

Abstract
Mammalian histone deacetylases (HDACs) are a class of enzymes that play important roles in biological pathways. Existing HDAC inhibitors target multiple HDACs without much selectivity. Inhibitors that target one particular HDAC will be useful for investigating the biological functions of HDACs and for developing better therapeutics. Here, we report the development of HDAC11-specific inhibitors using an activity-guided rational design approach. The enzymatic activity and biological function of HDAC11 have been little known, but recent reports suggest that it has efficient defatty-acylation activity and inhibiting it could be useful for treating a variety of human diseases, including viral infection, multiple sclerosis, and metabolic diseases. Our best inhibitor, SIS17, is active in cells and inhibited the demyristoylation of a known HDAC11 substrate, serine hydroxymethyl transferase 2, without inhibiting other HDACs. The activity-guided design may also be useful for the development of isoform-specific inhibitors for other classes of enzymes.
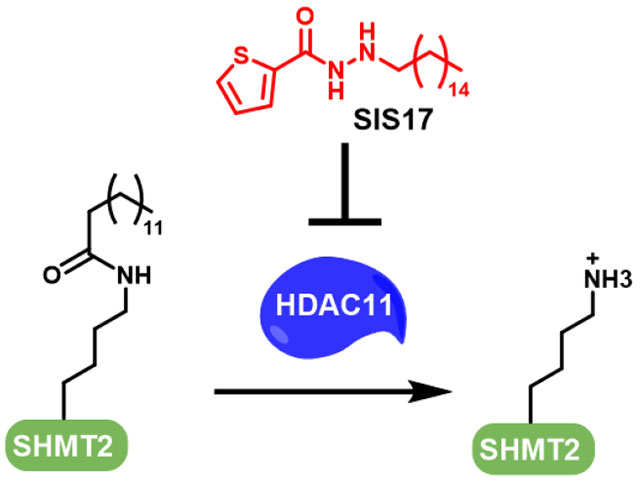
Graphical Abstract
Histone deacetylases are a class of zinc-dependent amide hydrolases with important biological functions.1,2 They catalyze the deacetylation of numerous substrate proteins, including histones, transcription factors, and signaling proteins and thus regulate various biological processes.1,2 They are the targets of important anticancer natural products, such as trapoxin and trichostatin, and clinically used synthetic anticancer drugs, such as Vorinostat and Romidepsin.1,3,4 Because of their clinical relevance, many small molecule inhibitors are commercially available. However, most of these inhibitors target multiple HDACs and thus the biological effect of targeting a specific HDAC is not clear. It is highly possible that small molecule inhibitors that selectively inhibit only one HDAC are better therapeutics due to lower toxicity. Developing small molecules that target only one of the HDACs is key to addressing these questions.
HDAC11 is the only Class IV HDAC and its biological function remains largely unknown. Three research teams independently discovered that HDAC11 works as a defatty-acylase instead of a deacetylase (Figure 1A).5–7 One physiological substrate identified is serine hydroxymethyl transferase 2 (SHMT2), an enzyme important for one-carbon metabolism.7 Interestingly, HDAC11 and lysine fatty acylation does not affect the enzymatic activity if SHMT2.7 Instead, they affect the ability of SHMT2 to deubiquitinate type I interferon receptor. Inhibiting HDAC11 therefore has the potential to treat diseases that involve type I interferon signaling. Along this line, there have been reports suggesting that suppression of HDAC11 could be beneficial for treating cancer,8–10 multiple sclerosis,11 viral infection,7 and metabolic disease.12,13 We therefore set out to develop HDAC11-specific inhibitors utilizing its unique activity profile, the preference for long-chain fatty acyl lysine.
Figure 1.

The design of HDAC11 specific inhibitors based on its catalytic activity. (A) HDAC11 hydrolyzes long chain fatty acyl lysine efficiently, but not acetyl lysine. (B) Modification of existing HDAC inhibitors SAHA and UF010 to obtain potential HDAC11 inhibitors.
Although there has been no reported X-ray crystal structure of HDAC11, HDAC11 is similar to Class I HDACs, sharing 30% sequence identity.14 Therefore, our HDAC11 inhibitors were designed based on known Class I HDAC inhibitors. Known HDAC inhibitors typically contain three structural motifs, a zinc chelation group, a linker, and a surface recognition group.1,4,15 Because HDAC11 prefers long-chain fatty acyl groups, we imagined that installation of a long hydrophobic moiety to known HDAC inhibitors could confer HDAC11 selectivity. We chose two simple HDAC inhibitors to test this hypothesis, Vorinostat (SAHA) and UF010 (Figure 1B).16,17 Neither SAHA or UF010 could inhibit HDAC11. We modified this structure by adding a long alkyl group while maintaining the zinc chelation group, linker, and the surface recognition group (Figure 1). The compounds (SIS4 and SIS15) were synthesized following the procedure described in the Supporting Information. We then tested them using an HPLC-based assay with a myristoyl-H3K9 peptide. Interestingly, the SAHA-derived SIS15 showed no inhibition of HDAC11 up to 100 μM, while the UF010-derived SIS4 showed modest inhibition of HDAC11 with an IC50 value about 35 μM (Figure 1B).
Given that UF010 did not inhibit HDAC11 while the derivative SIS4 did, this indicated that our strategy could work. We then decided to further optimize SIS4 by carrying out structure-activity relationship (SAR) studies. Changing the substituents on the benzene ring showed that replacing the bromo substituent with t-butyl (SIS2) or dimethylamino (SIS7) improved the IC50 values to 8.0 μM and 0.91 μM, respectively, when assayed using myristoyl-H3K9 peptide as the substrate. In contrast, changing the bromo substituent to fluoro (SIS5) or trifluoromethyl (SIS21) did not improve inhibition. Replacing the benzene ring with a butyryl group also led to loss of HDAC11 inhibition (SIS18). Thus, it seems that aromatic rings with an electron-donating group are preferred (Figure 2A).
Figure 2.

In vitro assay with HDAC11 inhibitors. (A) Optimization of HDAC11 inhibitors based on SIS4. Optimization of HDAC11 inhibitors based on SIS4. IC50 values were obtained by using myristoyl-H3K9 peptide as the substrate except values shown in parentheses for SIS7 and SIS17, which are measured using myristoyl-SHMT2 peptide as the substrate. Some of the IC50 curves are shown in Supplementary Figure 1. (B) Comparing SIS7 and SIS17 to FT895 on HDAC11 inhibition using myristoyl-H3K9. IC50 values using myristoyl-H3K9 derived from GraphPad Prism are presented as mean values from three independent experiments. (C) In vitro IC50 values against different HDACs and sirtuins with acyl-H3K9 peptides. [a] acyl-H3K9 peptides used. [b] FT895 inhibited 50% of HDAC4 activity at 25 μM but inhibition with higher concentration did not increase inhibition rate. [c] Deacetylation activity of enzyme tested. [d] Detrifluoroacetylation activity of enzyme tested. [e] Demyristoylation activity of enzyme tested. [f] Both deacetylation and demyristoylation activities of enzyme tested.
Based on this SAR observation, we then replaced the benzene ring with other electron-rich aromatic rings, thiophene (SIS17) and pyrrole (SIS39) rings. The thiophene-containing compound was similar to SIS7, with an IC50 value of 0.83 μM, but the pyrrole-containing compound SIS39 did not inhibit HDAC11. The pyrrole ring is likely less electron-donating compared to the thiophene ring due to stronger inductive effect of nitrogen atom. Alternatively, the NH of the pyrrole ring might be too polar to be accommodated by the HDAC11 active site.
Based on SIS17, we also explored the requirement of the carbohydrazide moiety, the presumed zinc chelating group, for HDAC11 inhibition. Replacing the carbohydrazide with an amide led to SIS40, which did not inhibit HDAC11. Thus, the zinc chelating carbohydrazide is crucial for HDAC11 inhibition.
The alkyl chain length on SIS17 also seemed to be important, as shortening it (SIS49, SIS50 and SIS65) or lengthening it (SIS52 and SIS66) decreased the HDAC11 inhibition potency. This was consistent with the previous HDAC11 enzymatic activity profile which showed that shorter or longer acyl chains were not good substrates of HDAC11.7
With the two potent HDAC11 inhibitors, SIS7 and SIS17, we then tested whether they are selective toward HDAC11. This is important as the purpose of our activity-based design was to obtain HDAC11-selective inhibitors. Furthermore, several other HDACs, including SIRT2, SIRT6, and HDAC8, have been shown to be able to remove long chain fatty acyl groups from protein lysine residues. Thus it is important to make sure that they are not inhibited by the HDAC11 inhibitors. 19–21 To examine the selectivity of SIS7 and SIS17, we picked representative HDACs from each class, Class I (HDAC1 and HDAC8), Class II (HDAC4), and the NAD-dependent Class III (sirtuins, SIRT1, SIRT2, SIRT3, and SIRT6). Satisfyingly, SIS7 and SIS17 did not show significant inhibition of these HDACs at 100 μM concentrations (Figure 2C). The selectivity of these two inhibitors highlights that our activity-guided design is effective.
In the literature, only a few HDAC11 inhibitors had been reported, including palmitic acid, Trapoxin A, and FT895.5,6,18 In our hands, palmitic acid did not show good inhibition on HDAC11 (no inhibition at 10 μM). TpxA could completely abolish HDAC11 activity at 1 μM but it is a pan-HDAC inhibitor. Forma Therapeutics recently reported an HDAC11-specific inhibitor, FT895.18 The reported in vitro IC50 value is 3 nM, but it was measured using a trifluoroacetyl lysine compound, not the physiological myristoyl lysine peptide. Therefore, we synthesized FT895 and measured its IC50 value using the myristoyl-H3K9 in the HPLC assay. FT895 inhibited HDAC11 with an IC50 value of 0.74 μM which is slightly better than SIS7 (IC50 0.91 μM) and SIS17 (IC50 0.83 μM). However, it is not as selective as SIS7 or SIS17 because it also inhibited HDAC4 (IC50 25 μM), and HDAC8 (IC50 9.2 μM) (Figure 2C). Thus, SIS7 and SIS17 are the most selective HDAC11 inhibitors known.
Because of the lack of physiological substrates for HDAC11, none of the known HDAC11 inhibitors had been tested in cellular assays. It was recently reported that HDAC11 defatty-acylates SHMT2, which in turn regulates type I interferon receptor signaling.7 We thus checked whether our HDAC11 inhibitors can increase the fatty acylation level of endogenous SHMT2 using a previously established detection method. We first tested whether SIS7 and SIS17 can inhibit the demyristoylation of a myristoyl-SHMT2 peptide by HDAC11 in vitro. Using a myristoyl-SHMT2 peptide as the substrate, SIS7 and SIS17 could still inhibit HDAC11 with IC50 values of 1.0 μM and 270 nM, respectively, which is slightly different from that measured with myristoyl-H3K9 peptides (Supplementary Figure 2).
We then tested whether SIS7 and SIS17 could inhibit HDAC11-mediated SHMT2 defatty-acylation in cells. We treated MCF7 cells with 50 μM of an alkyne-tagged palmitic acid analog, Alk14, along with 0, 12.5, 25.0, and 50.0 μM of SIS7 or SIS17 for 6 hours at 37oC. The labeled proteins were pulled down with streptavidin beads after installation of a biotin tag with click chemistry. The amount of acylated SHMT2 was then detected by Western blot (Figure 3). SIS17 increased the fatty acylation level of SHMT2 significantly in MCF7 cells down to 12.5 μM demonstrating that it is cell permeable and can inhibit HDAC11 in living cells. SIS7 was less effective than SIS17 in cells. (Figure 3). We believe this was mainly due to cell permeability or metabolic stability. Using and LC-MS based assay, we could readily detect SIS17 in cell lysate, but not SIS7 (Supplementary Table 1).
Figure 3.

Endogenous fatty acylation levels of SHMT2 in MCF7 cells treated with 12.5, 25.0 and 50.0 μM of SIS7, SIS17 and FT895. Representative Western blot images are shown. Signal intensity was quantified by ImageJ and signal of control group without inhibitor treatment set as 1.0. *, p value < 0.1, **, p value < 0.05, ***, p value < 0.01; ns, not statistically significant.
To further demonstrate the selectivity of these inhibitors in cells, we checked whether SIS7, SIS17, and FT895 could increase the acetylation level of α-tubulin and histone H3 (Figure 4). SIS7, SIS17 and FT895 up to 50 μM did not affect the acetylation levels of α-tubulin and histone H3. In contrast, the pan-HDAC inhibitor SAHA significantly increased the acetylation levels at 1 μM. Thus, the selectivity of SIS7 and SIS17 for HDAC11 is maintained in cells.
Figure 4.

Acetylation in cells is not affected by HDAC11 inhibitors. Immunoblots are shown for acetyl-α-tubulin (K40) and acetyl-histone H3 in HEK 293T cells treated with inhibitors for 6 hrs. Loading for each sample was checked by blue-stained membrane (Supplementary Figure 3).
In summary, using an activity-guided rational design approach, we have developed two potent and selective inhibitors, SIS7 and SIS17, for HDAC11. In vitro, SIS7 and SIS17 are slightly less potent against HDAC11 compared to FT895 but are more selective. In cells, SIS17 appears better than FT895 and SIS7 due to increased cell permeability and/or metabolic stability. To our best knowledge, SIS17 represents the first HDAC11-selective inhibitor to show cellular effects. Given the recent increased research interest in HDAC11, we believe SIS17 will be a useful tool compound to explore the biological function and therapeutic potential of HDAC11. Furthermore, we believe this activity-guided design approach could be generally useful for the development of isoform-selective inhibitors for other families of enzymes.
Supplementary Material
ACKNOWLEDGMENT
This work is supported by HHMI, Cornell University, and a grant from NIH/NIDDK DK107868. This work used the Cornell University NMR Facility, which is supported by the NSF through MRI award CHE-1531632.
Footnotes
Supporting Information
Full details of materials and methods, including reagents information, synthesis of each compounds, procedure for in cell experiments, 1H/ 13C NMR data and HR-MS of compounds, and experimental details were described in supporting information.
REFERENCES
- (1).Falkenberg KJ; Johnstone RW Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014, 13, 673–691. [DOI] [PubMed] [Google Scholar]
- (2).Yang XJ; Seto E The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008, 9, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Finnin MS; Donigian JR; Cohen A; Richon VM; Rifkind RA; Marks PA; Breslow R; Pavletich NP Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999, 401, 188–193. [DOI] [PubMed] [Google Scholar]
- (4).Lombardi PM; Cole KE; Dowling DP; Christianson DW Structure, Mechanism, and Inhibition of Histone Deacetylases and Related Metalloenzymes. Curr. Opin. Struct. Biol 2011, 21, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Moreno-Yruela C; Galleano I; Madsen AS; Olsen CA Histone Deacetylase 11 Is an ε-N-Myristoyllysine Hydrolase. Cell Chem. Biol 2018, 25, 849–856. [DOI] [PubMed] [Google Scholar]
- (6).Kutil Z; Novakova Z; Meleshin M; Mikesova J; Schutkowski M; Barinka C Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem Biol. 2018, 13, 685–693. [DOI] [PubMed] [Google Scholar]
- (7).Cao J; Sun L; Aramsangtienchai P; Spiegelman NA; Zhang X; Seto E; Lin H HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. PNAS, 2019, 116, 5487–5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Deubzer HE; Schier MC; Oehme I; Lodrini M; Haendler B; Sommer A; Witt O HDAC11 is a novel drug target in carcinomas. Int J Cancer. 2013, 132, 2200–2208. [DOI] [PubMed] [Google Scholar]
- (9).Buglio D; Khaskhely NM; Voo KS; Martinez-Valdez H; Liu YJ; Younes A HDAC11 plays an essential role in regulating OX40 ligand expression in Hodgkin lymphoma. Blood. 2011, 117, 2910–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Thole TM; Lodrini M; Fabian J; Wuenschel J; Pfeil S; Hielscher T; Kopp-Schneider A; Heinicke U; Fulda S; Witt O; Eggert A; Fischer M; Deubzer HE Neuroblastoma cells depend on HDAC11 for mitotic cell cycle progression and survival. Cell Death Disease 2017, 8, e2635–e2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sun L; Telles E; Karl M; Cheng F; Luetteke N; Sotomayor EM; Miller RH; Seto E Loss of HDAC11 ameliorates clinical symptoms in a multiple sclerosis mouse model. Life Science Alliance 2018. 1, e201800039–e201800039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bagchi RA; Ferguson BS; Stratton MS; Hu T; Cavasin MA; Sun L; Lin YH; Liu D; Londono P; Song K; Pino MF; Sparks LM; Smith SR; Scherer PE; Collins S; Seto E; McKinsey TA HDAC11 suppresses the thermogenic program of adipose tissue via BRD2. JCI Insight. 2018, 3, 120159–12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sun L; Marin de Evsikova C; Bian K; Achille A; Telles E; Pei H; Seto E Programming and Regulation of Metabolic Homeostasis by HDAC11. EBioMedicine. 2018, 33, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gao L; Cueto MA; Asselbergs F; Atadja P Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem. 2002, 277, 25748–25755. [DOI] [PubMed] [Google Scholar]
- (15).Hancock WW; Akimova T; Beier UH; Liu Y; Wang L HDAC inhibitor therapy in autoimmunity and transplantation. Ann. Rheum. Dis 2012, 71, i46–i54. [DOI] [PubMed] [Google Scholar]
- (16).Marks PA; Breslow R Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol 2007, 25, 84–90. [DOI] [PubMed] [Google Scholar]
- (17).Wang Y; Stowe RL; Pinello CE; Tian G; Madoux F; Li D; Zhao LY; Li JL; Wang Y; Wang Y; Ma H; Hodder P; Roush WR; Liao D Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem Biol. 2015, 22, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Martin MW; Lee JY; Lancia DR Jr.; Ng PY; Han B; Thomason JR; Lynes MS; Marshall CG; Conti C; Collis A; Morales MA; Doshi K; Rudnitskaya A; Yao L; Zheng X Discovery of novel N-hydroxy-2-arylisoindoline-4-carboxamides as potent and selective inhibitors of HDAC11. Bioorg Med Chem Lett. 2018, 28, 2143–2147. [DOI] [PubMed] [Google Scholar]
- (19).Jing H; Zhang X; Wisner SA; Chen X; Spiegelman NA; Linder ME; Lin H SIRT2 and lysine fatty acylation regulate the transforming activity of K-Ras4a, eLife, 2017, 6, e32436–e32468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhang X; Spiegelman NA; Nelson OD; Jing H; Lin H SIRT6 regulates Ras-related protein R-Ras2 by lysine defatty-acylation, eLife, 2017, 6, e25158–e25174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang X; Khan S; Hong J; Antonyak MA; Chen X; Spiegelman NA; Shrimp JH; Cerione RA; Lin H Identifying the functional contribution of the defatty-acylase activity of SIRT6, Nat. Chem. Biol, 2016, 12, 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Aramsangtienchai P; Spiegelman NA; He B; Miller SP; Dai L; Zhao Y; Lin H HDAC8 Catalyzes the Hydrolysis of Long Chain Fatty Acyl Lysine, ACS Chem. Biol, 2016, 11, 2685–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.