

Abstract
NF-κB signals through canonical transcription factor p65 (RelA)/p50 and noncanonical avian reticuloendotheliosis viral oncogene related B (RelB)/p52 pathways. The RelA/p50 is involved in basal and inflammatory lymphangiogenesis. However, the role of RelB/p52 in lymphatic vessel biology is unknown. Herein, we investigated changes in lymphatic vessels (LVs) in mice deficient in noncanonical NF-κB signaling and the function of RelB in lymphatic endothelial cells (LECs). LVs were examined in Relb−/−, p52−/−, or control mice, and the gene expression profiles in LECs with RelB knockdown. Relb−/−, but not p52−/−, mice exhibited multiple LV abnormalities. They include the following: i) increased capillary vessel diameter, ii) reduced smooth muscle cell (SMC) coverage of mature vessels, iii) leakage, and iv) loss of active and passive lymphatic flow. Relb−/− mature LVs had thinner vessel walls, more apoptotic LECs and SMCs, and fewer LEC junctions. RelB knockdown LECs had decreased growth, survival, and adhesion, and dysregulated signaling pathways involving these cellular events. These results suggest that Relb−/− mice have abnormal LVs, mainly in mature vessels with reduced SMC coverage, leakage, and loss of contractions. RelB knockdown in LECs leads to reduced growth, survival, and adhesion. RelB plays a vital role in LEC-mediated LV maturation and function.
The lymphatic vessel (LV) system plays a critical role in maintaining tissue fluid homeostasis and trafficking of immune cells into the blood circulation. It is also implicated in many disease processes, such as inflammation and tumor metastasis.1, 2, 3, 4, 5, 6 Dysfunctional LVs often lead to chronic edema and impaired immune responses.7,8 LVs are composed of primary capillaries and mature vessels, which have different morphologies, structural compositions, and functions. Lymphatic capillaries are blind ended and consist of a single-layer endothelial wall, which is responsible for the uptake of lymph from interstitial tissues. Endothelial cells of lymphatic capillaries express lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1) as a selective surface marker.9, 10, 11 Lymphatic capillaries connect to collecting lymphatic vessels, which are covered by smooth muscle cells (SMCs), and contain unidirectional bicuspid valves.12, 13, 14, 15 Contraction of SMCs leads to active lymph flow, and can be measured in vivo by near-infrared indocyanine green (NIR-ICG) imaging.16, 17, 18 Endothelial cells of collecting lymphatic vessels are LYVE-1 negative and form an integral endothelial wall with zipper junction, allowing lymph flow without leakage.19
Development and maturation of lymphatic vessels are controlled by a group of lymphatic-specific transcription factors, growth factors, and receptors.20,21 Vascular endothelial growth factor C (VEGF-C) and its receptor, VEGF receptor 3 (VEGFR-3), are essential for embryonic lymphangiogenesis.22, 23, 24 VEGF-C and VEGFR-3 also play important roles in pathologic lymphangiogenesis because their expression levels are markedly increased in inflamed tissues, which are considered to be a compensatory mechanism for the body to limit or resolve chronic inflammation.25 VEGF-C and VEGFR-3 are regulated by NF-κB.26,27 NF-κB signals through heterodimeric canonical RelA/p50 and noncanonical RelB/p52 transcription factor pathways. Activated NF-κB RelA/p50 signaling promotes the expression of VEGF-C and VEGF-D,28,29 as well as VEGFR-3,30 and coincides with increased numbers of VEGFR-3+ LVs during inflammation.30 These findings indicate that RelA/p50 contributes to inflammation-mediated lymphangiogenesis by regulating the expression of the LV growth factors VEGF-C and VEGFR-3.
The role of the noncanonical NF-κB signaling and its involvement in human disease are less studied. Relb−/− mice have a complex phenotype, including multiorgan inflammation, splenomegaly, lack of lymph nodes, and multifocal defects in immune responses.31, 32, 33 The p52−/− mice have abnormalities primarily in immune organs, including the following: i) an absence of B-cell follicles and follicular dendritic cell networks in secondary lymphoid organs; ii) lack of germinal centers and splenic marginal zone structures; iii) reduced expression of homing chemokines; and iv) impaired development of Peyer patch–organizing centers.34, 35, 36, 37 However, the role of the noncanonical NF-κB pathway in lymphangiogenesis has not been studied. We hypothesize that RelB and p52 proteins affect the functions of lymphatic endothelial cells (LECs), thereby influencing the development and maturation of LVs.
To test this hypothesis, structural and functional changes of LVs were examined in Relb−/− and p52−/− mice using combinations of morphologic and lymphatic imaging. Transcriptome profiling and pathway analysis of LECs, in which RelB was knocked down by shRNA, were also performed. Relb−/− mice exhibited multiple abnormalities, especially in mature LVs; and RelB knockdown LECs had decreased growth, survival, migration, and adhesion, as well as dysregulated signaling pathways involving cell survival and adhesion. These data indicate, for the first time, that NF-κB RelB plays an important role in LV maturation, partially by affecting LECs.
Materials and Methods
Animals
Relb−/− and p52−/− mice in mixed 129/C57BL6 background have been described previously.31,38 Mice, ages 6 to 8 weeks, were used. The littermate mice were used as controls. Animal experiments were performed using University of Rochester Medical Center (Rochester, NY) Institutional Animal Care and Use Committee–approved protocols.
NIR-ICG Lymphatic Imaging
NIR-ICG lymphatic imaging was performed as described previously.16, 17, 18 In brief, fur was removed from legs with hair removal lotion. ICG solution (0.1 mg/mL, 6 μL) was injected intradermally into footpads. The dynamics of ICG fluorescence over the entire leg were visualized under an NIR laser and recorded by a charge-coupled device camera. The ICG fluorescence in the region of interest over the footpad and leg was immediately recorded for 30 to 60 minutes. Sequential images were analyzed for ICG intensity using ImageJ software (ImageJ version 1.50i bundled with 64-bit Java 1.8.0_112; NIH, Bethesda, MD; https://imagej.nih.gov/ij/download.html), resulting in an outcome measure of LV contraction, which represents the ICG pulses that pass the region of interest within 100 seconds, as described previously.17
Whole-Mount Immunofluorescence Staining of Lymphatic Vessels
Antibodies used included rabbit polyclonal to LYVE-1 (catalog number ab14917; Abcam Inc., San Francisco, CA), phycoerythrin (PE)-conjugated rat anti-mouse CD31 (catalog number 553373; BD Pharmingen Inc., San Diego, CA), fluorescein isothiocyanate monoclonal anti–α-smooth muscle actin (α-SMA; catalog number F3777; Sigma-Aldrich, St. Louis, MO), Syrian hamster monoclonal to podoplanin (PDPN; catalog number ab11936; Abcam Inc.), Alexa 488–conjugated donkey against rabbit (catalog number A21206; Molecular Probes, Invitrogen, Carlsbad, CA), and Dylight 405–conjugated goat anti-hamster IgG (H + L) (catalog number NBP1-73013; Novus Biologicals, LLC, Littleton, CO). For whole-mount immunofluorescence staining, fur was removed with hair removal lotion and dorsal skin of the foot and dorsal ear skin were fixed in 10% formalin, then blocked with 3% milk in 0.3% Triton X-100. Tissues were incubated with the following: i) 1:1000 dilution of primary anti–LYVE-1 antibody at 4°C overnight, followed by 1:400 dilution Alexa 488–conjugated secondary antibody for staining LYVE-1+ lymphatic capillary vessels; ii) 1:400 dilution of fluorescein isothiocyanate–anti–α-SMA antibody and 1:80 dilution of PE–anti-CD31 antibody for staining collecting lymphatic vessels, as has been reported in numerous literature publications39, 40, 41; and iii) 1:400 dilution of fluorescein isothiocyanate–anti–α-SMA, 1:80 dilution of PE–anti-CD31, and 1:1000 dilution of hamster–anti-PDPN, followed by 1:400 dilution of Dylight–anti-hamster antibodies for staining mature LVs. Tissues were then mounted with glycerin and imaged with a fluorescence microscope (Olympus IX 71; Olympus, Tokyo, Japan).
Quantification of Diameter, Branch Points, Smooth Muscle Cell Coverage, and the Ratio of α-SMA/CD31 of Lymphatic Vessels in Whole-Mount Stained Sections
Diameters and branch points and the percentage of vessels covered by α-SMA+ SMCs were used to evaluate changes of lymphatic capillaries and collecting lymphatic vessels, respectively. These are commonly used outcome measures for lymphatic vessels in whole-mount stained sections.42, 43, 44 For lymphatic capillaries, LYVE-1–stained samples were imaged at low magnification (×4), resulting in an image containing 22 to 30 vessels. The diameters and branch points of LYVE-1+ vessels in an entire sample were measured using Image Pro Plus software version 5.0 (IPP 5.1 for Windows; Media Cybernetics, Silver Spring, MD). Briefly, double trace lines along both sides of a lymphatic vessel were generated, and the diameters were measured as the average thickness between two traces lines. The branch points of vessels were counted manually. For collecting lymphatic vessels, CD31/α-SMA double-stained samples were imaged at high magnification (×10). Four images at different sites were taken per sample. Collecting lymphatic vessels were defined as vessels that contained strong CD31+ valves and were covered with α-SMA+ SMCs. Vessels that did not contain CD31+ valves and had more concentrated α-SMA+ SMCs were considered as blood vessels.42, 43, 44 The percentage of SMC coverage was calculated as follows: (SMC coverage area/whole lymphatic vessel area) × 100%. CD31/α-SMA/PDPN trichrome-stained samples were scanned using an Olympus VS-110 whole-slide imaging system. The percentage of SMC coverage in CD31+ or PDPN+ vessels was determined as described above. The results from four to five images were combined as one data point.
Transmission Electron Microscopy
Lymphatic vessels draining the popliteal lymph node were identified by intradermal injection of 0.5% Evans Blue Dye (10 μL, diluted with 1% bovine serum albumin in 0.9% NaCl) into the footpad. The blue color–stained vessels were excised under anesthesia and fixed with 2.5% glutaraldehyde/4% formaldehyde in 0.1 mol/L sodium cacodylate buffer, pH 7.4, for 24 hours. The specimens were post-fixed in 1.0% buffered osmium tetroxide for 12 hours at 4°C, dehydrated through a graded series of ethanol, infiltrated/embedded into EPON Araldite epoxy resin (Electron Microscopy Sciences, Hatfield, PA), and polymerized at 60°C for 2 days. Sections (1 μm thick) were sliced and stained with toluidine blue to identify the lymphatic vessel. Then, sections (70 nm thin) were mounted onto 200 mesh carbon-coated nickel grids and stained with uranyl acetate and lead citrate for ultrastructural examination. The grids were examined and imaged using a Hitachi 7650 transmission electron microscope (Hitachi, Tokyo, Japan) with an attached Gatan 11 megapixel Erlangshen digital camera (Gatan, Pleasanton, CA).
Generation of RelB Knockdown LECs
A rat LEC cell line was provided by Dr. Sophia Ran (University of Illinois, Chicago, IL). To knock down RelB, three oligonucleotides encoding shRNA against RelB were designed, resulting in RelB-1 (5′-GGAAGATCCAGCTGGGAATTG-3′), RelB-2 (5′-GCTCTGATCCACATGGAATCG-3′), and RelB-3 (5′-GCCAACCTTGATCAGTCTTCC-3′). The shRNA without RelB knockdown was used as control shRNA, which contains 5′-GATTACCACGACTATGGATCC-3′ as forward sequence and 5′-GGATCCATAGTCGTGGTAATC-3′ as reverse sequence. Basic Local Alignment Search Tool (BLAST) searches were performed with the National Center for Biotechnology Information database to ensure that the shRNA constructs were targeting only mouse RelB. The oligonucleotides encoding RelB or control shRNA were annealed and cloned into the pll3.7 vector, as the manufacturer described. The Pll3.7 expression vector (4 μg), psPAX2 packaging plasmid (4 μg; Addgene, Watertown, MA), and pMD2.G envelope plasmid (2 μg, Addgene) were cotransfected into HEK293T cells by a calcium phosphate precipitation method. After 48 hours, lentiviruses containing RelB or control shRNA were collected and used to infect LECs. Knockdown efficiency was determined by real-time quantitative PCR (qPCR) and Western blot analysis.
RT-qPCR
Total RNA was extracted using an RNA Extraction Kit (TaKaRa Biotechnology, Dalian, China), according to the manufacturer's instructions. RNA (500 ng) was subjected to cDNA synthesis using TaKaRa PrimeScript RT reagent kit (TaKaRa Biotechnology). RT-qPCR was performed using SYBR Premix Ex Taq II (TaKaRa Biotechnology) in an iCycler (Bio-Rad, Hercules, CA) real-time PCR machine. The specificity of detected signals was confirmed by a dissociation curve consisting of a single peak. All samples were analyzed in triplicate. Fold changes were determined by dividing the value of each sample by the value from control shRNA–infected cells and using the latter value as 1. β-Actin was used as an internal control. The 2−ΔΔCT method was used to calculate the relative expression levels of each gene. Sequences of primers used in real-time PCR were listed in Table 1.
Table 1.
Sequences of Primers Used in Real-Time PCR
| Gene | Accession no.∗ | Sequence | Product size, bp |
|---|---|---|---|
| Atm | NM_001106821.1 | F: 5′-ACTCCAGCTTTAGAGAGGTGTGT-3′ | 169 |
| R: 5′-TCGCCATCATAACTGCCAGC-3′ | |||
| Gadd45α | NM_024127.2 | F: 5′-GCAGAGCTGTTGCTACTGGA-3′ | 77 |
| R: 5′-CAGTGTAAGTCCGGGGTCTG-3′ | |||
| β-Actin (Actb) | F: 5′-CCCGCGAGTACAACCTTCTT-3′ | 83 | |
| R: 5′-CGCAGCGATATCGTCATCCA-3′ | |||
| Cldn7 | NM_001193619.1 | F: 5′-TCCTGTACCTACCTGGTCCT-3′ | 139 |
| R: 5′-CGAGTTGGCCATTTCCTTGTC-3′ | |||
| Cd40 | NM_011611.2 | F: 5′-TTGTTGACAGCGGTCCATCT-3′ | 154 |
| R: 5′-CTGAGTCACATGGGTGGCAT-3′ | |||
| Icam1 | NM_010493.3 | F: 5′-GTCTGCACCCAGTGCTAGTG-3′ | 172 |
| R: 5′-GGAAGGCTTCTCTGGGATGG-3′ | |||
| Itgb7 | NM_013566.2 | F: 5′-CCTCTGTGGGTCAAGTAGCC-3′ | 200 |
| R: 5′-AGTCACAGTGGATGACAGGC-3′ | |||
| Relb | NM_001290457.1 | F: 5′-CCCTGTCACTAACGGTCTCC-3′ | 94 |
| R: 5′-ATCTCACTCAGCTGTGTCCC-3′ | |||
| Prox1 | NM_001107201.1 | F: 5′-TTGACTCGGGACACAACGAG-3′ | 191 |
| R: 5′-TGATAGCCCTTCATTGCGCT-3′ | |||
| Flt4 | NM_053652.1 | F: 5′-CATTGGGGGCCTCTCCATAC-3′ | 127 |
| R: 5′-CAACTCTGCATGATGTGGCG-3′ | |||
| Pdpn | NM_019358.1 | F: 5′-TTTCTGGAAGGTTCTCGCCC-3′ | 136 |
| R: 5′-TGGGCATTCCATGGGTCATC-3′ | |||
| Vegfb | NM_053549.1 | F: 5′-GCTCTATGACAGGGACAACGA-3′ | 90 |
| R: 5′-TCGTGTAAAGCCCGTGGAAA-3′ | |||
| Vegfc | NM_053653.1 | F: 5′-TTGTCTCTGGCGTGTTCCTT-3′ | 138 |
| R: 5′-TTCGAAACCCTTGACCTCGC-3′ | |||
| Vegfd | NM_031761.1 | F: 5′-AATGTATGGAGAGTGGGCCG-3′ | 128 |
| R: 5′-CCAACACAGACCGGGATGAT-3′ | |||
| Pdgfb | NM_031524.1 | F: 5′-ATGACCCGAGCACATTCTGG-3′ | 121 |
| R: 5′-ACACCTCTGTACGCGTCTTG-3′ | |||
| Igfbp3 | NM_012588.2 | F: 5′-CGCAAATAACGGTGCGCTTA-3′ | 117 |
| R: 5′-TTTTGAAAGCTGCCTCCCCT-3′ | |||
| Tie2 | NM_001290549.1 | F: 5′-CCAAGCCCTGCTGATACCAA-3′ | 179 |
| R: 5′-CATGGTCCACTCGCTCTTGT-3′ |
F, forward primer; R, reverse primer.
Data available at https://www.ncbi.nlm.nih.gov/nuccore.
Western Blot Analysis
Whole cell lysates were harvested, and samples (30 μg protein/lane) were fractionated by SDS-PAGE and transferred to nitrocellulose membranes. Immunoblotting was performed using antibodies to RelB (catalog number sc-226; Santa Cruz Biotechnology, Dallas, TX), claudin 7 (catalog number ab27487; Abcam Inc.), and β-actin (catalog number A2228; Sigma-Aldrich). Bands were visualized using enhanced chemiluminescence (catalog number P0018; BeyoECL Plus; Beyotime, Nanjing, China).
RNA Sequencing and Pathway Analysis
RelB knockdown and control LECs were used. Total RNA was isolated using the TRIzol reagent (Invitrogen) and purified using a polyA-based probe. An Agilent 2100 Bioanalyzer (Agilent Technologies, Ringwood, VIC, Australia) was used for RNA quality verification. The cDNA fragments were purified using a QIAquick PCR extraction kit and enriched by PCR to construct the final cDNA library. Library preparation and sequencing were performed at Shanghai Personal Biotechnology Co, Ltd (Shanghai, China). Sequencing was implemented using the next-generation sequencing technology on an Illumina (San Diego, CA) NextSeq500 platform (NextSeq control software version 1.2/Real Time Analysis version 2.1). For analysis of differentially expressed genes, the DESeq version 1.18.0 was used for differential expression (n = 3) with the gene symbol annotation (false discovery rate ≤ 0.001, log2 ratio ≥ 1.5). Enrichment analysis of differentially expressed genes was performed by using the Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis (http://www.genome.jp/kegg). Using the entire genome as background, hypergeometric distribution was used to calculate the significant enrichment pathways of differential genes. The raw sequence array data have been uploaded to the Bio Project of the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/bioproject; accession number PRJNA528148).
Cell Assays
RelB knockdown and control LECs were used.
Growth
Cells were plated at a density of 5 × 103 cells/well in 96-well plates in triplicate. At 24, 48, and 72 hours, cells were incubated with 10 μL MTT solution at 37°C for 4 hours, followed by 200 μL of dimethyl sulfoxide to terminate the reaction. The plates were read at 570 nm using a benchmark microplate reader (BioTek, Winooski, VT).
Apoptosis
Cells were stained by PE–annexin V and 7–aminoactinomycin D (catalog numbers 51-65875X and 51-68981E, respectively; BD Biosciences, Franklin Lakes, NJ). Flow cytometry analysis was conducted (BD FACSCalibur Flow Cytometer; BD Biosciences) using BD CellQuest Pro software version 3.3.
Migration
Cells (1 × 105/well) were loaded into the upper chamber of a transwell insert (8-μm; Corning Costar, Corning, NY) for 24 hours and cultured with Dulbecco’s modified Eagle’s medium containing 3% fetal bovine serum. The transwell inserts were immediately moved to wells of a 24-well tissue culture dish containing Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum. After 24 hours of incubation, the migrated cells in the bottom of the transwell inserts were stained with Crystal Violet Staining Solution (catalog number C0121; Beyotime).
Adhesion
The lymphatic smooth muscle cells (LSMCs) were isolated from rat mesenteric LVs and cultured, as previously described.45 The passage 3 to 6 LSMCs were labeled with 5 μmol/L eFluor 670 (Thermo Fisher Scientific, Waltham, MA) for 20 minutes and cultured for 48 hours to reach confluence. RelB knockdown and control LECs were inoculated onto LSMCs for 1 hour. Floating cells were washed away with phosphate-buffered saline, and the remaining cells were digested with trypsin for flow cytometry. The first channel detected green fluorescent protein, and the fourth detected the fluorescence of eFluor 670. The differences in cell numbers with respect to green fluorescent protein labeling were counted under one channel. High green fluorescent protein content represents high adhesion rate.
Statistical Analysis
Data are expressed as the means ± SD. Statistical analyses were performed with SPSS software version 16.0 (SPSS, Chicago, IL). Comparisons between two groups were analyzed using a two-tailed unpaired t-test. One-way analysis of variance and Dunnett's post hoc multiple comparisons were used for comparisons among three or more groups. P < 0.05 was considered statistically significant. Correction was determined by a linear regression assay.
Results
Relb−/−, but Not p52−/−, Mice Have Abnormal Lymphatic Capillary and Mature Vessels
To investigate if noncanonical NF-κB signaling plays a role in LV function, whole-mount immunofluorescence staining was performed to examine morphologic changes of ear LVs in Relb−/− and p52−/− mice and their wild-type (WT) littermates. The lymphatic capillary vessels were identified using anti–LYVE-1 antibody. In WT mice, LYVE-1+ vessels were present in a highly branched network with multiple branch points (Figure 1). Although p52−/− mice appeared to have similar vessel diameters and branch points as WT mice, LYVE-1+ vessels in Relb−/− mice were wider and more irregular, with fewer branch points than those of WT or p52−/− mice (Figure 1, A and B), which were confirmed using morphometric analysis (Figure 1C).
Figure 1.

Abnormal lymphatic capillary in ear soft tissues of Relb−/− mice. Whole-mount immunofluorescence staining for capillary lymphatic vessels was performed in ear soft tissues of p52−/−, Relb−/−, and wild-type control mice. A and B: Representative low (A) and high (B) magnification images showing lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1)+ lymphatic capillaries. White arrows point to mature lymphatic vessels that do not express LYVE-1. C: Diameter and branch points of LYVE-1+ vessels. One-way analysis of variance with Dunnett's test was performed. Data are expressed as means ± SD (C). n = 5 mice (10 legs; C). *P < 0.05 versus control mice. Original magnifications, ×4 (A); ×20 (B).
To examine whether RelB or p52 is required for LV maturation, anti-CD31 and anti–α-SMA antibodies were used to identify mature LVs, on the basis of previous reports.39,43,44 Because LVs have wide lumen and valves, CD31+/α-SMA+ vessels with wide lumen and valves were defined as lymphatic LVs in this study. To further confirm that CD31 and SMC double-stained LVs are also stained positively for PDPN, a marker expressed by LECs, triple fluorescent staining was performed on the ear skin of WT mice with fluorescein isothiocyanate–anti–α-SMA (green), PE–anti-CD31 (red), and hamster–anti-PDPN, followed by Dylight–anti-hamster (blue). CD31+ vessels with valves also stained positively for PDPN (Supplemental Figure S1A). The percentage of SMC coverage is similar between CD31+ LVs and PDPN+ LVs (Supplemental Figure S1B). The correlation between the percentage of SMC coverage in CD31+ LVs and PDPN+ LVs is significant (Supplemental Figure S1C). These data indicate that CD31+/α-SMA+ vessels with valves in Figure 2 are likely also stained positively for PDPN.
Figure 2.

Abnormal lymphatic mature vessels in ear soft tissues of Relb−/− mice. Whole-mount immunofluorescence staining for mature lymphatic vessels was performed in ear soft tissues of p52−/−, Relb−/−, and wild-type control mice. A: Representative images showing CD31+/α-smooth muscle actin (α-SMA)+ mature lymphatic vessels. Vessels covered by smooth muscle cells (SMCs) are indicated by white arrows, and vessels without SMC coverage are indicated by blue arrows. Fewer SMC-covered vessels are in Relb−/− mice. B: SMC coverage (%)/vessel. One-way analysis of variance with Dunnett's test was performed. Data are expressed as means ± SD (B). n = 5 mice (B). *P < 0.05 versus control mice. Original magnification, ×10.
SMC coverage is a well-accepted feature of mature vessels.39,43,44 CD31+ LVs in control and p52−/− mice were >80% covered by SMCs, whereas Relb−/− vessels had significantly less SMC coverage (Figure 2). These data indicate that RelB, but not p52, plays a role in LV maturation. Thus, additional experiments were not performed with p52−/− mice.
To determine whether the changes in lymphatic capillaries and mature vessels in the ear skin of Relb−/− mice also occur in other tissues, whole-mount immunofluorescence imaging of LVs was performed in foot skin, whose draining function can also be quantitatively assessed by in vivo NIR-ICG imaging.16, 17, 18 Similar to the changes in ear skin, Relb−/− lymphatic capillaries in the lower extremity exhibited increased diameter and reduced numbers of branch points (Figure 3, A and B), whereas their mature LVs had markedly decreased SMC coverage (Figure 3, C and D).
Figure 3.

Abnormal lymphatic capillary and mature vessels in foot area of Relb−/− mice. Whole-mount immunofluorescence staining for lymphatic vessels was performed in foot skin of Relb−/− and control mice. A: Representative images showing lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1)+ capillary lymphatic vessels. B: Diameter and branch points of LYVE-1+ vessels. C: Representative images showing CD31+/α-smooth muscle actin (α-SMA)+ mature lymphatic vessels (white arrows). D: Smooth muscle cell coverage (%) per vessel and the ratio of α-SMA (the green intensity)/CD31 (the red intensity; the amount of uncovered vascular space). Unpaired t-test was performed. Data are expressed as means ± SD (B and D). n = 5 mice (B and D). *P < 0.05 versus control mice. Original magnification: ×4 (A); ×10 (C).
Mature Lymphatic Vessels from Relb−/− Mice Have Impaired Draining Function and Abnormal Ultrastructure
LVs mediate lymph drainage by passive flow, which increases when interstitial fluid pressure is high and adjacent tissues compress LVs to force the lymph through unidirectional valves. This is augmented by contraction of LV SMCs.14 These processes can be quantified by NIR-ICG imaging,16, 17, 18 in which passive flow through LVs is measured by the signal intensity and contractions are detected by lymph pulses.46 Consistent with prior NIR-ICG studies,16, 17, 18 it was confirmed that WT mice have two dominant LVs that drain lymph from the footpad to the popliteal lymph node in the popliteal fossa, and these LVs contract one to two times per minute (Figure 4). In contrast, NIR imaging of ICG injected into the footpad of Relb−/− mice revealed massive leakage from distal lymphatic capillaries with drainage through collateral LVs that bypass the popliteal lymph node (Figure 4A). In addition, the dominant LVs afferent to popliteal lymph nodes had irregular contractions and had a lower ICG signal intensity than WT LVs (Figure 4, B and C), suggesting that the Relb−/− mice have defective active lymphatic flow. These collateral LVs had a similar ICG signal intensity as WT LVs afferent to popliteal lymph nodes, indicating normal passive lymphatic flows, but they did not contract (Figure 4B), suggesting normal passive, but no active, flow.
Figure 4.

Impaired draining function of mature lymphatic vessels (LVs) in Relb−/− mice. Near-infrared indocyanine green (NIR-ICG) lymphatic imaging was used in Relb−/− and control mice after injection of ICG intradermally into footpads. A: Representative NIR-ICG images showing mature LVs (red arrows, blue circle), upstream popliteal lymph node (PLN; yellow arrows), footpad area (black arrows), and region of interest (ROI) where the pulses were measured (red circle indicates ROI at collateral LVs that bypass the PLN). Marked ICG leakage (green arrows) is observed in a Relb−/− mouse leg. B: ICG signals in a dominant LV (blue) of one control mouse and a dominant LV (green) and a collateral LV (red) of one Relb−/− mice. C: Lymphatic pulses/minute of dominant LV. Unpaired t-test was performed. Data are expressed as means ± SD (C). n = 5 control mice (10 legs; C); n = 4 Relb−/− mice (8 legs; C). *P < 0.05 versus control mice.
To investigate the cellular mechanisms that might mediate the defects in mature lymphatic vessels in Relb−/− mice, mature LVs isolated from Relb−/− mice and WT littermates were examined using transmission electronic microscopy. Ultrastructural examination (Figure 5) showed that LVs from WT mice had regular endothelial cell layers, tight endothelial junctions, flat SMCs, empty lumina, and abundant interstitial tissue elements. In contrast, in lymphatic vessels of Relb−/− mice, macrophage-like cells were attached to endothelial cells, endothelial junctions were loose, and SMCs were severely deformed with condensed nuclear chromosome, consistent with apoptosis. SMCs were missing from some parts of the endothelial wall, and loosely arranged fibrous elements were present in the subendothelial tissues. In addition, the tips of Relb−/− lymphatic valves had vacuolar changes with endothelial protrusions and reduced numbers of collagen at their roots.
Figure 5.
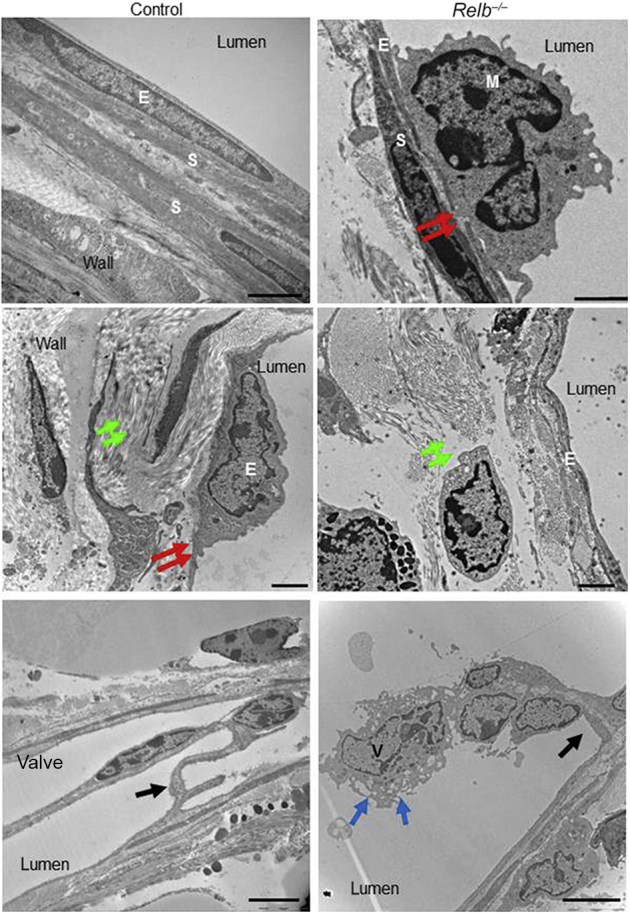
Ultrastructural features of mature lymphatic vessels isolated from Relb−/− and control mice. Mature lymphatic vessels draining the ankle/foot area were isolated from Relb−/− and littermate control mice and subjected to electron microscopy. Images of the walls and valves of lymphatic vessels from a control and Relb−/− mouse, in which lymphatic endothelial cells are indicated by E, smooth muscle cells by S, a macrophage by M, a lymphatic valve by V, tight junctions by red arrows, the interstitial elements by green arrows, an endothelial cell at a valve by blue arrows, and the root of valves by black arrows. Representative images were taken from three mice per genotype. Scale bars: 2 μm (top and middle panels); 5 μm (bottom panels).
RelB Knockdown in LECs Decreases Growth, Survival, Migration, and Adhesion
To assess direct physiological effects of RelB deficiency in LECs, RelB was knocked down in a rat LEC line using a lentivirus expressing shRNA against RelB. qPCR and Western blot analyses demonstrated that RelB-3 shRNA inhibited RelB expression with up to 80% efficiency (Figure 6A). Assessment of the growth and death of these LECs with approximately 80% RelB knocked down revealed similar cell numbers at 24 hours, but these decreased to 30% at 48 hours and 40% at 72 hours (Figure 6B). Apoptosis assays revealed that RelB knocked-down LECs had markedly increased apoptosis (17.7% ± 2.4% versus 1.5% ± 0.6% in control cells) (Figure 6C). Transwell migration assays indicated that RelB knocked-down LECs migrated more slowly than control cells (Figure 6D). In blood vessels, SMC coverage is an important component of vessel maturation in which SMCs attach to endothelial cells via cell adhesion.47, 48, 49 In WT mice, LECs may retain SMC attachment by producing adhesion molecules. In Relb−/− mice, the capacity of LECs to retain SMC attachment is reduced due to decreased expression of adhesion molecules, resulting in fewer areas of LVs that are covered by LSMCs. This possibility can be tested in the adhesion assay, in which RelB knocked-down LECs will have reduced capacity to attach LSMCs. Adhesion assays revealed that fewer RelB knocked-down LECs were attached to SMCs than control LECs (Figure 6E). These results suggest that RelB maintains LEC function by regulating their growth, survival, migration, and adhesion.
Figure 6.

Functional changes in lymphatic endothelial cells (LECs) with RelB knocked down. LECs were infected with lentiviruses containing shRNA against RelB or control shRNA. A: The expression levels of RelB by real-time quantitative PCR and Western blot analysis. B: Growth curves of LECs by MTT assays. Unpaired t-test was performed. The experiment was repeated three times. C: Cells were stained with annexin V–phycoerythrin (V-PE)/7–aminoactinomycin D (7-AAD) and analyzed by flow cytometry. The percentage of late (annexin V+/7-AAD+) apoptotic cells is shown in the upper right corner and early (annexin V+/7-AAD-) apoptotic cells is shown in the lower right corner. D: Cell migration by transwell assays. Unpaired t-test was performed. The experiment was repeated two times. E: Adhesion was examined by co-culturing eFluor 670–labeled lymphatic smooth muscle cells (LSMCs) and green fluorescent protein (GFP)–labeled LECs for 1 hour, washing off floating cells, and then analyzing the number of GFP+ LECs by flow cytometry. The percentage of GFP+ LECs per total cells is given. Unpaired t-test was performed. The experiment was repeated three times. Data are expressed as means ± SD (A, B, D, and E). n = 12 wells (A and B). n = 3 wells (D and E). *P < 0.05 versus control shRNA. Original magnification, ×4 (D).
RelB Regulates the Expression of Genes Related to Cell Survival and Adhesion in LECs
To explore the molecular mechanisms by which RelB may affect LECs, RNA sequencing and pathway analysis was performed using RelB knocked-down and control cells. A total of 934 genes were differentially expressed, including 368 up-regulated and 566 down-regulated genes. Kyoto Encyclopedia of Genes and Genomes pathway analysis revealed 192 dysregulated signaling pathways. The top 24 dysregulated pathways are listed in Figure 7 with information on false discovery rates and differentially expressed genes. Given the known role of RelB in tumor necrosis factor–mediated signaling in other cell types, the TNF signaling pathway had the highest differentially expressed gene numbers in RelB knocked-down cells. Consistent with the reduced survival and adhesion of RelB knocked-down cells, described in Figure 6, dysregulated cell adhesion and apoptosis signaling pathways were ranked the 22nd and 24th, respectively. Differentially expressed genes in tumor necrosis factor, cell adhesion, apoptosis, and tight junction signaling pathways are listed in Table 2. qPCR (Figure 8A) confirmed markedly decreased expression of genes related to adhesion, including Cd40, intercellular adhesion molecule 1 (Icam1), and integrin β 7 (Itgb7), in RelB knocked-down cells. Similarly, decreased expression of the survival gene growth arrest and DNA damage–inducible protein (Gadd45a) and increased expression of the proapoptotic gene ataxia telangiectasia mutated family protein (Atm) were also detected in RelB knocked-down cells. Because endothelial junctions of lymphatic vessels isolated from Relb−/− mice were loose (Figure 5), genes related to gap junctions within the RNA sequencing data set were searched for and low levels of the tight junction gene claudin 7 (Cldn7) were found in RelB knocked-down cells, which was confirmed by qPCR (Figure 8A) and Western blot analysis (Figure 8B). The expression levels of LEC regulators, including prospero homeobox 1 (Prox1), vascular endothelial growth factor receptor 3 (Vegfr3), also named as fms-related tyrosine kinase 4 (Flt4), podoplanin (Pdpn), tyrosine-protein kinase receptor Tie-2 (Tie2), vascular endothelial growth factor B (Vegfb), vascular endothelial growth factor C (Vegfc), and vascular endothelial growth factor D (Vegfd), were also examined by qPCR; and the expression of Prox1, Pdpn, Tie2, and Vegfb was found to be decreased, whereas the expression of Vegfr3 was increased, in RelB knockdown LECs (Figure 8C).
Figure 7.

Changes in expression levels of genes related to cell adhesion and survival by RNA sequencing. RelB knocked-down and control lymphatic endothelial cells were subjected to RNA sequencing. Differentially expressed genes (DEGs; changes equal to or larger than log2) were analyzed by the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. The top 24 dysregulated pathways are shown in a senior bubble chart. ECM, extracellular membrane; JAK, janus kinase; FoxO, forkhead box protein O1; PI3K, phosphatidylinositol 3-kinase; TNF, tumor necrosis factor.
Table 2.
Differentially Expressed Genes in TNF, Cell Adhesion, Apoptosis, and Tight Junction Signaling Pathways
| KEGG entry no. | Ensembl ID no. | Gene name | RelB shRNA/Ctl (fold) |
|---|---|---|---|
| TNF signaling pathway | |||
| K04734 | ENSRNOG00000007390 | NFKBIA; NF-κB inhibitor α | −1.69 |
| K09028 | ENSRNOG00000042838 | JUNB; transcription factor jun-B | −4.13 |
| K04430 | ENSRNOG00000003834 | MAP2K4, MKK4; mitogen-activated protein kinase kinase 4 (EC:2.7.12.2) | 0.50 |
| K11859 | ENSRNOG00000049517 | TNFAIP3, A20, OTUD7C; tumor necrosis factor-α-induced protein 3 (EC:3.4.19.12) | −1.67 |
| K04379 | ENSRNOG00000008015 | FOS; proto-oncogene protein c-fos | −2.89 |
| K05427 | ENSRNOG00000026805 | CSF2, GMCSF; granulocyte-macrophage colony-stimulating factor | −5.09 |
| K16366 | ENSRNOG00000014361 | EDN1; endothelin-1 | 0.55 |
| K14624 | ENSRNOG00000007159 | CCL2, MCP1; C-C motif chemokine 2 | −1.96 |
| K06490 | ENSRNOG00000020679 | ICAM1; intercellular adhesion molecule 1 | −1.74 |
| K05508 | ENSRNOG00000016326 | CX3CL1, NTT; C-X3-C motif chemokine 1 | −2.16 |
| K02649 | ENSRNOG00000000145 | PIK3R; phosphoinositide-3-kinase, regulatory subunit | 0.66 |
| K04374 | ENSRNOG00000017801 | ATF4, CREB2; cAMP-dependent transcription factor ATF-4 | −1.88 |
| K16060 | ENSRNOG00000005731 | BIRC2_3; baculoviral IAP repeat-containing protein 2/3 | −2.02 |
| K09258 | ENSRNOG00000043416 | BCL3; B-cell CLL/lymphoma protein 3 | −4.55 |
| K05419 | ENSRNOG00000007002 | LIF; leukemia inhibitory factor | −4.27 |
| K05505 | ENSRNOG00000002802 | CXCL1_2_3, GRO; C-X-C motif chemokine 1/2/3 | −1.70 |
| K05505 | ENSRNOG00000002792 | CXCL1_2_3, GRO; C-X-C motif chemokine 1/2/3 | −5.29 |
| K05405 | ENSRNOG00000010278 | IL6; IL-6 | −3.25 |
| K04696 | ENSRNOG00000002946 | SOCS3, CIS3; suppressor of cytokine signaling 3 | −2.16 |
| Cell adhesion molecules | |||
| K06590 | ENSRNOG00000012208 | ITGB7; integrin β 7 | −2.05 |
| K06087 | ENSRNOG00000001476 | CLDN; claudin | −1.93 |
| K06087 | ENSRNOG00000001419 | CLDN; claudin | −2.34 |
| K06539 | ENSRNOG00000019202 | PVR, NECL5; poliovirus receptor | −1.50 |
| K06496 | ENSRNOG00000002794 | SELP; selectin, platele | −1.83 |
| K06796 | ENSRNOG00000020129 | CDH3; cadherin 3, type 1, P-cadherin | 0.54 |
| K16359 | ENSRNOG00000013694 | NTNG2; netrin-G2 | −1.90 |
| K06490 | ENSRNOG00000020679 | ICAM1; intercellular adhesion molecule 1 | −1.74 |
| K06464 | ENSRNOG00000001224 | ITGB2; integrin β 2 | −2.28 |
| K06781 | ENSRNOG00000018778 | CADM1, IGSF4, NECL2, TSLC1; cell adhesion molecule 1 | 0.64 |
| K06495 | ENSRNOG00000002776 | SELL; selectin, lymphocyte | −2.74 |
| K16338 | ENSRNOG00000014297 | SDC4; syndecan 4 | −2.01 |
| K03160 | ENSRNOG00000018488 | TNFRSF5, CD40; TNF receptor superfamily member 5 | −2.21 |
| K06470 | ENSRNOG00000010283 | CD28; CD28 antigen | 0.58 |
| Apoptosis | |||
| K04734 | ENSRNOG00000007390 | NFKBIA; NF-κB inhibitor α | −1.69 |
| K04379 | ENSRNOG00000008015 | FOS; proto-oncogene protein c-fos | −2.89 |
| K04402 | ENSRNOG00000005615 | GADD45; growth arrest and DNA damage inducible protein | −2.01 |
| K02649 | ENSRNOG00000000145 | PIK3R; phosphoinositide-3-kinase, regulatory subunit | 0.66 |
| K04374 | ENSRNOG00000017801 | ATF4, CREB2; cAMP-dependent transcription factor ATF-4 | −1.88 |
| K04452 | ENSRNOG00000006789 | DDIT3, GADD153; DNA damage–inducible transcript 3 | −1.65 |
| K16060 | ENSRNOG00000005731 | BIRC2_3; baculoviral IAP repeat-containing protein 2/3 | −2.02 |
| K01379 | ENSRNOG00000020206 | CTSD; cathepsin D (EC:3.4.23.5) | −1.56 |
| K02582 | ENSRNOG00000016571 | NGFB; nerve growth factor, β | −1.60 |
| K04728 | ENSRNOG00000029773 | ATM, TEL1; ataxia telangiectasia mutated family protein (EC:2.7.11.1) | 0.59 |
| Tight junctions | |||
| K06087 | ENSRNOG00000001476 | CLDN; claudin | −1.93 |
| K06087 | ENSRNOG00000001419 | CLDN; claudin | −2.34 |
| K06108 | ENSRNOG00000008001 | RAB3B; Ras-related protein Rab-3B | 0.56 |
| K03456 | ENSRNOG00000010922 | PPP2R1; serine/threonine-protein phosphatase 2A regulatory subunit A | 0.65 |
| K06101 | ENSRNOG00000020386 | ASH1L; histone-lysine N-methyltransferase ASH1L (EC:2.1.1.43) | 0.66 |
Ctl, control; KEGG, Kyoto Encyclopedia of Genes and Genomes; TNF, tumor necrosis factor.
Figure 8.

Changes in expression levels of genes related to cell adhesion, survival, and critical lymphatic endothelial cell (LEC) regulators. A: Expression of genes related to cell adhesion and survival was examined by real-time quantitative PCR (qPCR) in RelB knocked-down and control LECs. Unpaired t-test was performed. The experiment was repeated two times. B: Expression of claudin 7 (CLDN7) proteins was examined by Western blot analysis. C: Expression of LEC regulators was examined by qPCR in RelB knocked-down and control LECs. Unpaired t-test was performed. The experiment was repeated two times. Data are expressed as means ± SD (A and C). n = 3 wells (A and C). *P < 0.05 versus control shRNA.
Discussion
This is first report describing a role for the noncanonical NF-κB protein, RelB, to maintain the structural integrity and function of LVs in adult mice. The mice deficient in RelB, but not in p52, have massive leakage of indocyanine green from their lymphatic capillaries and mature vessels after its injection into their footpads, associated with enhanced lymphatic drainage by collateral LVs, which has been observed after local administration of sildenafil [a phosphodiesterase 5 (PDE5) inhibitor], the active ingredient of sildenafil (Viagra; Pfizer, Groton, CT).25 Electron microscopy examination of mature lymphatic vessels from Relb−/− mice revealed poor lymphatic endothelial cell junctions and irregular endothelium on valves and macrophages on the lumen surfaces of endothelial cells, suggesting involvement of RelB in the structural integrity of the lymphatic endothelial wall and a repair response to LV damage and apoptosis.
Using ICG lymphatic imaging in tumor necrosis factor transgenic mice, a mouse model of inflammatory arthritis, sildenafil (a PDE5 inhibitor) treatment was found to open the collateral LVs in mouse legs. These collateral LVs do not have pulses despite the fact that they can transfer injected ICG from footpad to popliteal lymph nodes, indicating that collateral LVs passively transfer lymph. Sildenafil may reduce inflammation by transferring catabolic factors and cells via the collateral LVs. Some of the sildenafil study has been included in a recent publication.25 Because Relb−/− mice have opened collateral LVs, it may be why Relb−/− mice have no edema in foot despite lymphatic leakage. Because little is known about the regulation of collateral LVs, it will be interesting to investigate if RelB interacts with PDE5-mediated cellular events in LECs.
NF-κB signals regulate the tight junction complex and endothelial cell permeability of blood vessels.50, 51, 52 However, the mechanism by which RelB affects lymphatic endothelial junctions is not known. RelB may interfere with the composition and the organization of cell junctions in LVs by altering the expression of adhesion molecules and/or tight junction–associated genes.53 The RNA sequencing, qPCR, and Western blot data indicate that the expression of claudin, one of the most important components of tight junctions, is significantly decreased in RelB knockdown LECs. Claudins establish the paracellular barrier that controls the flow of molecules in the intercellular space between epithelial cells of organs.54 Whether claudins are involved in the leakiness of lymphatic vessels in Relb−/− mice remains unknown and necessitates further study.
Another feature of Relb−/− lymphatic vessels is reduced SMC coverage. This defect is probably involved in multiple mechanisms, one of which is decreased attachment between LECs and SMCs. Significantly decreased expression of the adhesion molecules ICAM1, CD40, and ITGβ7 in RelB knocked-down cells supports this hypothesis. Poor adhesion capacity of LECs also explains the presence of macrophages within the lumen of Relb−/− vessels because ICAM1 and CD40 contribute to leukocyte transendothelial migration.55,56 Dysregulated apoptosis pathway and significant changes of apoptosis-related genes in RelB knocked-down LECs strongly suggest that RelB regulates their survival, which is consistent with the previously known prosurvival role of RelB in other cell types.57 Increased death of LECs may lead to detachment of SMCs. It is also possible that RelB deficiency directly affects SMCs, which needs to be investigated.
The architecture and, therefore, the function of the lymphatic vasculature network is also affected by vessel branching, which occurs by the sprouting of new tubes from preexisting ones. Thus, the molecular mechanisms regulating sprouting are central to how a given branching system forms.58 Capillary lymphatic vessels from Relb−/− mice possess fewer branches than those of WT littermates. RNA sequencing results indicated reduced expression levels of semaphorin and netrin members, genes that regulate axon outgrowth and repulsion. Whether the reduction of those regulators contributes to the decreased branch points of capillary lymphatic vessels in Relb−/− mice merits further investigation.
It is well known that the p50 subunit of NF-κB also regulates lymphangiogenesis.59 RelB generates functional complexes with p50.60,61 Thus, it is possible that LV deficiencies in Relb−/− mice are due to inability to generate such functional complexes between p50 and RelB. NF-κB p50−/− mice have markedly decreased LVs, which is accompanied by decreased VEGFR-3 and prospero homeobox protein 1 levels.59 However, Relb−/− mice have reduced SMC-covered mature LVs but relatively normal lymphatic capillaries (Figure 1). Furthermore, LECs with RelB knockdown express higher levels Vegfr3 than control cells (Figure 8C). Thus, lymphatic defect in Relb−/− mice is unlikely due to decreased p50/RelB complex. However, more study is needed to examine the possible alternation of NF-κB signal pathway in the absence of RelB in LECs and its impact on LV integrity.
In the current study, RelB global knockout mice were used under a basal condition. A more important question will be the role of RelB during inflammation, where LV remodeling rather than development may be affected. Therefore, mice deficient in RelB specifically in LECs should be generated and inflammation induced to study the requirement of LEC RelB for LV homeostasis and remodeling during inflammation. On the basis of the data from RelB knockdown LECs (Figures 7 and 8), it is possible that the absence of RelB in LECs will make mice more susceptible to inflammation due to dysfunctional LECs. Another possibility is that inflammation may alter the balance between RelB/p52- and RelA/p50-mediated signal pathways in LECs, thereby affecting LV remodeling. These possibilities need to be investigated in the future.
In summary, LV morphology and functions were examined in mice deficient in noncanonical NF-κB signaling; and it was found that Relb−/− mice, but not p52−/− mice, exhibited multiple abnormalities, especially in mature LVs. Relb−/− mature LVs had thinner vessel walls, apoptotic appearance of LECs and SMCs, loss of LEC junctions, severe leakage, and reduced contractions. Collateral LVs that mediated passive, but not active, lymphatic flow were also observed. RelB knockdown LECs demonstrated decreased growth, survival, migration, and adhesion, as well as dysregulated signaling pathways involving cell survival and adhesion. NF-κB RelB plays an important role in lymphatic vessel maturation, partially by affecting LECs.
Acknowledgments
We thank Dr. Sophia Ran (University of Illinois, Chicago, IL) for providing a rat lymphatic endothelial cell line; Jinman Chen for performing the staining and taking images of Supplemental Figure S1A; Xuefei Li and Zixin Zhuang (Shanghai University of Traditional Chinese Medicine, China) for performing histomorphometric analysis of Supplemental Figure S1, B and C; and Yan Xiu (University of Rochester Medical Center, Rochester, NY) for performing the phenotypic identification of mice.
Footnotes
Supported by National Natural Science Foundation of China research grants 81822050 and 81673990 (Q.L.) and 81220108027 (Y.J.W.); NIH research grants AR063650 and AR069789 (L.P.X.); the Lymphatic Malformation Institute (L.P.X.); the Program for Innovative Research Team of Ministry of Science and Technology of China research grant 2015RA4002 (Y.J.W.); and Shanghai Traditional Chinese Medicine Medical Center of Chronic Disease research grant 2017ZZ01010 (Y.J.W.).
Q.L. and L.Z. contributed equally to this work.
Disclosures: None declared.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2019.08.009.
Contributor Information
Yongjun Wang, Email: 0000002722@shutcm.edu.cn.
Lianping Xing, Email: lianping_xing@urmc.rochester.edu.
Supplemental Data
Supplemental Figure S1.

CD31+ lymphatic vessels (LVs) are stained positively for podoplanin (PDPN). Whole-mount triple fluorescent staining was performed on the ear skin of wild-type mice with fluorescein isothiocyanate–anti–α-smooth muscle actin (α-SMA; green), phycoerythrin (PE)–anti-CD31 (red), and hamster–anti-PDPN, followed by Dylight–anti-hamster (blue). A: Representative images showing a large LV with valve (arrowhead) that is stained positively for CD31 and PDPN and partially for α-SMA. B: Smooth muscle cell (SMC) coverage in CD31+ LVs and SMC coverage in PDPN+ LVs. Unpaired t-test was performed. C: Correlation analysis of SMC coverage in CD31+ LVs and in PDPN+ LVs. Data are expressed as means ± SD (B). n = 5 mice (B). Scale bar = 50 μm (A).
References
- 1.Adams R.H., Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- 2.Alitalo K., Tammela T., Petrova T.V. Lymphangiogenesis in development and human disease. Nature. 2005;438:946–953. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- 3.Oliver G., Detmar M. The rediscovery of the lymphatic system: old and new insights into the development and biological function of the lymphatic vasculature. Genes Dev. 2002;16:773–783. doi: 10.1101/gad.975002. [DOI] [PubMed] [Google Scholar]
- 4.He Y., Karpanen T., Alitalo K. Role of lymphangiogenic factors in tumor metastasis. Biochim Biophys Acta. 2004;1654:3–12. doi: 10.1016/j.bbcan.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Cueni L.N., Detmar M. New insights into the molecular control of the lymphatic vascular system and its role in disease. J Invest Dermatol. 2006;126:2167–2177. doi: 10.1038/sj.jid.5700464. [DOI] [PubMed] [Google Scholar]
- 6.Baluk P., McDonald D.M. Markers for microscopic imaging of lymphangiogenesis and angiogenesis. Ann N Y Acad Sci. 2008;1131:1–12. doi: 10.1196/annals.1413.001. [DOI] [PubMed] [Google Scholar]
- 7.Ribera J., Pauta M., Melgar-Lesmes P., Tugues S., Fernandez-Varo G., Held K.F., Soria G., Tudela R., Planas A.M., Fernandez-Hernando C., Arroyo V., Jimenez W., Morales-Ruiz M. Increased nitric oxide production in lymphatic endothelial cells causes impairment of lymphatic drainage in cirrhotic rats. Gut. 2013;62:138–145. doi: 10.1136/gutjnl-2011-300703. [DOI] [PubMed] [Google Scholar]
- 8.Petrova T.V., Karpanen T., Norrmen C., Mellor R., Tamakoshi T., Finegold D., Ferrell R., Kerjaschki D., Mortimer P., Yla-Herttuala S., Miura N., Alitalo K. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat Med. 2004;10:974–981. doi: 10.1038/nm1094. [DOI] [PubMed] [Google Scholar]
- 9.Johnson L.A., Banerji S., Lawrance W., Gileadi U., Prota G., Holder K.A., Roshorm Y.M., Hanke T., Cerundolo V., Gale N.W., Jackson D.G. Dendritic cells enter lymph vessels by hyaluronan-mediated docking to the endothelial receptor LYVE-1. Nat Immunol. 2017;18:762–770. doi: 10.1038/ni.3750. [DOI] [PubMed] [Google Scholar]
- 10.Platonova N., Miquel G., Regenfuss B., Taouji S., Cursiefen C., Chevet E., Bikfalvi A. Evidence for the interaction of fibroblast growth factor-2 with the lymphatic endothelial cell marker LYVE-1. Blood. 2013;121:1229–1237. doi: 10.1182/blood-2012-08-450502. [DOI] [PubMed] [Google Scholar]
- 11.Rudno-Rudzinska J., Kielan W., Grzebieniak Z., Dziegiel P., Donizy P., Mazur G., Knakiewicz M., Frejlich E., Halon A. High density of peritumoral lymphatic vessels measured by D2-40/podoplanin and LYVE-1 expression in gastric cancer patients: an excellent prognostic indicator or a false friend? Gastric Cancer. 2013;16:513–520. doi: 10.1007/s10120-012-0216-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore J.E., Jr., Bertram C.D. Lymphatic system flows. Annu Rev Fluid Mech. 2018;50:459–482. doi: 10.1146/annurev-fluid-122316-045259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabine A., Davis M.J., Bovay E., Petrova T.V. Characterization of mouse mesenteric lymphatic valve structure and function. Methods Mol Biol. 2018;1846:97–129. doi: 10.1007/978-1-4939-8712-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y., Jin Y., Mae M.A., Zhang Y., Ortsater H., Betsholtz C., Makinen T., Jakobsson L. Smooth muscle cell recruitment to lymphatic vessels requires PDGFB and impacts vessel size but not identity. Development. 2017;144:3590–3601. doi: 10.1242/dev.147967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scallan J.P., Zawieja S.D., Castorena-Gonzalez J.A., Davis M.J. Lymphatic pumping: mechanics, mechanisms and malfunction. J Physiol. 2016;594:5749–5768. doi: 10.1113/JP272088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo R., Zhou Q., Proulx S.T., Wood R., Ji R.C., Ritchlin C.T., Pytowski B., Zhu Z., Wang Y.J., Schwarz E.M., Xing L. Inhibition of lymphangiogenesis and lymphatic drainage via vascular endothelial growth factor receptor 3 blockade increases the severity of inflammation in a mouse model of chronic inflammatory arthritis. Arthritis Rheum. 2009;60:2666–2676. doi: 10.1002/art.24764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Q., Wood R., Schwarz E.M., Wang Y.J., Xing L. Near-infrared lymphatic imaging demonstrates the dynamics of lymph flow and lymphangiogenesis during the acute versus chronic phases of arthritis in mice. Arthritis Rheum. 2010;62:1881–1889. doi: 10.1002/art.27464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Q., Guo R., Wood R., Boyce B.F., Liang Q., Wang Y.J., Schwarz E.M., Xing L. Vascular endothelial growth factor C attenuates joint damage in chronic inflammatory arthritis by accelerating local lymphatic drainage in mice. Arthritis Rheum. 2011;63:2318–2328. doi: 10.1002/art.30421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tammela T., Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 20.Jha S.K., Rauniyar K., Jeltsch M. Key molecules in lymphatic development, function, and identification. Ann Anat. 2018;219:25–34. doi: 10.1016/j.aanat.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 21.Zheng W., Aspelund A., Alitalo K. Lymphangiogenic factors, mechanisms, and applications. J Clin Invest. 2014;124:878–887. doi: 10.1172/JCI71603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ward M.C., Cunningham A.M. Developmental expression of vascular endothelial growth factor receptor 3 and vascular endothelial growth factor C in forebrain. Neuroscience. 2015;303:544–557. doi: 10.1016/j.neuroscience.2015.04.063. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L., Zhou F., Han W., Shen B., Luo J., Shibuya M., He Y. VEGFR-3 ligand-binding and kinase activity are required for lymphangiogenesis but not for angiogenesis. Cell Res. 2010;20:1319–1331. doi: 10.1038/cr.2010.116. [DOI] [PubMed] [Google Scholar]
- 24.Kreuger J., Nilsson I., Kerjaschki D., Petrova T., Alitalo K., Claesson-Welsh L. Early lymph vessel development from embryonic stem cells. Arterioscler Thromb Vasc Biol. 2006;26:1073–1078. doi: 10.1161/01.ATV.0000217610.58032.b7. [DOI] [PubMed] [Google Scholar]
- 25.Bouta E.M., Bell R.D., Rahimi H., Xing L., Wood R.W., Bingham C.O., 3rd, Ritchlin C.T., Schwarz E.M. Targeting lymphatic function as a novel therapeutic intervention for rheumatoid arthritis. Nat Rev Rheumatol. 2018;14:94–106. doi: 10.1038/nrrheum.2017.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du Q., Jiang L., Wang X., Wang M., She F., Chen Y. Tumor necrosis factor-alpha promotes the lymphangiogenesis of gallbladder carcinoma through nuclear factor-kappaB-mediated upregulation of vascular endothelial growth factor-C. Cancer Sci. 2014;105:1261–1271. doi: 10.1111/cas.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flister M.J., Wilber A., Hall K.L., Iwata C., Miyazono K., Nisato R.E., Pepper M.S., Zawieja D.C., Ran S. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1. Blood. 2010;115:418–429. doi: 10.1182/blood-2008-12-196840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ristimaki A., Narko K., Enholm B., Joukov V., Alitalo K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem. 1998;273:8413–8418. doi: 10.1074/jbc.273.14.8413. [DOI] [PubMed] [Google Scholar]
- 29.Iwata C., Kano M.R., Komuro A., Oka M., Kiyono K., Johansson E., Morishita Y., Yashiro M., Hirakawa K., Kaminishi M., Miyazono K. Inhibition of cyclooxygenase-2 suppresses lymph node metastasis via reduction of lymphangiogenesis. Cancer Res. 2007;67:10181–10189. doi: 10.1158/0008-5472.CAN-07-2366. [DOI] [PubMed] [Google Scholar]
- 30.Baluk P., Tammela T., Ator E., Lyubynska N., Achen M.G., Hicklin D.J., Jeltsch M., Petrova T.V., Pytowski B., Stacker S.A., Yla-Herttuala S., Jackson D.G., Alitalo K., McDonald D.M. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest. 2005;115:247–257. doi: 10.1172/JCI22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burkly L., Hession C., Ogata L., Reilly C., Marconi L.A., Olson D., Tizard R., Cate R., Lo D. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 32.Weih F., Carrasco D., Durham S.K., Barton D.S., Rizzo C.A., Ryseck R.P., Lira S.A., Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 33.Weih F., Warr G., Yang H., Bravo R. Multifocal defects in immune responses in RelB-deficient mice. J Immunol. 1997;158:5211–5218. [PubMed] [Google Scholar]
- 34.Caamano J.H., Rizzo C.A., Durham S.K., Barton D.S., Raventos-Suarez C., Snapper C.M., Bravo R. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–196. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Franzoso G., Carlson L., Poljak L., Shores E.W., Epstein S., Leonardi A., Grinberg A., Tran T., Scharton-Kersten T., Anver M., Love P., Brown K., Siebenlist U. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poljak L., Carlson L., Cunningham K., Kosco-Vilbois M.H., Siebenlist U. Distinct activities of p52/NF-kappa B required for proper secondary lymphoid organ microarchitecture: functions enhanced by Bcl-3. J Immunol. 1999;163:6581–6588. [PubMed] [Google Scholar]
- 37.Weih D.S., Yilmaz Z.B., Weih F. Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J Immunol. 2001;167:1909–1919. doi: 10.4049/jimmunol.167.4.1909. [DOI] [PubMed] [Google Scholar]
- 38.Franzoso G., Carlson L., Xing L., Poljak L., Shores E.W., Brown K.D., Leonardi A., Tran T., Boyce B.F., Siebenlist U. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou F., Chang Z., Zhang L., Hong Y.K., Shen B., Wang B., Zhang F., Lu G., Tvorogov D., Alitalo K., Hemmings B.A., Yang Z., He Y. Akt/protein kinase B is required for lymphatic network formation, remodeling, and valve development. Am J Pathol. 2010;177:2124–2133. doi: 10.2353/ajpath.2010.091301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dellinger M., Hunter R., Bernas M., Gale N., Yancopoulos G., Erickson R., Witte M. Defective remodeling and maturation of the lymphatic vasculature in angiopoietin-2 deficient mice. Dev Biol. 2008;319:309–320. doi: 10.1016/j.ydbio.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baluk P., Yao L.C., Feng J., Romano T., Jung S.S., Schreiter J.L., Yan L., Shealy D.J., McDonald D.M. TNF-alpha drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J Clin Invest. 2009;119:2954–2964. doi: 10.1172/JCI37626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bouvree K., Brunet I., Del Toro R., Gordon E., Prahst C., Cristofaro B., Mathivet T., Xu Y., Soueid J., Fortuna V., Miura N., Aigrot M.S., Maden C.H., Ruhrberg C., Thomas J.L., Eichmann A. Semaphorin3A, Neuropilin-1, and PlexinA1 are required for lymphatic valve formation. Circ Res. 2012;111:437–445. doi: 10.1161/CIRCRESAHA.112.269316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jurisic G., Maby-El Hajjami H., Karaman S., Ochsenbein A.M., Alitalo A., Siddiqui S.S., Ochoa Pereira C., Petrova T.V., Detmar M. An unexpected role of semaphorin3a-neuropilin-1 signaling in lymphatic vessel maturation and valve formation. Circ Res. 2012;111:426–436. doi: 10.1161/CIRCRESAHA.112.269399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lutter S., Xie S., Tatin F., Makinen T. Smooth muscle-endothelial cell communication activates Reelin signaling and regulates lymphatic vessel formation. J Cell Biol. 2012;197:837–849. doi: 10.1083/jcb.201110132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang Q., Ju Y., Chen Y., Wang W., Li J., Zhang L., Xu H., Wood R.W., Schwarz E.M., Boyce B.F., Wang Y., Xing L. Lymphatic endothelial cells efferent to inflamed joints produce iNOS and inhibit lymphatic vessel contraction and drainage in TNF-induced arthritis in mice. Arthritis Res Ther. 2016;18:62. doi: 10.1186/s13075-016-0963-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leahy A.A., Esfahani S.A., Foote A.T., Hui C.K., Rainbow R.S., Nakamura D.S., Tracey B.H., Mahmood U., Zeng L. Analysis of the trajectory of osteoarthritis development in a mouse model by serial near-infrared fluorescence imaging of matrix metalloproteinase activities. Arthritis Rheumatol. 2015;67:442–453. doi: 10.1002/art.38957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foo S.S., Turner C.J., Adams S., Compagni A., Aubyn D., Kogata N., Lindblom P., Shani M., Zicha D., Adams R.H. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell. 2006;124:161–173. doi: 10.1016/j.cell.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 48.Scheppke L., Murphy E.A., Zarpellon A., Hofmann J.J., Merkulova A., Shields D.J., Weis S.M., Byzova T.V., Ruggeri Z.M., Iruela-Arispe M.L., Cheresh D.A. Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basement membrane. Blood. 2012;119:2149–2158. doi: 10.1182/blood-2011-04-348706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brudno Y., Ennett-Shepard A.B., Chen R.R., Aizenberg M., Mooney D.J. Enhancing microvascular formation and vessel maturation through temporal control over multiple pro-angiogenic and pro-maturation factors. Biomaterials. 2013;34:9201–9209. doi: 10.1016/j.biomaterials.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aveleira C.A., Lin C.M., Abcouwer S.F., Ambrosio A.F., Antonetti D.A. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59:2872–2882. doi: 10.2337/db09-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown R.C., Mark K.S., Egleton R.D., Huber J.D., Burroughs A.R., Davis T.P. Protection against hypoxia-induced increase in blood-brain barrier permeability: role of tight junction proteins and NFkappaB. J Cell Sci. 2003;116:693–700. doi: 10.1242/jcs.00264. [DOI] [PubMed] [Google Scholar]
- 52.Kisseleva T., Song L., Vorontchikhina M., Feirt N., Kitajewski J., Schindler C. NF-kappaB regulation of endothelial cell function during LPS-induced toxemia and cancer. J Clin Invest. 2006;116:2955–2963. doi: 10.1172/JCI27392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baluk P., Fuxe J., Hashizume H., Romano T., Lashnits E., Butz S., Vestweber D., Corada M., Molendini C., Dejana E., McDonald D.M. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. 2007;204:2349–2362. doi: 10.1084/jem.20062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kirschner N., Rosenthal R., Furuse M., Moll I., Fromm M., Brandner J.M. Contribution of tight junction proteins to ion, macromolecule, and water barrier in keratinocytes. J Invest Dermatol. 2013;133:1161–1169. doi: 10.1038/jid.2012.507. [DOI] [PubMed] [Google Scholar]
- 55.Ramirez S.H., Fan S., Dykstra H., Reichenbach N., Del Valle L., Potula R., Phipps R.P., Maggirwar S.B., Persidsky Y. Dyad of CD40/CD40 ligand fosters neuroinflammation at the blood-brain barrier and is regulated via JNK signaling: implications for HIV-1 encephalitis. J Neurosci. 2010;30:9454–9464. doi: 10.1523/JNEUROSCI.5796-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schnoor M. Endothelial actin-binding proteins and actin dynamics in leukocyte transendothelial migration. J Immunol. 2015;194:3535–3541. doi: 10.4049/jimmunol.1403250. [DOI] [PubMed] [Google Scholar]
- 57.Guerin S., Baron M.L., Valero R., Herrant M., Auberger P., Naquet P. RelB reduces thymocyte apoptosis and regulates terminal thymocyte maturation. Eur J Immunol. 2002;32:1–9. doi: 10.1002/1521-4141(200201)32:1<1::AID-IMMU1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 58.Horowitz A., Simons M. Branching morphogenesis. Circ Res. 2008;103:784–795. doi: 10.1161/CIRCRESAHA.108.181818. [DOI] [PubMed] [Google Scholar]
- 59.Flister M.J., Volk L.D., Ran S. Characterization of Prox1 and VEGFR-3 expression and lymphatic phenotype in normal organs of mice lacking p50 subunit of NF-kappaB. Microcirculation. 2011;18:85–101. doi: 10.1111/j.1549-8719.2010.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bhardwaj R., Yester J.W., Singh S.K., Biswas D.D., Surace M.J., Waters M.R., Hauser K.F., Yao Z., Boyce B.F., Kordula T. RelB/p50 complexes regulate cytokine-induced YKL-40 expression. J Immunol. 2015;194:2862–2870. doi: 10.4049/jimmunol.1400874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gasparini C., Foxwell B.M., Feldmann M. RelB/p50 regulates TNF production in LPS-stimulated dendritic cells and macrophages. Cytokine. 2013;61:736–740. doi: 10.1016/j.cyto.2012.12.029. [DOI] [PubMed] [Google Scholar]