

Abstract
Background:
Cigarette smoke (CS)-induced lung inflammation and Chronic Obstructive Pulmonary disease (COPD) involves mitochondrial dysfunction. Mesenchymal stem cells (MSC) and MSC-derived exosomes (EXO) are reported to show therapeutic effects in many animal models of inflammation and injury. In the present study, we determined the role of MSC and EXO intervention in CS-induced lung inflammation with focus on mitochondrial dysfunction.
Methods:
EXO were characterized using Western blot for exosomal markers, tunable resistive pulse sensing by qNano and transmission electron microscopy (TEM). Mitochondrial reporter mice (Mt-Keima and mito-QC) were exposed to air or CS for 10 days. mt-Keima mice were treated with intraperitoneal injections of MSC or EXO or MSC and EXO (MSC+EXO) for 10 days. Total cell counts, differential cell counts were performed using automated cell counter and flow cytometry respectively. Further, the levels of pro-inflammatory mediators in bronchoalveolar lavage (BAL) fluid were measured using ELISA. Western blot analysis, quantitative PCR, confocal microscopy were used in the current study to determine the effects in the lungs of CS exposed mice. Seahorse flux analyzer was used to measure the oxidative-phosphorylation (OXPHOS) in the BEAS2B cells and BEAS2B+mMSC co-culture experiments.
Results:
CS exposure increased the inflammatory cellular infiltrations in the lungs of the mt-Keima mice. MSC+EXO treatment showed protection compared to individual treatments (MSC or EXO alone). There were no changes in the mitophagy proteins like Pink1 and Parkin, which was also found using the mitoQC mice. CS exposure led to significant increase in the mitochondrial fission protein DRP1 and other DAMPs pathway mediators like S100A4 and S100A8, HMGB1, RAGE and AGE. MSC+EXO treatment increased the gene expression of (fusion genes) like mfn1, mfn2 and opa1. Additionally, the rhot1 gene expression was increased in MSC+EXO treatment group compared to Air- and CS exposed groups. BEAS2B-MSC co-cultures showed protective response against the CSE-altered mitochondrial respiration parameters, confirming the beneficial effect of MSC towards human bronchial lung epithelial cells.
Conclusion:
CS affects some of early mitochondrial genes involved in the fission/fusion process, enhancing the damage response along with altered cytokine levels. MSC+EXO combination treatment showed their protective effects. MSC+EXO combination treatment may act against these early events caused by CS exposure owing to its anti-inflammatory and other mitochondrial transfer mechanisms.
Keywords: Mitochondria, Cellular Senescence, Mesenchymal stem cells, Exosomes, COPD
1. Introduction
Cigarette smoke (CS) is the main etiological risk factor involved in the pathogenesis of Chronic Obstructive Pulmonary Disease (COPD) [1]. CS contains several free radicals and chemicals, which cause lung inflammation [2, 3]. Earlier, we have characterized that acute CS exposure causes lung inflammation and changes in some of the early cytokine responses inducing lung damage [4], and CS extract caused mitochondrial dysfunction in lung epithelial cells [5]. Mesenchymal stem cells (MSC) and MSC-derived exosomes (EXO) are gaining wide attention as therapeutic intervention in various inflammatory models of disease [6–9]. MSC showed its beneficial effect in several acute lung injury models [10]. Further, MSC shows its therapeutic benefits in multitude ways in which transfer of mitochondria is also reported as one of the potential mechanisms [11]. MSCs also exert paracrine effect by releasing the vesicles-like exosomes and other mediators which carry the information and signaling for the cellular protection [12]. However their role in the acute CS-induced lung inflammation and injury is not known. On the other hand exosomal cargo includes but not limited to DNA, mRNA, miRNA, lncRNA and proteins, which play a crucial role in the cell signaling and response [13]. Exosomes were also reported to carry the functional mitochondrial elements and communicate [14].
The role of MSC and exosomes were shown in several cellular and animal models of lung injury [9, 10, 15, 16]. Recently, we have shown that plasma-derived extracellular vesicles (EVs) contain distinct protein signature among smoker and COPD subjects, compared to never smoking group [17]. Both MSC and EXO were reported to have anti-inflammatory effect in several animal models [18]. Recently, it was reported that intraperitoneal injection of EXO prevented bronchopulmonary dysplasia (BPD) in rats [19]. Porzionato et al., found that intratracheal injection of EVs showed better activity compared to MSC in a rat model of BPD [20]. Recently it was shown that mesenchymal stromal cell exosomes prevent the severity of pulmonary fibrosis induced by bleomycin in mice [21]. Previously, it was reported that CS exposure induces lung inflammation, along with the altered levels of cytokines in the BAL fluid of different mouse strains [3, 4]. Keeping in view the beneficial effects of both MSC and EXO, here we have investigated the combination effects of bone marrow-derived MSC and EXO (MSC+EXO) in acute CS-induced (10 days) lung inflammation model in mitochondrial reporter mt-Keima mice. We hypothesized that exosomes given along with the MSC may have better beneficial outcomes compared to individual treatments (MSC or EXO alone) in acute CS-induced lung inflammation. We have determined the effect of this treatment on 1) inflammatory cytokines/related pathways, 2) key mitochondrial proteins involved in biogenesis, dynamics (fusion and fission process) and mitophagy, and 3) damage-associated molecular patterns (DAMPs).
2. Materials and methods
All the key biological and/or chemical resources that are used in this study were validated and authenticated (methods and resources) and are of scientific standard from commercial sources.
Ethical approval
All the animal protocols and procedures described in this study were approved by the University Committee on Animal Resources of the University of Rochester, Rochester, NY.
2.1. Animals and treatment
mt-Keima mice (4-7 months) was used in the present investigation to observe the effects of MSC and EXO in the lung inflammation induced by acute CS exposure as previously reported [4]. The mice were sacrificed 2 hours after the last day of the CS exposure. The mice (n=6/group) were divided into the following, group-1; air exposed group (Air), in which the animals were air exposed and given intraperitoneal dose of saline, group-2; cigarette smoke group (CS), mice under this group were exposed to 10 days of cigarette smoke, group-3,4 and 5 were similar to the group-2 in terms of CS exposure, but received MSC (~1x106 cells, one day prior to CS and on 6th day of CS exposure, designated as MSC), EXO (~15 μg, were given daily through intraperitoneal route after the CS exposure, designated as EXO) or both MSC and EXO (designated as MSC+EXO). mito-QC (heterozygous, 6-7 months) were also used in this study and were exposed to either air or CS for 10 days, as described above to determine the mitochondrial/mitophagy parameters. In the current experiment intraperitoneal route was selected based on its advantages as reported earlier [22] and since there are multiple or daily treatments with MSC and EXO, this route was chosen over intravenous route. Further, the dose of EXO was selected based on prior reports that showed beneficial effects in vivo in mouse [19,23].
2.2. MSC procurement and culture
Mouse MSC were purchased from Cyagen (#MUBMX-01001) received with passage number 6 and used within passage 10 (mostly 7-8) as per the manufacturer’s instructions. The MSC are positive for CD34, CD44, Sca-1 and negative for CD117 by flow cytometry analysis. Mouse MSC were grown in the Mesencult expansion medium (Stem Cell Technologies, 05513) and later maintained in Iscove’s Modified Dulbecco’s Medium (Gibco, 12440) with 10% exosome-free FBS (Fetal Bovine Serum, exosome-depleted, One Shot, A2720803).
2.3. Exosome isolation and characterization
Exosomes from mouse MSC conditioned media was isolated as per the manufacturer’s instructions (Norgen Biotek kit for cell culture media-exosomes-midi preparation # 60500). The isolated EXO were characterized for the presence of markers like CD63, flotillin and alix and the purity using anti-calnexin antibody. EXO were quantified using tunable resistive pulse sensing by qNano (Izon). TEM imaging was performed to observe the size and morphology of the isolated EXO. EXO protein was quantified using micro BCA kit (ThermoFisher).
2.4. Cell count analysis and pro-inflammatory mediators in the BAL
Total and differential cell counts in BAL fluid was performed as described earlier [24], except the detection was done using Guava easyCyte (Millipore). ELISA and Mouse cytokine array (R&D systems) analysis were performed to detect different cytokines in the BAL fluid [24].
2.5. BEAS2B and mMSC co-culture and measurement of OXPHOS
Seahorse XFp Cell Mito Stress Test kit (103010-100) was used to determine the effect of cigarette smoke extract (CSE) on BEAS2B, mMSC and mMSC-BEAS2B (bronchial epithelial cell) co-culture in XFp plate using Seahorse XFp analyzer. Cells (BEAS2B alone, mMSC alone and BEAS2B+mMSC co-culture) were treated for 24 hours using 0.5% CSE prepared as described previously [25]. Various OXPHOS parameters were evaluated using the WAVE software version 2.6.0 (Agilent).
2.6. Protein quantification in whole lung homogenate and mitochondrial fraction using Western blotting
Lung tissue homogenates were used to quantify various levels of mitochondrial, inflammatory and DAMPs markers across all the groups. Based on the initial observations in Air, CS and MSC+EXO groups, the study was expanded to determine other protein markers in rest of the groups. The following antibodies were procured from abeam parkin (ab15954, 1:1000), PGC1α (ab72230, 1:1000), AGE (ab23722, 1:1000), S100A4 (ab27957, 1:500), OXPHOS for rodent (MS604-300, 1:500), MMP9 (ab38898, 1:1000), MMP12 (ab39876, 1:1000), flotillin (ab1927, 1:250) and β-actin (ab20272, 1:1000); Cell signaling technologies pink1 (6946S, 1:500), OPA1 (80471S, 1:1000), pP65-NfκB (3036S, 1:1000), HMGB1 (6893S, 1:1000); Santa Cruz NF-κB p50 (SC-114, 1:1000), RAGE (SC-365154, 1:500), CD63 (SC-15363, 1:500), Aik (SC-11397, 1:250), Calnexin (SC-53540, 1:250); DRP1 (Novus biological, NB110-55288,1:1000), Tfam (EMD Millipore, ABE483, 1:1000), TSG6 (Sigma, SAB4502987, 1:1000) to determine the relative expression of various proteins as described earlier [4].
The mitochondrial fraction from lungs of mice were isolated according to the manufacturer’s protocol (ThermoFisher, kit #89801) and used to check for the protein expressions as described above.
2.7. Quantitative real-time PCR
Total RNA was extracted from the lungs using Direct-zol RNA Kits (Zymo Research, #R2072) and this was subsequently used to prepare cDNA, and following this gene expression analysis were performed as described previously [25]. The following primers were used in the current study rhot1 F- TGGGCAGCACTGATAGAATAGA, R- GCAAAGACCGTAGCACCAAAG; mfn1 F- CCTACTGCTCCTTCTAACCCA, R- AGGGACGCCAATCCTGTGA; mfn2 F- CCAACTCCAAGTGTCCGCTC, R- GTCCAGCTCCGTGGTAACATC; opa1 F- TGGAAAATGGTTCGAGAGTCAG, R- ATTCCGTCTCTAGGTTAAAGCG; fis1 F- GCCCCTGCTACTGGACCAT, R- CCCTGAAAGCCTCACACTAAGG and gapdh F- TTCCCGTTCAGCTCTGGG, R- CCCTGCATCCACTGGTGC. All the primers were obtained from integrated DNA technologies.
2.8. Confocal imaging
Mitochondria reporter (mito-QC) mouse lungs were OCT fixed, cryo-sectioned and kept in PBS for two intervals of 5 min each and mounted with the help of prolong anti-fade containing DAPI. These sections were observed for GFP, mCherry and DAPI under suitable wavelengths using FV1000 Olympus confocal microscope with a 100x oil immersion objective.
2.9. Statistical analysis
Two-tail unpaired student’s t-test or One-way analysis of variance (ANOVA) with Tukey’s post hoc analysis were used to compare the differences in the mean across various groups using GraphPad Prism 7. All the values are represented as mean ± SEM and P < 0.05 was considered to be statistically significant.
3. Results
3.1. Isolation and characterization of MSC-derived exosomes
Exosomes were isolated using the Norgen Biotek kit based method and immunoblot analysis was performed to observe for various exosomal markers. The mMSC exosomes were found to be positive for CD63 (using slot-blot analysis), flotillin and Alix as shown in Fig. 1A. The isolated exosomes were not contaminated by the endoplasmic reticulum as the calnexin bands were not detected but showed positive for the presence of the complex V (mitochondrial OXPHOS component) protein (data not shown). The isolated exosomes were observed for their size and morphology using TEM as shown in the Fig. 1A. Tunable resistive pulse sensing (TRPS) by qNano was used to determine the particle size, distribution and count/concentration of the isolated exosomes. The mean diameter was found to be around 115 ± 5 nm as determined using the NP250 membrane and CPC100 calibration beads.
Fig. 1.
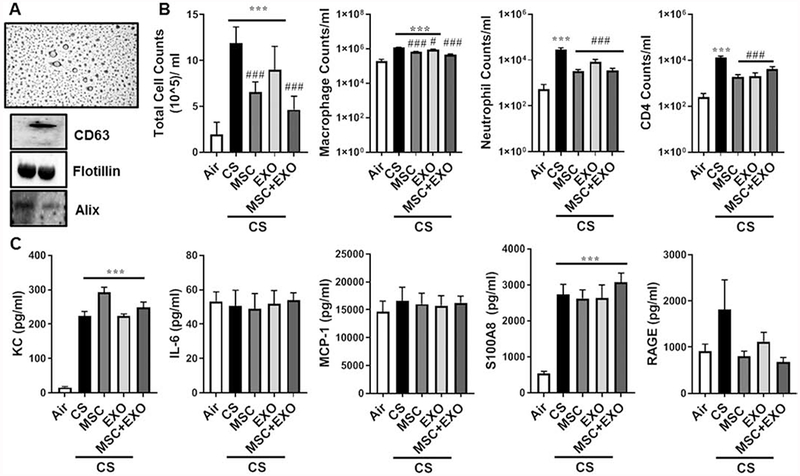
Characterization of mMSC-derived exosomes and the effect of their combination MSC+EXO on various pro-infammatory mediators in BAL fluid of acute 10days CS exposed mice. A) Isolation and characterization of mMSC-derived EXO using TEM, qNano and Western blotting. B) Intraperitoneal injection of MSC and EXO combination decreased lung cell infiltrations in mice exposed to acute CS estimated in the BAL fluid. C) Effect of MSC and EXO on different pro-inflammatory mediators in response to acute CS. Data were shown as mean ± SEM (n=6/group). ***P < 0.001 vs. air; # P < 0.05, ### P < 0.001 vs. CS.
3.2. Effect of different treatments on the cell counts in BAL of CS exposed mice
CS exposure led to a significant increase in the levels of total cell counts. Further, differential cell count analysis showed increase in the macrophage, neutrophil and CD4+ lymphocyte counts in the BAL fluid. Treatment with MSC, EXO and MSC+EXO significantly decreased the levels of the cell counts, macrophage, neutrophils and CD4+ cell population in the BAL as shown in the Fig. 1B implying some protective role during the acute CS-induced lung inflammation.
3.3. Effect of MSC and EXO treatment on various cytokine levels in BAL of CS exposed mice
CS exposure for 10 days significantly increased the levels of KC and S100A8, among the estimated inflammatory mediators using ELISA. However, the MSC and EXO showed no effect in decreasing these levels. Among other estimated mediators IL-6, MCP-1 and RAGE levels were unchanged across all the groups compared to air exposed control (Fig. 1C). While cytokine profiler has showed increased levels of Adiponectin, KC, MPO, BAFF, MPO, IGFBP-2, RAGE, CCL6/C10 and CCL17 in the CS exposed mice. MSC+EXO treatment was found to be beneficial in restoring these changes as shown in the Fig. 2.
Fig. 2.
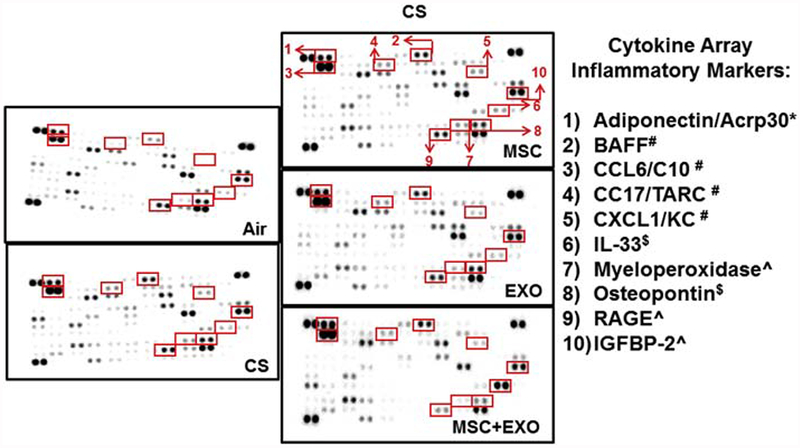
Estimation of various pro-inflammatory cytokines in the BAL fluid following acute CS exposure by mouse cytokine array. Representative images of the cytokine arrays with the red arrows indicating the position of particular cytokines which was significantly altered among the different treatment groups. The list of significantly altered cytokines along with their original positions in the cytokine array is clearly indicated. * Cytokines increased in all the treatment groups except CS exposed group; # Cytokines increased in all the CS exposed groups (MSC, EXO and MSC+EXO); $ Cytokines osteopontin was decreased and IL-33 increased only in CS exposed MSC+EXO group; ^ Cytokines increased in all the CS exposed groups (except decreased in MSC+EXO).
3.4. Effect of different treatments on the gene expression and protein levels in lungs of CS exposed mice
Initial gene expression analysis showed increase in the rhot1 (plays crucial role in the transfer of mitochondria and mitophagy) levels in MSC+EXO combination. Further, the expression levels of mitochondrial fusion genes mfn1, mfn2 and opa1 were significantly decreased in the CS group and MSC+EXO combination significantly improved these levels. However, the fis1 expression levels remain unaltered (Fig. 3). Protein expressions of PINK1 (mitophagy related), TFAM (mitochondria biogenesis and damage related), BECN1 (autophagy marker) remain unaltered, while PGC1α (mitochondria biogenesis) was alone found to be decreased in MSC+EXO group compared to air alone (Fig. 4). The protein expression analysis of DAMPs markers revealed that MSC+EXO significantly reduced the levels of S100A4 as compared to CS group, while RAGE and AGE remain unaltered (Fig. 4). Furthermore, protein analysis among all the groups showed that CS exposure increased the levels of DRP1 (mitochondrial fission related) with no changes in OPA1 (fusion) and parkin (mitophagy) in all the treatment groups (Fig. 5). Significant increase was observed in the levels of other inflammatory and DAMPs markers like MMP9 and HMGB1 in the CS group, whereas MSC+EXO significantly decreased these levels. However, no changes were observed in the MMP12 (data not shown) and TSG6 levels (Fig. 5). MSC treatment alone showed changes in the p65 NF-κB levels compared to MSC+EXO treatment group (Fig. 5). The protein expressions of parkin, OPA1 and TFAM remain unaltered in the mitochondrial protein fractions analyzed by Western blotting (Fig. 6). OXPHOS immunoblot analysis for various mitochondrial OXPHOS complexes remains unaltered in the current study (Fig. 7).
Fig. 3.
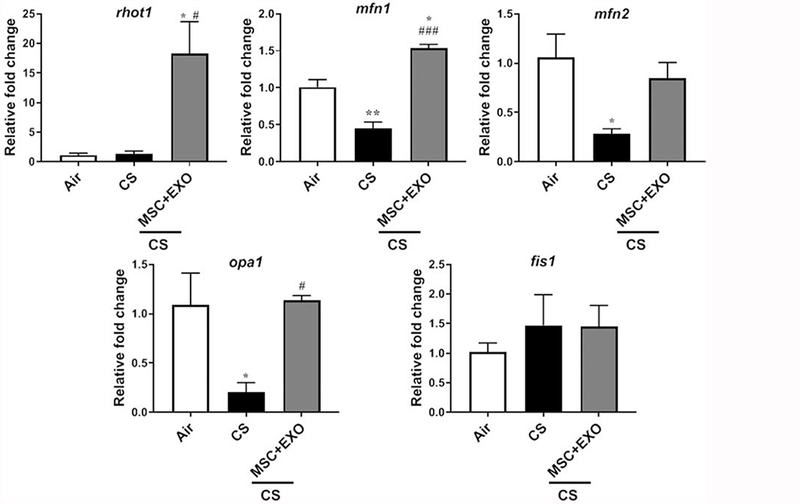
Effect of intraperitoneal treatment of MSC and EXO combination on different mitochondrial trafficking and dynamics genes assessed by qPCR in acute CS exposed mouse lungs. mt-Keima mice was exposed to acute CS and total RNA isolated from mouse lung were used for gene expression analysis by qPCR. Gene expression of mitochondrial trafficking and dynamics target genes such as rhot1, mfn1, mfn2, opa1 and fis1 were qPCR using the 2−ΔΔCt method. Bar graphs represent the mean normalized expression of samples in air, CS and CS exposed MSC+EXO treatment groups. Data were normalized using the endogenous housekeeping gene gapdh as reference and air as calibrators. Data were shown as mean ± SEM (n=3/group). * P < 0.05 vs. air; # P < 0.05, ### P < 0.001 vs. CS.
Fig. 4.
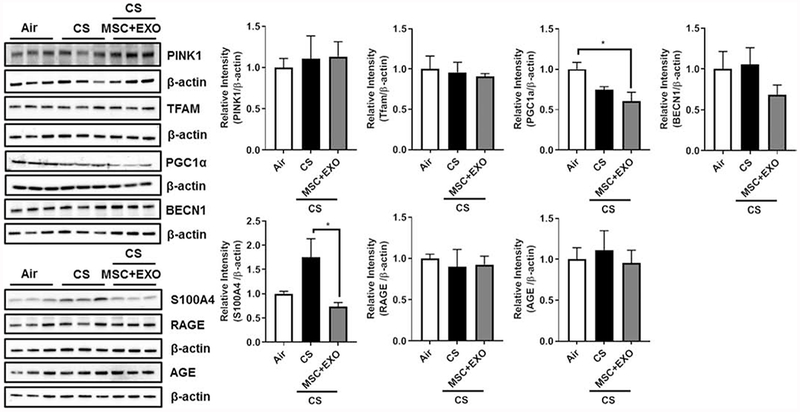
Effect of intraperitoneal treatment of MSC and EXO on mitochondrial and DAMPs protein expression in acute CS exposed mouse lung. Whole lung homogenates were used for Western blot analysis against mitochondrial markers using anti-Pink1, anti-TFAM, anti-PGC1α, and anti-BECN1; DAMPs markers using anti-S100A4, anti-RAGE and anti-AGE antibodies. The protein abundance of mitochondrial and DAMPs markers was measured following acute CS exposure in mouse lungs. The band intensity was measured by densitometry, and data are shown as relative intensity to β-actin as loading control. Data were shown as mean ± SEM (n=3/group). * represent P value < 0.05.
Fig. 5.

Effect of intraperitoneal treatment of MSC and EXO combination on mitochondrial, DAMPs and inflammation protein expression in acute CS exposed mouse lung. Whole lung homogenates were used for Western blot analysis against mitochondrial (anti-parkin, anti-Drp1, and anti-Opa1), DAMPs (anti-MMP9 and anti-HMGB1), and inflammation (anti-TSG6, anti-105, anti-p50 and anti-pP65) markers using antibodies. The protein abundance of mitochondrial, DAMPs and inflammation markers was measured following acute CS exposure in mouse lungs. The band intensity was measured by densitometry, and data are shown as relative intensity to β-actin as loading control. Data were shown as mean ± SEM (n=3/group). * P < 0.05.
Fig. 6.
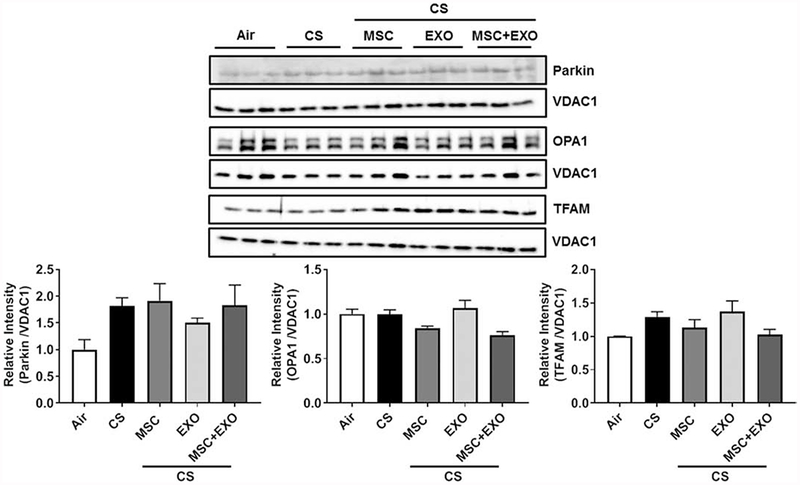
Effect of intraperitoneal treatment of MSC and EXO combination on the key mitochondrial proteins using the mitochondrial fraction in acute CS exposed mouse lung. Mitochondrial extracts were used for Western blot analysis against mitochondrial markers (anti-parkin, anti-Opa1 and anti-TFAM antibodies) and anti-VDAC1 antibody for mitochondrial loading control. The protein abundance of mitochondrial markers was measured following acute CS exposure using the mitochondrial fraction isolated from the mouse lung. The band intensity was measured by densitometry, and data are shown as relative intensity to VDAC1 as loading control. Data were shown as mean ± SEM (n=3/group).
Fig. 7.
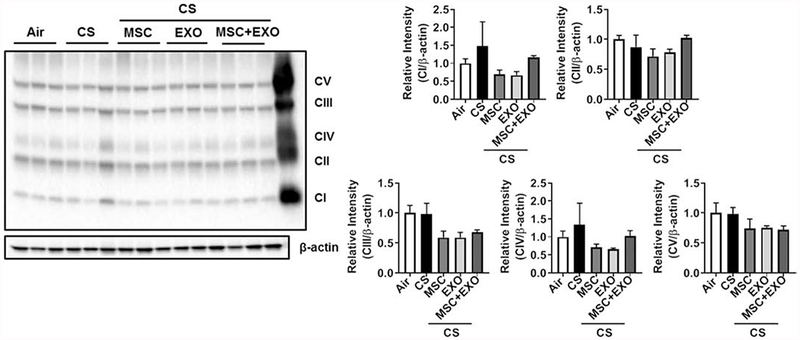
Effect of intraperitoneal treatment of MSC and EXO combination on mitochondrial OXPHOS related complex proteins in acute CS exposed mouse lung. Representative Western blot for Total OXPHOS showing levels of mitochondrial Complex I (NDUFB8), Complex II (SDHB), Complex III (UQCRC2), Complex IV (MTCO1) and Complex V (ATP5A) in whole lung homogenates. Densitometry of the corresponding mitochondrial complexes (Complexes I–V) was normalized with β-actin as loading control. Last lane in the image represents positive control sample from rat heart mitochondria provided for location and identification of various OXPHOS protein complexes. Data were shown as mean ± SEM (n=3/group).
3.5. Co-culture of MSC with lung epithelial cells protects from the mitochondrial stress
BEAS2B cells when exposed to 0.5% CSE for 24 hrs decreased many OXPHOS parameters like basal respiration, spare respiratory capacity, mitochondrial ATP production and maximal respiration. On the other hand, mMSC when exposed to 0.5% CSE for 24 hrs increased the maximal respiration and spare respiratory capacity. BEAS2B+ mMSC (at 1:1 ratio) co-culture when exposed to 0.5 % CSE for 24 hrs has no change in any of the parameters measured when compared their respective untreated controls (Fig. 8).
Fig. 8.
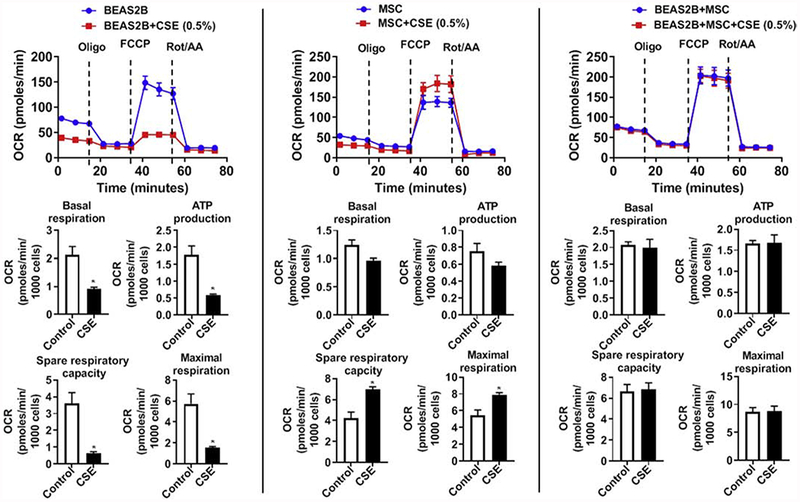
Effect of CSE treatment on the mitochondrial function in BEAS2B and MSC (1:1) co-culture estimated using Seahorse XFp flux analyzer. Individual BEAS2B or MSC alone or co-culture (BEAS2B+MSC) were treated with 0.5% CSE for 24 hrs and oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using Seahorse XFp Mito Stress Test kit at the indicated time points as shown. CSE treatment induced changes in basal respiration, ATP production, spare respiratory capacity, and maximal respiration were quantified. Data were shown as mean ± SEM (n=3/group) and were normalized to the total cell count and expressed per 1000 cells. * P < 0.05 vs control.
3.6. Effect of CS exposure on mitophagy in the lungs of mitoQC mice
mito-QC mice were exposed to either air or acute CS exposure for 10 days, similar to that of mt-Keima mice as mentioned earlier. In mito-QC mice, the mitochondrial fluorescence both red and green under basal/steady condition, but under mitophagy conditions, the mCherry fluorescence remains stable with diminished GFP signal, which can be seen as the puncta in the merged images and are indicative of mitophagy. Air group has basal mitophagy as evident by the presence of both GFP and mCherry signals. CS exposure has a slight increase in the number of mCherry puncta (Fig. 9A). Protein analysis revealed no significant changes in the whole lung homogenates of the mito-QC mice, affirming the results as observed in the mt-Keima mice (Fig. 9B).
Fig. 9.
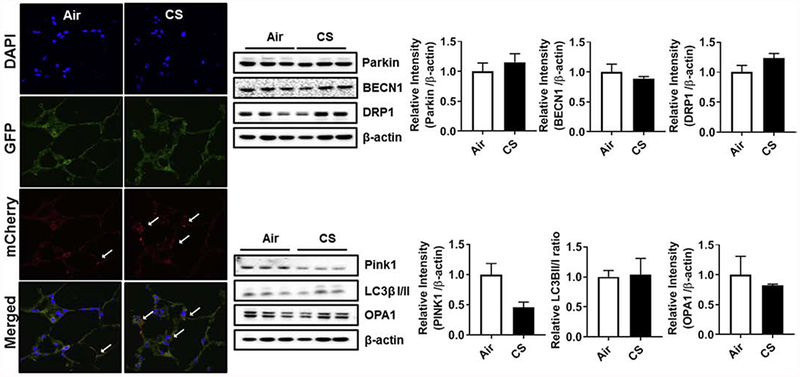
Mitophagy and mitochondrial architecture in alveolar lung epithelium and mitochondrial/mitophagy markers in acute CS exposed mitoQC mouse lung. (A) Representative images from cryo-sectioned OCT-fixed mitoQC mouse lung following acute 10 days CS exposure were analyzed by confocal microscopy. mCherry+ and GFP+ signal indicates no mitophagy and mCherry+ and GFP− signal co-localization indicates occurrence of mitophagy. The increased mCherry signals is indicated by white arrows. All the images were captured using a 100 x oil objective. (B) Mitochondrial and mitophagy protein abundance in whole lung homogenates of acute CS exposed mito-QC mice. Western blot analysis against mitochondrial/mitophagy markers (anti-parkin, anti-BECN1, anti-Drp1, anti-Pink1, anti-LC3β-I/II and anti-OPA1 antibodies) was measured in whole lung homogenates. The band intensity was measured by densitometry, and data are shown as relative intensity to β-actin as loading control. Data were shown as mean ± SEM (n=3/group).
4. Discussion
CS exposure induced mitochondrial gene alterations, structure and function in several types of lung cells in vitro and animal models in vivo as reported previously [26–30]. Several of the mitochondrial targeting compounds show beneficial effect in CS-induced lung/cellular dysfunction [31–33]. In the present study, we used the Seahorse XFp flux analyzer to determine the mitochondrial bio-energetics in control and CSE treated human bronchial epithelial cells and MSC. Our data showed that CSE-treated BEAS2B and mMSC co-cultures showed protective response compared to their individual CSE exposure profiles, implying that MSC has potential to protect against CS-induced lung epithelial damage. Exosomal transfers of whole mitochondrial genome as well as functional mitochondria are reported, emphasizing on the importance of novel cellular communication mediated through exosomes [14, 34]. Mitochondrial reporter, mt-Keima mice exposed to CS were treated to determine the possible ameliorative effects of MSC, EXO and MSC+EXO on CS-induced lung inflammation, with emphasis on the mitochondrial damage responses. CS exposure increased cellular infiltrations in the BAL fluid of mt-Keima mice. MSC, EXO and MSC+EXO treatment significantly restored these levels to some extent, implying a plausible protective role towards early damage responses evoked by the inflammatory cells. The increase in the neutrophil levels was reflected in the increased levels of chemokine KC in the BAL of the mice exposed to CS. Along with this S100A8 was also significantly increased by CS treatment indicating the involvement of DAMPs in CS-induced lung inflammation as previously reported in several CS exposure models [35, 36]. Furthermore, cytokine protein profiler showed increase in various pro-inflammatory markers which were earlier reported to be increased by CS exposure in various organ systems implying the CS-induced inflammatory effects on multiple organ markers [36].
The role of mitochondria accounting for the lung damage in the pathogenesis of lung diseases is gaining wide importance for therapeutic interventions [37]. Lung epithelial cells are rich in mitochondria, owing to their function in maintaining various physiological activities [38]. Mitochondria maintain their dynamics using various processes among which the balance between fission and fusion are widely studied. Various cellular responses including but not limited to CS were reported to cause changes in these process, by mainly altering the crucial proteins involved in either fusion process like MFN1, MFN2 and OPA1 or fission like DRP1 and FIS1. We and others have reported the role of CS in causing changes in these proteins, which ultimately lead to the cellular damage responses/cell death or mitophagy [39, 40]. On the other hand, mitophagy process also plays an important role in counteracting various cellular insults by recruiting/processing the damaged mitochondria to lysosomes and prevent from further cell damage [39]. PINK1 and parkin plays an important role in initiating the mitophagy process. CS was reported to have varying effects in inducing the mitophagy depending upon the duration of the exposure [31]. Here we have found no change in these protein levels, among all the groups compared to air exposed mice. mito-QC and mt-Keima mice were developed to easily monitor/visualize the process of mitophagy in various organs. Mitochondrial reporter mice mito-QC has advantage with respect to tissue fixation, giving stable signals as compared to the pH-dependent mt-Keima mice, which cannot be fixed and needs live imaging of tissues immediately after the experiments. For the first time, we have used mito-QC (heterozygous) mice to observe the changes caused by acute CS exposure in the lung mitophagy. CS exposure has no much effect in inducing the mitophagy changes as visualized by GFP/mCherry signals (although there was a non-significant increase in the mCherry signal by the CS), which is in accordance with the mt-Keima protein expression data mentioned above. Even though there is no change in the mitophagy initiation markers like PINK1 and parkin, we have found that CS increased the levels of mitochondrial fission protein DRP, indicating the initiation of the mitochondrial dysfunction. Previous studies observed changes in the p-DRP1 levels in the mice lungs exposed to CS for 3 weeks, without any changes in the autophagy related proteins [31], implying that fission/fusion plays an important role in counter acting the acute CS-induced damage response in vivo. Further CS exposure decreased the mitochondrial fusion genes like mfn1, mfn2 and opa1, implying the initiation of mitochondrial damage responses, which may be later followed by the phenomena of mitophagy. Further the MSC+EXO treatment reversed these changes, indicating its therapeutic potential in counteracting these damage responses. MSC were reported to show their protective action by direct mitochondrial transfer involving some of the crucial genes like rhot1 (miro1) or by secreting vesicles (exosomes) containing mitochondrial elements/protein to confer its protection [41, 42]. We found that MSC+EXO treatment increased the Rhot1 levels compared to air and CS groups. However, further protein analysis and specific Rhot1 related mechanisms need to confirm this initial observation. Studies are in progress to further dissect the exact role of rhot1 gene in these transfer mechanism, employing cell-specific knockout mice. In parallel with the mitochondrial markers, DAMPs pathway is also taken into account in this study, since DAMPs are reported to connect with the mitochondrial damage process [43]. CS exposure increased some of the DAMPs markers like RAGE, HMGB1, S100A4 and S100A8. MSC+EXO treatment was found to be beneficial in bringing down these levels in some of these markers. Our data was in accordance with some of the observations with respect to increase in markers like MMP9 and S100A8. MSC+EXO decreased MMP9 levels in the lungs, but not S100A8.
In conclusion, intraperitoneal injection of MSC+EXO combination showed protective response against acute CS-induced lung inflammation in mice. This study further emphasizes the beneficial role of MSC+EXO treatment in short-term CS exposure in mice. The CS exposure and treatments targeted the DAMPs and mitochondrial genes, implying the crucial role played by the mitochondria towards the progression of the COPD. Further studies employing different sources/routes of exosomes needs to be characterized to better understand the cell/vesicle based therapeutics in CS-induced models of COPD/emphysema.
Research Highlights.
The protective role of MSC and EXO in CS-induced lung mitochondrial dysfunction was determined.
MSC + EXO treatment showed much protection compared to individual treatments.
CS exposure led to significant increase in the mitochondrial fission protein and DAMPs.
BEAS2B-MSC co-culture showed protection against the CSE-altered mitochondrial respiration.
MSC + EXO treatment protect against CS-mediated against mitochondrial dysfunction.
Acknowledgement
The authors would like to thank Qixin Wang, Ph.D., for his assistance in analyzing the flow data.
Funding This study was supported by the NIH R01 HL135613, R21 ES028006, R01 HL133404, and R01 HL137738.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interests
The authors declare no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
5. References
- [1].Churg A, Cosio M, Wright JL, Mechanisms of cigarette smoke-induced COPD: insights from animal models, Am J Physiol-Lung C, 294 (2008) L612–L631. [DOI] [PubMed] [Google Scholar]
- [2].Sundar IK, Yao H, Rahman IJA, r. signaling, Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases, 18 (2013) 1956–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vlahos R, Bozinovski S, Jones J, Powell J, Gras J, Lilja A, Hansen M, Gualano R, Irving L, Anderson G.J.A.J.o.P.-L.C., M. Physiology, Differential protease, innate immunity, and NF-κB induction profiles during lung inflammation induced by subchronic cigarette smoke exposure in mice, 290 (2006) L931–L945. [DOI] [PubMed] [Google Scholar]
- [4].Sundar IK, Rashid K, Sellix MT, Rahman IJB, b.r. communications, The nuclear receptor and clock gene REV-ERBα regulates cigarette smoke-induced lung inflammation, 493 (2017) 1390–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sundar IK, Maremanda KP, Rahman I, Mitochondrial dysfunction is associated with Miro1 reduction in lung epithelial cells by cigarette smoke, Toxicol Lett, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].D’Agostino B, Sullo N, Siniscalco D, De Angelis A, Rossi F, Mesenchymal stem cell therapy for the treatment of chronic obstructive pulmonary disease, Expert Opin Biol Ther, 10 (2010) 681–687. [DOI] [PubMed] [Google Scholar]
- [7].Kennelly H,Mahon BP, English K, Human mesenchymal stromal cells exert HGF dependent cytoprotective effects in a human relevant pre-clinical model of COPD, Sci Rep, 6 (2016) 38207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Alipoor SD, Mortaz E, Garssen J, Movassaghi M, Mirsaeidi M, Adcock IM, Exosomes and Exosomal miRNA in Respiratory Diseases, Mediat Inflamm, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Willis GR, Fernandez-Gonzalez A, Anastas J, Vitali SH, Liu XL, Ericsson M, Kwong A, Mitsialis SA, Kourembanas S, Mesenchymal Stromal Cell Exosomes Ameliorate Experimental Bronchopulmonary Dysplasia and Restore Lung Function through Macrophage Immunomodulation, Am J Resp Crit Care, 197 (2018) 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Matthay MA, Goolaerts A, Howard JP, Lee JW, Mesenchymal stem cells for acute lung injury: Preclinical evidence, Crit Care Med, 38 (2010) S569–S573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li X, Zhang Y, Yeung SC, Liang Y, Liang X, Ding Y, Ip MS, Tse H-F, Mak JC, Lian Q.J.A.j.o.r.c., m. biology, Mitochondrial transfer of induced pluripotent stem cell–derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke–induced damage, 51(2014)455–465. [DOI] [PubMed] [Google Scholar]
- [12].Pierro M, Ionescu L, Montemurro T, Vadivel A, Weissmann G, Oudit G, Emery D, Bodiga S, Eaton F, Péault BJT, Short-term, long-term and paracrine effect of human umbilical cord-derived stem cells in lung injury prevention and repair in experimental bronchopulmonary dysplasia, 68 (2013)475–484. [DOI] [PubMed] [Google Scholar]
- [13].Conigliaro A, Fontana S, Raimondo S, Alessandro R, Exosomes: Nanocarriers of Biological Messages, Adv Exp Med Biol, 998 (2017) 23–43. [DOI] [PubMed] [Google Scholar]
- [14].Hough KP, Trevor JL, Strenkowski JG, Wang Y, Chacko BK, Tousif S, Chanda D, Steele C, Antony VB, Dokland T, Ouyang X Zhang J Duncan SR, Thannickal VJ, Darley-Usmar VM, Deshane JS, Exosomal transfer of mitochondria from airway myeloid-derived regulatory cells to T cells, Redox Biol, 18 (2018) 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA, Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice, J Immunol, 179 (2007) 1855–1863. [DOI] [PubMed] [Google Scholar]
- [16].Ortiz LA, Gambelli F, McBride C, Gaupp D, Baddoo M, Kaminski N, Phinney DG, Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects, P Natl Acad Sci USA, 100(2003) 8407–8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sundar IK, Li D, Rahman I, Proteomic Analysis of Plasma-Derived Extracellular Vesicles in Smokers and Patients with Chronic Obstructive Pulmonary Disease, ACS Omega, 4 (2019) 10649–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim SH, Bianco N, Menon R, Lechman ER, Shufesky WJ, Morelli AE, Robbins PD, Exosomes derived from genetically modified DC expressing FasL are anti-inflammatory and immunosuppressive, Mol Ther, 13 (2006) 289–300. [DOI] [PubMed] [Google Scholar]
- [19].Braun RK, Chetty C, Balasubramaniam V, Centanni R, Haraldsdottir K, Hematti P, Eldridge MW, Intraperitoneal injection of MSC-derived exosomes prevent experimental bronchopulmonary dysplasia, Biochem Bioph Res Co, 503 (2018) 2653–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Porzionato A, Zaramella P, Dedja A, Guidolin D, Van Wemmel K, Macchi V, Jurga M, Perilongo G, De Caro R, Baraldi E, Muraca M, Intratracheal administration of clinical-grade mesenchymal stem cell-derived extracellular vesicles reduces lung injury in a rat model of bronchopulmonary dysplasia, Am J Physiol-Lung C, 316 (2019) L6–L19. [DOI] [PubMed] [Google Scholar]
- [21].Mansouri N, Willis GR, Fernandez-Gonzalez A, Reis M, Nassiri S, Mitsialis A, Kourembanas S, Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes, JCI Insight, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bazhanov N, Ylostalo JH, Bartosh TJ, Tiblow A, Mohammadipoor A, Foskett A, Prockop DJ, Intraperitoneally infused human mesenchymal stem cells form aggregates with mouse immune cells and attach to peritoneal organs, Stem Cell Res Ther, 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dougherty JA, Mergaye M, Kumar N, Chen CA, Angelos MG, Khan M, Potential Role of Exosomes in Mending a Broken Heart: Nanoshuttles Propelling Future Clinical Therapeutics Forward, Stem Cells Int, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rashid K, Sundar IK, Gerloff J, Li DM, Rahman I, Lung cellular senescence is independent of aging in a mouse model of COPD/emphysema, Sci Rep-Uk, 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sundar IK, Rahman I, Gene expression profiling of epigenetic chromatin modification enzymes and histone marks by cigarette smoke: implications for COPD and lung cancer, Am J Physiol-Lung C, 311 (2016) L1245–L1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S, Dijk F, Kalicharan D, Kelders M, Gosker H.R.J.R.r., Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells, 14 (2013) 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].van der Toorn M, Slebos D-J, de Bruin HG, Leuvenink HG, Bakker SJ, Gans RO, Koëter GH, van Oosterhout AJ, Kauffman H.F.J.A.J.o.P.-L.C., M. Physiology, Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis, 292 (2007) L1211–L1218. [DOI] [PubMed] [Google Scholar]
- [28].Ballinger SW, Bouder TG, Davis GS, Judice SA, Nicklas JA, Albertini RJ.J.C.r., Mitochondrial genome damage associated with cigarette smoking, 56 (1996) 5692–5697. [PubMed] [Google Scholar]
- [29].Hara H, Araya J, Ito S, Kobayashi K, Takasaka N, Yoshii Y, Wakui H, Kojima J, Shimizu K, Numata T.J.A.J.o.P.-L.C., M. Physiology, Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence, 305 (2013) L737–L746. [DOI] [PubMed] [Google Scholar]
- [30].Aravamudan B, Kiel A, Freeman M, Delmotte P, Thompson M, Vassallo R, Sieck GC, Pabelick CM, Prakash YJAJOP-LC, M. Physiology, Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle, 306 (2014) L840–L854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko G.R.J.T.J.o.c.i., Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD, 124(2014) 3987–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Son ES, Kim S-H, Ryter SW, Yeo E-J, Kyung SY, Kim YJ, Jeong SH Lee CS, Park J.-W.J.T.i.v., Quercetogetin protects against cigarette smoke extract-induced apoptosis in epithelial cells by inhibiting mitophagy, 48 (2018) 170–178. [DOI] [PubMed] [Google Scholar]
- [33].Ahmad T, Sundar IK, Lemer CA, Gerloff J, Tormos AM, Yao H, Rahman IJTFJ, Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease, 29(2015)2912–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, Stepanova A, Iommarini L, Mastroleo C, Daly L, Galkin A, Thakur BK, Soplop N, Uryu K, Hoshino A, Norton L, Bonafe M, Cricca M, Gasparre G, Lyden D, Bromberg J Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer, Proc Natl Acad Sci U S A, 114 (2017) E9066–E9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Heijink IH, Pouwels SD, Leijendekker C, de Bruin HG, Zijlstra GJ, van der Vaart H, ten Hacken NH, van Oosterhout AJ, Nawijn MC, van der Toorn M.J.A.j.o.r.c., m. biology, Cigarette Smoke–Induced Damage-Associated Molecular Pattern Release from Necrotic Neutrophils Triggers Proinflammatory Mediator Release, 52 (2015)554–562. [DOI] [PubMed] [Google Scholar]
- [36].Lee H, Park JR, Kim WJ, Sundar IK, Rahman I, Park SM, Yang SR Blockade of RAGE ameliorates elastase-induced emphysema development and progression via RAGE-DAMP signaling Faseb J, 31 (2017)2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cloonan SM, Choi AMK, Mitochondria in lung disease, J Clin Invest, 126 (2016) 809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Carraway MS, Suliman HB, Kliment C, Welty-Wolf KE, Oury TD, Piantadosi CAJA, r. signaling Mitochondrial biogenesis in the pulmonary vasculature during inhalational lung injury and fibrosis, 10 (2008) 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ahmad T, Sundar IK, Lerner CA, Gerloff J Tormos AM, Yao HW, Rahman I, Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease, Faseb J, 29 (2015) 2912–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Aoshiba K, Nagai A, Oxidative stress, cell death, and other damage to alveolar epithelial cells induced by cigarette smoke, Tob Induc Dis, 1 (2003) 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Las G, Shirihai OS, Miro1: New wheels for transferring mitochondria, Embo J, 33 (2014) 939–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, Rehman R, Tiwari BK, Jha KA, Barhanpurkar AP, Wani MR, Roy SS, Mabalirajan U, Ghosh B, Agrawal A, Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue effcacy, Embo J, 33 (2014) 994–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Heijink IH, Pouwels SD, Leijendekker C, de Bruin HG, Zijlstra GJ, van der Vaart H, ten Hacken NH, van Oosterhout AJ, Nawijn MC, van der Toorn M, Cigarette smoke-induced damage-associated molecular pattern release from necrotic neutrophils triggers proinflammatory mediator release, Am J Respir Cell Mol Biol, 52 (2015) 554–562. [DOI] [PubMed] [Google Scholar]