue, stained by DAPI), and overlay (scale bar = 10 μm). (C) Tumor-free C57BL/6 mice were treated with DOX or amph-DOX (10 mg/kg equivalent doxorubicin) at days 5, 8, and 11, and a final dose of 20 mg/kg on day 16. Body weight of mice was monitored (n = 8).
CONCLUSIONS
The physicochemical properties appear to have important consequences for the behavior of anthracyclines in biological systems. In this work, we described a simple molecular approach to deliver doxorubicin to tumor mitochondria in vivo. We showed that in mice, diacyl lipid conjugation on doxorubicin linked with a PEG linker uniquely achieves tissue-, cellular-, and mitochondria-selective accumulation of doxorubicin and significantly enhances the antitumor efficacy of the drug. This new type of molecular anticancer drug conjugate features several favorable advantages as therapeutic options in cancer therapy: (i) this approach uses a simple molecular conjugate to achieve multiple levels of targeting in vivo. First, the amphiphilic DOX can reach and penetrate solid tumor by hitchhiking on albumin protein.40,41 Compared with soluble DOX, albumin–drug complex exhibits increased hydrodynamic size, prolongs DOX’s circulating half-life, and retargets the drug to the tumor by both passive and active targeting mechanisms.36-39 Second, amphiphilic DOX accumulates in mitochondria following tumor cell uptake through a yet unknown mechanism. Several long circulating doxorubicin formulations exist in clinical or preclinical studies (e.g., liposomal DOX: Doxil; DOX–albumin covalent conjugate: aldoxorubicin).61,62 However, none of these formulations is able to selectively target mitochondria. Unlike many of the previous mitochondriotropic ligands, which are concentrated in mitochondria in response to negative transmembrane potentials,7-15,19-21 our amphiphilic drug conjugate has a completely different structure. Our approach thus challenges current paradigms in mitochondria targeting, providing a new mechanism to potentiate the efficacy and safety for future mitochondria drug design. (ii) Our molecular approach is carrier free. Amphiphilic DOX relies on endogenous albumin protein for tumor targeting and intracellular sorting mechanisms for mitochondria targeting. Anticancer drug delivered via endogenous protein particles has the potential to hold the key advantages while completely avoiding the side effects (e.g., immunogenicity) associated with exogenous carriers. (iii) Targeting doxorubicin to mitochondria enables a mechanism to overcome the drug efflux-mediated resistance by delivering doxorubicin to intracellular organelle where the drug efflux protein cannot access.9 (iv) Compared with proteins or nanoparticles, the molecular conjugate is fully synthetic, which is favored in production, cost, stability, safety and in principle could be readily translated to clinical cancer chemotherapy. Altogether, the results presented here demonstrate that amphiphilic modification on doxorubicin, which targets doxorubicin to mitochondria, is an effective approach to simultaneously enhance the drug’s potency and safety. This approach might be applicable to many other anthracyclines in cancer chemotherapy.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported in part by Wayne State University President’s Research Enhancement Program, and NSF CAREER Award (1750607) to H.L.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.8b17399.
Synthesis and characterization of amph-DOX; characterization of self-assemble and albumin-binding properties’; lysosome colocalization confocal images; intracellular quantification of amph-DOX in B16F10 cells; mitochondria accumulation in 4T1 tumor cells; intracellular quantification of amph-DOX in NCI/ADR-RES cells; uptake and intracellular distribution of DOX-loaded DSPE-PEG2000 micelles; uptake mechanisms of amph- DOX; effect of serum on amph-DOX uptake and subcellular location; intracellular distribution of amph-DOX with a pH-sensitive hydrozone linkage; intracellular distribution of amph-Fluorescein; intracellular distribution of cholesterol-DOX; cytotoxic effect of DSPE-PEG2000-NHS; photograph of tumor before and after isolation; H&E images of heart sections in mice treated with DOX or amph-DOX (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Minotti G; Menna P; Salvatorelli E; Cairo G; Gianni L Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharmacol. Rev 2004, 56, 185–229. [DOI] [PubMed] [Google Scholar]
- (2).Chabner BA; Roberts TG Jr. Chemotherapy and the War on Cancer. Nat. Rev. Cancer 2005, 5, 65. [DOI] [PubMed] [Google Scholar]
- (3).Holohan C; Van Schaeybroeck S; Longley DB; Johnston PG Cancer Drug Resistance: An Evolving Paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [DOI] [PubMed] [Google Scholar]
- (4).DeVita VT Jr.; Chu E A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653. [DOI] [PubMed] [Google Scholar]
- (5).Pinto AC; Moreira JN; Simões S Combination Chemotherapy in Cancer: Principles, Evaluation and Drug Delivery Strategies In Current Cancer Treatment-Novel beyond Conventional Approaches; InTech, 2011. [Google Scholar]
- (6).Horobin RW; Trapp S; Weissig V Mitochondriotropics: A Review of Their Mode of Action, and Their Applications for Drug and DNA Delivery to Mammalian Mitochondria. J. Controlled Release 2007, 121, 125–136. [DOI] [PubMed] [Google Scholar]
- (7).D’Souza GG; Wagle MA; Saxena V; Shah A Approaches for Targeting Mitochondria in Cancer Therapy. Biochim. Biophys. Acta, Bioenerg 2011, 1807, 689–696. [DOI] [PubMed] [Google Scholar]
- (8).Fulda S; Galluzzi L; Kroemer G Targeting Mitochondria for Cancer Therapy. Nat. Rev. Drug Discovery 2010, 9, 447. [DOI] [PubMed] [Google Scholar]
- (9).Chamberlain GR; Tulumello DV; Kelley SO Targeted Delivery of Doxorubicin to Mitochondria. ACS Chem. Biol 2013, 8, 1389–1395. [DOI] [PubMed] [Google Scholar]
- (10).Buondonno I; Gazzano E; Jean SR; Audrito V; Kopecka J; Fanelli M; Salaroglio IC; Costamagna C; Roato I; Mungo E; et al. Mitochondria-Targeted Doxorubicin: A New Therapeutic Strategy against Doxorubicin-Resistant Osteosarcoma. Mol. Cancer Ther 2016, 15, 2640–2652. [DOI] [PubMed] [Google Scholar]
- (11).Riganti C; Rolando B; Kopecka J; Campia I; Chegaev K; Lazzarato L; Federico A; Fruttero R; Ghigo D Mitochondrial-Targeting Nitrooxy-Doxorubicin: A New Approach to Overcome Drug Resistance. Mol. Pharmaceutics 2013, 10, 161–174. [DOI] [PubMed] [Google Scholar]
- (12).Han M; Vakili MR; Soleymani Abyaneh H; Molavi O; Lai R; Lavasanifar A Mitochondrial Delivery of Doxorubicin Via Triphenylphosphine Modification for Overcoming Drug Resistance in Mda-Mb-435/Dox Cells. Mol. Pharmaceutics 2014, 11, 2640–2649. [DOI] [PubMed] [Google Scholar]
- (13).Modica-Napolitano JS; Weissig V Treatment Strategies That Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents. Int. J. Mol. Sci 2015, 16, 17394–17421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jung K; Reszka R Mitochondria as Subcellular Targets for Clinically Useful Anthracyclines. Adv. Drug Delivery Rev 2001, 49, 87–105. [DOI] [PubMed] [Google Scholar]
- (15).Lu P; Bruno BJ; Rabenau M; Lim CS Delivery of Drugs and Macromolecules to the Mitochondria for Cancer Therapy. J. Controlled Release 2016, 240, 38–51. [DOI] [PubMed] [Google Scholar]
- (16).Zhang Y; Shen Y; Teng X; Yan M; Bi H; Morais PC Mitochondria-Targeting Nanoplatform with Fluorescent Carbon Dots for Long Time Imaging and Magnetic Field-Enhanced Cellular Uptake. ACS Appl. Mater. Interfaces 2015, 7, 10201–10212. [DOI] [PubMed] [Google Scholar]
- (17).Zhang Y; Zhang C; Chen J; Liu L; Hu M; Li J; Bi H Trackable Mitochondria-Targeting Nanomicellar Loaded with Doxorubicin for Overcoming Drug Resistance. ACS Appl. Mater. Interfaces 2017, 9, 25152–25163. [DOI] [PubMed] [Google Scholar]
- (18).Wang H; Gao Z; Liu X; Agarwal P; Zhao S; Conroy DW; Ji G; Yu J; Jaroniec CP; Liu Z; Lu X; Li X; He X Targeted Production of Reactive Oxygen Species in Mitochondria to Overcome Cancer Drug Resistance. Nat. Commun 2018, 9, No. 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Smith RA; Porteous CM; Gane AM; Murphy MP Delivery of Bioactive Molecules to Mitochondria in Vivo. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 5407–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Modica-Napolitano JS; Aprille JR Delocalized Lipophilic Cations Selectively Target the Mitochondria of Carcinoma Cells. Adv. Drug Delivery Rev 2001, 49, 63–70. [DOI] [PubMed] [Google Scholar]
- (21).Murphy MP Selective Targeting of Bioactive Compounds to Mitochondria. Trends Biotechnol. 1997, 15, 326–330. [DOI] [PubMed] [Google Scholar]
- (22).Galluzzi L; Zamzami N; de La Motte Rouge T; Lemaire C; Brenner C; Kroemer G Methods for the Assessment of Mitochondrial Membrane Permeabilization in Apoptosis. Apoptosis 2007, 12, 803–813. [DOI] [PubMed] [Google Scholar]
- (23).Yousif LF; Stewart KM; Kelley SO Targeting Mitochondria with Organelle-Specific Compounds: Strategies and Applications. ChemBioChem 2009, 10, 1939–1950. [DOI] [PubMed] [Google Scholar]
- (24).Millard M; Pathania D; Shabaik Y; Taheri L; Deng J; Neamati N Preclinical Evaluation of Novel Triphenylphosphonium Salts with Broad-Spectrum Activity. PLoS One 2010, 5, No. e13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Porteous CM; Logan A; Evans C; Ledgerwood EC; Menon DK; Aigbirhio F; Smith RA; Murphy MP Rapid Uptake of Lipophilic Triphenylphosphonium Cations by Mitochondria in Vivo Following Intravenous Injection: Implications for Mitochondria-Specific Therapies and Probes. Biochim. Biophys. Acta 2010, 1800, 1009–1017. [DOI] [PubMed] [Google Scholar]
- (26).Propper DJ; Braybrooke JP; Taylor DJ; Lodi R; Styles P; Cramer JA; Collins WCJ; Levitt NC; Talbot DC; Ganesan TS; Harris AL Phase I Trial of the Selective Mitochondrial Toxin Mkt 077 in Chemo-Resistant Solid Tumours. Ann. Oncol 1999, 10, 923–927. [DOI] [PubMed] [Google Scholar]
- (27).Kelso GF; Porteous CM; Coulter CV; Hughes G; Porteous WK; Ledgerwood EC; Smith RAJ; Murphy MP Selective Targeting of a Redox-Active Ubiquinone to Mitochondria within Cells - Antioxidant and Antiapoptotic Properties. J. Biol. Chem 2001, 276, 4588–4596. [DOI] [PubMed] [Google Scholar]
- (28).Yousif LF; Stewart KM; Horton KL; Kelley SO Mitochondria-Penetrating Peptides: Sequence Effects and Model Cargo Transport. ChemBioChem 2009, 10, 2081–2088. [DOI] [PubMed] [Google Scholar]
- (29).Neupert W; Herrmann JM Translocation of Proteins into Mitochondria. Annu. Rev. Biochem 2007, 76, 723–749. [DOI] [PubMed] [Google Scholar]
- (30).Vestweber D; Schatz G DNA-Protein Conjugates Can Enter Mitochondria Via the Protein Import Pathway. Nature 1989, 338, 170–172. [DOI] [PubMed] [Google Scholar]
- (31).Horton KL; Stewart KM; Fonseca SB; Guo Q; Kelley SO Mitochondria-Penetrating Peptides. Chem. Biol 2008, 15, 375–382. [DOI] [PubMed] [Google Scholar]
- (32).Maiti KK; Lee WS; Takeuchi T; Watkins C; Fretz M; Kim DC; Futaki S; Jones A; Kim KT; Chung SK Guanidine-Containing Molecular Transporters: Sorbitol-Based Transporters Show High Intracellular Selectivity toward Mitochondria. Angew. Chem 2007, 119, 5984–5988. [DOI] [PubMed] [Google Scholar]
- (33).Han K; Zhu JY; Jia HZ; Wang SB; Li SY; Zhang XZ; Han HY Mitochondria-Targeted Chimeric Peptide for Trinitarian Overcoming of Drug Resistance. ACS Appl. Mater. Interfaces 2016, 8, 25060–25068. [DOI] [PubMed] [Google Scholar]
- (34).Han K; Lei Q; Wang S-B; Hu J-J; Qiu W-X; Zhu J-Y; Yin W-N; Luo X; Zhang X-Z Dual-Stage-Light-Guided Tumor Inhibition by Mitochondria-Targeted Photodynamic Therapy. Adv. Funct. Mater 2015, 25, 2961–2971. [Google Scholar]
- (35).Marrache S; Dhar S Engineering of Blended Nanoparticle Platform for Delivery of Mitochondria-Acting Therapeutics. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 16288–16293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Weissig V DQAsomes as the Prototype of Mitochondria-Targeted Pharmaceutical Nanocarriers: Preparation, Characterization, and Use In Mitochondrial Medicine: Volume II, Manipulating Mitochondrial Function; Humana Press, 2015; pp 1–11. [DOI] [PubMed] [Google Scholar]
- (37).Pathak RK; Kolishetti N; Dhar S Targeted Nanoparticles in Mitochondrial Medicine. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 2015, 7, 315–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Patel NR; Hatziantoniou S; Georgopoulos A; Demetzos C; Torchilin VP; Weissig V; D’Souza GG Mitochondria-Targeted Liposomes Improve the Apoptotic and Cytotoxic Action of Sclareol. J. Liposome Res 2010, 20, 244–249. [DOI] [PubMed] [Google Scholar]
- (39).Anselmo AC; Mitragotri S Nanoparticles in the Clinic. Bioeng. Transl. Med 2016, 1, 10–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kratz F Albumin as a Drug Carrier: Design of Prodrugs, Drug Conjugates and Nanoparticles. J. Controlled Release 2008, 132, 171–183. [DOI] [PubMed] [Google Scholar]
- (41).Larsen MT; Kuhlmann M; Hvam ML; Howard KA Albumin-Based Drug Delivery: Harnessing Nature to Cure Disease. Mol. Cell. Ther 2016, 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Liu H; Moynihan KD; Zheng Y; Szeto GL; Li AV; Huang B; Van Egeren DS; Park C; Irvine DJ Structure-Based Programming of Lymph-Node Targeting in Molecular Vaccines. Nature 2014, 507, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kastantin M; Missirlis D; Black M; Ananthanarayanan B; Peters D; Tirrell M Thermodynamic and Kinetic Stability of Dspe-Peg(2000) Micelles in the Presence of Bovine Serum Albumin. J. Phys. Chem. B 2010, 114, 12632–12640. [DOI] [PubMed] [Google Scholar]
- (44).Castelletto V; Krysmann M; Kelarakis A; Jauregi P Complex Formation of Bovine Serum Albumin with a Poly(Ethylene Glycol) Lipid Conjugate. Biomacromolecules 2007, 8, 2244–2249. [DOI] [PubMed] [Google Scholar]
- (45).Yu C; An M; Li M; Liu H Immunostimulatory Properties of Lipid Modified Cpg Oligonucleotides. Mol. Pharm 2017, 14, 2815–2823. [DOI] [PubMed] [Google Scholar]
- (46).Inui O; Teramura Y; Iwata H Retention Dynamics of Amphiphilic Polymers Peg-Lipids and Pva-Alkyl on the Cell Surface. ACS Appl. Mater. Interfaces 2010, 2, 1514–1520. [DOI] [PubMed] [Google Scholar]
- (47).Castelletto V; Krysmann M; Kelarakis A; Jauregi P Complex Formation of Bovine Serum Albumin with a Poly(Ethylene Glycol) Lipid Conjugate. Biomacromolecules 2007, 8, 2244–2249. [DOI] [PubMed] [Google Scholar]
- (48).Hwang T; Han HD; Song CK; Seong H; Kim JH; Chen X; Shin BC In Anticancer Drug-Phospholipid Conjugate for Enhancement of Intracellular Drug Delivery, Macromolecular symposia; Wiley Online Library, 2007; pp 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Marczak A; Kowalczyk A; Wrzesień-Kus A; Robak T; Jóźwiak Z Interaction of Doxorubicin and Idarubicin with Red Blood Cells from Acute Myeloid Leukaemia Patients. Cell Biol. Int 2006, 30, 127–132. [DOI] [PubMed] [Google Scholar]
- (50).Arancia G; Molinari A; Crateri P; Calcabrini A; Silvestri L; Isacchi G Adriamycin-Plasma Membrane Interaction in Human Erythrocytes. Eur. J. Cell Biol 1988, 47, 379–387. [PubMed] [Google Scholar]
- (51).Suwalsky M; Hernández P; Villena F; Aguilar F; Sotomayor CP The Anticancer Drug Adriamycin Interacts with the Human Erythrocyte Membrane. Z. Naturforsch., C: J. Biosci 1999, 54, 271–277. [DOI] [PubMed] [Google Scholar]
- (52).Awasthi S; Sharma R; Awasthi YC; Belli JA; Frenkel EP The Relationship of Doxorubicin Binding to Membrane Lipids with Drug Resistance. Cancer Lett. 1992, 63, 109–116. [DOI] [PubMed] [Google Scholar]
- (53).Soengas MS; Lowe SW Apoptosis and Melanoma Chemoresistance. Oncogene 2003, 22, 3138–3151. [DOI] [PubMed] [Google Scholar]
- (54).Liu H; Zhu Z; Kang H; Wu Y; Sefan K; Tan W DNABased Micelles: Synthesis, Micellar Properties and Size-Dependent Cell Permeability. Chemistry 2010, 16, 3791–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Thomas H; Coley HM Overcoming Multidrug Resistance in Cancer: An Update on the Clinical Strategy of Inhibiting P-Glycoprotein. Cancer Control 2003, 10, 159–165. [DOI] [PubMed] [Google Scholar]
- (56).Ke W; Yu P; Wang J; Wang R; Guo C; Zhou L; Li C; Li K Mcf-7/Adr Cells (Re-Designated Nci/Adr-Res) Are Not Derived from Mcf-7 Breast Cancer Cells: A Loss for Breast Cancer Multidrug-Resistant Research. Med. Oncol 2011, 28, 135–141. [DOI] [PubMed] [Google Scholar]
- (57).Kopecka J; Salzano G; Campia I; Lusa S; Ghigo D; De Rosa G; Riganti C Insights in the Chemical Components of Liposomes Responsible for P-Glycoprotein Inhibition. Nanomedicine 2014, 10, 77–87. [DOI] [PubMed] [Google Scholar]
- (58).Rajendran L; Udayar V; Goodger ZV Lipid-Anchored Drugs for Delivery into Subcellular Compartments. Trends Pharmacol. Sci 2012, 33, 215–222. [DOI] [PubMed] [Google Scholar]
- (59).Gill KK; Kaddoumi A; Nazzal S Peg-Lipid Micelles as Drug Carriers: Physiochemical Attributes, Formulation Principles and Biological Implication. J. Drug Targeting 2015, 23, 222–231. [DOI] [PubMed] [Google Scholar]
- (60).Sand KMK; Bern M; Nilsen J; Noordzij HT; Sandlie I; Andersen JT Unraveling the Interaction between Fcrn and Albumin: Opportunities for Design of Albumin-Based Therapeutics. Front. Immunol 2014, 5, 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kratz F Doxo-Emch (Inno-206): The First Albumin-Binding Prodrug of Doxorubicin to Enter Clinical Trials. Expert Opin. Invest. Drugs 2007, 16, 855–866. [DOI] [PubMed] [Google Scholar]
- (62).Gabizon A; Shmeeda H; Barenholz Y Pharmacokinetics of Pegylated Liposomal Doxorubicin - Review of Animal and Human Studies. Clin. Pharmacokinet 2003, 42, 419–436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.