

Abstract
Kidney fibrosis occurs in almost every type of chronic kidney disease. We found that microRNA (miR)-26a was decreased in the kidney, muscle, and exosomes of unilateral ureteral obstruction (UUO) mice. We hypothesized that exogenous miR-26 could suppresses renal fibrosis and muscle wasting in obstructive kidney disease. For this purpose, we generated exosomes that encapsulated miR-26, then injected these into skeletal muscle of UUO mice. The expression of miR-26a was elevated in serum exosomes from UUO mice following exosome–miR-26a injection. In these mice, muscle wasting has been ameliorated as evidenced by increased muscle weights. In addition, a muscle atrophy marker, myostatin, is increased in UUO muscle; provision of miR-26a abolished this increase. We detected a remote effect of exosomes containing miR-26a in UUO-induced renal fibrosis. The intervention of miR-26a attenuated UUO-induced renal fibrosis as determined by immunohistological assessment of α-smooth muscle actin and Masson’s trichrome staining. Furthermore, exogenous miR-26a decreased the protein levels of 2 profibrosis proteins, connective tissue growth factor (CTGF) and TGF-β1, in UUO kidney. Our data showed that exosomes containing miR-26a prevented muscle atrophy by inhibiting the transcription factor forkhead box O1. Likewise, the exosome-carried miR-26a limited renal fibrosis by directly suppressing CTGF. Our findings provide an experimental basis for exosome-mediated therapy of muscle atrophy and renal fibrosis.—Zhang, A., Wang, H., Wang, B., Yuan, Y., Klein, J. D., Wang, X. H. Exogenous miR-26a suppresses muscle wasting and renal fibrosis in obstructive kidney disease.
Keywords: exosome, UUO, muscle atrophy, CTGF, TGF-β1
Chronic kidney disease (CKD) is an escalating health problem with a high economic burden (1). Renal fibrosis represents a common pathway underlying the progression of CKD that finally leads to end-stage renal disease. Unilateral ureteral obstruction (UUO) has been used extensively as a representative model of renal fibrosis. It has the advantage of inducing a rapid development of renal fibrosis while also mimicking cellular processes in the progression of interstitial fibrosis (2). Furthermore, muscle wasting, or atrophy, defined as a decrease in the muscle mass accompanied by loss of muscle strength, is frequently seen in patients with CKD and is associated with a poor prognosis (3). A useful marker for muscle atrophy is myostatin protein production (4, 5). Myostatin is a member of the TGF-β superfamily of secreted proteins and functions as a negative regulator of muscle mass (6). However, at present, there is no single therapy that treats both muscle wasting and kidney fibrosis in CKD (7).
MicroRNAs (miRs) are important regulators of gene expression that function by targeting mRNAs that hinder translation or enhance mRNA degradation (8). Multiple studies have demonstrated that miRs play critical roles in the regulation of many biologic processes, including renal fibrosis and muscle atrophy (9–11). A number of miRs have been shown to influence renal fibrotic processes; these include miR-21, miR-26, miR-141, miR-29, and miR-200 (12, 13). Among the miRs investigated, miR-26 has been reported to exert an antifibrotic effect by inhibiting proliferation, differentiation, and collagen secretion in lung fibroblasts. This is accomplished by directly targeting connective tissue growth factor (CTGF) (14), a key mediator of tissue fibrosis (15). Previous studies have demonstrated that F-box protein 32 (atrogin-1) and tripartite motif containing 63 (muscle ring finger 1) are the transcriptional targets of forkhead box O (FoxO) (16, 17). FoxO depletion in skeletal muscles reduces the muscle protein loss and muscle functional decline that results from fasting and denervation (18, 19). FoxO1, which has been shown to serve a critical role in CKD-induced muscle atrophy, can be regulated by several miRs (8, 11). Our laboratory recently reported that FoxO1 was a target of miR-26 (20). However, the effect of miR-26 in UUO-induced renal fibrosis and muscle atrophy is still unproven. We will provide data to clarify this relationship in this paper.
Exosomes are small (30–100 nm in diameter) extracellular, membrane-enclosed vesicles released by all cell types and found in all bodily fluids, including blood, saliva, and urine (21). Exosomes deliver growth factors, proteins, miR, mRNA, noncoding RNA, and lipids throughout the body (9, 10, 22). Because of this capability, they play an important role in intercellular communication. Exosomally transferred miRs are regarded as novel regulators in cellular function (9, 10, 20). It has been reported that exosomes from mesenchymal stem cells are able to selectively transfer miR-let7c to damaged kidney cells, resulting in attenuated renal fibrosis (23). In addition, exosomes from human endothelial cells have been shown to transfer miR-486 to reduce ischemic kidney injury (24).
In the present study, we hypothesized that in a UUO mouse model, exogenous delivery of exosomes containing miR-26a to muscle could inhibit muscle atrophy by targeting FoxO1 and also move to the kidney, where they could down-regulate CTGF and ameliorate renal fibrosis. The results of the present study suggest a basis for the use of exosomes for the treatment of renal fibrosis and muscle atrophy in a clinical setting.
MATERIALS AND METHODS
Animal model
Male C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Mice were maintained at 19–21°C on a 12-h light/dark cycle, were fed a standard rodent diet, and were allowed free access to drinking water. UUO was induced as previously described in ref. 10. Briefly, under general anesthesia, the left ureter was ligated at the ureter-pelvic junction with 4-0 silk through a left flank incision. Kidney tissue was harvested 14 d after UUO or sham surgery. Exosomes with miR-26a harvested from conditioned medium of cultured human embryonic kidney 293 (HEK293) cells were injected into tibialis anterior (TA) muscle of mice on the day the UUO model was established.
Generation of exosomes with high levels of miR-26a from HEK293 cells and cell culture
The gene for miR-26a precursor was cloned into a pLamp2 vector, which contains an exosomal membrane protein gene, lysosomal-associated membrane protein 2b (Lamp2b). HEK293 cells were grown to 60% confluence in DMEM containing 10% fetal bovine serum (25). To create the exosome-generating cell line, we transfected the Lamp2b–miR-26a-5p vector into HEK293 cells using the Effectene transfection reagent (Qiagen, Germantown, MD, USA). In parallel, we prepared a control cell line that was transfected with Lamp2b-Flag. We used G418 (Geneticin; Thermo Fisher Scientific, Waltham, MA, USA) to select for successfully transfected cells. To harvest exosomes, we exchanged growth medium with exosome-free medium and allowed exosome secretion for 48 h. Finally, the miR-26a–enriched exosomes and control (Flag-carrying) exosomes were isolated from the conditioned medium using a series of centrifugations and resuspended in PBS. The size and concentration of the exosomes were analyzed using NanoSight (Malvern Panalytical, Malvern, United Kingdom) instrument measurement and electron microscopy (20). The identity of the exosomes was confirmed by showing exosomal marker protein TSG101 by Western blot (9). Exosomes were labeled with a fluorescent tag DiR (1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide; D12731; Thermo Fisher Scientific, USA). Exosome distribution images were taken with the Bruker small animal optical imaging system (In Vivo Xtreme II; Bruker, Billerica, MA, USA). To determine if the mice responded to the exosome injection alone, we injected exosome-carried Flag into TA muscle of normal mice, isolated protein from skeletal muscle and kidney of these mice and mice that were not injected, and compared the abundance of FoxO1, myostatin, TGF-β, and SMAD2 in these 2 groups of mice. There was no significant difference in protein abundances between exosome-carried Flag–injected and noninjected normal mice (Supplemental Fig. S1). Primary cultures of muscle satellite cells were prepared using a Skeletal Muscle Dissociation Kit (130-098-305, Macs; Miltenyi Biotec, Bergisch Gladbach, Germany) and a Satellite Cell Isolation Kit (130-104-267, Macs; Miltenyi Biotec) according to the manufacturer’s directions. The detailed procedure is as previously described in Hu et al. (26).
Real-time quantitative PCR
To measure miR, total RNA was extracted using Tri-Reagent (Molecular Research Center, Cincinnati, OH, USA). To synthesize cDNA, 10 ng of total extracted RNA with verified small RNA content was reverse transcribed using the NCode miR cDNA Synthesis Kit (Qiagen). The expression of miR was measured as described in Su et al. (19). Primers were purchased from Qiagen. The mouse U6 gene was used as the standard for evaluation of the tissue expression of individual miR. miR-103 was used to evaluate serum miR levels. The response or change in expression levels of individual miRs was calculated as the difference between the threshold values of genes of sham-treated mice and UUO mice (ΔΔCq) (27, 28).
miRNA library preparation and sequencing was performed by the Genomics Core of Yerkes National Primate Research Center (Emory University). Qualitative and quantitative analyses of the total RNA were performed using the Thermo Nanodrop 2000 (Thermo Fisher Scientific) and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), respectively. Small RNA libraries were prepared using the SeqMatic Tailormix miR Sample Preparation Kit (SeqMatic, Fremont, CA, USA) according to the manufacturer’s instructions. Briefly, 100 ng of total RNA was used for library preparation. Small RNAs were ligated with Illumina-compatible adapters, and each sample was tagged with a unique barcode to allow multiplexing. The adapter-ligated libraries were then enriched using PCR amplification followed by gel enrichment for mature miR library. The amplified library was validated using a High Sensitivity DNA chip on the Agilent Bioanalyzer. The libraries were further quantified on Qubit 2.0 Fluorometer (Thermo Fisher Scientific) using the High Sensitivity dsDNA Assay. Libraries from all the samples were multiplexed and run in a single lane of Illumina 3K Flowcell (Illumina, San Diego, CA, USA). PhiX was used as an internal control on each lane to monitor the error statistics. Cluster generation was performed on the v3 flowcell on the Illumina cBot. The clustered flowcell was sequenced on the Illumina HiSeq3000 System as a 100-cycle single-read multiplexed run.
Western blotting and antibodies
Western blot was performed as previously described in Wang et al. (29). The total proteins were isolated from frozen muscle or kidney tissue specimens using the gentle lysis buffer (10 mM Tris-HCl, 10 mM NaCl, 2 mM EDTA, 0.5% NP-40, 1% glycerol; fresh added: 1 mM Na3VO4, 10 μg/ml PMSF, 5 μg/ml aprotinin, 1 μg/ml leupeptin) with phosphatase inhibitors cocktail 1 and 2 (MilliporeSigma, Burlington, MA, USA). Equal amounts of proteins were separated by SDS-PAGE and transferred to a PVDF membrane. Immunoblotting was performed with the following primary antibodies: anti–TGF-β1 (ab179695) and anti–growth differentiation factor 8 (myostatin, ab71808) from Abcam (Cambridge, MA, USA); anti-FoxO1 (C29H4), anti–glycogen synthase kinase (GSK; D5C5Z), anti-Smad2 (D43B4), and anti–glyceraldehyde 3-phosphate dehydrogenase (GAPDH; D16H11) antibodies Cell Signaling Technology (Danvers, MA, USA); and anti-CTGF (L-20) from Santa Cruz Biotechology (Dallas, TX, USA). The secondary antibodies that we used were Alexa Fluor 680 goat anti-rabbit IgG or goat anti-mouse IgG from Thermo Fisher Scientific. Protein bands were scanned and quantified using the Li-Cor Odyssey Infrared Scanning System (Li-Cor Biosciences, Lincoln, NE, USA).
Kidney histology, immunofluorescence, and Masson’s trichrome staining
Kidneys were fixed in 3.7% formaldehyde-PBS (pH 7.4) and dehydrated, paraffin embedded, and sectioned. Masson’s trichromatic staining was performed with a Masson Modified International Medical Equipment (IMEB) Stain Kit (K7298, IMEB, San Marcos, CA, USA). Images were visualized with an Olympus 1X 51 inverted fluorescence microscope and captured with an Olympus DP73-1-51-17MP Color Camera (Olympus, Tokyo, Japan) (9). Collagen (blue color) in kidney was measured using the CellSens Dimension 1.9 Software (Olympus), and color density was calculated as the mean from 10 individual fields. The kidneys used for immunofluorescence analysis were fixed for 15 min in 4% paraformaldehyde on ice, rinsed once in PBS for 10 min, and embedded in tissue-freezing medium (H-TFM; Thermo Fisher Scientific). Frozen sections were incubated with anti–α-smooth muscle actin (α-SMA, A2547) and anti-fibronectin (F3648), which were obtained from MilliporeSigma.
Luciferase reporter assay and transfection
Because CTGF was a predicted target of miR-26a, (TargetScan: http://www.targetscan.org/vert_72/; PITA: https://genie.weizmann.ac.il/pubs/mir07/mir07_data.html and miRanda http://www.microrna.org/microrna/home.do), the luciferase reporter constructs in which the luciferase coding sequence was fused to the 3′-UTRs of CTGF (pLuc.miR-26a–CTGF-3′UTR) were generated by the Emory Integrated Genomics Core. The transfection procedure was as previously described in ref. 9. The firefly and Renilla luciferase activities were measured consecutively by dual-luciferase assays (Promega, Madison, WI, USA) using a TD-20/20 Luminometer (Turner Designs, Sunnyvale, CA, USA). We calculated the results as the ratio of firefly luciferase to Renilla luciferase.
Statistical analysis
All values are expressed as means ± sem. For statistical significance, to identify significant differences between 2 groups, comparisons were made by using a Student’s t test. Multiple comparisons among 3 or more groups were performed using 1-way ANOVA followed by Bonferroni’s multiple comparison when appropriate. A value of P < 0.05 was considered to be statistically significant.
RESULTS
The expression of miR-26 was decreased in the serum exosomes of UUO mice
To identify therapeutic and diagnostic markers, we analyzed serum exosomes from UUO mice and sham-treated mice by NanoSight instrument measurement. The size of the exosomes in UUO mice was larger than in sham-treated mice 7 d after UUO surgery; however, the concentration of exosomes was significantly lower in UUO mice vs. sham-treated mice (Fig. 1A). miR deep sequencing was performed in serum exosomes isolated from sham-treated control mice or UUO mice 28 d after surgery. We found that 51 miRs were significantly altered in exosomes of UUO mice vs. sham-treated mice (Fig. 2). A decrease in miR-26a-5p and miR-26b-5p particularly caught our attention because we previously saw that these miRs were decreased in skeletal and cardiac muscle of CKD mice (20). Using quantitative PCR (qPCR), we confirmed that miR-26a-5p was 51% decreased and that miR-26b-5p was 18% decreased in the exosomes from UUO mice compared with sham-treated mice (Fig. 1B). To verify whether decreased miR-26 affected translation of targeted proteins (20), Western blot was used to test GSK-3β, FoxO1, and CTGF, and we found that these 3 proteins were significantly increased in exosomes of UUO mice compared with levels in sham-treated mice (Fig. 1C). TGF-β1 was also higher in UUO exosomes. Because GSK-3β and FoxO1 are related to insulin resistance and CTGF and TGF-β1 are related to tissue fibrosis, these results suggest that a decrease in miR-26a in the circulating exosomes of UUO mice could have multiorgan impact.
Figure 1.

The expression of miR-26 was decreased in serum exosomes of UUO mice. A) Exosomes were isolated from the serum of UUO mice and sham-treated mice. The NanoSight instrument was used to measure the size and concentration of exosomes. The top image shows the size (x axis: diameter in nanometers) and concentration (y axis: particles/ml) of exosomes. The bar graph compares these 2 parameters in control vs. UUO exosomes (bars: means ± se; n = 3/group). *P < 0.05 vs. sham-treated mice. B) Total RNA was extracted from serum exosomes of UUO mice or sham-treated mice. The expression of miR-26a-5p and miR-26b-5p were assayed by real-time qPCR. The bar graph shows miR from the exosomes of UUO mice compared with levels in sham-treated mice mice (represented by 1-fold). Results are normalized to miR-103 (bars: means ± se; n = 6/group). *P < 0.05 vs. sham-treated mice. C) Serum exosomes were boiled with Western blot loading buffer; 30 μg protein was used for loading SDS-PAGE gel. The proteins GSK-3β, FoxO1, TGF-β1, and CTGF were measured by Western blotting in UUO mice and sham-treated mice. The bottom point graphs show the change of each protein band normalized to GAPDH (data represent means ± se; n = 6/group). #P < 0.05 vs. sham-treated mice.
Figure 2.
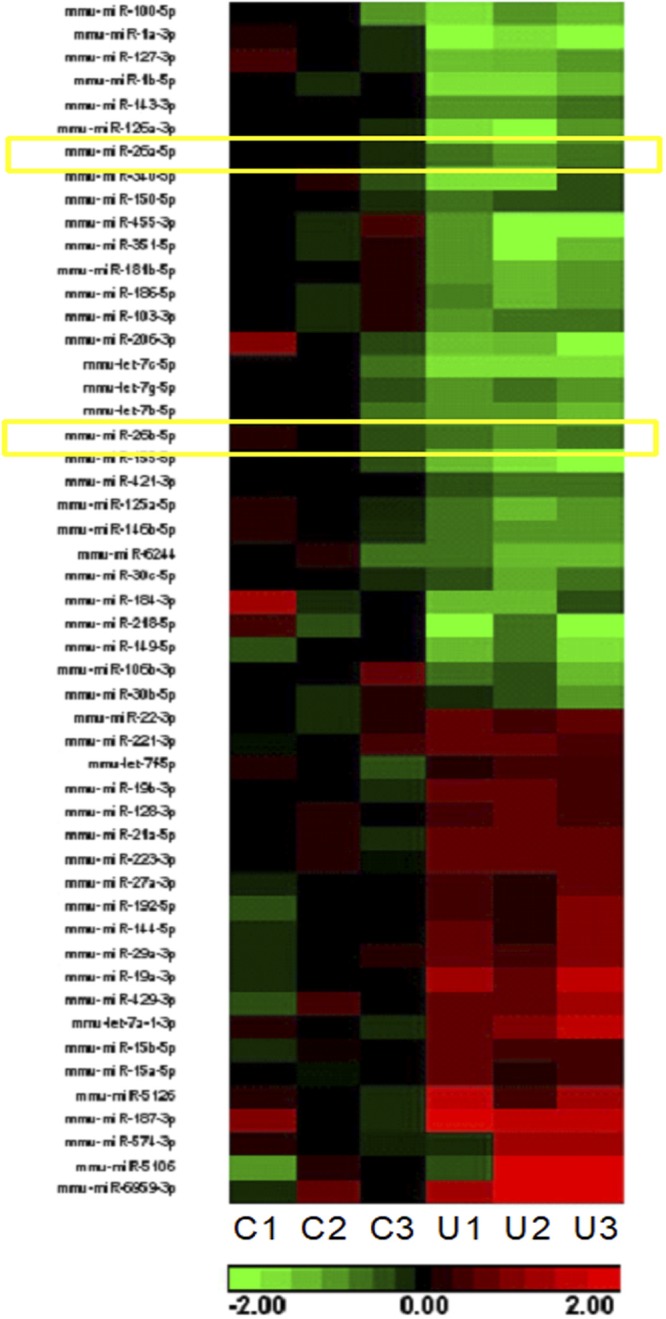
Fifty-one miRs are altered in the serum exosomes from UUO mice. Total RNA was isolated from serum exosomes of UUO mice and sham-treated mice. Small RNA libraries were prepared using the SeqMatic tailormix miR Sample Preparation Kit. miR deep sequencing was performed. The heat map showed that there are 51 miRs that are altered in the serum exosomes from UUO mice. Left 3 columns (C1–C3) show levels in sham-treated mice; right 3 columns (U1–U3) show levels in UUO mice. Green indicates decrease; red indicates increase. The yellow squares indicate the change of miR-26a and miR-26b.
The expression of miR-26 was decreased in skeletal muscle and kidney of UUO mice
To identify possible impacts resulting from the decrease in miR-26a revealed by the deep sequencing, we isolated RNA from skeletal muscle and kidney. We found the expression of miR-26a-5p also significantly decreased in the muscle and kidney of UUO mice at 28 d (Fig. 3A, B). These results were further confirmed in a cell culture model. We treated skeletal muscle satellite cells with exosomes (100 μg/ml) isolated from sham-treated mice and UUO mice. We found that UUO exosome treatment resulted in 20% lower miR-26a-5p expression than control exosome-treated skeletal muscle satellite cells (Fig. 3C). Provision of exosomes of UUO mice also reduced miR-26a-5p expression by 37% vs. levels in sham-treated mice in kidney HEK293 cells (Fig. 3D). We evaluated the impact of the lower miR-26a levels on transcription factors related to muscle protein metabolism in the cultured muscle satellite cells by Western blots. UUO exosomes increased FoxO1 and SMAD2 protein amounts over control levels. Myostatin, a protein that promotes muscle catabolism, also significantly increased (Fig. 3E). Treatment of HEK293 cells with UUO exosomes increased the prefibrotic protein CTGF. The transcription factors FoxO1 and SMAD2 were also increased (Fig. 3F). These data suggested that UUO exosomes, possibly through a decrease in miR-26a, up-regulate protein catabolic and fibrosis signaling in cultured skeletal muscle and kidney cells.
Figure 3.

The expression of miR-26 was decreased in skeletal muscle and kidney of UUO mice. A, B) Total RNA was extracted from skeletal muscle (A) and kidney (B) of UUO mice and sham-treated mice. The expression of miR-26a-5p was assayed by real-time qPCR at 28 d after UUO surgery. The bar graph shows miR from the gastrocnemius muscle (A) or whole kidney (B) of UUO mice compared with levels in sham-treated mice (defined as 1-fold). Results are normalized to U6 (bars: means ± se; n = 6/group). #P < 0.05 vs. sham-treated mice. C, D) Total RNA was extracted from satellite cells (C) or HEK293 cells (D) that were treated with exosomes isolated from UUO serum (100 μg/ml) for 24 h. The expression of miR-26a-5p in the cell extracts was assayed by real-time qPCR. The bar graph shows miR-26a-5p from the UUO serum-treated group compared with the level in the control serum-treated group (represented by 1-fold). Results are normalized to U6 (bars: means ± se; n = 8/group). #P < 0.05 vs. control serum. E) Satellite cells were treated for 24 h with 100 μg/ml exosomes isolated from UUO serum. Protein was isolated from the satellite cells. Myostatin, FoxO1, and SMAD2 in cell lysates were measured by Western blot. The point graphs show the change of each protein band normalized to GAPDH. Results are reported as means ± se; n = 8/group. #P < 0.05 vs. serum from sham-treated mice. F) HEK293 cells treated for 24 h with 100 μg/ml exosomes isolated from UUO serum. Protein was isolated from the HEK293 cells. CTGF, FoxO1, and SMAD2 in cell lysates were measured by Western blots. The point graphs show the change of each protein band normalized to GAPDH. Results are reported as means ± se; n = 8/group). #P < 0.05 vs. sham-treated mice. Ctrl, control.
Exogenous exosome–carried miR-26 suppressed UUO-induced muscle atrophy
Our previous publication proved that an increase in miR-26a attenuated muscle wasting in a mouse model of CKD (20). To hypothesize that miR-26 could benefit obstructive kidney disease, we generated a stable cell line that produces the exosome-carried miR-26 for treatment of muscle wasting and renal fibrosis. The gene for miR-26a precursor was cloned into a vector that contains the exosomal membrane protein gene Lamp2b. Because Lamp2b is ubiquitously expressed on the surface of exosomes, we anticipated that Lamp2b would bring the miR-26a into the exosome, then through the circulation to other organs, such as kidney. Exosomes harvested from conditioned medium of HEK293 cells, stably transfected with the Lamp2b–miR-26 vector, contained higher levels of miR-26a than exosomes from cells transfected with the Lamp2b-Flag vector (Fig. 4A). Then, harvested exosomes containing miR-26a were injected into TA muscle of UUO mice and sham-treated mice. As shown in Fig. 4B, miR-26a was decreased in the muscle of UUO mice compared with levels in sham-treated mice, and intramuscular injection of exosomes containing miR-26a reversed this decrease. The muscle atrophy marker myostatin was significantly increased in UUO muscle. Provision of miR-26a attenuated the UUO-induced increase in myostatin abundance (Fig. 4C). Increasing miR-26a also ablated the up-regulation of GSK-3β, FoxO1, and SMAD2 by UUO. These results suggested that exosome-delivered miR-26a could inhibit muscle atrophy by limiting myostatin and insulin resistance in the muscle of UUO mice.
Figure 4.

Exogenous exosome–carried miR-26 suppressed UUO-induced muscle atrophy. A) Exosomes were harvested from conditioned medium from established HEK293–miR-26a or HEK293-Flag cell lines. Total RNA was isolated from these 2 groups. The expression of miR-26a-5p in exosomes was assayed by real-time qPCR. The bar graph shows expression levels of miR-26a in the HEK293–miR-26a group compared with the level in the HEK293-Flag group (designated as 1-fold). Results are normalized to U6. Results are reported as means ± se; n = 6/group. *P < 0.001 vs. HEK293-Flag. B) Total RNA was isolated from the TA muscle of mice given a sham intramuscular injection with exosome-carried Flag (sham/F), mice given a sham treatment with exosome-carried miR-26a (sham/26), UUO mice treated with exosome-carried Flag (UUO/F), and UUO mice treated with exosome-carried miR-26 (UUO/26). The expression of miR-26a-5p was assayed by real-time qPCR. The bar graph shows miR-26a expression from each group compared with levels in the sham/F group (represented at 1-fold). Results are normalized to U6. Results are reported as means ± se; n = 6/group. *P < 0.05 vs. sham/F; #P < 0.05 vs. UUO/F. C) The proteins were isolated from the TA muscle of sham/F, sham/26, UUO/F, and UUO/26 mice. The proteins GSK-3β, FoxO1, myostatin, and SMAD2 were measured by Western blotting in different groups of mice. Top: representative immunoblots. Bottom: densitometry analysis. The point graphs show the change of each protein band normalized to GAPDH. Results reported as means ± se; n = 6/group. *P < 0.05 vs. sham/F; #P < 0.05 vs. UUO/F.
Exogenous exosome–carried miR-26 attenuated UUO-induced renal fibrosis
There is growing evidence that exosomes are crucial in cell-cell communication (30). Thus, we further determined whether intramuscular injection of exosome-carried miR-26a would transfer miR-26a to kidney. As in muscle, miR-26a expression was also decreased in kidney after UUO. Intramuscular injection of exosomes with high miR-26 content enhanced the expression of miR-26a in kidney (Fig. 5A). We then examined possible mechanisms for exosome-derived miR-26a effects in UUO-induced renal fibrosis. Masson’s trichrome staining revealed a significant reduction in collagen deposit in the UUO kidney after intramuscular injection of exosome-carried miR-26a, compared with injection of exosome-carried Flag (Fig. 5B). Consistent with this observation, the amounts of α-SMA, a myofibroblast marker, and fibronectin, a glycoprotein of the extracellular matrix, were sharply increased in UUO kidney (Fig. 5C, D). Provision of miR-26a limited these changes. These results strongly suggested that exosome-derived miR-26a protected against renal fibrosis in UUO mice.
Figure 5.

Exogenous exosome–carried miR-26 attenuated UUO-induced renal fibrosis by limiting CTGF. A) Total RNA was isolated from the whole kidney of mice given a sham intramuscular injection with exosome-carried Flag (sham/F), mice given a sham treatment with exosome-carried miR-26a (sham/26), UUO mice treated with exosome-carried Flag (UUO/F), and UUO mice treated with exosome-carried miR-26 (UUO/26). The expression of miR-26a-5p was assayed by real-time qPCR. The bar graph shows miR-26a expression from each group compared with levels in sham with sham/F group (represented at 1-fold). Results are normalized to U6. Results are reported as means ± se; n = 6/group. *P < 0.05 vs. sham with sham/F; #P < 0.05 vs. UUO with UUO/F. B) Shown are representative Masson’s trichrome staining of paraffin sections from the kidney of mice given a sham intramuscular injection with exosome-carried Flag (sham/F or sham/Ex-Flag), mice given a sham treatment with exosome-carried miR-26a (sham/26 or sham/Ex-miR26), UUO mice treated with exosome-carried Flag (UUO/F or UUO/Ex-Flag), and UUO mice treated with exosome-carried miR-26 (UUO/26 or UUO/Ex-miR-26) (original magnification, ×200). The bar graph shows collagen amount in Masson’s trichrome staining (bars: means ± se; n = 6/group). *P < 0.05 vs. sham/F; #P < 0.05 vs. UUO/F. C) Representative cryosections of kidney were immunostained with α-SMA antibody in 4 different treatment groups (original magnification, ×200). D) Representative cryosections of kidney were immunostained with fibronectin antibody in 4 different treatment groups to detect interstitial fibrosis (original magnification, ×200).
Exosomes originating at the muscle can target the kidney
Previous studies have shown that miR-26a negatively regulated CTGF at the posttranscriptional level in vivo and in vitro (20, 31). We investigated whether miR-26a inhibits the CTGF protein level in our UUO mice. As shown in Fig. 6A, exosome-derived miR-26a significantly reduced CTGF protein abundance. It also reduced the increase in TGF-β and SMAD2 that is seen in the UUO mice. Taken together, these results indicated that exosome-derived miR-26a might inhibit renal fibrosis by targeting CTGF and limiting TGF-β expression in UUO. To trace the exosome-carried miR-26 in vivo, we labeled exosome-carried miR-26a with fluorescent lipophilic DiR, then injected into TA muscles of sham-treated mice and UUO mice. Twenty-eight days after injection, the intensity of fluorescence was still highly increased in the injected muscle. Most interestingly, when UUO mice were injected intramuscularly with exosome-carried miR-26a, fluorescent intensity was greater in the UUO kidney compared with the unobstructed, contralateral kidney (Fig. 6B). Minimal and background fluorescence was seen in other organs. To rule out the possibility that nonspecific spreading of the dye was responsible for the increased fluorescence in the kidney, we injected the fluorescent lipophilic DiR without exosome-carried miR-26a into TA muscle and looked for fluorescence throughout the body. Fluorescence was detected in muscle at 7 d and persisted up to 21 d after DiR injection. Other organs examined both in vivo and ex vivo showed no fluorescence signal resulting from injection of fluorescent lipophilic DiR (Supplemental Fig. S2). Because CTGF was predicted as a target of miR-26a-5p by in silico search we performed a luciferase reporter analysis in cultured HEK293 cells using a reporter construct (pLuc.miR-26a–CTGF-3′UTR) in which the luciferase coding sequence was fused to the 3′-UTRs (position 571–577 nt) of mouse CTGF. The luciferase activity was markedly repressed by miR-26a in cells transfected with pLuc.miR-26a–CTGF-3′UTR. Mutation of the miR-26a target sites abrogated miR-26a–induced repression of luciferase activity (Fig. 6C). To further confirm that miR-26a inhibits CTGF, we transfected miR-26a mimic into HEK293 cells and found the amount of CTGF protein was decreased (Fig. 6D). These data suggest that CTGF is a direct target of miR-26a.
Figure 6.

Exosomes originating at the muscle can target the kidney. A) The proteins were isolated from the kidney of mice given a sham intramuscular injection with exosome-carried Flag (sham/F), mice given a sham treatment with exosome-carried miR-26a (sham/26), UUO mice treated with exosome-carried Flag (UUO/F), and UUO mice treated with exosome-carried miR-26 (UUO/26). The proteins TGF-β1, CTGF, and SMAD2 were measured by Western blotting in different groups of mice. Top: representative immunoblots. Bottom: densitometry analysis. The point graphs show the change of each protein band normalized to GAPDH. Values are means ± se; n = 6/group. *P < 0.05 vs. sham/F; #P < 0.05 vs. UUO/F. B) The exosomes that are enriched with miR-26a were harvested from culture conditioned medium. Exosome was labeled with with 1-μM fluorescent lipophilic tracer DiR. Labeled exosome-carried miR-26a was injected into the left TA muscle of sham-treated mice or UUO mice. The control was no injection. The fluorescence was assessed at 28 d after injection. A Bruker small animal optical imaging system acquired representative fluorescent organ images. Organs from top to bottom: brain, lung (left) and heart (right), liver (left) and spleen (right), and kidneys and TA muscle. C) HEK293 cells were transfected with luciferase pLuc control vector (pLuc/ctrl) or the vector containing the 3′-UTR of CTGF (pMIR/CTGF) or a vector containing a mutated 3′-UTR of CTGF (pMIR-mut/CTGF). Cells were cotransfected with Renilla luciferase as a transfection control and with miR-control (miR-c) or miR-26 mimic (miR26). The firefly luciferase (FFL) results were normalized by Renilla luciferase (RL) activity. Activity in cells that received pLuc control with miR-control was designated as the 100% control. The other bars show the response as a percent of the control. Triplicate determinations were made in each condition, and each experiment was repeated a total of 3 times. Data are represented as means ± se; n = 9. *P < 0.05 vs. pMIR/CTGF + miR-ctrl. D) Proteins CTGF, FoxO1, and GAPDH were measured by Western blotting in HEK293 cells transfected with miR-control (miR-c), miR-26, or miR-26 inhibitor (miR-26/in or 26-inhi) or not transfected (no treat). The point graphs show the change of each protein band normalized to GAPDH (bars: means ± se; n = 4/group). *P < 0.05 vs. control.
DISCUSSION
In this study, we demonstrated that muscle injection of exosomes loaded with miR-26a could enhanced the expression of miR-26a in both muscle and kidney, which protected against muscle atrophy by regulating FoxO1 and GSK-3β and reduced renal fibrosis by modulating CTGF and TGF-β1 in a mouse model of UUO. Our study provides a potential strategy for the treatment of muscle atrophy and renal fibrosis in obstructive kidney disease by using exosome-derived miR.
Muscle atrophy is prevalent among patients with CKD (7, 32). An emerging body of evidence suggests that miRs are involved in the regulation of CKD-induced muscle atrophy (8). Previously, we examined a profile of miRs in muscles from mice with CKD and found that miR-29 was significantly decreased (25). Overexpressing miR-29 directly inhibits the myogenesis-suppressing transcription factor, ying-yang 1, leading to increase myogenesis (25). Our current study shows that miR-26a is decreased in muscle of UUO mice and finds that enhanced expression of miR-26a could inhibit UUO-induced muscle atrophy. The mechanism by which miR-26a reduces muscle wasting involves the direct targeting of the transcription factor FoxO1 (20). FoxO1 promotes the expression of 2-ubiquitin E3 ligases, tripartite motif containing 63 (muscle ring finger 1) and F-box protein 32 (atrogin-1), causing muscle atrophy by increasing protein degradation in CKD mice (33, 34). Here we showed that provision of miR-26a could suppress UUO-induced FoxO1 expression in skeletal muscle, leading to decreased muscle loss. Another possible mechanism by which miR-26a can limit muscle loss is by directly targeting GSK-3β. Activation of GSK-3β induces insulin resistance, which induces muscle loss in metabolic disease, so targeting by miR-26a would reduce GSK-3β and increase insulin sensitivity (35). In agreement with our findings, other investigators have shown that a decrease of miR-26a impairs glucose homeostasis and increases insulin resistance, whereas an overexpression of miR-26a inhibits GSK-3β activation, leading to increased insulin sensitivity (36, 37).
Renal fibrosis is the inevitable consequence of excessive accumulation of extracellular matrix and occurs in virtually every type of CKD (38). miR-26a has previously been linked with changes in fibrosis in the eye. It was reported that miR-26a significantly inhibited proliferation, migration, epithelial-to-mesenchymal transition of lens epithelial cells, and lens fibrosis in vitro and in vivo (39). In kidney, our results showed that miR-26a levels were reduced with UUO, a model of increased fibrosis, and when miR-26a was provided to UUO mice, renal fibrosis was decreased. Other investigators have reported that miR-26a is a potent suppressor of CTGF and TGF-β1 (40–42). CTGF is a critical fibrogenic mediator of TGF-β1 and a target of miR-26a (12). We found that CTGF was increased by UUO and that providing miR-26a to UUO mice decreased CTGF expression in kidney. Suppression of CTGF by miR-26a could, therefore, contribute to renal protection. CTGF is a major downstream profibrotic factor of TGF-β1, and CTGF activation could feed back to enhance TGF-β–SMAD signaling (40). TGF-β–induced collagen production is attenuated by a CTGF-neutralizing antibody or antisense oligonucleotide targeting CTGF (42). The current study found that provision of miR-26a suppressed CTGF and TGF-β1, which are the key mediators of renal fibrosis.
The difficulties involved in delivering RNA to damaged tissue have hindered the use of RNA therapy (43); however, exosome-carried RNA appears to be an effective method to modulate cellular signaling and biologic processes in recipient cells (44). One article reported that exosome-carried growth factor receptor mRNA can accelerate the regeneration of cisplatin-injured renal proximal tubular cells (45). Another article demonstrated that exosomes derived from sunitinib-resistant cells contained long noncoding RNA that was able to confer a resistance phenotype to recipient renal cell carcinoma cells in vivo (46). In the present study, we show that injection of exosomes with high levels of miR-26a into muscle enhanced the expression of miR-26a in both uninjected muscle and kidney, which suggests that exosome-carried mature miR could be taken up by adjacent muscles and might be transferred to remote organs. Evidence for remote organ effects of by exosome transfer exists in the form of an induced protection of heart by a phenomenon known as remote ischemic preconditioning, which appears to involve exosomes as carriers of the cardioprotective factor released by remote ischemic preconditioning (47). In addition, we found that damaged organs, such as UUO kidneys, accumulate more exosome-carried miR-26a than noninjured organs after intramuscular injection (Fig. 6B). The reason needs further investigation. We posit that the damaged tissue creates a hierarchy for recruitment of exosomes and that the injured kidney induces secretion of inflammatory cytokines, leading to increased capillary permeability, which could be the reason for more exosome uptake (48). The tendency of exosomes to accumulate in damaged tissue suggests, clinically, that treatment with therapeutic exosomes could result in higher levels of therapeutic miRs concentrated at damaged kidney, which could aid in recovery from fibrotic lesions.
We believe that the increase in miR-26a in the kidney may result from 2 separate pathways. The first is that when the exosomes are injected into the muscle, some of them fuse to the cellular membrane and are endocytosed into the muscle fiber, which helps to ameliorate muscle wasting. However, in the muscle fiber, miR-26 could reassemble into multivesicular bodies and be secreted by muscle into the circulation as exosome-carried miR-26. It is also possible that some of the exosome-carried miR-26 that was injected could directly enter the circulation present in the muscle. Either way, the circulating exosome-carried miR-26 could be taken up by the kidney, where the miR-26 exerts its influence on CTGF, collagen 1, and TGF-β to reduce fibrosis in the kidney. Whether the exosome-carried miR-26 goes to the kidney directly by entering the muscle vasculature or indirectly through muscle multivesicular body exocytosis, the result is reduction of kidney fibrosis and limiting of muscle loss. Understanding this process in greater detail will require further investigation.
In summary, our data demonstrated that exosome-carried miR-26a prevented muscle atrophy by inhibiting FoxO1 and limited renal fibrosis by suppressing CTGF in a mouse model of UUO. Our results provided novel insights into the intercommunication between muscle and kidney tissue via exosomes. Our data suggest that exosome-derived miR-26a might be a therapeutic option to suppress muscle atrophy and renal fibrosis.
ACKNOWLEDGMENTS
The authors thank Dr. Matthew Wood and Dr. Yiqi Seow (University of Oxford, Oxford, United Kingdom) for providing the pLamp2b-Flag vector. This research was supported by the U.S. National Institutes of Health (NIH) National Institute of Arthritis and Musculoskeletal and Skin Diseases (Grant R01 AR060268), American Heart Association Discover and Innovation grants supported by the Bayer Group (17IBDG33780000 to X.W.), the National Natural Science Foundation of China (81670628 to Y.Y.), and the Jangsu Province Science Foundation of China (BK20161071) and Nanjing Medical Science and Technique Foundation (YKK17213 to A.Z.). This research was also supported, in part (microRNA deep sequencing), by the Genomics Core of Yerkes National Primate Research Center [NIH Office of Research Infrastructure Programs/Office of the Director (ORIP/OD) P51OD011132] and, in part (luciferase constructs), by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities (UL1TR000454). The content is solely the responsibility of the authors and does not necessarily reflect the official views of the NIH or the U.S. Government. The authors declare no conflicts of interest.
Glossary
- α-SMA
α-smooth muscle actin
- CKD
chronic kidney disease
- CTGF
connective tissue growth factor
- DiR
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide
- FoxO
forkhead box O
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GSK
glycogen synthase kinase
- HEK293
human embryonic kidney 293
- Lamp2b
lysosomal-associated membrane protein 2b
- miR
microRNA
- qPCR
quantitative PCR
- TA
tibialis anterior
- UUO
unilateral ureteral obstruction
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
X. H. Wang designed the research; A. Zhang, H. Wang, and B. Wang conducted the research; A. Zhang, H. Wang, and Y. Yuan analyzed the data and performed statistical analysis; A. Zhang, Y. Yuan, J. D. Klein, and X. H. Wang wrote the manuscript and had responsibility for its final content; and X. H. Wang is the guarantor of this work, has complete access to all the data in the study, and takes ultimate responsibility for the study design and integrity of data analysis.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Hill N. R., Fatoba S. T., Oke J. L., Hirst J. A., O’Callaghan C. A., Lasserson D. S., Hobbs F. D. (2016) Global prevalence of chronic kidney disease - a systematic review and meta-analysis. PLoS One 11, e0158765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Y. L., Xie J., An S. W., Oliver N., Barrezueta N. X., Lin M. H., Birnbaumer L., Huang C. L. (2017) Inhibition of TRPC6 channels ameliorates renal fibrosis and contributes to renal protection by soluble klotho. Kidney Int. 91, 830–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanatani S., Izumiya Y., Araki S., Rokutanda T., Kimura Y., Walsh K., Ogawa H. (2014) Akt1-mediated fast/glycolytic skeletal muscle growth attenuates renal damage in experimental kidney disease. J. Am. Soc. Nephrol. 25, 2800–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang L., Rajan V., Lin E., Hu Z., Han H. Q., Zhou X., Song Y., Min H., Wang X., Du J., Mitch W. E. (2011) Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 25, 1653–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang L., Pan J., Dong Y., Tweardy D. J., Dong Y., Garibotto G., Mitch W. E. (2013) Stat3 activation links a C/EBPδ to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 18, 368–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee S. J. (2004) Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 20, 61–86 [DOI] [PubMed] [Google Scholar]
- 7.Wang X. H., Mitch W. E. (2014) Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 10, 504–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X. H. (2013) MicroRNA in myogenesis and muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 16, 258–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang A., Li M., Wang B., Klein J. D., Price S. R., Wang X. H. (2018) miRNA-23a/27a attenuates muscle atrophy and renal fibrosis through muscle-kidney crosstalk. J. Cachexia Sarcopenia Muscle 9, 755–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H., Wang B., Zhang A., Hassounah F., Seow Y., Wood M., Ma F., Klein J. D., Price S. R., Wang X. H. (2019) Exosome-mediated mir-29 transfer reduces muscle atrophy and kidney fibrosis in mice. Mol. Ther. 27, 571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang B., Zhang C., Zhang A., Cai H., Price S. R., Wang X. H. (2017) MicroRNA-23a and microRNA-27a mimic exercise by ameliorating CKD-induced muscle atrophy. J. Am. Soc. Nephrol. 28, 2631–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng Z., Guan M., Jia Y., Wang D., Pang R., Lv F., Xiao Z., Wang L., Zhang H., Xue Y. (2016) The coordinated roles of miR-26a and miR-30c in regulating TGFβ1-induced epithelial-to-mesenchymal transition in diabetic nephropathy. Sci. Rep. 6, 37492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H., Wang B., Zhang A., Hassounah F., Seow Y., Wood M., Ma F., Klein J. D., Price S. R., Wang X. H. (2019) Exosome-mediated miR-29 transfer reduces muscle atrophy and kidney fibrosis in mice. Mol. Ther. 27, 571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang H., Xu C., Pan Z., Zhang Y., Xu Z., Chen Y., Li T., Li X., Liu Y., Huangfu L., Lu Y., Zhang Z., Yang B., Gitau S., Lu Y., Shan H., Du Z. (2014) The antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol. Ther. 22, 1122–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phanish M. K., Winn S. K., Dockrell M. E. (2010) Connective tissue growth factor-(CTGF, CCN2)--a marker, mediator and therapeutic target for renal fibrosis. Nephron, Exp. Nephrol. 114, e83–e92 [DOI] [PubMed] [Google Scholar]
- 16.Sandri M., Sandri C., Gilbert A., Skurk C., Calabria E., Picard A., Walsh K., Schiaffino S., Lecker S. H., Goldberg A. L. (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X. H., Du J., Klein J. D., Bailey J. L., Mitch W. E. (2009) Exercise ameliorates chronic kidney disease-induced defects in muscle protein metabolism and progenitor cell function. Kidney Int. 76, 751–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milan G., Romanello V., Pescatore F., Armani A., Paik J. H., Frasson L., Seydel A., Zhao J., Abraham R., Goldberg A. L., Blaauw B., DePinho R. A., Sandri M. (2015) Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 6, 6670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su Z., Hu L., Cheng J., Klein J. D., Hassounah F., Cai H., Li M., Wang H., Wang X. H. (2016) Acupuncture plus low-frequency electrical stimulation (Acu-LFES) attenuates denervation-induced muscle atrophy. J. Appl. Physiol. (1985) 120, 426–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang B., Zhang A., Wang H., Klein J. D., Tan L., Wang Z. M., Du J., Naqvi N., Liu B. C., Wang X. H. (2019) miR-26a limits muscle wasting and cardiac fibrosis through exosome-mediated microRNA transfer in chronic kidney disease. Theranostics 9, 1864–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin J., Li J., Huang B., Liu J., Chen X., Chen X. M., Xu Y. M., Huang L. F., Wang X. Z. (2015) Exosomes: novel biomarkers for clinical diagnosis. ScientificWorldJournal 2015, 657086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ha D., Yang N., Nadithe V. (2016) Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm. Sin. B 6, 287–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang B., Yao K., Huuskes B. M., Shen H. H., Zhuang J., Godson C., Brennan E. P., Wilkinson-Berka J. L., Wise A. F., Ricardo S. D. (2016) Mesenchymal stem cells deliver exogenous microRNA-let7c via exosomes to attenuate renal fibrosis. Mol. Ther. 24, 1290–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Viñas J. L., Burger D., Zimpelmann J., Haneef R., Knoll W., Campbell P., Gutsol A., Carter A., Allan D. S., Burns K. D. (2016) Transfer of microRNA-486-5p from human endothelial colony forming cell-derived exosomes reduces ischemic kidney injury. Kidney Int. 90, 1238–1250 [DOI] [PubMed] [Google Scholar]
- 25.Wang X. H., Hu Z., Klein J. D., Zhang L., Fang F., Mitch W. E. (2011) Decreased miR-29 suppresses myogenesis in CKD. J. Am. Soc. Nephrol. 22, 2068–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Z., Klein J. D., Mitch W. E., Zhang L., Martinez I., Wang X. H. (2014) MicroRNA-29 induces cellular senescence in aging muscle through multiple signaling pathways. Aging (Albany N.Y.) 6, 160–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu L., Klein J. D., Hassounah F., Cai H., Zhang C., Xu P., Wang X. H. (2015) Low-frequency electrical stimulation attenuates muscle atrophy in CKD--a potential treatment strategy. J. Am. Soc. Nephrol. 26, 626–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su Z., Klein J. D., Du J., Franch H. A., Zhang L., Hassounah F., Hudson M. B., Wang X. H. (2017) Chronic kidney disease induces autophagy leading to dysfunction of mitochondria in skeletal muscle. Am. J. Physiol. Renal Physiol. 312, F1128–F1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X. H., Zhang L., Mitch W. E., LeDoux J. M., Hu J., Du J. (2010) Caspase-3 cleaves specific 19 S proteasome subunits in skeletal muscle stimulating proteasome activity. J. Biol. Chem. 285, 21249–21257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Camussi G., Deregibus M. C., Bruno S., Cantaluppi V., Biancone L. (2010) Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 78, 838–848 [DOI] [PubMed] [Google Scholar]
- 31.Koga K., Yokoi H., Mori K., Kasahara M., Kuwabara T., Imamaki H., Ishii A., Mori K. P., Kato Y., Ohno S., Toda N., Saleem M. A., Sugawara A., Nakao K., Yanagita M., Mukoyama M. (2015) MicroRNA-26a inhibits TGF-β-induced extracellular matrix protein expression in podocytes by targeting CTGF and is downregulated in diabetic nephropathy. Diabetologia 58, 2169–2180 [DOI] [PubMed] [Google Scholar]
- 32.Wang X. H., Mitch W. E. (2013) Muscle wasting from kidney failure-a model for catabolic conditions. Int. J. Biochem. Cell Biol. 45, 2230–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J., Li R., Workeneh B., Dong Y., Wang X., Hu Z. (2012) Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 82, 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S. W., Dai G., Hu Z., Wang X., Du J., Mitch W. E. (2004) Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J. Am. Soc. Nephrol. 15, 1537–1545 [DOI] [PubMed] [Google Scholar]
- 35.Wang Q. M., Fiol C. J., DePaoli-Roach A. A., Roach P. J. (1994) Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J. Biol. Chem. 269, 14566–14574 [PubMed] [Google Scholar]
- 36.Fu X., Dong B., Tian Y., Lefebvre P., Meng Z., Wang X., Pattou F., Han W., Wang X., Lou F., Jove R., Staels B., Moore D. D., Huang W. (2015) MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. J. Clin. Invest. 125, 2497–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohamed J. S., Lopez M. A., Boriek A. M. (2010) Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3β. J. Biol. Chem. 285, 29336–29347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang D., Sun L., Xian W., Liu F., Ling G., Xiao L., Liu Y., Peng Y., Haruna Y., Kanwar Y. S. (2010) Low-dose paclitaxel ameliorates renal fibrosis in rat UUO model by inhibition of TGF-beta/Smad activity. Lab. Invest. 90, 436–447 [DOI] [PubMed] [Google Scholar]
- 39. Chen, X., Xiao, W., Chen, W., Liu, X., Wu, M., Bo, Q., Luo, Y., Ye, S., Cao, Y., Liu, Y. (2017) MicroRNA-26a and -26b inhibit lens fibrosis and cataract by negatively regulating Jagged-1/Notch signaling pathway. Cell Death Differ.24, 1431–1442; erratum: 1990. [DOI] [PMC free article] [PubMed]
- 40.Parada C., Li J., Iwata J., Suzuki A., Chai Y. (2013) CTGF mediates Smad-dependent transforming growth factor β signaling to regulate mesenchymal cell proliferation during palate development. Mol. Cell. Biol. 33, 3482–3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abreu J. G., Ketpura N. I., Reversade B., De Robertis E. M. (2002) Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell Biol. 4, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duncan M. R., Frazier K. S., Abramson S., Williams S., Klapper H., Huang X., Grotendorst G. R. (1999) Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by cAMP. FASEB J. 13, 1774–1786 [PubMed] [Google Scholar]
- 43.Qu Y., Zhang Q., Cai X., Li F., Ma Z., Xu M., Lu L. (2017) Exosomes derived from miR-181-5p-modified adipose-derived mesenchymal stem cells prevent liver fibrosis via autophagy activation. J. Cell. Mol. Med. 21, 2491–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melo S. A., Sugimoto H., O’Connell J. T., Kato N., Villanueva A., Vidal A., Qiu L., Vitkin E., Perelman L. T., Melo C. A., Lucci A., Ivan C., Calin G. A., Kalluri R. (2014) Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 26, 707–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomasoni S., Longaretti L., Rota C., Morigi M., Conti S., Gotti E., Capelli C., Introna M., Remuzzi G., Benigni A. (2013) Transfer of growth factor receptor mRNA via exosomes unravels the regenerative effect of mesenchymal stem cells. Stem Cells Dev. 22, 772–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qu L., Ding J., Chen C., Wu Z. J., Liu B., Gao Y., Chen W., Liu F., Sun W., Li X. F., Wang X., Wang Y., Xu Z. Y., Gao L., Yang Q., Xu B., Li Y. M., Fang Z. Y., Xu Z. P., Bao Y., Wu D. S., Miao X., Sun H. Y., Sun Y. H., Wang H. Y., Wang L. H. (2016) Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell 29, 653–668 [DOI] [PubMed] [Google Scholar]
- 47.Lawson C., Vicencio J. M., Yellon D. M., Davidson S. M. (2016) Microvesicles and exosomes: new players in metabolic and cardiovascular disease. J. Endocrinol. 228, R57–R71 [DOI] [PubMed] [Google Scholar]
- 48.Bivol L. M., Iversen B. M., Hultström M., Wallace P. W., Reed R. K., Wiig H., Tenstad O. (2016) Unilateral renal ischaemia in rats induces a rapid secretion of inflammatory markers to renal lymph and increased capillary permeability. J. Physiol. 594, 1709–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.