

Abstract
Recent works highlight the therapeutic potential of targeting cyclic guanosine monophosphate (cGMP)-dependent pathways in the context of brain ischemia/reperfusion injury (IRI). Although cGMP-dependent protein kinase I (cGKI) has emerged as a key mediator of the protective effects of nitric oxide (NO) and cGMP, the mechanisms by which cGKI attenuates IRI remain poorly understood. We used a novel, conditional cGKI knockout mouse model to study its role in cerebral IRI. We assessed neurological deficit, infarct volume, and cerebral perfusion in tamoxifen-inducible vascular smooth muscle cell-specific cGKI knockout mice and control animals. Stroke experiments revealed greater cerebral infarct volume in smooth muscle cell specific cGKI knockout mice (males: 96 ± 16 mm3; females: 93 ± 12 mm3, mean±SD) than in all control groups: wild type (males: 66 ± 19; females: 64 ± 14), cGKI control (males: 65 ± 18; females: 62 ± 14), cGKI control with tamoxifen (males: 70 ± 8; females: 68 ± 10). Our results identify, for the first time, a protective role of cGKI in vascular smooth muscle cells during ischemic stroke injury. Moreover, this protective effect of cGKI was found to be independent of gender and was mediated via improved reperfusion. These results suggest that cGKI in vascular smooth muscle cells should be targeted by therapies designed to protect brain tissue against ischemic stroke.
Keywords: Cerebral blood flow, cGMP-dependent protein kinase I, ischemia–reperfusion injury, middle cerebral artery occlusion-reperfusion model, stroke
Introduction
Cyclic guanosine monophosphate (cGMP) is a cyclic nucleotide derived from guanosine triphosphate (GTP). cGMP acts as a second messenger via activation of intracellular protein kinases. cGMP regulates ion channel conductance, glycogenolysis, cellular apoptosis, relaxes smooth muscle tissues, particularly vascular smooth muscles, leading to vasodilation.1 The cGMP signaling pathway has been identified as a potential target of protective therapies for cerebral ischemia/reperfusion injury.2,3 Cyclic GMP-dependent protein kinases (cGKs) (aka protein kinases G) are serine/threonine-specific protein kinases that are activated by cGMP and regulate smooth muscle relaxation,4 cell differentiation,5,6 platelet activation,7 nociception,8 and memory formation.9 Two cGK subtypes have been identified, cGKI and cGKII. These cGKs are products of different genes and exhibit differential expression patterns in cells. For example, cGKI is highly expressed in smooth muscle cells and platelets7,10 while cGKII is most abundant in renal,11,12 neuronal parenchymal,13,14 and intestinal mucosa cells.15 cGKI is expressed as two isoforms, cGKIα and cGKIβ. These isoforms are encoded by the same gene, but differ in their first ∼100 amino acids. This NH2-terminal region of the cGKI isoforms contains a leucine zipper-like motif, which is responsible for their homodimerization and differential interaction with anchoring proteins and phosphorylation targets.16 Both isoforms are expressed together in various SMCs.17,18 These isozymes interact with different proteins and affect SMC relaxation through different mechanisms.19–22 cGKIα interacts with MYPT1 (myosin-interacting subunit of myosin phosphatase 1)20 increasing myosin phosphatase activity23–25 and with RGS-2 (regulator of G protein signaling 2) increasing the inactivation of phospholipases C through increased hydrolysis of GαqGTP to GαqGDP,19,26,27 whereas cGKIβ shows specificity for inositol 1′,4′,5′-triphosphate receptor-associated G kinase substrate (IRAG).21,22 Phosphorylation of IRAG by cGKIβ decreases the release of Ca2+ from the 1′,4′,5′ triphosphate stores and thereby may reduce cytosolic increases in [Ca2+]i and SMC tone.28 Weber with coauthors29 showed that both cGKI isozymes can compensate for each other in the regulation of vascular tone in the awake mouse and that individual SMC-specific expression of the cGKIα as well as the cGKIβ isozyme was sufficient to support basic regulatory functions of the NO/cGMP signaling cascade. Their results indicate that the functional outcome of either isoform-dependent signaling in SMCs is similar regardless of the specific cGKI targets involved and that both isozymes of cGKI contribute significantly to the regulation of SM tone by NO and cGMP.29
Although cGKI has a protective role in ischemia/reperfusion injury,30,31 the underlying mechanisms remain incompletely understood. Several studies indicate that cGKI has an important effect on nitric oxide signaling in the vasculature. For instance, cGKI has been found to be critical for the nitroglycerin-induced relaxation of aortic rings in rats.32 Moreover, studies employing genetic ablation of cGKI demonstrated that it is required for NO/cGMP-mediated relaxation of smooth muscle cells in murine aortic rings.4 However, other work indicates that NO-mediated vascular relaxation can also be independent of this kinase.33,34 Since it has been suggested that NO and cGMP might signal independent of cGKs, further investigation of the mechanisms by which cGKI regulates pathways that are relevant for IRI are of great importance.33
Because of the central role that cGKI plays in homeostasis, global cGKI knockouts in rodents is associated with early mortality.4,10,35 Therefore, studies of the physiological role of cGKI have been conducted using inhibitors such as KT5823,36,37 H8,38 or H89.39 However, there is no cGK inhibitor available that is entirely specific and generally applicable, as they decrease cGKI activity in a variety of cell types, and likely have off-target effects. Recently, cell-specific tamoxifen-induced cGKI knockout mice have been generated that can provide important information about cGKI mechanisms in vivo.40 In particular, an induced, smooth muscle cell-specific cGKI knockout mouse model provides an opportunity to explore the specific role of cGKI in blood vessels, while avoiding early lethality and confounding influences of cGKI deletion in neurons, cardiomyocytes, and other cell types of global cGKI knockout mice.10,33 In this study, for the first time, we explore the role of smooth muscle cell cGKI in a model of cerebral IRI using smooth muscle cell-specific induced cGKI knockout mice. Moreover, because sex differences can modulate stroke outcomes,41–50 we tested the role of smooth muscle cell cGKI in both male and female mice.
Materials and methods
Animals
We used an efficient, tamoxifen-induced smooth muscle cell-specific cGKI knockout mouse model. The model is based on a conditional loxP-flanked (“floxed”) cGKI allele,51 flanking DNA encoded in both cGKI isoforms, combined with a SMA-CreERT2 transgene expressing the CreERT2 recombinase under the control of the smooth muscle alpha-actin promoter.52 SMA-CreERT2_cGKIfloxflox mice that were treated with 1 mg tamoxifen (TAM) i.p. each day for five consecutive days40 at the age of five to six weeks were used as experimental animals (cGKI SMKO mice). All experiments were conducted five to seven weeks after the final tamoxifen injection. For the middle cerebral artery occlusion (MCAO)/reperfusion model, we studied three types of control animals to take into consideration the potential effects of tamoxifen treatment and baseline Cre protein expression in smooth muscle cells: (1) C57BL/6 wild type (WT) mice not treated with tamoxifen; (2) litter mate SMA-CreERT2_cGKIfloxflox mice not treated with tamoxifen (cGKI control); and (3) litter mate cGKIfloxflox mice (without SMA-CreERT2 transgene) treated with tamoxifen (cGKI control TAM). Each group subjected to the MCAO/reperfusion was further divided into male (n = 12 animals in WT, cGKI control, and cGKI SMKO groups, n = 10 in cGKI control TAM group) and female (n = 12 animals in each group) subgroups. All procedures were performed in accordance with the recommendations outlined in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, approved by the Massachusetts General Hospital Subcommittee on Research and Animal Care, and reported in compliance with the ARRIVE guidelines.
Characterization of inducible smooth muscle cell-specific cGKI knockout mice
The floxed cGKI allele was genotyped by PCR using the following primers: 5′-CCT GGC TGT GAT TTC ACT CCA -3′ and 5′-GTC AAG TGA CCA CTA TG-3′. The SMA-CreERT2 transgene was genotyped by PCR using the following primers detecting the Cre sequence: 5′-GCT GCC ACG ACC AAG TGA CAG CAA TG -3′ and 5′-GTA GTT ATT CGG ATC ATC AGC TAC AC -3′. PCR was performed using GoTaq ® Flexi DNA Polymerase (Promega). PCR products were examined on 2% agarose gels. The results were interpreted as follows: cGKI flox – 338 bp, WT – 284 bp; SMA-CreERT2 – 402 bp.
For immunohistochemistry, 6-μm-thick coronal sections of formaldehyde-fixed, paraffin-embedded mouse brains were deparaffinized and rehydrated before being subjected to antigen retrieval, using 10 mM sodium citrate containing 0.05% Tween 20 (pH 6) solution and a pressure cooker. Subsequently, the sections were permeabilized using 0.1% Triton X-100 in phosphate-buffered saline, blocked using goat serum in phosphate-buffered saline, and then exposed to either an anti-cGKI rabbit polyclonal antibody that targets the COOH-terminal portion of the molecule (Enzo, #ADI-KAP-PK005-F), which is shared in the cGKI isoforms, an anti-smoothelin rabbit polyclonal antibody (Santa Cruz Biotechnology, #sc-28562), or control rabbit IgG (Abcam, #ab37416). The specificity of the anti-cGKI antibody used in our study was characterized previously by a pre-adsorption study using recombinant cGKI.53 After extensive washing to remove unbound antibodies, the sections were exposed to biotinylated horse anti-rabbit antibodies, avidin-conjugated alkaline phosphatase (VECTASTAIN® ABC-AP Staining Kit, Vector Labs), and a colorimetric substrate (VECTOR Red, Vector Labs) before being counterstained with hematoxylin, dehydrated, mounted, and coverslipped. Brightfield microscopy images were captured using an integrated microscope and charge-coupled camera system (Nikon TiE and DS-Ri1, respectively).
For Western blotting, aortic tissues were homogenized in an SDS lysis buffer, and 3 µg of protein extracts were subjected to reducing polyacrylamide gel electrophoresis. Proteins were electro-transferred to polyvinylidene difluoride membranes, reacted with antibodies, and detected using enhanced chemiluminescence and a charge-coupled molecular imaging device. Polyclonal rabbit antibodies to cGKI were developed in Dr. R. Feil's laboratory54; antibodies to GADPH were obtained commercially (Cell Signaling #2118).
Chemicals
All chemicals were obtained from Sigma Chemicals (St. Louis, MO, U.S.A.) unless otherwise indicated.
Surgical preparation and cerebral reperfusion model
Thirty minute MCAO followed by 48-h reperfusion model was performed as previously described.55 In short, the transient MCAO model was performed under general anesthesia (1.5% isoflurane in 30% O2 and 70% N2O). We monitored body temperature with a thermal probe inserted into the rectum and maintained temperature between 36.5 ℃ and 37.0 ℃ with a heating pad. A fiber optic probe was affixed to the skull over the area supplied by the middle cerebral artery (1 mm posterior and 7 mm lateral from bregma) for relative cerebral blood flow (CBF) measurements via laser Doppler flowmetry (LDF) during the surgery. MCAO was performed by inserting a 7–0 nylon filament covered by silicon (Doccol) into the external carotid artery (ECA) and advancing it into the internal carotid artery and middle cerebral artery for 30 min of ischemia with subsequent reperfusion for 48 h.55,56 After MCAO, reperfusion was performed in two steps: 15 min of partial reperfusion through the Willis circle (after withdrawal of filament from middle cerebral artery while ipsilateral common carotid artery was closed), followed by the total reperfusion after closing the ECA and opening the ipsilateral common carotid artery. After confirming total reperfusion by LDF and closure of the surgical wounds, mice were placed in postsurgical cages for recovery.
Evaluation of neurological deficit
We performed neurological evaluations after 48 h of reperfusion using a 5-point scale, as described previously.56 Normal motor function was scored as 0, flexion of the contralateral torso and forearm upon lifting the animal by the tail as 1, circling to the contralateral side as 2, leaning to the contralateral side at rest as 3, and no spontaneous motor activity as 4.
Evaluation of infarct volume
After neurological evaluation at 48 h of reperfusion, mice were euthanized, and brains were cut into 2-mm-thick coronal sections and stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) diluted in phosphate-buffered saline for 1 h at 37 ℃ in the dark.57 The area of infarction in each section was expressed as a fraction of the non-ischemic part of the ipsilateral hemisphere (indirect volume of infarct).57 Brain images were analyzed by a single operator blinded to information about the group of animals. Mice died after surgery were excluded from the infarct volume calculations.
Laser speckle contrast imaging
To determine whether the differences in stroke size and neurological outcome were associated with differences in cerebral perfusion defect during ischemia, we mapped the cortical CBF deficit using laser speckle contrast imaging during distal MCAO. Since we did not find difference between any control and gender group in infarct volume, neurological outcome and CBF by LDF, we compared male cGKI SMKO mice with male WT and male cGKI control mice in these advanced studies. Mice were anesthetized (1% of isoflurane in 30% O2 and 70% N2O), intubated, paralyzed (pancuronium bromide, 0.4 mg/kg intraperitoneally), mechanically ventilated (SAR-830; CWE), and placed in a stereotaxic frame (Kopf) as described previously.56 Arterial blood pressure was continuously monitored, and arterial pO2, CO2 and pH were measured. A burr hole (2-mm diameter) was drilled over the middle cerebral artery above the zygomatic arch, and the middle cerebral artery was occluded using a microvascular clip (Zen clip; Oswa). A laser diode (780 nm) was used to illuminate the intact skull surface. Raw speckle images were acquired with a video camera (Cohu, San Diego) and used to compute speckle contrast. Ten consecutive raw speckle images were acquired at 15 Hz and processed to speckle contrast using a sliding grid of 7 × 7 pixels. Contrast images were converted to correlation time values, and relative cerebral blood flow images were calculated by following the procedure outlined in literature.58,59 Perfusion images were obtained 30 min after distal MCAO and after 5 min of reperfusion. The areas of severe (0%–20% residual blood flow, representing core ischemic areas), moderate (21% to 30% residual blood flow) and mild (31% to 40% residual blood flow) blood flow deficits were quantified using a thresholding paradigm as we previously described.58
For assessment of the cerebral vascular reactivity to NO in cGKI SMKO and WT mice, the spontaneously breathing animals were anesthetized (1.5 % isoflurane in 30% O2 and 70% N2O) and placed in a stereotaxic frame. Arterial blood pressure, pO2, CO2 and pH were measured. The LSCI was performed by using the CMOS camera (acA1300-200 µm, Basler). For each time point, three sequences of 100 consecutive raw speckle images were acquired (image width = 1280, image height = 1024, exposure time = 5 ms, frame rate = 100 fps, frames/sequence = 100, number of sequences = 3, acquisition of three sequences was performed in <15 s). The data were processed by following the same procedure outlined in the dMCAO experiment above. The speckle images were acquired in WT and cGKI SMKO mice (n = 4 in each group) at baseline and 10 min after intravenous (femoral) injection (0.1 mg/kg) of the NO donor MAHMA NONOate (Cayman Chemical), and relative blood flow change was estimated between these two time points.
Statistical analysis
All results are expressed as mean ± SD. Statistical analysis was performed with one-way repeated measures analysis of variance (ANOVA) with Bonferroni post hoc test, or Kruskal–Wallis one-way analysis of variance on ranks (neurological deficit). LSCI results of distal MCAO were analyzed using two-way ANOVA followed by the two-stage set-up method of Benjamini, Krieger, and Yekutieli. The LSCI results of the cerebral vascular reactivity to NO were analyzed by using the two-tailed t-test. Differences of p < 0.05 were considered statistically significant.
Results
Immunohistochemistry and Western blot analysis confirm efficient cGK1 ablation in smooth muscle cells
Efficient, smooth muscle cell-specific ablation of cGKI protein expression in the posterior communicating artery was confirmed by immunohistochemistry (Figure 1(a)) in cGKI SMKO mice. In contrast, cGKI immunoreactivity was detected in the posterior communicating artery of control mice. In addition, cGKI immunoreactivity was detected in the subcommissural organ (SCO) of cGKI SMKO mice, which indicates that the cGKI was knocked out specifically in smooth muscle cells in these mice. We described the expression of the cGKI in the SCO previously.60
Figure 1.

(a) Cerebrovascular expression of cGKI in cGKI SMKO and control mice. Posterior communicating arteries exhibit cGKI immunoreactivity in cGKI control and WT mouse in vascular smooth muscle cells (left panels, red color), which are identified by their location within the vessel wall and their reaction with an anti smoothelin antibody in subsequent brain sections (right panels, red color). However, cGKI expression was not detected in smooth muscle cells of cGKI SMKO mice; the anti-cGKI antibody reactivity in smooth muscle cells of these vessels is similar to that detected in cells in sections exposed to non-immune IgG (lower panel). Immunostaining of the SCO shows cGKI immunoreactivity in cGKI SMKO mice, demonstrating the smooth muscle cell-specificity of the cGKI knockout in the mice. Counterstaining was performed with hematoxylin. Original magnification, × 60; the bar is 40 µm-long. (b) Ablation of cGKI in aorta of cGKI SMKO mice. Representative Western blot demonstrating control levels of cGKI in aortae of cGKI control TAM mice and lower levels of cGKI in cGKI SMKO mice.
We demonstrated efficient ablation of cGKI protein (cGKIα isoform ≈76 kDa, cGKIβ isoform ≈78 kDa61,62) expression in aorta by Western blot analysis (Figure 1(b)) in cGKI SMKO mice, finding regular expression of the protein in control group. The residual expression of cGKI detected in the cGKI SMKO shown in the Figure 1(b) is consistent with persistent expression of the protein in non-smooth muscle cells (e.g. fibroblasts).
cGKI deficiency in smooth muscle cells decreases cerebral reperfusion
The MCAO model allowed us to implement ischemic stroke in mice followed by reperfusion, utilize CBF measurements using laser Doppler flowmetry to monitor the ischemia, and to confirm the reperfusion values. Although intraischemic core CBF did not differ significantly among groups, the CBF, monitored by LDF above the core of the middle cerebral artery territory during reperfusion, was significantly greater in WT, cGKI control, and cGKI control TAM mice than in cGKI SMKO mice. Similar results were obtained in male (Figure 2(a)) and female (Figure 2(b)) mice.
Figure 2.

Cerebral blood flow by LDF (%): common carotid artery ligation (CCAL), internal carotid artery ligation (ICAL), middle cerebral artery occlusion (MCAO), partial reperfusion through the Willis circle (Rf WC), the reperfusion through the ipsilateral common carotid artery (Rf CCA) (*p < 0.05; **p < 0.01, WT, cGKI control, and cGKI control TAM mice vs. cGKI SMKO mice).
cGKI deficiency increases cerebral infarct volume
To evaluate the stroke volumes, we harvested the brains and measured infarct size 48 h after reperfusion. Stroke experiments revealed significantly greater cerebral infarct volume in cGKI SMKO (males: 96 ± 16 mm3; females: 93 ± 12 mm3, mean±SD) than in WT (males: 66 ± 19; females: 64 ± 14), cGKI control (males: 65 ± 18; females: 62 ± 14), and cGKI control TAM (males: 70 ± 8; females: 68 ± 10) mice (Figure 3(c)) for both males (Figure 3(a)) and females (Figure 3(b)) subgroups.
Figure 3.

Greater infarct volume in cGKI SMKO mice as compared to all control groups in both males and females subgroups. (a, b) analysis of infarct volumes at 48 h after reperfusion in male and female subgroups (**p < 0.001). (c) TTC staining of representative brain coronal sections at 48 h of reperfusion after 30 min filament MCAO.
Neurological deficit
In accordance with our results depicting greater infarct volume in cGKI SMKO mice, after 30 min of MCAO via filament with 48 h of reperfusion, the neurological deficit according to the 5-point scale was greater in cGKI SMKO mice than in WT, cGKI control, and cGKI control TAM in both males (Figure 4(a)) and females (Figure 4(b)) subgroups.
Figure 4.

Greater neurological deficit in cGKI SMKO mice than in control mice in both males and females subgroups (*p < 0.05, **p < 0.01).
Altogether, MCAO followed by reperfusion experiments showed that cGKI SMKO mice, having lower level of reperfusion, subsequently developed larger infarct volume and more severe neurological deficit, which emphasizes the importance of the neuroprotective properties of cGKI in cerebral IRI.
The mortality rate was higher in cGKI SMKO group (36.8% males, 25% females) than in WT (14.3% males and females), cGKI control (7.7% males, 0% females), and cGKI control TAM (0% males and females) groups.
We did not detect statistically significant differences in the infarct volume, CBF, and neurological outcomes between male and female mice in any of the mouse lines tested.
Laser speckle contrast imaging shows lower reperfusion in cGKI SMKO mice
Blood pressure, blood pH, pCO2 and pO2 were similar in WT, cGKI control and cGKI SMKO mice (Table 1). During distal MCAO, we mapped the cortical CBF deficit using laser speckle contrast imaging. Although intraischemic CBF did not differ among groups (Figure 5(a) and (b)), after 5 min of reperfusion, the core CBF was significantly greater in cGKI control (58 ± 23%) and WT mice (67 ± 22 %) than in cGKI SMKO mice (40 ± 20%) (p < 0.05, n = 8–9/group) (Figure 5(c)).
Table 1.
Systemic physiological parameters in dMCAO experiments.
| Mouse strain | Mean arterial pressure, onset, mm Hg | Mean arterial pressure intraischemic, mm Hg | Intraoperative blood pH | Intraoperative blood pCO2, mm Hg | Intraoperative blood pO2, mm Hg |
|---|---|---|---|---|---|
| WT | 88 ± 17 | 77 ± 11 | 7.32 ± 0.04 | 37 ± 5 | 132 ± 22 |
| cGKI control | 84 ± 8 | 81 ± 7 | 7.34 ± 0.08 | 34 ± 6 | 146 ± 20 |
| cGKI SMKO | 81 ± 8 | 79 ± 6 | 7.33 ± 0.05 | 35 ± 4 | 148 ± 13 |
Note: Mean arterial blood pressure before and during distal MCAO, arterial pH, arterial pCO2, and arterial pO2 during distal MCAO in WT (n = 8), cGKI control (n = 9) and cGKI SMKO mice (n = 9) (Mean ± SD). p – not significant.
Figure 5.
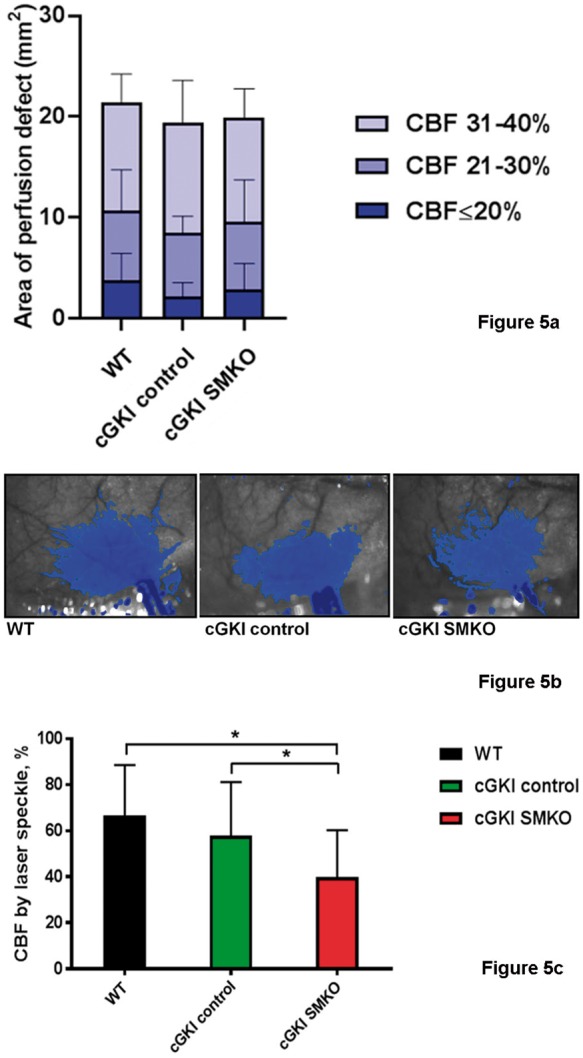
(a) The area of perfusion defect measured using laser speckle flowmetry. The number of pixels displaying three different severities of intraischemic CBF reduction was quantified using a thresholding paradigm and then converted to area (mm2) by the known scale factor. There was no statistically significant difference among the groups for any of the CBF segments. (b) Representative laser speckle flowmetry images in WT, cGKI control and cGKI SMKO mice 30 min after distal MCAO. Superimposed blue pixels indicate regions with ≤40% residual CBF. Imaging field dimensions are 6 × 8 mm. Imaging field is positioned over the right hemisphere. The long blue towards the bottom of each image is the clip artifact. (c) CBF after 5 min of reperfusion by laser speckle. After reperfusion by removing the microvascular clip occluding the distal middle cerebral artery, CBF was restored to a higher level in WT (n = 8) and cGKI control (n = 9) than cGKI SMKO (n = 9) mice (*p < 0.05).
Laser speckle contrast imaging shows decreased vascular reactivity in cGKI SMKO mice
Blood gases were similar in WT (pH 7.39 ± 0.13; pCO2 35 ± 11 mmHg; pO2 171 ± 36 mmHg) and cGKI SMKO (pH 7.44 ± 0.10; pCO2 29 ± 6 mmHg; pO2 167 ± 11 mmHg, n = 4 in each group, p-NS) mice. After injection of NO donor, mean arterial blood pressure decreased slightly and transiently by 10 to 15 mmHg for less than 10 min. The mean arterial blood pressure (mmHg) was not different between WT (88 ± 5) and CGKI SMKO (90 ± 6) mice before and 10 min after injection of NO donor (90 ± 6 vs. 89 ± 10). The mean relative blood flow increase (%) 10 min after injection of the MAHMA NONOate was significantly smaller in the cGKI SMKO mice (2 ± 4) than in the WT mice (14 ± 9, n = 4 in each group, *p < 0.05) (Figure 6).
Figure 6.
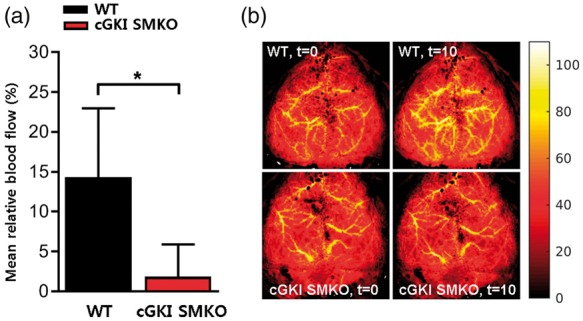
NO-dependent cerebrovascular responses in WT and cGKI SMKO mice. (a) MAHMA NONOate administered intravenously (0.1 mg/kg) increased the mean relative cerebral blood flow (%) in WT mice but the mean relative cerebral blood flow of cGKI SMKO mice was significantly lower than in WT mice. (b) Representative relative blood flow images before (t=0 ) and 10 min (t=10) after injection of MAHMA NONOate (0.1 mg/kg, i.v.) in WT and cGKI SMKO mice. These panels represent relative blood flow images. For each mouse, the max blood flow at t=0 was scaled to 100% and the same scaling factor was applied to the blood flow map at t=10 min after injection of the NO donor. Values are mean ± SD. *p < 0.05 for comparison between WT and cGKI SMKO mice (n = 4 for each group).
Discussion
Recent works have identified important roles of NO and cGMP signaling in mechanisms that regulate ischemia/reperfusion injury. cGKI has emerged as a key influencer of the protective effects of NO and is produced by several cell types within the brain.60,63 cGKI is known to be expressed by smooth muscle cells within the cerebral vasculature,60 but the specific role of cGKI residing within those cells in regulating cerebral ischemia/reperfusion injury is not entirely understood. In the present study, we employed a recently discovered40 mouse model of smooth muscle cell-specific cGKI deficiency to investigate the role of vascular cGMP signaling in stroke outcome.
Since the “bottleneck” of the study was to obtain sufficient ablation of smooth muscle cell cGKI using tamoxifen injections, we used immunohistochemical and Western blot assays to confirm specific and efficient ablations of smooth muscle cell cGKI gene expression in the mouse model. After tamoxifen treatment, cGKI immunoreactivity was decreased in the posterior communicating arteries of the conditional knockout mice, while its expression was retained in other cerebral cells, such as those partitioned in the subcommissural organ.
To exclude off target outcomes, we studied a potential effect of the described regiment of tamoxifen treatment and baseline Cre protein expression in smooth muscle cells for the stroke outcome of our induced knockout mice. Neuroprotective effects of tamoxifen have been reported in rodents: tamoxifen renders its neuroprotective mechanism likely by acutely alleviating cerebral ischemia injury, at least in part via an antioxidant mechanism and independent of any cerebral blood flow changes.64–66 However, the pharmacokinetics of tamoxifen in mice (t1/2 = 11.9 hours67) allows us to use it in our model while avoiding confounding effects on stroke outcome. Experimental models utilizing Cre-lox systems with similar tamoxifen administration schemes for conditional knockout mice generation were already utilized in previous MCAO studies.68,69 It was important to demonstrate that expression of Cre protein in smooth muscle cells, and our regimen, of tamoxifen treatment, by itself did not have influence on stroke outcome in our mice and MCAO model. These results allow us to reduce the amount of control groups of mice in further studies.
We studied three types of control animals: WT mice not treated with tamoxifen were studied as a standard control; litter mate SMA-CreERT2_cGKIfloxflox mice not treated with tamoxifen were studied as a control for baseline Cre protein expression in smooth muscle cells; and litter mate cGKIfloxflox mice (without SMA-CreERT2 transgene) treated with tamoxifen were studied as a control for tamoxifen treatment. Our data suggest that smooth muscle cell cGKI expression was required to demonstrate neuroprotection and to improve stroke outcomes. Subsequently, in the temporal MCAO stroke model, infarct volumes were larger, neurological outcomes were worse, and levels of reperfusion were lower in cGKI SMKO mice than in all control groups.
To obtain quantitative cerebral reperfusion data, we performed additional advanced studies using laser speckle CBF measurements. This method quantitatively measures both hypoperfused and ischemic zones, allowing for a wider evaluation of the perfusion deficiency in an acute cerebral ischemia model under the control of physiologic parameters. During laser speckle contrast imaging, we assessed two-dimensional CBF with high temporal and spatial resolution in real-time over a large area of brain – qualities in which LDF lacks. The size of the imaging field is adjustable, making it possible to study CBF in the large brain area without sacrificing spatial resolution. Laser speckle contrast imaging has a temporal resolution approaching real-time, which allows for monitoring of rapid changes in blood flow, such as arterial occlusion. In these studies, we used a non-surviving acute stroke model with distal temporal occlusion of the MCA via clip under artificial ventilation. In contrast to LDF monitoring of CBF in the filament model of MCAO, the acute clip model allows for the control of physiological parameters (blood gases and arterial blood pressure) under artificial ventilation. In the distal MCAO model, laser speckle contrast imaging can quantify the ischemic effect and its time course.58 Our results demonstrated that the hypoperfused areas were larger in cGKI SMKO mice than in cGKI control mice and WT mice. We also showed that cGKI SMKO mice have significantly reduced cerebral vascular reactivity to NO in comparison with WT mice, proving that cGKI mediates cerebral vascular response to NO.
Our data on neurological deficit and infarct volume in mice, devoid of cGKI in vascular smooth muscle, prove that the presence of cGKI in the vascular smooth muscle is a prerequisite for better stroke outcome.
We demonstrated an important role of cGKI in the regulation of CBF by smooth muscle cells during reperfusion by two different methods (LDF and laser speckle), and by two different approaches (MCAO by filament and distal MCAO by clip). Both approaches confirmed that reperfusion was lower in cGKI SMKO mice than in control mice, proving that cGKI in SMC is crucial for adequate reperfusion, which subsequently contributes to smaller infarct volume.
We described previously that decreased reperfusion in the MCAO model was caused, at least in part, by impaired vascular reactivity.56,70 Impaired vascular relaxation in eNOS knockout mice has been suggested to affect CBF during cerebral ischemia and to influence stroke outcome.56 Drugs targeting the NO-cGMP signaling pathway, initially identified as vasorelaxants, were shown to improve cerebrovascular reactivity and to reduce the infarct volume in rat stroke models.71 Furthermore, treatment with phosphodiesterase type 5 inhibitors, which elevates the level of cGMP by blocking its metabolism, also decreases cerebral infarct size72 and improves functional recovery73 in a rat model of MCAO. These findings suggest a central role of vascular relaxation in stroke outcome. cGKI is an important effector of the NO-cGMP signaling cascade.74 Upon stimulation by cGMP, cGKI in smooth muscle cells can phosphorylate a variety of cytosolic proteins that cause relaxation of the cells by regulating the intracellular calcium concentration and the cytoskeleton.31 cGKI is colocalized with the large conductance Ca2+-activated K+(BK) channels and controls the extent of an increase in BK current caused by an activation of NO/cGMP signaling in cerebral arterial smooth muscle. Thus, dynamic regulation of BK channel activity by the co-localized cGKI may contribute to NO-evoked dilation of cerebral resistance arteries.75
We studied both male and female mice for our stroke surviving experiments because previous studies suggested the existence of sex differences in stroke outcomes.41–50,76 We demonstrated that smooth muscle cell cGKI protected the brain against stroke injury associated with cerebral ischemia and reperfusion in a gender-independent manner. Females generally showed a tendency for better neurologic outcome in each group, which reflects the well-known protective effect of estrogens against ischemic stroke,41,77,78 but the sex difference was not statistically significant in our model.
Langhauser et al.79 found that the activity of the NO receptor, soluble guanylyl cyclase (NO-GC), which produces cGMP upon stimulation by NO, also protects against ischemic stroke. These results are in line with our data, that cGKI SMKO have worst stroke outcome and, together, disclose a protective NO-cGMP-cGKI pathway in ischemic stroke. In contrast to our study, authors used drugs to activate NO-GC rather than cell type-specific knockout mice and made no conclusions about the cell-type specificity of found effects. It is conceivable that beneficial effects of these small molecules are mediated in part by direct neuroprotection independent of vascular effects.79
Interestingly, cGKI activity is abnormal in spontaneously hypertensive rats (SHRs) in which high blood pressure is one of key factors contributing to ischemic stroke.80 The low expression level of cGKI in these animals underlies the lack of NO/cGMP-dependent regulation of intracellular calcium transient in aortic smooth muscle cells from SHRs. Decreased cGKI expression may contribute also to the development of hypertension and explain suboptimal responses to nitroglycerin and other NO-releasing molecules in patients.81
The neuroprotective effects of the NO-cGMP-cGKI signaling pathway may be mediated by mechanisms beyond vasorelaxation. NO-cGMP-cGKI signaling cascade has been shown to be involved in control of inflammation, metabolism, and cell proliferation in different cell and disease models.6,82–86 Accumulating evidence indicates that cGKI can regulate smooth muscle cell phenotype through mechanisms that are independent of its effect on intracellular calcium concentration and the cytoskeleton.5,83,87 Upon cGMP stimulation, smooth muscle cell cGKI has been observed to localize to the nucleus and phosphorylate transcription factors. In these cells, cGMP stimulates cGKI proteolysis, caused in part by proprotein convertases residing within the endomembrane system,53,88 releasing a COOH-terminal, constitutively active kinase fragment.86 This cGKI fragment migrates into the nucleus,86 through classical nuclear, pore-regulated mechanisms,6 and transactivates gene expression and regulates cell phenotype. Studies have determined that cGKI regulates smooth muscle cell phenotype by stimulating its nuclear and not cytosolic targets.6
The translation of data generated by using cGKI inhibitors in animals, or genetically modified animal models, to humans is challenging; however, sooner or later the systemic administration of pharmacological modulators of NO-cGMP-cGKI signaling pathway would become feasible. In this regard, it is essential to study complicated non-vascular effects of cGKI modulation. For instance, genetic deletion of cGKI in non-neuronal cells results in a complex metabolic phenotype, including liver inflammation and fasting hyperglycemia.82 cGMP-elevating compounds and ischemic conditioning provide cardioprotection against ischemia and reperfusion injury via cardiomyocyte-specific BK channels.89 Platelet cGKI but not endothelial or smooth muscle cGKI is essential to prevent intravascular adhesion and aggregation of platelets after ischemia,90 and NO-cGMP signaling in platelets was recently shown to be highly shear-dependent and limit thrombosis.91 It was demonstrated that cGKI protects against renal IRI partially through inhibiting inflammatory cell infiltration into the kidney, reducing kidney inflammation, and inhibiting tubular cell apoptosis.92 The overall profile of the cell type- and tissue-specific functions of cGKI is rather encouraging as cGKI seems to mostly play a protective role at a systemic level. However, future discovery of the approaches to the cell type-specific modulation of cGKI in humans would be quite promising.
In conclusion, our data suggest that cGKI in smooth muscle cells plays an important role in regulating cerebral blood flow during reperfusion and improves stroke outcome by a sex-independent mechanism, at least in part through the modulation of reperfusion. Our results suggest that cGKI in smooth muscle cells could be targeted to protect brain tissue against stroke. Future studies are required to further explore the neuroprotective potential of cGKI modulation in stroke treatment and prevention.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institutes of Health [NINDS R01 NS-096237 to Dmitriy N. Atochin, HL-125715 to Jesse D. Roberts Jr., P01CA080124, R01CA208205, R35CA197743, U01CA224173 to Dai Fukumura] and grants from the Deutsche Forschungsgemeinschaft [FOR 2060 projects FE 438/5-2 and FE 438/6-2, Projektnummer 374031971 – TRR 240 to Robert Feil].
Authors' contributions
MS, DNA, and PLH designed the study. MS performed statistical analysis and wrote the manuscript. RF and DF were responsible for conditional knockout mice generation and breeding. MS, PR and MML performed stroke surgeries, neurologic evaluation of mice after stroke, brain harvesting and staining, image analysis. JDR Jr., DNA and MS performed immunohistochemical analysis and tissue processing. RF performed Western blot analysis. CA and TS performed laser speckle dMCAO experiments. HHK, SS, İŞ, and GL performed and analyzed laser speckle measurements of cerebral vascular reactivity. MS, DNA, JDR Jr., RF, MML and ESB performed literature sesarch. ESB and PLH provided texnical support and critically revised the manuscript. All authors reviewed and approved the final version of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Francis SH, Corbin JD. Cyclic nucleotide-dependent protein kinases: intracellular receptors for cAMP and cGMP Action. Crit Rev Clin Lab Sci 1999; 36: 275–328. [DOI] [PubMed] [Google Scholar]
- 2.Zhang R, Wang L, Zhang L, et al. Nitric oxide enhances angiogenesis via the synthesis of vascular endothelial growth factor and cGMP after stroke in the rat. Circ Res 2003; 92: 308–313. [DOI] [PubMed] [Google Scholar]
- 3.Chen X, Wang N, Liu Y, et al. Yonkenafil: a novel phosphodiesterase type 5 inhibitor induces neuronal network potentiation by a cGMP-dependent Nogo-R axis in acute experimental stroke. Exp Neurol 2014; 261: 267–277. [DOI] [PubMed] [Google Scholar]
- 4.Pfeifer A, Klatt P, Massberg S, et al. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J 1998; 17: 3045–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pilz RB, Broderick KE. Role of cyclic GMP in gene regulation. Front Biosci 2005; 10: 1239–1268. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Roberts JD., Jr cGMP-dependent protein kinase I gamma encodes a nuclear localization signal that regulates nuclear compartmentation and function. Cell Signal 2014; 26: 2633–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walter U. Physiological role of cGMP and cGMP-dependent protein kinase in the cardiovascular system. Rev Physiol Biochem Pharmacol 1989; 113: 41–88. [DOI] [PubMed] [Google Scholar]
- 8.Feil R, Hofmann F, Kleppisch T. Function of cGMP-dependent protein kinases in the nervous system. Rev Neurosci 2005; 16: 23–41. [DOI] [PubMed] [Google Scholar]
- 9.Paul C, Schoberl F, Weinmeister P, et al. Signaling through cGMP-dependent protein kinase I in the amygdala is critical for auditory-cued fear memory and long-term potentiation. J Neurosci 2008; 28: 14202–14212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feil R, Lohmann SM, de Jonge H, et al. Cyclic GMP-dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ Res 2003; 93: 907–916. [DOI] [PubMed] [Google Scholar]
- 11.Wagner C, Pfeifer A, Ruth P, et al. Role of cGMP-kinase II in the control of renin secretion and renin expression. J Clin Invest 1998; 102: 1576–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gambaryan S, Häusler C, Markert T, et al. Expression of type II cGMP-dependent protein kinase in rat kidney is regulated by dehydration and correlated with renin gene expression. J Clin Invest 1996; 98: 662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vente J, Asan E, Gambaryan S, et al. Localization of cGMP-dependent protein kinase type II in rat brain. Neuroscience 2001; 108: 27–49. [DOI] [PubMed] [Google Scholar]
- 14.El-Husseini AE-D, Bladen C, Vincent SR. Molecular characterization of a type II cyclic GMP-dependent protein kinase expressed in the rat brain. J Neurochem 2002; 64: 2814–2817. [DOI] [PubMed] [Google Scholar]
- 15.Wang R, Kwon I-K, Thangaraju M, et al. Type 2 cGMP-dependent protein kinase regulates proliferation and differentiation in the colonic mucosa. Am J Physiol Liver Physiol 2012; 303: G209–G219. [DOI] [PubMed] [Google Scholar]
- 16.Khatri JJ, Joyce KM, Brozovich F V, et al. Role of myosin phosphatase isoforms in cGMP-mediated smooth muscle relaxation. J Biol Chem 2001; 276: 37250–37257. [DOI] [PubMed] [Google Scholar]
- 17.Keilbach A, Ruth P, Hofmann F. Detection of cGMP dependent protein kinase isozymes by specific antibodies. Eur J Biochem 1992; 208: 467–473. [DOI] [PubMed] [Google Scholar]
- 18.Geiselhöringer A, Gaisa M, Hofmann F, et al. Distribution of IRAG and cGKI-isoforms in murine tissues. FEBS Lett 2004; 575: 19–22. [DOI] [PubMed] [Google Scholar]
- 19.Tang M, Wang G, Lu P, et al. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat Med 2003; 9: 1506–1512. [DOI] [PubMed] [Google Scholar]
- 20.Surks HK, Mochizuki N, Kasai Y, et al. Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Ialpha. Science 1999; 286: 1583–1587. [DOI] [PubMed] [Google Scholar]
- 21.Schlossmann J, Ammendola A, Ashman K, et al. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Iβ. Nature 2000; 404: 197–201. [DOI] [PubMed] [Google Scholar]
- 22.Ammendola A, Geiselhöringer A, Hofmann F, et al. Molecular determinants of the interaction between the Inositol 1,4,5-trisphosphate receptor-associated cGMP kinase substrate (IRAG) and cGMP kinase Iβ. J Biol Chem 2001; 276: 24153–24159. [DOI] [PubMed] [Google Scholar]
- 23.Huang QQ, Fisher SA, Brozovich FV. Unzipping the role of myosin light chain phosphatase in smooth muscle cell relaxation. J Biol Chem 2004; 279: 597–603. [DOI] [PubMed] [Google Scholar]
- 24.Wooldridge AA, MacDonald JA, Erdodi F, et al. Smooth muscle phosphatase is regulated in vivo by exclusion of phosphorylation of threonine 696 of MYPT1 by phosphorylation of serine 695 in response to cyclic nucleotides. J Biol Chem 2004; 279: 34496–34504. [DOI] [PubMed] [Google Scholar]
- 25.Bonnevier J, Fässler R, Somlyo AP, et al. Modulation of Ca2+ sensitivity by cyclic nucleotides in smooth muscle from protein kinase G-deficient mice. J Biol Chem 2004; 279: 5146–5151. [DOI] [PubMed] [Google Scholar]
- 26.Obst M, Tank J, Plehm R, et al. NO-dependent blood pressure regulation in RGS2-deficient mice. Am J Physiol Integr Comp Physiol 2006; 290: R1012–R1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun X, Kaltenbronn KM, Steinberg TH, et al. RGS2 is a mediator of nitric oxide action on blood pressure and vasoconstrictor signaling. Mol Pharmacol 2004; 67: 631–639. [DOI] [PubMed] [Google Scholar]
- 28.Geiselhöringer A, Werner M, Sigl K, et al. IRAG is essential for relaxation of receptor-triggered smooth muscle contraction by cGMP kinase. EMBO J 2004; 23: 4222–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weber S, Bernhard D, Lukowski R, et al. Rescue of cGMP kinase I knockout mice by smooth muscle–specific expression of either isozyme. Circ Res 2007; 101: 1096–1103. [DOI] [PubMed] [Google Scholar]
- 30.Lukowski R, Krieg T, Rybalkin SD, et al. Turning on cGMP-dependent pathways to treat cardiac dysfunctions: boom, bust, and beyond. Trends Pharmacol Sci 2014; 35: 404–413. [DOI] [PubMed] [Google Scholar]
- 31.Hofmann F. A concise discussion of the regulatory role of cGMP kinase I in cardiac physiology and pathology. Basic Res Cardiol 2018; 113: 31. [DOI] [PubMed] [Google Scholar]
- 32.Satake N, Fujimoto S, Shibata S. The potentiation of nitroglycerin-induced relaxation by PKG inhibition in rat aortic rings. Gen Pharmacol 1996; 27: 701–705. [DOI] [PubMed] [Google Scholar]
- 33.Hofmann F, Feil R, Kleppisch T, et al. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev 2006; 86: 1–23. [DOI] [PubMed] [Google Scholar]
- 34.Hess DT, Matsumoto A, Kim S-O, et al. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 2005; 6: 150–166. [DOI] [PubMed] [Google Scholar]
- 35.Ruth P. Cyclic GMP-dependent protein kinases: understanding in vivo functions by gene targeting. Pharmacol Ther 1999; 82: 355–372. [DOI] [PubMed] [Google Scholar]
- 36.Kim WM, Yoon MH, Cui JH. Role of PKG-L-type calcium channels in the antinociceptive effect of intrathecal sildenafil. J Vet Sci 2010; 11: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andersen A, Povlsen JA, Johnsen J, et al. sGC–cGMP–PKG pathway stimulation protects the healthy but not the failing right ventricle of rats against ischemia and reperfusion injury. Int J Cardiol 2016; 223: 674–680. [DOI] [PubMed] [Google Scholar]
- 38.Gao Y, Dhanakoti S, Tolsa J-F, et al. Role of protein kinase G in nitric oxide- and cGMP-induced relaxation of newborn ovine pulmonary veins. J Appl Physiol 1999; 87: 993–998. [DOI] [PubMed] [Google Scholar]
- 39.Song J, Cheon SY, Lee WT, et al. PKA Inhibitor H89 (N-[2-p-bromocinnamylamino-ethyl]-5-isoquinolinesulfonamide) attenuates synaptic dysfunction and neuronal cell death following ischemic injury. Neural Plast 2015; 2015: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feil S, Valtcheva N and Feil R. Inducible Cre mice. In: Wurst W and Kühn R (eds) Gene Knockout Protocols. Methods in molecular biology (Methods and Protocols). Clifton, N.J.: Humana Press, 2009, pp. 343–363. [DOI] [PubMed]
- 41.Koellhoffer EC, McCullough LD. The Effects of Estrogen in Ischemic Stroke. Transl Stroke Res 2013; 4: 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Broughton BRS, Brait VH, Kim HA, et al. Sex-dependent effects of g protein-coupled estrogen receptor activity on outcome after ischemic stroke. Stroke 2014; 45: 835–841. [DOI] [PubMed] [Google Scholar]
- 43.Davis CM, Fairbanks SL, Alkayed NJ. Mechanism of the sex difference in endothelial dysfunction after stroke. Transl Stroke Res 2013; 4: 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roy-O'Reilly M, McCullough LD. Sex differences in stroke: the contribution of coagulation. Exp Neurol 2014; 259: 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fairbanks SL, Young JM, Nelson JW, et al. Mechanism of the sex difference in neuronal ischemic cell death. Neuroscience 2012; 219: 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim T, Chelluboina B, Chokkalla AK, et al. Age and sex differences in the pathophysiology of acute CNS injury. Neurochem Int 2019; 127: 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim T-H, Vemuganti R. Effect of Sex and Age Interactions on Functional Outcome after Stroke. CNS Neurosci Ther 2015; 21: 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nowak TS, Mulligan MK. Impact of C57BL/6 substrain on sex-dependent differences in mouse stroke models. Neurochem Int 2019; 127: 12–21. [DOI] [PubMed] [Google Scholar]
- 49.Zhao L, Mulligan MK, Nowak TS. Substrain- and sex-dependent differences in stroke vulnerability in C57BL/6 mice. J Cereb Blood Flow Metab 2019; 39: 426–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bushnell CD, Chaturvedi S, Gage KR, et al. Sex differences in stroke: challenges and opportunities. J Cereb Blood Flow Metab 2018; 38: 2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wegener JW, Nawrath H, Wolfsgruber W, et al. cGMP-dependent protein kinase I mediates the negative inotropic effect of cGMP in the murine myocardium. Circ Res 2002; 90: 18–20. [DOI] [PubMed] [Google Scholar]
- 52.Wendling O, Bornert J-M, Chambon P, et al. Efficient temporally-controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis 2009; 47: 14–18. [DOI] [PubMed] [Google Scholar]
- 53.Kato S, Chen J, Cornog KH, et al. The Golgi apparatus regulates cGMP-dependent protein kinase I compartmentation and proteolysis. Am J Physiol Physiol 2015; 308: C944–C958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valtcheva N, Nestorov P, Beck A, et al. The commonly used cGMP-dependent protein kinase type I (cGKI) inhibitor Rp-8-Br-PET-cGMPS can activate cGKI in vitro and in intact cells. J Biol Chem 2009; 284: 556–562. [DOI] [PubMed] [Google Scholar]
- 55.Atochin DN, Schepetkin IA, Khlebnikov AI, et al. A novel dual NO-donating oxime and c-Jun N-terminal kinase inhibitor protects against cerebral ischemia–reperfusion injury in mice. Neurosci Lett 2016; 618: 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atochin DN, Wang A, Liu VWT, et al. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. J Clin Invest 2007; 117: 1961–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Atochin DN, Murciano JC, Gursoy-Ozdemir Y, et al. Mouse model of microembolic stroke and reperfusion. Stroke 2004; 35: 2177–2182. [DOI] [PubMed] [Google Scholar]
- 58.Ayata C, Dunn AK, Gursoy-Özdemir Y, et al. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab 2004; 24: 744–755. [DOI] [PubMed] [Google Scholar]
- 59.Boas DA, Dunn AK. Laser speckle contrast imaging in biomedical optics. J Biomed Opt 2010; 15: 011109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feil S, Zimmermann P, Knorn A, et al. Distribution of cGMP-dependent protein kinase type I and its isoforms in the mouse brain and retina. Neuroscience 2005; 135: 863–868. [DOI] [PubMed] [Google Scholar]
- 61.Waldkirch ES, Ückert S, Langnäse K, et al. Immunohistochemical distribution of cyclic GMP-dependent protein kinase-1 in human prostate tissue. Eur Urol 2007; 52: 495–502. [DOI] [PubMed] [Google Scholar]
- 62.Gerzanich V, Ivanov A, Ivanova S, et al. Alternative splicing of cGMP-dependent protein kinase I in angiotensin-hypertension. Circ Res 2003; 93: 805–812. [DOI] [PubMed] [Google Scholar]
- 63.Uchida H, Matsumura S, Katano T, et al. Two isoforms of cyclic GMP-dependent kinase-I exhibit distinct expression patterns in the adult mouse dorsal root ganglion. Mol Pain 2018; 14: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wakade C, Khan MM, De Sevilla LM, et al. Tamoxifen neuroprotection in cerebral ischemia involves attenuation of kinase activation and superoxide production and potentiation of mitochondrial superoxide dismutase. Endocrinology 2008; 149: 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Milatovic D, Aschner M, et al. Neuroprotection by tamoxifen in focal cerebral ischemia is not mediated by an agonist action at estrogen receptors but is associated with antioxidant activity. Exp Neurol 2007; 204: 819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mehta SH, Dhandapani KM, De Sevilla LM, et al. Tamoxifen, a selective estrogen receptor modulator, reduces ischemic damage caused by middle cerebral artery occlusion in the ovariectomized female rat. Neuroendocrinology 2003; 77: 44–50. [DOI] [PubMed] [Google Scholar]
- 67.Robinson SP, Langan-Fahey SM, Johnson DA, et al. Metabolites, pharmacodynamics, and pharmacokinetics of tamoxifen in rats and mice compared to the breast cancer patient. Drug Metab Dispos 1991; 19: 36–43. [PubMed] [Google Scholar]
- 68.Begum G, Song S, Wang S, et al. Selective knockout of astrocytic Na+ /H+ exchanger isoform 1 reduces astrogliosis, BBB damage, infarction, and improves neurological function after ischemic stroke. Glia 2018; 66: 126–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sato H, Ishii Y, Yamamoto S, et al. PDGFR-β plays a key role in the ectopic migration of neuroblasts in cerebral stroke. Stem Cells 2016; 34: 685–698. [DOI] [PubMed] [Google Scholar]
- 70.Atochin DN, Yuzawa I, Li Q, et al. Soluble guanylate cyclase 1 1 limits stroke size and attenuates neurological injury. Stroke 2010; 41: 1815–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sallustio F, Diomedi M, Centonze D, et al. Saving the ischemic penumbra: potential role for statins and phosphodiesterase inhibitors. Curr Vasc Pharmacol 2007; 5: 259–265. [DOI] [PubMed] [Google Scholar]
- 72.Gao F, Sugita M, Nukui H. Phosphodiesterase 5 inhibitor, zaprinast, selectively increases cerebral blood flow in the ischemic penumbra in the rat brain. Neurol Res 2005; 27: 638–643. [DOI] [PubMed] [Google Scholar]
- 73.Zhang L, Zhang Z, Zhang RL, et al. Tadalafil, a long-acting type 5 phosphodiesterase isoenzyme inhibitor, improves neurological functional recovery in a rat model of embolic stroke. Brain Res 2006; 1118: 192–198. [DOI] [PubMed] [Google Scholar]
- 74.Hofmann F and Wegener JW. cGMP-dependent protein kinases (cGK). In: Krieg T and Lukowski R (eds) Guanylate Cyclase and Cyclic GMP. Methods in Molecular Biology (Methods and Protocols). Totowa, N.J.: Humana Press, 2013, pp. 17–50. [DOI] [PubMed]
- 75.Kyle BD, Mishra RC, Braun AP. The augmentation of BK channel activity by nitric oxide signaling in rat cerebral arteries involves co-localized regulatory elements. J Cereb Blood Flow Metab 2017; 37: 3759–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Spychala MS, Honarpisheh P, McCullough LD. Sex differences in neuroinflammation and neuroprotection in ischemic stroke. J Neurosci Res 2017; 95: 462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suzuki S, Brown CM, Wise PM. Neuroprotective effects of estrogens following ischemic stroke. Front Neuroendocrinol 2009; 30: 201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murphy SJ, McCullough LD, Smith JM. Stroke in the female: role of biological sex and estrogen. ILAR J 2004; 45: 147–59. [DOI] [PubMed] [Google Scholar]
- 79.Langhauser F, Casas AI, Dao V-T-V, et al. A diseasome cluster-based drug repurposing of soluble guanylate cyclase activators from smooth muscle relaxation to direct neuroprotection. npj Syst Biol Appl 2018; 4: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boehme AK, Esenwa C, Elkind MSV. Stroke risk factors, genetics, and prevention. Circ Res 2017; 120: 472–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Di Cesare Mannelli L, Nistri S, Mazzetti L, et al. Altered nitric oxide calcium responsiveness of aortic smooth muscle cells in spontaneously hypertensive rats depends on low expression of cyclic guanosine monophosphate-dependent protein kinase type I. J Hypertens 2009; 27: 1258–1267. [DOI] [PubMed] [Google Scholar]
- 82.Lutz SZ, Hennige AM, Feil S, et al. Genetic ablation of cGMP-dependent protein kinase type I causes liver inflammation and fasting hyperglycemia. Diabetes 2011; 60: 1566–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lehners M, Dobrowinski H, Feil S, et al. cGMP signaling and vascular smooth muscle cell plasticity. J Cardiovasc Dev Dis 2018; 5: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Haas B, Mayer P, Jennissen K, et al. Protein kinase g controls brown fat cell differentiation and mitochondrial biogenesis. Sci Signal 2009; 2: ra78–ra78. [DOI] [PubMed] [Google Scholar]
- 85.Chiche JD, Schlutsmeyer SM, Bloch DB, et al. Adenovirus-mediated gene transfer of cGMP-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J Biol Chem 1998; 273: 34263–34271. [DOI] [PubMed] [Google Scholar]
- 86.Sugiura T, Nakanishi H, Roberts JD. Proteolytic processing of cGMP-dependent protein kinase I mediates nuclear cGMP signaling in vascular smooth muscle cells. Circ Res 2008; 103: 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lincoln TM, Dey N, Sellak H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J Appl Physiol 2001; 91: 1421–1430. [DOI] [PubMed] [Google Scholar]
- 88.Kato S, Zhang R, Roberts JD. Proprotein convertases play an important role in regulating PKGI endoproteolytic cleavage and nuclear transport. Am J Physiol Cell Mol Physiol 2013; 305: L130–L140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Frankenreiter S, Bednarczyk P, Kniess A, et al. cGMP-Elevating compounds and ischemic conditioning provide cardioprotection against ischemia and reperfusion injury via cardiomyocyte-specific BK channels. Circulation 2017; 136: 2337–2355. [DOI] [PubMed] [Google Scholar]
- 90.Massberg S, Sausbier M, Klatt P, et al. Increased adhesion and aggregation of platelets lacking cyclic guanosine 3',5'-monophosphate kinase I. J Exp Med 1999; 189: 1255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wen L, Feil S, Wolters M, et al. A shear-dependent NO-cGMP-cGKI cascade in platelets acts as an auto-regulatory brake of thrombosis. Nat Commun 2018; 9: 4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li Y, Tong X, Maimaitiyiming H, et al. Overexpression of cGMP-dependent protein kinase I (PKG-I) attenuates ischemia-reperfusion-induced kidney injury. Am J Physiol Physiol 2012; 302: F561–F570. [DOI] [PMC free article] [PubMed] [Google Scholar]