

Abstract
Voltage-gated potassium (K+) channel subfamily B member 1 (KCNB1, Kv2.1) and integrin-α5 form macromolecular complexes—named integrin-α5-KCNB1 complexes (IKCs)—in the human brain, but their function was poorly understood. Here we report that membrane depolarization triggered IKC intracellular signals mediated by small GTPases of the Ras subfamily and protein kinase B (Akt) to advance the development of filopodia and lamellipodia in Chinese hamster ovary cells, stimulate their motility, and enhance neurite outgrowth in mouse neuroblastoma Neuro2a cells. Five KCNB1 mutants (L211P, R312H G379R, G381R, and F416L) linked to severe infancy or early-onset epileptic encephalopathy exhibited markedly defective conduction. However, although L211P, G379R, and G381R normally engaged Ras/Akt and stimulated cell migration, R312H and F416L failed to activate Ras/Akt signaling and did not enhance cell migration. Taken together, these data suggest that IKCs modulate cellular plasticity via Ras and Akt signaling. As such, defective IKCs may cause epilepsy through mechanisms other than dysregulated excitability such as, for example, abnormal neuronal development and resulting synaptic connectivity.—Yu, W., Shin, M. R., Sesti, F. Complexes formed with integrin-α5 and KCNB1 potassium channel wild type or epilepsy-susceptibility variants modulate cellular plasticity via Ras and Akt signaling.
The canonical function of K+ channels in the nervous system is to modulate the firing patterns that are required for processing sensory information and generating motor outputs. However, in addition to shaping neuronal excitability, K+ channels can participate in intracellular signaling by interacting with receptors and proteins of different classes and functions (1). In this context, integrins are common partners of K+ channels (2). One of those macromolecular complexes results from the assembly of integrin-α5 (α5) with voltage-gated K+ channel subfamily B member 1 (KCNB1; also known as Kv2.1) in the neurons of the human and murine brains [integrin-α5-KCNB1 complexes (IKCs)] (3, 4). Oxidative stress that develops as a consequence of brain trauma or in Alzheimer disease leads to the formation of disulfide bridges that cross-link KCNB1 subunits to each other (KCNB1 oligomers) (3, 5, 6). Oligomerization of the channel in IKC is translated by the integrin partner into a signaling cascade mediated by focal adhesion kinase (FAK), proto-oncogene tyrosine-protein kinase (Src) tyrosine kinases, and JNKs that induce more oxidative stress and ultimately apoptosis (4, 7). It is plausible that α5 interprets the formation of KCNB1 oligomers—which tend to accumulate in the plasma membrane—as detachment from the extracellular matrix and, because this is a general requirement for cell survival, elicits apoptosis in response (7, 8).
However, the physiologic role of IKCs remained largely unexplored. Integrins connect the cell to the extracellular matrix. They mediate cellular processes as varied as growth, division, motility, survival, apoptosis, and axon repair in the nervous system, primarily through interacting with the actin cytoskeleton (9, 10). This suggests that IKCs might be involved in the regulation of some of those cellular processes. Most importantly, because KCNB1 plays a well-established causative role in epilepsy—specifically in infancy or early-onset epileptic encephalopathy (EOEE), a particularly critical pathology often associated with severe developmental delay (SDD) in children—this further implies that aside from promoting neurodegeneration, IKCs may be implicated in epileptic disorders (11–17).
To gain insight into the function of IKCs we investigated their properties in heterologous expression systems. Our results indicate that IKCs sense the electrical activity at the membrane and transduce it into signals that modulate dynamic processes in the cell such as migration and neuritogenesis. IKCs achieve this modulation by targeting actin, via a signaling pathway that includes small GTPases of the Ras subfamily and protein kinase B (Akt). Certain EOEE-susceptibility variants disrupt this signaling modality and consequently impair the plasticity of the cell. Furthermore, under conditions of oxidative stress, IKCs engage the same signaling molecules, with the exception that Akt is inhibited, to presumably allow for the development of apoptotic programs.
MATERIALS AND METHODS
Reagents
Anti-Kv2.1 (clone K89/34) also called anti-KCNB1 here, was purchased from NeuroMab (University of California–Davis, Davis, Davis, CA; NIH, Bethesda, MD, USA). Anti-integrin-α5 (clone sc-10729) and Akt inhibitor class IV were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Cyclo(-RGDfK) was purchased from APExBIO (Houston, TX, USA). Farnesyl thiosalicylic acid (FTA), 3-isobutyl-1-methylxanthine (IBMX), and forskolin were purchased from Cayman Chemicals (Ann Arbor, MI, USA). Arp2/3 complex inhibitor class I was purchased from MilliporeSigma (Burlington, MA, USA). Pierce Cell Surface Isolation kit, Akt total, phospho-S473 ELISA kits, and Lipofectamine 2000 were purchased from Thermo Fisher Scientific (Waltham, MA, USA). PfuUltra II polymerase was purchased from Avantor (Radnor, PA, USA). Ras-pull down Activation Assay was purchased from Cytoskeleton (Denver, CO, USA). All other reagents were purchased from MilliporeSigma.
Molecular biology
KCNB1 mutants were constructed by site-directed mutagenesis using Pfu polymerase and the human KCNB1–influenza hemagglutinin (HA) plasmid as template (5). All sequences were confirmed by automated DNA sequencing. Transcripts were quantified by spectroscopy and compared with control samples separated by agarose gel electrophoresis stained with ethidium bromide. The clones have been donated to the Addgene repository and can be obtained from them.
Biochemistry
Chinese hamster ovary (CHO)-K1 cells were grown in DMEM medium and transfected in 6 cm-dishes with Lipofectamine 2000. For each transfection, 20 μl of transfection reagent and a total of 4 μg of plasmid DNA were used.
Twenty-four hours after transfection, cells were lysed with 1 ml of RIPA buffer (10 mM Tris-Cl pH 8.0, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 1 mM PMSF) and protease cocktail inhibitors (MilliporeSigma). Cell lysates were centrifuged for 20 min at 4°C, and the protein in the supernatant was assessed with the Bradford colorimetric assay (MilliporeSigma). Twenty micrograms of protein was resolved by SDS-PAGE (8% for KCNB1 or 12% for all other proteins) and transferred onto a PVDF membrane. After blocking in a 5% solution of nonfat milk in 0.1% Tween 20-PBS (PBST), membranes were probed with a primary antibody (1/500 dilution for KCNB1 or 1/1000 for the other antibodies) at 4°C overnight. After washing with PBST for 20 min, peroxidase labeled secondary antibodies (1/3000 dilution; MilliporeSigma) were applied for 1 h at room temperature, washed with PBST and incubated with peroxidase-based (POD) chemiluminescence substrates (Roche, Basel, Switzerland) and exposed.
Ras activity was measured using the Ras-pull down Activation Assay according to the manufacturer’s instructions.
ELISA was performed using the Akt total and phospho-S473 ELISA kits according to the manufacturer’s instructions and measured using an Infinite pro F200 (Tecan, Männedorf, Switzerland) plate reader.
Membrane biotinylation was performed using the Pierce Cell Surface Isolation kit according to the manufacturer’s instruction, and proteins were resolved by 8% SDS-PAGE and transferred onto a PVDF membrane for blotting.
Neurite outgrowth
Neuro2a (N2A) cells at an ∼60% confluence were transfected with wild type (WT) or DN and green fluorescent protein (GFP) cDNA or, for control, GFP alone and incubated with 200 μM IBMX and 30 μM forskolin. Forty-eight hours later they were photographed with a Zeiss Axiovert 200M microscope equipped with GFP lamp (Carl Zeiss GmbH, Oberkochen, Germany). The neurite (the longer one in multineuritic cells) of GFP-fluorescent cells was measured using ImageJ 1.52a software (National Institutes of Health, Bethesda, MD, USA). Histograms were calculated and fitted to a gaussian distribution:
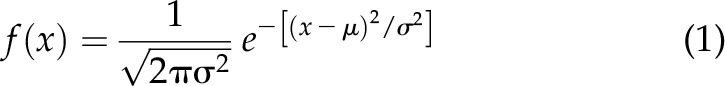 |
where μ is the median and σ is the sd, using Igor pro 6.37 software (Wavemetrics, Lake Oswego, OR, USA).
Wound healing assay
CHO cells were seeded into 6-well tissue culture plates at a density that after 24 h of growth would typically reach ∼80% confluence. Twenty-four hours after seeding, the cells were transfected as already described. The monolayer was perpendicularly scratched twice with a 200 μl (yellow) pipette tip. The well was washed with DMEM to remove the detached cells, and the scratches were photographed with a Zeiss Axiovert 200M microscope at ×10 magnification. After 24 h, the scratches were rephotographed. The gap covered in 24 h was measured using ImageJ 1.52a software (National Institutes of Health, Bethesda, MD, USA). Gap distance was calculated as follows:
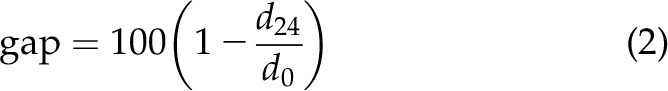 |
where d0 and d24 are the gap widths at the time of the scratch and 24 h later (2 technical replicates/time point/experiment).
Electrophysiology
Data were recorded with an Axopatch 200B (Molecular Devices, Sunnyvale, CA, USA), a personal computer (Dell, Round Rock, TX, USA), and Clampex software (Molecular Devices) and filtered at fc = 1 kHz and sampled at 2.5 kHz. Bath solution was (in mM) 4 KCl, 100 NaCl, 10 Hepes (pH = 7.5 with NaOH), 1.8 CaCl2, and 1.0 MgCl2. Pipette solution: 100 KCl, 10 Hepes [pH = 7.5 with potassium hydroxide (KOH)], 1.0 MgCl2, 1.0 CaCl2, 10 EGTA (pH = 7.5 with KOH).
Pipettes (∼5 MΩ) were obtained by pulling borosilicate glass with a Sutter P-97 puller (Sutter Instruments, Novato, CA, USA). Whole-cell currents were evoked by 1 s voltage sweeps from a holding potential of −80 mV to +100 mV in 20 mV increments, and leak subtraction was performed digitally using Clampfit software (Molecular Devices). Macroscopic conductance (G) curves were calculated as follows:
 |
where I is the macroscopic current at steady state (at the end of the voltage pulse) and Vrev is the calculated equilibrium potential = −95.7 mV. Offset potentials, including series resistance, were estimated to be ≤5 mV and were not compensated for when generating current-voltage relationships. G/GMax curves were fitted to the Boltzmann function:
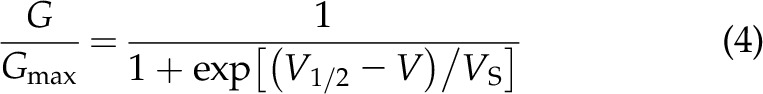 |
where V is the membrane voltage, V1/2 is the value of the voltage at which Eq. 4 is equal to 0.5 and VS is the slope coefficient (in mV).
Confocal microscopy
CHO cells transfected with Lifeact-GFP (18) and either mock (pCI-neo vector), WT, or nonconducting double-mutant, W369C-Y384T (19) (DN) cDNA were analyzed and photographed 24 h later with an Olympus FV1000MPE (Orangeburg, NY, USA) multiphoton microscope with full capacity for confocal microscopy and dedicated software.
Statistical analysis
Quantitative data are presented as means ± sem. The level of significance, assumed at the 95% confidence limit or greater (P < 0.05), was estimated using 1-way ANOVA with a Tukey post hoc test (http://astatsa.com/OneWay_Anova_with_TukeyHSD/).
RESULTS
IKCs engage Ras GTPases
In the human and murine brains and in CHO cells, oxidation/oligomerization of KCNB1 in IKCs promotes the activation of FAK and Src tyrosine kinases via phosphorylation of specific residues (3, 4, 7). Small GTPases of the Ras subfamily are one among the substrates of integrin-FAK-Src signaling; therefore, we sought to determine whether KCNB1 oxidation/oligomerization in IKCs would lead to Ras activation. Following an oxidative insult, significantly larger amounts of GTP-bound (activated) Ras GTPases were detected in lysates from CHO cells transfected with the WT KCNB1 channel compared with cells transfected with vector alone (mock) or cells transfected with a KCNB1 mutant, bearing a Cys73 to Ala replacement (C73A) that does not oligomerize (5) (Fig. 1A, B). The levels of activated Ras proteins were also significantly higher in mock-transfected cells compared to C73A-transfected cells, indicating that there are receptors in CHO cells that in the presence of oxidative conditions, can activate Ras GTpases independently of KCNB1. If KCNB1 oligomerization can trigger Ras signaling through α5, it is likely it can also turn on Ras during normal activity. To test this idea, we subjected the cells to KCl-induced depolarization, which promotes KCNB1 opening and conduction. This manipulation was associated with activation of Ras GTPases in cells transfected with WT but not in mock-transfected cells. Inhibiting integrin signaling by Cyclo(-RGDfK), a potent inhibitor of integrin αVβx but also of α5βx in the 10–100 nM range (20), prevented the activation of the GTPases (Fig. 1A, C). Thus, both oxidation-induced oligomerization of KCNB1 and the normal activity of the channel can lead to Ras activation via their integrin partners.
Figure 1.

IKCs activate Ras GTPases. A) Representative Western blots of activated (GTP-bound) Ras GTPases and for control, actin, from CHO cells transfected with the indicated cDNAs and incubated in DMEM + 25 μM 2,2′-dithiodipyridine for 5 min or 30 mM KCl for 15 min, prior to lysis. Cyclo(-RGDfK) (200 nM) and TEA (15 mM) were added 10 min prior to KCl and maintained until lysis. B) Densitometric quantification of the of amounts of activated Ras protein normalized to actin in response to an oxidative challenge. N = 6 biologic experiments (dots). C) Densitometric quantification of activated Ras protein normalized to actin in response to KCl-induced depolarization. For each bar, N ≥ 3 biologic experiments (dots). Densitometry analysis was performed using ImageJ 1.52a software. *P < 0.05, **P < 0.01. Pair-wise comparisons are referred to WT.
IKCs activate Ras mainly through non-ionic mechanisms
KCNB1 oligomers do not conduct current (it has been suggested that oxidation of Cys73 may obstruct the mutual movement of the N terminus and C terminus of the channel, an important step toward conduction) (5, 21), and this raised the question as to whether K+ current has a role in the activation of Ras GTPases by IKCs. Blocking KCNB1 with tetraethylammonium (TEA) prevented Ras activation (Fig. 1A, C). TEA causes conformational changes in the selectivity filter (22) that might have impeded structural rearrangements in KCNB1. Therefore, we next asked whether K+ current can lead to Ras recruitment, regardless of IKCs, by expressing Caenorhabditis elegans K+ voltage-sensitive channel subunit 1 (KVS-1) and K+ channel for habituation to tap 1 (KHT-1) that conduct robust K+ currents in CHO cells (23, 24). Both failed to induce Ras activation (Fig. 1A, C). IKCs might turn on Ras via non-ionic mechanisms that involve structural rearrangements of the channel—for example, displacements of the voltage sensor—that are translated into intracellular signals by α5. However, a dominant-negative, nonconducting double-mutant, W369C-Y384T (19), here named DN, did not lead to activation of Ras (Fig. 1A, C). Because CHO cells do not express endogenous K+ channels and DN does not conduct, it is plausible that KCl was ineffective at generating membrane depolarization and, consequently, provoking structural changes in the mutant. Indeed, when we coexpressed KVS-1 with DN, this manipulation led to partial activation of Ras GTPases in the presence of KCl (Fig. 1A, C).
IKCs differentially modulate Akt
The PI3K/Akt pathway is one of the major vehicles through which integrins convey signals important for survival, growth, proliferation, and cell migration (25–27) [Src kinases alone can also activate PI3K/Akt (28)]. This argues that in order to induce apoptosis, KCNB1 oxidation must be associated with PI3K/Akt silencing. Accordingly, following an oxidative challenge, the fraction of Akt protein phosphorylated at Ser473 (pAkt) was maximal in cells expressing C73A, intermediate in mock-transfected cells, and minimal in cells expressing WT KCNB1, a result largely expected because the latter undergo substantial apoptosis under those stressful conditions (4, 5, 7, 29) (Fig. 2A). The kinase was also more active in cells transfected with WT that were subjected to KCl-induced depolarization compared with mock-transfected or cells expressing the DN variant or KVS-1 or incubated with Cyclo(-RGDfK) or TEA (Fig. 2B). In all the latter conditions, the fraction of pAkt/Akt ranged between ∼20 and 30%, revealing the existence of basal Akt activity independent of IKCs.
Figure 2.

IKCs promote Akt phosphorylation at Ser473. A) ELISA quantification of pAkt normalized to total Akt in CHO cells incubated in DMEM + 25 μM 2,2’-dithiodipyridine for 5 min prior to lysis. B) As in A, for cells incubated in DMEM + 30 mM KCl for 15 min prior to lysis. Cyclo(-RGDfK) (200 nM) and TEA (15 mM) were added 10 min prior to KCl and maintained until lysis. N = 3 biologic experiments (dots) each with 2 technical replicates/experiment. *P < 0.05, **P < 0.01. Pair-wise comparisons are referred to WT (B).
KCNB1 enhances neurite outgrowth
Integrins play a major role in neuritogenesis (30) and other cellular processes relying on actin and its cytoskeleton (9, 10). For example, neurite formation and growth proceed through changes in the actin cytoskeleton driven by external cues (31–35). On the other hand, K+ channels have been implicated in IBMX/forskolin-induced neuritogenesis in mouse neuroblastoma N2A cells (36), implying that IKCs may affect neurite outgrowth. To test this possibility, we measured neurite lengths in cells transfected with GFP alone or with WT or DN (representative images are shown in Fig. 3). Neurite lengths were normally distributed (Eq. 1) with medians equal to 38.7, 63.3, and 32.5 arbitrary units for GFP alone, WT+GFP, and DN+GFP, respectively (Fig. 3A–C). Mean neurite lengths were 80.4 ± 4.5, 95.8 ± 4.4, and 76.9 ± 4.3 arbitrary units, respectively (WT+GFP vs. GFP P < 0.05, WT+GFP vs. DN+GFP P < 0.01, and DN+GFP vs. GFP not statistically significant; N = 108, 105, and 103 cells for GFP, WT+GFP, and DN+GFP, respectively).
Figure 3.

IKCs stimulate neurite outgrowth in N2A cells. Representative images of N2A cells transfected with GFP alone (A), WT+GFP (1:5) (B), and DN+GFP (1:5) (C) and distributions of neurite length in arbitrary units (Au). Examples of how neurites were measured are shown in yellow color. Histograms were calculated and fitted to a gaussian distribution (Eq. 1) with medians, μ = 38.7, 63.3 and 32.5 Au and sd, σ = 65.2, 76.6 and 73.8 Au, respectively, using Igor software. Images were analyzed with ImageJ 1.52a. Scale bar, 50 μm. N = 3 biologic experiments. Number of samples = 108, 105, and 103 for GFP, WT+GFP, and DN+GFP, respectively.
IKCs affect cell motility
Because our experiments in N2A cells indicated that KCNB1 can impact neurite outgrowth, we next qualitatively probed whether KCNB1 could induce changes in F-actin, which was detected under confocal microscopy by Lifeact-GFP, a 17-aa-peptide that stains F-actin without interfering with actin dynamics (18). An increase in the concentration of external K+ triggered the development of lamellipodia and filopodia projections, which are primarily composed of actin, in CHO cells transfected with KCNB1 (Fig. 4A) but not with DN or mock cDNA (unpublished results). Cells use lamellipodia and filopodia projections to migrate. Accordingly, in the wound healing assay (37), gap closure (Eq. 2) was significantly enhanced in CHO cells transfected with WT compared with mock cells (Fig. 4B) in agreement with the work of Wei et al. (38), who showed that KCNB1 promotes cell polarization and accelerates cell motility by stimulating integrin-mediated FAK activation. Furthermore, the DN variant or KVS-1 failed to enhance gap closure compared to control. To determine whether Ras-Akt signaling is responsible for the effects of IKCs on gap closure, cells transfected with mock cDNA or WT were incubated with 20 μM Ras inhibitor FTA or, separately, 1.5 μM Akt inhibitor class IV. Twenty-four hours incubation with FTA did not alter the motility of mock-transfected cells but significantly inhibited the motility of WT cells to mock levels (Fig. 4B). Conversely, inhibition of Akt activity almost completely suppressed the motility of both mock- and WT-transfected cells. These results are consistent with previous biochemical measurements of Ras and Akt activities that indicated that Ras GTPases are primarily active in cells expressing functional IKCs (Fig. 1), whereas Akt is also active in cells lacking functional IKCs (Fig. 2). Because cell motility relies on actin, we pharmacologically targeted the Arp2/3 complex, which promotes actin polymerization through the creation of a nucleation core. Likewise, for Akt, inhibition of Arp2/3 complex activity by class I inhibitor suppressed the motility of mock- and WT-transfected cells (Fig. 4B).
Figure 4.

IKCs enhance cell motility. A) Representative confocal images of CHO cells transfected with WT and Lifeact-GFP incubated in DMEM+30 mM KCl starting at t = 0 min. Representative changes in lamellipodia and filopodia structures are indicated by arrows in the insets. Scale bar, 10 μm. B) Mean distance covered in 24 h are expressed as percents of the original distance (gap, Eq. 2) in CHO cells transfected with the indicated cDNAs. Ras inhibitor FTA (20 μM), Akt inhibitor class IV (1.5 μM), and Arp2/3 complex inhibitor class I (15 μM) were applied at the time of the scratch. Inset representative pictures of gap distance in mock and WT transfected cells at 0 and 24 h. Scale bar, 100 μm. Images were analyzed with ImageJ 1.52a. For each bar, N ≥ 3 biologic experiments (dots) with 2 technical replicates/experiment. *P < 0.05, **P < 0.01 for statistical significance (pair-wise) compared to WT, #P < 0.05 for statistical significance compared to mock.
Biochemical characterization of KCNB1 variants associated with severe epilepsy
Missense mutations in the KCNB1 gene have been linked to infancy or EOEEs, one of the most severe forms of epilepsy that are often associated with progressive psychomotor disorders (13, 15, 16, 39). We analyzed 5 KCNB1 variants (L211P, R312H, G379R, G381R, and F416L) that cause severe clinical phenotypes (13). Thus, L211P, which occurs in the putative S1-S2 linker, was first detected in a child with developmental delay and dyspraxia. The R312H mutation is located in the putative voltage sensor domain (S4) and was initially found in 2 children both exhibiting SDDs. One stopped walking at 5 yr of age and never developed language. The other could not articulate active speech at 8 yr of age and walked on tiptoe. G379R and G381R occur in the selectivity filter (GYG) and were initially discovered in children with SDD in both motor and language function. F416L occurs in the putative S6 transmembrane domain and was initially identified in 2 children affected by SDD. We expressed these variants in CHO cells and characterized their biochemical properties. In 2 experiments, all 5 variants expressed total amounts of protein comparable to WT. Surface expression was also comparable to WT for all mutants (Fig. 5). Our results are in agreement with the work of Torkamani et al. (16), who previously reported normal surface expression of G379R.
Figure 5.
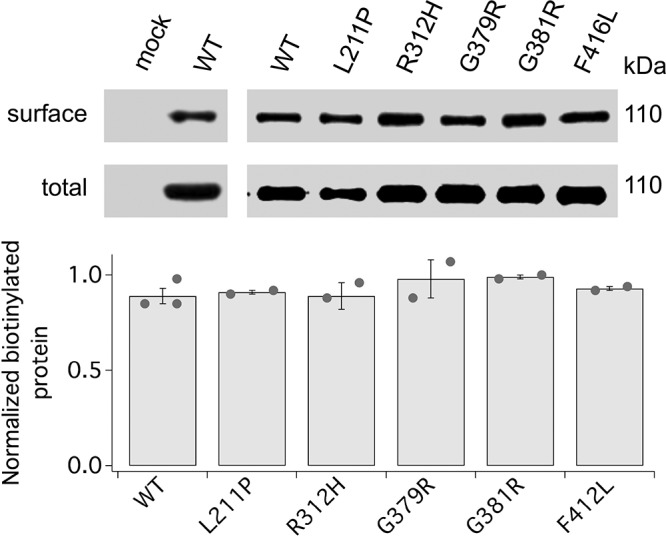
Surface expression of EOEE-susceptibility variants. Representative Western blots of surface and total expression of mock, WT, and EOEE-susceptibility KCNB1 variants and densitometric quantification of N = 2–3 experiments (dots). Normalization to total CHO cells were incubated with a membrane impermeant biotin analog before lysis. Western blot visualization was performed with anti-KCNB1 mAb.
Electrophysiological characterization of KCNB1 EOEE variants
Representative whole-cell currents recorded in cells that were transfected with mock, WT cDNA alone (WT/WT), or R312H cDNA alone (R312H/R312H), and, to mimic the case of patients heterozygous in R312H, a mixture of WT and R312H cDNA in a 1:1 ratio (WT/R312H) are illustrated in Fig. 6A. Homomeric WT channels conducted robust currents exhibiting typical slow activation and late inactivation. In contrast, homomeric R312H channels produced smaller currents (Fig. 6B) that activated at more depolarizing voltages, consistent with the fact that the R to H replacement occurs in the voltage sensor domain (V1/2 = 14.4 ± 2.3 mV for WT/WT and V1/2 = 51.8 ± 2.1 mV for R312H/R312H P < 0.01; Vs = 10.5 ± 0.8 mV and Vs = 13.5 ± 0.9 mV, respectively, Eqs. 3, 4, N = 9 cells each, Fig. 6C). Heteromeric channels formed by WT and R312H subunits exhibited intermediate characteristics between those of homomeric WT and R312H channels (V1/2 = 32.9 ± 5.3 mV, P < 0.01 vs. both WT/WT and R312H/R312H; Vs = 15.6 ± 1.9 mV; N = 9 cells). The other mutants did not express detectable currents. Torkamani and colleagues (16) reported small, nonselective, G379R currents with linear current-voltage relationships but we did not record similar currents.
Figure 6.

R312H channels exhibit altered gating. A) Representative whole-cell currents in CHO cells transfected with mock, WT (WT/WT), R312H (R312H/R312H), or a 1:1 mixture of WT and R312H (WT/R312H) cDNAs. Inset: voltage protocol. B) Current-voltage relationships for WT/WT (circles), R312H/R312H (squares), and WT/R312H (triangles). C) Normalized macroscopic steady-state conductance-V relationships (G/GMax, Eq. 3) for WT/WT (circles), R312H/R312H (squares), and WT/R312H (triangles). Data are fitted to the Boltzmann function (Eq. 4) with V1/2 = 16.8 mV and Vs = 10.2 mV for WT/WT; V1/2 = 50.5 mV and Vs = 12.9 mV for R312H/R312H and V1/2 = 29.5 mV and Vs = 13.9 mV for WT/R312H. N = 9 cells for each group. *P < 0.05, **P < 0.01, for pair-wise comparisons between WT/WT and R312H/R312H; #P < 0.05 for pair-wise comparisons between WT/WT and WT/R312H.
KCNB1 EOEE variants affect cell motility via Ras-Akt signaling
Figure 7A shows activated Ras protein following KCl-induced depolarization in CHO cells transfected with WT and EOEE-susceptibility variants. Thus, L211P, G379R, and G381R were associated with levels of activated Ras comparable to WT. In contrast, the amounts of activated Ras GTPases were negligible in cells transfected with R312H and F416L. ELISA quantification of the fraction of pAkt is illustrated in Fig. 7B. Similar to Ras, in cells expressing L211P, G379R, and G381R pAkt/Akt was comparable to WT but was markedly decreased in cells expressing R312H and F416L. When tested in the wound healing assay, L211P, G379R, and G381R enhanced gap closure similar to WT, whereas R312H and F416L did not (Fig. 7C). In summary, L211P, G379R, and G381R activated Ras-Akt signaling and accelerated cell migration, whereas R312H and F416L did not activate Ras-Akt and had no effect on motility.
Figure 7.

EOEE-susceptibility variants affect Ras/Akt signaling and cell motility. A) Representative Western blot of activated Ras protein and for control, actin, and mean fractions of activated Ras protein (normalized to actin) in CHO cells transfected with WT or the indicated EOEE-susceptibility KCNB1 variants. Cells were incubated in DMEM + 30 mM KCl for 15 min prior to lysis. N = 3 biologic experiments (dots). B) ELISA quantification of pAkt normalized to total Akt. N = 2 biologic experiments (dots) with 2 technical replicates/experiment. C) Mean gap distances (Eq. 2) in CHO cells transfected with WT or the indicated KCNB1 variants. For each bar, N ≥ 5 biologic experiments (dots) with 2 technical replicates/experiment. Images were analyzed with ImageJ 1.52a. *P < 0.05, **P < 0.01.
DISCUSSION
In this study, we shed light on the function of complexes formed by integrin-α5 and KCNB1 K+ channels that were recently discovered (4). Our results provide a model that predicts that IKCs act to transduce cellular excitability into intracellular signals that affect actin and consequently play a role in the modulation of cellular plasticity. However, when the conditions of the cell worsen and oxidative stress ensues, IKCs become toxic and activate a pathway that results in the production of more reactive oxygen species (ROS) and ultimately apoptosis. In both pathways, IKCs signal through the same molecules, namely FAK (4, 38), Src tyrosine kinases (4), Ras GTPases, and Akt. Consistent, with the well-established role of Ras proteins controlling cell migration and apoptosis through Akt signaling (40, 41), the GTPases were specifically activated by IKCs subjected to KCl-induced depolarization or oxidation because DN, Cyclo(-RGDfK), TEA, or C73A failed to activate them. Akt plays a major role in cell survival and motility. Accordingly, Akt was “on” when IKCs were stimulated by KCl and “off” in the presence of robust KCNB1 oxidation because this leads to apoptosis (29), whereas in cells expressing C73A that survive the oxidative injury, Akt was active. In summary, considering that oxidation of KCNB1 is a physiologic process associated with aging and neurodegeneration (3, 42), these data underscore a previously unknown pleiotropic nature of IKCs of macromolecules that are beneficial early in life and become deleterious late in life through mechanisms that share key molecular components. The findings reported here also raise intriguing questions that await future studies. For example, through which molecular steps do IKCs translate events occurring at the plasma membrane into intracellular signals? Evidence points out to a non-ionic mechanism centered around structural rearrangements of the KCNB1 channel that are sensed by α5 but it cannot be ruled out at this stage that K+ current may have a role in the process. Furthermore, it is not known how the recruitment of Akt does proceed because both Ras GTPases (40, 41) and IKCs can directly engage the kinase.
These findings are relevant to epilepsy, the fourth common neurologic disorder. Seizures are due to abnormal neuronal electrical activity, and K+ channels are one of the most likely culprits given their central role in modulating excitability. Indeed, a specific term, channelepsy, has been coined (43). However, as our understanding of epilepsy advances, it has become clear that a picture in which K+ channels cause hyperexcitability and seizures by conducting poorly is too simplistic, and dysfunctional K+ channels might contribute to epileptic etiologies through processes other than dysregulated excitability, such as, for example, abnormal neuronal development and resulting synaptic connectivity (44–46). The findings reported here support this idea. All 5 EOEE-susceptibility variants that were tested exhibited impaired conduction, which was consistent with hyperexcitability (R312H is probably dominant because WT/R312H channels activate at more depolarizing voltages than WT channels). However, although 3 variants (L211P, G379R, and G381R) activated Ras-Akt and accelerated gap closure; 2 variants, F416L and R312H, failed to activate Ras-Akt and did not affect healing. Hence, the fact that R312H and F416L impaired cell motility may suggest that certain defective KCNB1 channels may also impinge on the development of neurons and of their networks in patients with epilepsy and that these effects anticipate and probably overwhelm the alterations of excitability due to defective conduction. This idea is consistent with the observation that one of the end points of IKC signaling is actin, which plays a major role in neuritogenesis (30). In CHO cells, WT, but not the DN variant, were associated with the development of lamellipodia and filopodia structures. Consistent with a role in the regulation of actin, first the WT channel, but not the DN variant enhanced neurite outgrowth and cell migration; second, pharmacological inhibition of Arp2/3 complex activity suppressed the effects on gap closure mediated by WT. Interactions of the actin cytoskeleton with K+ channels including Ca2+-activated K+ channels, inwardly rectifying K+ channels, ATP-sensitive K+ channels, 2-pore K+ channels, and voltage-dependent K+ channels, including KCNB1, are pervasive in animals and plants (38, 47–57). Generally, the actin cytoskeletal network provides a regulatory mechanism of the channels by influencing their gating, trafficking, and organization to the plasma membrane and stretch sensitivity. For example, the organization of actin filaments controls clustering of large conductance calcium-activated potassium channels [big potassium (BK)] at synapses via a mechanism that depends on the activity of PI3 kinase, an obligate activator of Akt (58). A recent study has implicated actin filaments in the modulation of the inactivation mechanism of KCNB1 (57). Cases in which the channels affect actin are less frequent but are documented. Piwkowska et al. (59) suggested that the activity of BK channels in podocytes may control glomerular filtration barrier via their indirect regulation of the actin cytoskeleton. Further, interaction of potassium 2 pore domain channel subfamily K member 2 with the actin cytoskeleton has been shown to influence both synaptogenesis and neuronal electrogenesis (60) and a genetic mutation in KCNC3 (Kv3.3) resulting in adult-onset spinocerebellar ataxia 13 leads to growth cones with deficient actin veils in stem cell–derived neurons (47). We and others have shown that KCNB1 and synapsin-1 physically interact in the mouse brain where moderate oxidation of KCNB1 during aging correlates with a decrease of the latter in presynaptic structures (42, 61). The maturation and plasticity of the synapses depend upon the remodeling of the actin cytoskeleton. Integrins play a central role in these dynamic processes, but also synapsin-1 interacts with actin and its cytoskeleton (62–66). One major mechanism of learning and memory in the hippocampus is long-term potentiation, which requires the involvement of both Ras and Akt signaling (64, 67, 68). At the level of the single synapse, long-term potentiation formation results in larger spine volume and a greater ratio of F-actin to G-actin (69, 70). IKCs are expressed in proximal dendrites and synapses (71), and this suggests that they may act to transduce membrane excitability into intracellular signals that are important for spine plasticity. Future studies will address these questions.
ACKNOWLEDGMENTS
The authors thank Drs. Mladen-Roko Rasin and Huaye Zhang (Robert Wood Johnson Medical School, Rutgers University) for help with the confocal microscope and Drs. Kiram Madura and Li Chen (Robert Wood Johnson Medical School, Rutgers University) for help with the plate reader. The Lifeact-GFP construct was provided by the Addgene repository. This work was supported by National Science Foundation (NSF) Grant 1456675 and U.S. National Institutes of Health (NIH) Grants R01AG060919 (National Institute on Aging) and R21NS0966 (National Institute of Neurological Disorders and Stroke) to F.S. The authors declare no conflicts of interest.
Glossary
- Akt
protein kinase B
- CHO
Chinese hamster ovary
- EOEE
early-onset epileptic encephalopathy
- FAK
focal adhesion kinase
- FTA
farnesyl thiosalicylic acid
- GFP
green fluorescent protein
- IBMX
3-isobutyl-1-methylxanthine
- IKC
integrin-α5-KCNB1 complex
- KCNB1
voltage-gated potassium (K+) channel subfamily B member 1
- N2A
Neuro2a
- pAkt
Akt phosphorylated at Ser473
- PBST
Tween 20-PBS
- SDD
severe developmental delay
- Src
proto-oncogene tyrosine-protein kinase
- TEA
tetraethylammonium
- WT
wild type
AUTHOR CONTRIBUTIONS
W. Yu performed research and analyzed data; M. R. Shin performed research; and F. Sesti designed research, performed electrophysiological research, analyzed data, and wrote the manuscript.
REFERENCES
- 1.Kaczmarek L. K. (2006) Non-conducting functions of voltage-gated ion channels. Nat. Rev. Neurosci. 7, 761–771 [DOI] [PubMed] [Google Scholar]
- 2.Becchetti A., Pillozzi S., Morini R., Nesti E., Arcangeli A. (2010) New insights into the regulation of ion channels by integrins. Int. Rev. Cell Mol. Biol. 279, 135–190 [DOI] [PubMed] [Google Scholar]
- 3.Wei Y., Shin M. R., Sesti F. (2018) Oxidation of KCNB1 channels in the human brain and in mouse model of Alzheimer’s disease. Cell Death Dis. 9, 820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu W., Gowda M., Sharad Y., Singh S. A., Sesti F. (2017) Oxidation of KCNB1 potassium channels triggers apoptotic integrin signaling in the brain. Cell Death Dis. 8, e2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cotella D., Hernandez-Enriquez B., Wu X., Li R., Pan Z., Leveille J., Link C. D., Oddo S., Sesti F. (2012) Toxic role of K+ channel oxidation in mammalian brain. J. Neurosci. 32, 4133–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu W., Parakramaweera R., Teng S., Gowda M., Sharad Y., Thakker-Varia S., Alder J., Sesti F. (2016) Oxidation of KCNB1 potassium channels causes neurotoxicity and cognitive impairment in a mouse model of traumatic brain injury. J. Neurosci. 36, 11084–11096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu X., Hernandez-Enriquez B., Banas M., Xu R., Sesti F. (2013) Molecular mechanisms underlying the apoptotic effect of KCNB1 K+ channel oxidation. J. Biol. Chem. 288, 4128–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frisch S. M., Francis H. (1994) Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 124, 619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vicente-Manzanares M., Choi C. K., Horwitz A. R. (2009) Integrins in cell migration--the actin connection. J. Cell Sci. 122, 199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brakebusch C., Fässler R. (2003) The integrin-actin connection, an eternal love affair. EMBO J. 22, 2324–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marini C., Romoli M., Parrini E., Costa C., Mei D., Mari F., Parmeggiani L., Procopio E., Metitieri T., Cellini E., Virdò S., De Vita D., Gentile M., Prontera P., Calabresi P., Guerrini R. (2017) Clinical features and outcome of 6 new patients carrying de novo KCNB1 gene mutations. Neurol. Genet. 3, e206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calhoun J. D., Vanoye C. G., Kok F., George A. L., Jr., Kearney J. A. (2017) Characterization of a KCNB1 variant associated with autism, intellectual disability, and epilepsy. Neurol. Genet. 3, e198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Kovel C. G. F., Syrbe S., Brilstra E. H., Verbeek N., Kerr B., Dubbs H., Bayat A., Desai S., Naidu S., Srivastava S., Cagaylan H., Yis U., Saunders C., Rook M., Plugge S., Muhle H., Afawi Z., Klein K. M., Jayaraman V., Rajagopalan R., Goldberg E., Marsh E., Kessler S., Bergqvist C., Conlin L. K., Krok B. L., Thiffault I., Pendziwiat M., Helbig I., Polster T., Borggraefe I., Lemke J. R., van den Boogaardt M. J., Møller R. S., Koeleman B. P. C. (2017) Neurodevelopmental disorders caused by de novo variants in KCNB1 genotypes and phenotypes. JAMA Neurol. 74, 1228–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thiffault I., Speca D. J., Austin D. C., Cobb M. M., Eum K. S., Safina N. P., Grote L., Farrow E. G., Miller N., Soden S., Kingsmore S. F., Trimmer J. S., Saunders C. J., Sack J. T. (2015) A novel epileptic encephalopathy mutation in KCNB1 disrupts Kv2.1 ion selectivity, expression, and localization. J. Gen. Physiol. 146, 399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saitsu H., Akita T., Tohyama J., Goldberg-Stern H., Kobayashi Y., Cohen R., Kato M., Ohba C., Miyatake S., Tsurusaki Y., Nakashima M., Miyake N., Fukuda A., Matsumoto N. (2015) De novo KCNB1 mutations in infantile epilepsy inhibit repetitive neuronal firing. Sci. Rep. 5, 15199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torkamani A., Bersell K., Jorge B. S., Bjork R. L., Jr., Friedman J. R., Bloss C. S., Cohen J., Gupta S., Naidu S., Vanoye C. G., George A. L., Jr., Kearney J. A. (2014) De novo KCNB1 mutations in epileptic encephalopathy. Ann. Neurol. 76, 529–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Latypova X., Matsumoto N., Vinceslas-Muller C., Bézieau S., Isidor B., Miyake N. (2017) Novel KCNB1 mutation associated with non-syndromic intellectual disability. J. Hum. Genet. 62, 569–573; erratum: 585 [DOI] [PubMed] [Google Scholar]
- 18.Riedl J., Crevenna A. H., Kessenbrock K., Yu J. H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T. A., Werb Z., Sixt M., Wedlich-Soldner R. (2008) Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5, 605–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malin S. A., Nerbonne J. M. (2002) Delayed rectifier K+ currents, IK, are encoded by Kv2 alpha-subunits and regulate tonic firing in mammalian sympathetic neurons. J. Neurosci. 22, 10094–10105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kapp T. G., Rechenmacher F., Neubauer S., Maltsev O. V., Cavalcanti-Adam E. A., Zarka R., Reuning U., Notni J., Wester H. J., Mas-Moruno C., Spatz J., Geiger B., Kessler H. (2017) A comprehensive evaluation of the activity and selectivity profile of ligands for RGD-binding integrins. Sci. Rep. 7, 39805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He K., McCord M. C., Hartnett K. A., Aizenman E. (2015) Regulation of pro-apoptotic phosphorylation of Kv2.1 K+ channels. PLoS One 10, e0129498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lenaeus M. J., Vamvouka M., Focia P. J., Gross A. (2005) Structural basis of TEA blockade in a model potassium channel. Nat. Struct. Mol. Biol. 12, 454–459 [DOI] [PubMed] [Google Scholar]
- 23.Bianchi L., Kwok S. M., Driscoll M., Sesti F. (2003) A potassium channel-MiRP complex controls neurosensory function in Caenorhabditis elegans. J. Biol. Chem. 278, 12415–12424 [DOI] [PubMed] [Google Scholar]
- 24.Cai S. Q., Wang Y., Park K. H., Tong X., Pan Z., Sesti F. (2009) Auto-phosphorylation of a voltage-gated K+ channel controls non-associative learning. EMBO J. 28, 1601–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song G., Ouyang G., Bao S. (2005) The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 9, 59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frisch S. M., Vuori K., Ruoslahti E., Chan-Hui P. Y. (1996) Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 134, 793–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ilić D., Almeida E. A., Schlaepfer D. D., Dazin P., Aizawa S., Damsky C. H. (1998) Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 143, 547–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang T., Qiu Y. (2003) Interaction between Src and a C-terminal proline-rich motif of Akt is required for Akt activation. J. Biol. Chem. 278, 15789–15793 [DOI] [PubMed] [Google Scholar]
- 29.Datta S. R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M. E. (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241 [DOI] [PubMed] [Google Scholar]
- 30.Da Silva J. S., Dotti C. G. (2002) Breaking the neuronal sphere: regulation of the actin cytoskeleton in neuritogenesis. Nat. Rev. Neurosci. 3, 694–704 [DOI] [PubMed] [Google Scholar]
- 31.Bentley D., Toroian-Raymond A. (1986) Disoriented pathfinding by pioneer neurone growth cones deprived of filopodia by cytochalasin treatment. Nature 323, 712–715 [DOI] [PubMed] [Google Scholar]
- 32.Forscher P., Smith S. J. (1988) Actions of cytochalasins on the organization of actin filaments and microtubules in a neuronal growth cone. J. Cell Biol. 107, 1505–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin C. H., Forscher P. (1993) Cytoskeletal remodeling during growth cone-target interactions. J. Cell Biol. 121, 1369–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Connor T. P., Bentley D. (1993) Accumulation of actin in subsets of pioneer growth cone filopodia in response to neural and epithelial guidance cues in situ. J. Cell Biol. 123, 935–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meberg P. J., Bamburg J. R. (2000) Increase in neurite outgrowth mediated by overexpression of actin depolymerizing factor. J. Neurosci. 20, 2459–2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leung Y. M., Huang C. F., Chao C. C., Lu D. Y., Kuo C. S., Cheng T. H., Chang L. Y., Chou C. H. (2011) Voltage-gated K+ channels play a role in cAMP-stimulated neuritogenesis in mouse neuroblastoma N2A cells. J. Cell. Physiol. 226, 1090–1098 [DOI] [PubMed] [Google Scholar]
- 37.Liang C. C., Park A. Y., Guan J. L. (2007) In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2, 329–333 [DOI] [PubMed] [Google Scholar]
- 38.Wei J. F., Wei L., Zhou X., Lu Z. Y., Francis K., Hu X. Y., Liu Y., Xiong W. C., Zhang X., Banik N. L., Zheng S. S., Yu S. P. (2008) Formation of Kv2.1-FAK complex as a mechanism of FAK activation, cell polarization and enhanced motility. J. Cell. Physiol. 217, 544–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Kovel C. G., Brilstra E. H., van Kempen M. J., Van’t Slot R., Nijman I. J., Afawi Z., De Jonghe P., Djémié T., Guerrini R., Hardies K., Helbig I., Hendrickx R., Kanaan M., Kramer U., Lehesjoki A. E., Lemke J. R., Marini C., Mei D., Møller R. S., Pendziwiat M., Stamberger H., Suls A., Weckhuysen S., Koeleman B. P.; EuroEPINOMICS RES Consortium (2016) Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients. Mol. Genet. Genomic Med. 4, 568–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campbell P. M., Der C. J. (2004) Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol. 14, 105–114 [DOI] [PubMed] [Google Scholar]
- 41.Cox A. D., Der C. J. (2003) The dark side of Ras: regulation of apoptosis. Oncogene 22, 8999–9006 [DOI] [PubMed] [Google Scholar]
- 42.Yu W., Zhang H., Shin M. R., Sesti F. (2019) Oxidation of KCNB1 potassium channels in the murine brain during aging is associated with cognitive impairment. Biochem. Biophys. Res. Commun. 512, 665–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D’Adamo M. C., Catacuzzeno L., Di Giovanni G., Franciolini F., Pessia M. (2013) K(+) channelepsy: progress in the neurobiology of potassium channels and epilepsy. Front. Cell. Neurosci. 7, 134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parent J. M., Yu T. W., Leibowitz R. T., Geschwind D. H., Sloviter R. S., Lowenstein D. H. (1997) Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J. Neurosci. 17, 3727–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scott B. W., Wojtowicz J. M., Burnham W. M. (2000) Neurogenesis in the dentate gyrus of the rat following electroconvulsive shock seizures. Exp. Neurol. 165, 231–236 [DOI] [PubMed] [Google Scholar]
- 46.Rubboli G., Plazzi G., Picard F., Nobili L., Hirsch E., Chelly J., Prayson R. A., Boutonnat J., Bramerio M., Kahane P., Dibbens L. M., Gardella E., Baulac S., Møller R. S. (2018) Mild malformations of cortical development in sleep-related hypermotor epilepsy due to KCNT1 mutations. Ann. Clin. Transl. Neurol. 6, 386–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y., Zhang X. F., Fleming M. R., Amiri A., El-Hassar L., Surguchev A. A., Hyland C., Jenkins D. P., Desai R., Brown M. R., Gazula V. R., Waters M. F., Large C. H., Horvath T. L., Navaratnam D., Vaccarino F. M., Forscher P., Kaczmarek L. K. (2016) Kv3.3 channels bind Hax-1 and Arp2/3 to assemble a stable local actin network that regulates channel gating. Cell 165, 434–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hwang J. U., Suh S., Yi H., Kim J., Lee Y. (1997) Actin filaments modulate both stomatal opening and inward K+-channel activities in guard cells of Vicia faba L. Plant Physiol. 115, 335–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furukawa T., Yamane T., Terai T., Katayama Y., Hiraoka M. (1996) Functional linkage of the cardiac ATP-sensitive K+ channel to the actin cytoskeleton. Pflugers Arch. 431, 504–512 [DOI] [PubMed] [Google Scholar]
- 50.Nakahira K., Matos M. F., Trimmer J. S. (1998) Differential interaction of voltage-gated K+ channel beta-subunits with cytoskeleton is mediated by unique amino terminal domains. J. Mol. Neurosci. 11, 199–208 [DOI] [PubMed] [Google Scholar]
- 51.Huang H., Rao Y., Sun P., Gong L. W. (2002) Involvement of actin cytoskeleton in modulation of Ca(2+)-activated K(+) channels from rat hippocampal CA1 pyramidal neurons. Neurosci. Lett. 332, 141–145 [DOI] [PubMed] [Google Scholar]
- 52.Piao L., Ho W. K., Earm Y. E. (2003) Actin filaments regulate the stretch sensitivity of large-conductance, Ca2+-activated K+ channels in coronary artery smooth muscle cells. Pflugers Arch. 446, 523–528 [DOI] [PubMed] [Google Scholar]
- 53.Kim E. Y., Suh J. M., Chiu Y. H., Dryer S. E. (2010) Regulation of podocyte BK(Ca) channels by synaptopodin, Rho, and actin microfilaments. Am. J. Physiol. Renal Physiol. 299, F594–F604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brady P. A., Alekseev A. E., Aleksandrova L. A., Gomez L. A., Terzic A. (1996) A disrupter of actin microfilaments impairs sulfonylurea-inhibitory gating of cardiac KATP channels. Am. J. Physiol. 271, H2710–H2716 [DOI] [PubMed] [Google Scholar]
- 55.Yokoshiki H., Katsube Y., Sunugawa M., Seki T., Sperelakis N. (1997) Disruption of actin cytoskeleton attenuates sulfonylurea inhibition of cardiac ATP-sensitive K+ channels. Pflugers Arch. 434, 203–205 [DOI] [PubMed] [Google Scholar]
- 56.Ehrhardt A. G., Frankish N., Isenberg G. (1996) A large-conductance K+ channel that is inhibited by the cytoskeleton in the smooth muscle cell line DDT1 MF-2. J. Physiol. 496, 663–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delgado-Ramirez M., Rodriguez-Menchaca A. A. (2019) Cytoskeleton disruption affects Kv2.1 channel function and its modulation by PIP2. J. Physiol. Sci. 69, 513–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Malley D., Irving A. J., Harvey J. (2005) Leptin-induced dynamic alterations in the actin cytoskeleton mediate the activation and synaptic clustering of BK channels. FASEB J. 19, 1917–1919 [DOI] [PubMed] [Google Scholar]
- 59.Piwkowska A., Rogacka D., Audzeyenka I., Kasztan M., Angielski S., Jankowski M. (2015) Insulin increases glomerular filtration barrier permeability through PKGIα-dependent mobilization of BKCa channels in cultured rat podocytes. Biochim. Biophys. Acta 1852, 1599–1609 [DOI] [PubMed] [Google Scholar]
- 60.Lauritzen I., Chemin J., Honoré E., Jodar M., Guy N., Lazdunski M., Jane Patel A. (2005) Cross-talk between the mechano-gated K2P channel TREK-1 and the actin cytoskeleton. EMBO Rep. 6, 642–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maiya R., Ponomarev I., Linse K. D., Harris R. A., Mayfield R. D. (2007) Defining the dopamine transporter proteome by convergent biochemical and in silico analyses. Genes Brain Behav. 6, 97–106 [DOI] [PubMed] [Google Scholar]
- 62.Webb D. J., Zhang H., Majumdar D., Horwitz A. F. (2007) alpha5 integrin signaling regulates the formation of spines and synapses in hippocampal neurons. J. Biol. Chem. 282, 6929–6935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shi Y., Ethell I. M. (2006) Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J. Neurosci. 26, 1813–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kumar V., Zhang M. X., Swank M. W., Kunz J., Wu G. Y. (2005) Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 25, 11288–11299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Greengard P., Benfenati F., Valtorta F. (1994) Synapsin I, an actin-binding protein regulating synaptic vesicle traffic in the nerve terminal. Adv. Second Messenger Phosphoprotein Res. 29, 31–45 [DOI] [PubMed] [Google Scholar]
- 66.Bloom O., Evergren E., Tomilin N., Kjaerulff O., Löw P., Brodin L., Pieribone V. A., Greengard P., Shupliakov O. (2003) Colocalization of synapsin and actin during synaptic vesicle recycling. J. Cell Biol. 161, 737–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Akama K. T., McEwen B. S. (2003) Estrogen stimulates postsynaptic density-95 rapid protein synthesis via the Akt/protein kinase B pathway. J. Neurosci. 23, 2333–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lynch M. A. (2004) Long-term potentiation and memory. Physiol. Rev. 84, 87–136 [DOI] [PubMed] [Google Scholar]
- 69.Fukazawa Y., Saitoh Y., Ozawa F., Ohta Y., Mizuno K., Inokuchi K. (2003) Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 38, 447–460 [DOI] [PubMed] [Google Scholar]
- 70.Bramham C. R. (2008) Local protein synthesis, actin dynamics, and LTP consolidation. Curr. Opin. Neurobiol. 18, 524–531 [DOI] [PubMed] [Google Scholar]
- 71.Du J., Tao-Cheng J. H., Zerfas P., McBain C. J. (1998) The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience 84, 37–48 [DOI] [PubMed] [Google Scholar]