

Abstract
Radical S-adenosylmethionine (SAM) enzymes are a superfamily of enzymes that use SAM and reduced [4Fe-4S] cluster to generate a 5′-deoxyadenosyl radical to catalyze numerous challenging reactions. We have reported a type of non-canonical radical SAM enzymes in the diphthamide biosynthesis pathway. These enzymes also use SAM and reduced [4Fe-4S] clusters, but generate a 3-amino-3-carboxypropyl (ACP) radical to modify the substrate protein, translation elongation factor 2. The regioselective cleavage of a different C-S bond of the sulfonium center of SAM in these enzymes comparing to canonical radical SAM enzymes is intriguing. Here, we highlight some recent findings in the mechanism of this type of enzymes, showing that the diphthamide biosynthetic radial SAM enzymes bound SAM with a distinct geometry. In this way, the unique iron of the [4Fe-4S] cluster in the enzyme can only attack the carbon on the ACP group to form an organometallic intermediate. The homolysis of the organometallic intermediate releases the ACP radical and generates the EF2 radial.
Graphical Abstract
Radical S-adenosylmethionine (SAM) enzymes are a superfamily of enzymes that use SAM and [4Fe-4S] clusters to carry out diverse and chemically challenging reactions.1 Examples of radical SAM enzymes include lysine 2,3-aminomutase,2 biotin synthase,3 lipoyl synthase,4 and pyruvate-formate lyase activating enzyme (PFL-AE).5 These radical SAM enzymes typically have a conserved CxxxCxxC motif that provides the three cysteine residues to bind the [4Fe-4S] cluster.1 Three of the four Fe atoms in the [4Fe-4S] cluster are coordinated by the three cysteine residues, and the fourth Fe (also called the unique iron) is used to bind SAM. SAM binds the fourth Fe with the amino and carboxylate groups, and the binding positions the sulfonium group of SAM near the fourth iron.6 Such an arrangement allows electron transfer from the reduced [4Fe-4S]1+ cluster to SAM, generating a deoxyadenosyl radical, which is responsible for initiating downstream chemistry that happens to the substrate.
In the last ten years, a different type of radical SAM enzymes have been identified, which are involved in the biosynthesis of diphthamide, a post-translationally modified histidine residue on archaeal and eukaryotic translation elongation factor 2 (EF2).7, 8 This unique type of radical SAM enzymes has revealed some interesting chemistry, which is different from, but helps to understand, the chemistry of canonical radical SAM enzymes. Here, we briefly summarize the diphthamide biosynthetic radical SAM enzymes and provide several perspectives about their chemistry by comparing them to canonical radical SAM enzymes.
Diphthamide biosynthesis pathway and proteins required
Diphthamide is the target of diphtheria toxin, which selectively ADP-ribosylates diphthamide and shuts down protein synthesis of the host cells.9 The structure of the modification was determined in 1980.10–12 Seven genes are required for the biosynthesis of diphthamide13–17 and their protein products are responsible for the four-step biosynthesis (Fig. 1).15–19 Dph1–4 are required for the addition of 3-amino-3-carboxypropyl (ACP) group from SAM to histidine in EF2.20, 21 The ACP modified EF2 is then methylated by Dph5, which add four methyl groups to form methylated diphthine.20, 21 Dph7 is responsible for hydrolyzing the methyl ester from methylated diphthine.21 The last step of amidation is catalyzed by Dph6 to produce diphthamide.18
Figure 1.

Diphthamide biosynthesis pathway in eukaryotes.
The first step of diphthamide biosynthesis is a complicated step involving at least four different proteins, among which the radical SAM enzyme Dph1-Dph2 heterodimer directly transfers ACP group from SAM to EF2. Dph3 serves as an electron carrier that is needed for reducing the [4Fe-4S]2+ in Dph1-Dph2. Dph3 is a versatile small protein that binds Fe with four conserved cysteine residues in a so called CSL zinc finger domain. Dph3 also participates in tRNA wobble uridine modification.22 tRNA wobble uridine modification requires a different radical SAM enzyme in the Elongator complex.23 Dph3 likely serve as electron carrier for Elongator complex as well. Yeast Cbr1 can utilize NADH to reduce Dph3.24 Deletion of Cbr1 slightly affects diphthamide biosynthesis but significantly affects wobble uridine biosynthesis, likely reflecting the relative amount of the two modifications and the different demands for the reducing equivalents.
The exact function of Dph4 is unknown. Dph4 contains a DnaJ domain25 and shares a similar CSL zinc finger domain with Dph3. Unlike Dph3, Dph4 cannot serve as the electron carrier needed to reduce Dph1-Dph226. The zinc finger domain prefers to bind iron over zinc.27 Its exact function in diphthamide biosynthesis remains to be elucidated.
Recently, another protein, Killer toxin insensitive gene 13 (Kti13), is reported to be important for the first step of diphthamide biosynthesis and Kti13 deletion decreases diphthamide modification. Kti13 is also required for tRNA wobble uridine modification.28–30 It was found to co-purify with Dph3.31 The binding of Dph3 to Kti13 inhibits the reduction of Dph3. The structure of Dph3 and Kti13 heterodimer was solved.32, 33 From the published structure, Kti13 restricts the reduction of Dph3 by blocking the access to the iron atom in Dph3. As Dph3 is the electron carrier important for reducing the Dph1-Dph2 complex, it is counterintuitive why Kti13 is important for diphthamide modification. This paradox suggests that much remain to be understood about the first step of diphthamide biosynthesis.
Dph5 was first identified as a methyl transferase in diphthamide biosynthesis pathway.15, 20 It was further confirmed by in vitro reconstitution of the second step diphthamide biosynthesis with diphthine synthetase in Pyrococcus horikoshii.34 PhDph5 can transfer three methyl groups to ACP modified EF2 and the trimethyl amino group can be eliminated.34 Saccharomyces cerevisiae Dph5, in contrast, transfer four methyl groups into ACP modified EF2 to form methylated diphthine.21 Why yeast dph5 transfer four, instead of three, methyl groups is not known. It is especially intriguing as the methyl added to the carboxylate needs to be removed later by another enzyme, Dph7.21
Dph7 was initially identified as a gene involved in the first step of diphthamide biosynthesis in a human haploid cell genetic screening.35 However, it functions in a later step in the biosynthesis.19 Using mass spectrometry to study the biosynthesis product in Dph7 deletion yeast cell, Dph7 was identified as a methylesterase that convert methylated diphthine to diphthine, a result that was further confirmed by in vitro reconstitution.21 Furthermore, Dph7 is the first identified WD40 domain with an enzymatic activity. Its catalytic mechanism is currently unknown. The last step of diphthamide biosynthesis is catalyzed by the ATP-dependent enzyme Dph6, which was discovered by three different labs independently.19, 36, 37
Catalytic mechanism of the radical SAM enzyme involved in diphthamide biosynthesis
The first step of diphthamide biosynthesis is the transfer of an ACP group from SAM to a histidine residue of EF2. The enzyme that performs this reaction is a Dph2 homodimer in archaea, such as Pyrococcus horikoshii (PhDph2)7 or a Dph1-Dph2 heterodimer in eukaryotes.8 Initial characterization of this non-canonical radical SAM enzyme was done using PhDph2, but in later studies, yeast Dph1-Dph2 heterodimer was found to be similar. PhDph2 binds an air sensitive [4Fe-4S] cluster with three cysteine residues, similar to canonical radical SAM enzymes.7 However, the three cysteine residues are separated from each other by more than 100 amino acids, which is different from that in canonical radical SAM enzymes.7 PhDph2 also binds SAM. When reduced by dithionite, the [4Fe-4S] of PhDph2 reductively cleaves the Cγ,Met-S bond of SAM and transfer the ACP group to PhEF2. An ACP radical was proposed to be involved due to the following three observations: 1) A reduced [4Fe-4S] cluster is required to catalyze the reaction; 2) Proton and dithionite radical quenched products, 2-aminobutyric acid and homocysteine sulfinic acid, were detected in the reaction without substrate protein, PhEF2 (Figure 2);7 3) When a carboxyallyl SAM analogue, SAMCA,38 was used in the reaction, dithionite-quenched carboxyallyl radical products were detected (Figure 2). However, no radical intermediate was directly observed in the reaction with SAM, and how it reacts with the imidazole ring on EF2 to form product was unaddressed.
Figure 2.

PhDph2-catalyzed reactions with SAM and SAMCA.
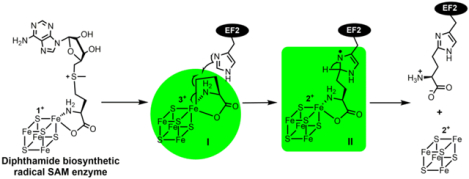
Recently, rapid freeze-quench (RFQ) was used to trap catalytic intermediates involved in PhDph2 or Dph1-Dph2 catalyzed reaction.39 Electron paramagnetic resonance (EPR) and electron-nuclear double resonance (ENDOR) spectroscopy were used to characterize the trapped intermediates. These studies used the yeast Dph1-Dph2 system because the enzyme is active at room temperature, which is more convenient to use in the RFQ experiments. Interestingly, two EPR-active intermedates (I and II) were detected. Both intermediate I and II are kinetically competent, suggesting that they are intermediates in the reaction pathway. Intermeidate I has a signal with g = 2.036, 2.005, which was detected by EPR at 12 K. This species was not an ACP radical, but instead represents an organometallic complex involving the [4Fe-4S] cluster. This intermediate is similar to the organometallic intermediate recently reported for PFL-AE, which was assigned to organometallic complex with deoxyadenosyl forms a Fe-C5′ bond with the unique cluster iron.40 Further 57Fe and (methionine-13C5)-SAM labeling experiments support that intermediate I involves both the ACP group and the Fe-S cluster.
In the presence of the substrate protein EF2, intermediate I forms first and then decays. As Intermediate I signal decreases, Intermediate II signal increases. Intermediate II appears to be an organic radical with g = 2 and a large hyperfine splitting (A = 120 MHz). The signal was saturated at 35 K, but readily observed at 70K. The organic radical involves the His699 residue of EF2 because with the EF2 His699Ala mutant, intermediate II signal was not detected.
The intermediate II organic radical contains both His699 of EF2 and ACP from SAM based on isotope EPR analysis. The EPR-resolved doublet collapsed to a single peak of the organic radical trapped with EF2 protein containing 2H5-His. When Intermediate II was trapped with (methionine-13C5)-SAM, the X-band EPR spectrum showed additional splitting and broadening compared to that of the natural-abundance SAM.
The EPR spectrum of the organic radical nicely parallel the cross-linked protein-nucleic acid radicals formed in the reaction catalyzed by the radical SAM enzymes, Cfr41 and RImN.42 Based on these and the structure of the final diphthamide product, an imidazole-derived organic radical was proposed (Figure 3).
Figure 3.

The proposed reaction mechanism of the first step of diphthamide biosynthesis.
Another interesting question that needs to be addressed is how the diphthamide biosynthetic radical SAM enzymes achieve the cleavage of a C-S bond in SAM that is different from all other canonical radical SAM enzymes. It has been suggested that for canonical radical SAM enzymes, SAM binds the [4Fe-4S] cluster in a way that allow the unique cluster Fe to carry out a backside attack on the C5′,Ade-SMet in a radical displacement reaction.43 Thus, it is easy to imagine that for the diphthamide biosynthetic radical SAM enzymes, SAM binds in a different orientation to allow a similar radical displacement reaction on a different C-S bond. Interestingly, recent X-ray crystal structures of archaeal Dph2 (PhDph2 and Candidatus Methanoperedens nitroreducens Dph2 or CmnDph2) in complex with SAM provide a different answer. In both structures, the amino and carboxyl groups of SAM interacted with the unique Fe of the Fe-S cluster, similar to canonical radical SAM enzymes. Perhaps not surprisingly, SAM in both PhDph2 and CmnDph2 binds in a quite different orientation compared to that in canonical radical SAM enzymes such as PFL-AE(PDB 3CB8). However, the surprising finding was that in the PhDph2 and CmnDph2 structures, the average Cγ,Met distance to the unique Fe is 3.7 Å, closer than SMet to the Fe (4.6 Å). In contrast, in the PFL-AE structure, the SMet is closer to the unique Fe than C5′,Ade, with an SMet-Fe distance of 3.2 Å. This distinct binding mode of SAM in the PhDph2 and CmnDph2 structures provides important insights into the mechanism of the reaction and how a different C-S bond in SAM get cleaved. The proximity of the Cγ,Met and the unique Fe suggests that during the Cγ,Met-SMet bond cleavage, the electron is transferred from the Fe-S cluster via Cγ,Met to the σ* orbital of the Cγ,Met-SMet bond, while for PFL-AE and other canonical radical SAM enzymes, the electron is generally believed to be transferred from the Fe-S cluster via SMet.1 When the electron is transferred to Cγ,Met, the Cγ,Met-SMet bond is the only weak bond that can be broken, thus explaining the selectivity of the bond cleavage.
On the basis of the two captured intermediates and structural observations, a reaction mechanism for the first step of diphthamide biosynthesis was proposed (Figure 3). The unique iron of [4Fe-4S]+ cluster in PhDph2 or Dph1-Dph2 transfers two electrons (either a one-step two-electron transfer or two one-electron transfers) to the γ carbon of the methionine in SAM, generating a 3-amino-3-carboxypropyl-[4Fe-4S]3+ organometallic intermediate I and MTA. Intermediate I serves as a stabilized ACP radical. In the presence of the substrate EF2, the homolysis of the Fe-C bond releases the ACP radical, which reacts with the imidazole ring of His699 of EF2 and generates the organic radical intermediate II. Intermediate II then loses a proton and an electron to form an ACP-modified histidine.
It should be noted that even though the organometallic intermediate I is similar to that reported for PFL-AE (see Figure 4B),40 the formation mechanism may be different. For the formation of the PFL-AE organometallic intermediate, because the electron is transferred to the S atom, the deoxyadenosyl radical forms first and then recombines with the [4Fe-4S] cluster (two one-electron transfers). For the diphthamide biosynthetic radical SAM enzymes, because electrons are directly transferred to Cγ,Met, it is possible that Intermediate I is formed via a one-step two-electron transfer process instead of a radical mechanism that is similar to PFL-AE. However, the two mechanisms cannot be experimentally differentiated now.
Figure 4.

Examples of organometallic intermediates serving as radical sources in biology. A, adenosylcobalamin; B, organometallic intermediate in PFL-AE; C, organometallic intermediate in Dph1-Dph2.
Organometallic complex as stabilized radical intermediate could be a general strategy in biology.
Organometallic complexes are widely applied in chemistry, such as Grignard reagent, ferrocene, orgnaozinc reagents and intermediates of transition metal catalyzed coupling reactions. In biology, several organometallic catalysts are reported: the Fe, Fe-Fe and Ni-Fe hydrogenase,44 nitrogenases,45 Ni-C bond in acetyl-CoA synthase46 and the well-studied vitamin B12.47, 48 The cobalt atom in B12 can form Co-C bonds with adenosyl, methyl, and cyano groups in adenosylcobalamin, methylcobalamin and cyanocobalamin, respectively.
Homolysis of Co-C bond in enzyme bonded adenosylcobalamin can generate a 5′-deoxyadenosyl (5′-dA) radical and catalyze several difficult reactions, like in amino mutase, ribonucleotide reducase, and dehydratase.48 The homolytic cleavage of Co-C bond relies on the weak Co-C bond and the existence of three different oxidation states of Co (I/II/III). Similarly, radial SAM enzymes use a [4Fe-4S] cluster and SAM to generate a 5′-dA radical and catalyze numerous radical involved reactions. Recent study shows a Fe-C5′ organometallic intermediate in a canonical radical SAM enzyme PFL-AE.40 Similar to Co-C bond in AdoCbl, there is a Fe-C bond between adenosyl and the unique iron of [4Fe-4S] cluster (Figure 4). The homolysis of this Fe-C5′ bond generates the 5′-dA radical, which extracts a proton from the substrate and produces a glycyl radial. Now the work on the radial SAM enzymes in diphthamide biosynthesis also shows the presence of an organometallic intermediate, which acts as the ACP radical source and reacts with EF2 substrate to generate an EF2 radial. In this process, the [4Fe-4S] cluster also experiences three different oxidation states (I/II/III).
Overall, adenosylcobalamin and Fe-C organometallic intermediates in PFL-AE and dipthamide biosynthesis all serve as stabilized form of the highly reactive primary organic radicals. The homolysis of the organometallic intermediates release 5′-dA or ACP radicals and catalyze the downstream reactions. Such organometallic intermediates likely act to ‘tame’ the reactive radicals generated by these enzymes. This may be a general strategy that nature uses to control radical reactions.
Funding
This work is supported by NIH (GM 088276 to H.L.)
Footnotes
The authors declare no competing financial interest.
REFERENCES
- [1].Broderick JB, Duffus BR, Duschene KS, and Shepard EM (2014) Radical S-adenosylmethionine enzymes, ChemRev 114, 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wang SC, and Frey PA (2007) Binding Energy in the One-Electron Reductive Cleavage of S-Adenosylmethionine in Lysine 2,3-Aminomutase, a Radical SAM Enzyme, Biochemistry 46, 12889–12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ugulava NB, Sacanell CJ, and Jarrett JT (2001) Spectroscopic Changes during a Single Turnover of Biotin Synthase: Destruction of a [2Fe-2S] Cluster Accompanies Sulfur Insertion, Biochemistry 40, 8352–8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Miller JR, Busby RW, Jordan SW, Cheek J, Henshaw TF, Ashley GW, Broderick JB, Cronan JE, and Marletta MA (2000) Escherichia coli LipA Is a Lipoyl Synthase: In Vitro Biosynthesis of Lipoylated Pyruvate Dehydrogenase Complex from Octanoyl-Acyl Carrier Protein, Biochemistry 39, 15166–15178. [DOI] [PubMed] [Google Scholar]
- [5].Henshaw TF, Cheek J, and Broderick JB (2000) The [4Fe-4S]1+ Cluster of Pyruvate Formate-Lyase Activating Enzyme Generates the Glycyl Radical on Pyruvate Formate-Lyase: EPR-Detected Single Turnover, J. Am. Chem. Soc 122, 8331–8332. [Google Scholar]
- [6].Chen D, Walsby C, Hoffman BM, and Frey PA (2003) Coordination and Mechanism of Reversible Cleavage of S-Adenosylmethionine by the [4Fe-4S] Center in Lysine 2,3-Aminomutase, J. Am.Chem. Soc 125, 11788–11789. [DOI] [PubMed] [Google Scholar]
- [7].Zhang Y, Zhu X, Torelli AT, Lee M, Dzikovski B, Koralewski RM, Wang E, Freed J, Krebs C, Ealick SE, and Lin H (2010) Diphthamide biosynthesis requires an organic radical generated by an iron-sulphur enzyme, Nature 465, 891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dong M, Su X, Dzikovski B, Dando EE, Zhu X, Du J, Freed JH, and Lin H (2014) Dph3 is an electron donor for Dph1-Dph2 in the first step of eukaryotic diphthamide biosynthesis, JAm.Chem.Soc 136, 1754–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Honjo T, Nishizuka Y, and Hayaishi O (1968) Diphtheria toxin-dependent adenosine diphosphate ribosylation of aminoacyl transferase II and inhibition of protein synthesis, J.Biol.Chem 243, 3553–3555. [PubMed] [Google Scholar]
- [10].Van Ness BG, Howard JB, and Bodley JW (1978) Isolation and properties of the trypsin-derived ADP-ribosyl peptide from diphtheria toxin-modified yeast elongation factor 2, J. Biol.Chem 253, 8687–8690. [PubMed] [Google Scholar]
- [11].Van Ness BG, Howard JB, and Bodley JW (1980) ADP-ribosylation of elongation factor 2 by diphtheria toxin. Isolation and properties of the novel ribosyl-amino acid and its hydrolysis products, J. Biol.Chem 255, 10717–10720. [PubMed] [Google Scholar]
- [12].Van Ness BG, Howard JB, and Bodley JW (1980) ADP-ribosylation of elongation factor 2 by diphtheria toxin. NMR spectra and proposed structures of ribosyl-diphthamide and its hydrolysis products, J. Biol. Chem 255, 10710–10716. [PubMed] [Google Scholar]
- [13].Dunlop PC, and Bodley JW (1983) Biosynthetic labeling of diphthamide in Saccharomyces cerevisiae, J.Biol. Chem 258, 4754–4758. [PubMed] [Google Scholar]
- [14].Chen JY, Bodley JW, and Livingston DM (1985) Diphtheria toxin-resistant mutants of Saccharomyces cerevisiae, Mol. Cell. Biol 5, 3357–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen JY, and Bodley JW (1988) Biosynthesis of diphthamide in Saccharomyces cerevisiae. Partial purification and characterization of a specific S-adenosylmethionine:elongation factor 2 methyltransferase, J. Biol. Chem 263, 11692–11696. [PubMed] [Google Scholar]
- [16].Moehring TJ, Danley DE, and Moehring JM (1984) In vitro biosynthesis of diphthamide, studied with mutant Chinese hamster ovary cells resistant to diphtheria toxin, Mol.Cell. Biol 4, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu S, Milne GT, Kuremsky JG, Fink GR, and Leppla SH (2004) Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2, Mol.Cell. Biol 24, 9487–9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Su X, Chen W, Lee W, Jiang H, Zhang S, and Lin H (2012) YBR246W is required for the third step of diphthamide biosynthesis, J.Am.Chem.Soc 134, 773–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Su X, Lin Z, Chen W, Jiang H, Zhang S, and Lin H (2012) Chemogenomic approach identified yeast YLR143W as diphthamide synthetase, Proc. Natl. Acad. Sci. U. S. A 109, 19983–19987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mattheakis LC, Shen WH, and Collier RJ (1992) DPH5, a methyltransferase gene required for diphthamide biosynthesis in Saccharomyces cerevisiae, Mol. Cell.Biol 12, 4026–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lin Z, Su X, Chen W, Ci B, Zhang S, and Lin H (2014) Dph7 catalyzes a previously unknown demethylation step in diphthamide biosynthesis, J.Am. Chem. Soc 136, 6179–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bar C, Zabel R, Liu S, Stark MJ, and Schaffrath R (2008) A versatile partner of eukaryotic protein complexes that is involved in multiple biological processes: Kti11/Dph3, Mol Microbiol 69, 1221–1233. [DOI] [PubMed] [Google Scholar]
- [23].Huang B, Johansson MJ, and Bystrom AS (2005) An early step in wobble uridine tRNA modification requires the Elongator complex, RNA 11, 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lin Z, Dong M, Zhang Y, Lee EA, and Lin H (2016) Cbr1 is a Dph3 reductase required for the tRNA wobble uridine modification, Nat.Chem. Biol 12, 995–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Webb TR, Cross SH, McKie L, Edgar R, Vizor L, Harrison J, Peters J, and Jackson IJ (2008) Diphthamide modification of eEF2 requires a J-domain protein and is essential for normal development, J. Cell. Sci 121, 3140–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dong M, Su X, Dzikovski B, Dando EE, Zhu X, Du J, Freed JH, and Lin H (2014) Dph3 is an electron donor for Dph1-Dph2 in the first step of eukaryotic diphthamide biosynthesis, J Am Chem Soc 136, 1754–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Thakur A, Chitoor B, Goswami AV, Pareek G, Atreya HS, and D’Silva P (2012) Structure and mechanistic insights into novel iron-mediated moonlighting functions of human J-protein cochaperone, Dph4, J.Biol.Chem 287, 13194–13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fichtner L, and Schaffrath R (2002) KTI11 and KTI13, Saccharomyces cerevisiae genes controlling sensitivity to G1 arrest induced by Kluyveromyces lactis zymocin, Mol. Microbiol 44, 865–875. [DOI] [PubMed] [Google Scholar]
- [29].Frohloff F, Fichtner L, Jablonowski D, Breunig KD, and Schaffrath R (2001) Saccharomyces cerevisiae Elongator mutations confer resistance to the Kluyveromyces lactis zymocin, Embo. J 20, 1993–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Karlsborn T, Tukenmez H, Mahmud AK, Xu F, Xu H, and Bystrom AS (2014) Elongator, a conserved complex required for wobble uridine modifications in eukaryotes, RNA Biol 11, 1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zabel R, Bar C, Mehlgarten C, and Schaffrath R (2008) Yeast alpha-tubulin suppressor Ats1/Kti13 relates to the Elongator complex and interacts with Elongator partner protein Kti11, Mol.Microbiol 69, 175–187. [DOI] [PubMed] [Google Scholar]
- [32].Glatt S, Zabel R, Vonkova I, Kumar A, Netz DJ, Pierik AJ, Rybin V, Lill R, Gavin AC, Balbach J, Breunig KD, and Muller CW (2015) Structure of the Kti11/Kti13 heterodimer and its double role in modifications of tRNA and eukaryotic elongation factor 2, Structure 23, 149–160. [DOI] [PubMed] [Google Scholar]
- [33].Kolaj-Robin O, McEwen AG, Cavarelli J, and Seraphin B (2015) Structure of the Elongator cofactor complex Kti11/Kti13 provides insight into the role of Kti13 in Elongator-dependent tRNA modification, FEBS J 282, 819–833. [DOI] [PubMed] [Google Scholar]
- [34].Zhu X, Kim J, Su X, and Lin H (2010) Reconstitution of diphthine synthase activity in vitro, Biochemistry 49, 9649–9657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, and Brummelkamp TR (2009) Haploid genetic screens in human cells identify host factors used by pathogens, Science 326, 1231–1235. [DOI] [PubMed] [Google Scholar]
- [36].Uthman S, Bar C, Scheidt V, Liu S, ten Have S, Giorgini F, Stark MJ, and Schaffrath R (2013) The amidation step of diphthamide biosynthesis in yeast requires DPH6, a gene identified through mining the DPH1-DPH5 interaction network, PLoS Genet 9, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].de Crecy-Lagard V, Forouhar F, Brochier-Armanet C, Tong L, and Hunt JF (2012) Comparative genomic analysis of the DUF71/COG2102 family predicts roles in diphthamide biosynthesis and B12 salvage, Biol Direct 7, 1745–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dong M, Horitani M, Dzikovski B, Pandelia ME, Krebs C, Freed JH, Hoffman BM, and Lin H (2016) Organometallic Complex Formed by an Unconventional Radical S-Adenosylmethionine Enzyme, J.Am. Chem. Soc 138, 9755–9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dong MKV; Fenwick MK; Torelli AT; Zhang Y; Caranto JD; Dzikovski B; Sharma A; Lancaster KM; Freed JH; Ealick SE; Hoffman BM; Lin H (2018) Organometallic and radical intermediates reveal mechanism of diphthamide biosynthesis, Science, 10.1126/science.aao65955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Horitani M, Shisler K, Broderick WE, Hutcheson RU, Duschene KS, Marts AR, Hoffman BM, and Broderick JB (2016) Radical SAM catalysis via an organometallic intermediate with an Fe-[5’-C]-deoxyadenosyl bond, Science 352, 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Grove TL, Livada J, Schwalm EL, Green MT, Booker SJ, and Silakov A (2013) A substrate radical intermediate in catalysis by the antibiotic resistance protein Cfr, Nat. Chem. Biol 9, 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Silakov A, Grove TL, Radle MI, Bauerle MR, Green MT, Rosenzweig AC, Boal AK, and Booker SJ (2014) Characterization of a Cross-Linked Protein–Nucleic Acid Substrate Radical in the Reaction Catalyzed by RlmN, J. Am. Chem.Soc 136, 8221–8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kampmeier JA (2010) Regioselectivity in the Homolytic Cleavage of S-Adenosylmethionine, Biochemistry 49, 10770–10772. [DOI] [PubMed] [Google Scholar]
- [44].Lubitz W, Ogata H, Rüdiger O, and Reijerse E (2014) Hydrogenases, ChemRev 114, 4081–4148. [DOI] [PubMed] [Google Scholar]
- [45].Wiig JA, Hu Y, Lee CC, and Ribbe MW (2012) Radical SAM-Dependent Carbon Insertion into the Nitrogenase M-Cluster, Science 337, 1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Can M, Giles LJ, Ragsdale SW, and Sarangi R (2017) X-ray Absorption Spectroscopy Reveals an Organometallic Ni–C Bond in the CO-Treated Form of Acetyl-CoA Synthase, Biochemistry 56, 1248–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Brown KL (2005) Chemistry and Enzymology of Vitamin B12, Chemical reviews 105, 2075–2150. [DOI] [PubMed] [Google Scholar]
- [48].Bridwell-Rabb J, and Drennan CL (2017) Vitamin B12 in the spotlight again, Current.Opinion in Chemical Biology 37, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]