

SUMMARY
Immune responses are essential for pathogen elimination but also cause tissue damage, leading to disease or death. However, it is unclear how the host immune system balances control of infection and protection from the collateral tissue damage. Here, we show that PD-1-mediated restriction of immune responses is essential for durable control of chronic LCMV infection in mice. In contrast to responses in the chronic phase, PD-1 blockade in the subacute phase of infection paradoxically results in viral persistence. This effect is associated with damage to lymphoid architecture and subsequently decreases adaptive immune responses. Moreover, this tissue damage is type I interferon dependent, as sequential blockade of the interferon receptor and PD-1 pathways prevents immunopathology and enhances control of infection. We conclude that PD-1-mediated suppression is required as an immunoregulatory mechanism for sustained responses to chronic viral infection by antagonizing type-I interferon-dependent immunopathology.
In Brief
Using stage-specific PD-1 blockade in LCMV-infected mice, Raju et al. uncover the requirement for PD-1-mediated suppression of CD8 T cells for durable immune response to chronic viral infection, as well as the requirement for IFNAR signaling in programming of CD8 T cells toward effectors that cause immunopathology.
Graphical Abstract
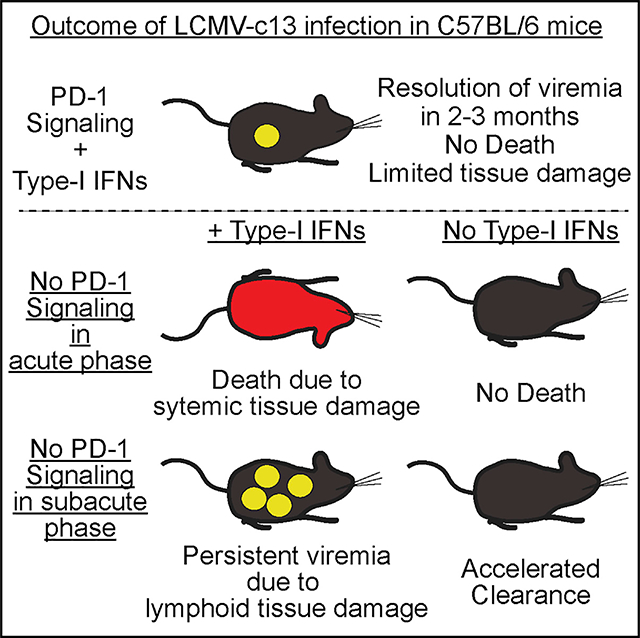
INTRODUCTION
The host immune system responds to invading pathogens through a variety of effector mechanisms that not only control infection but also cause host damage (Rouse and Sehrawat, 2010). Reduction of pathogen burden occurs primarily through immune effector mechanisms, including expression of cytokines and cytotoxic molecules (Kägi et al., 1994; Samuel, 2001). However, elimination of pathogens often causes collateral tissue damage, referred to as immunopathology, leading to significant disease or death (Doherty and Zinkernagel, 1974; Graham et al., 2005; Zinkernagel, 2005). Several regulatory mechanisms prevent excessive immune activation and development of immunopathology, including expression of inhibitory receptors and cytokines, as well as the generation of regulatory immune cells (Chen and Flies, 2013; Josefowicz et al., 2012; Moore et al., 2001). Although immunoregulatory factors restrict effector responses, the long-term consequence of attenuated activity on pathogen control is unclear.
The inhibitory receptor PD-1 is rapidly upregulated following exposure of T cells to antigen (Ag) and remains high during experimental infection of mice with rapidly replicating Lymphocytic choriomeningitis virus (LCMV) strains, such as clone 13 (Ahn et al., 2018; Barber et al., 2006). High Ag load results in decreased CD8 T cell function, termed T cell “exhaustion,” and a long-term viremia that is eventually resolved in a majority of immunocompetent mice (Matloubian et al., 1994; Moskophidis et al., 1993). A mechanistic link between PD-1-mediated inhibition of T cells and viral persistence is exemplified by the inhibition of the PD-1:PD-L1 interaction, which leads to “reinvigoration” of CD8 T cells and accelerates control of viremia (Barber et al., 2006; Lee et al., 2015). Thus, engagement of PD-1 signaling compromises viral control through attenuation of effector responses after CD8 T cells are exposed to high viral load. In contrast, infection of Pdcd1−/− or Cd274(encoding PD-L1)−/− mice with LCMVc-13 results in lethal immunopathology within several days after infection (Barber et al., 2006; Frebel et al., 2012), indicating that PD-1-dependent immune regulation is essential to restrict host damage. Thus, PD-1-mediated attenuation of antiviral responses by CD8 T cells is thought to be primarily an immunoregulatory mechanism by which the infected host minimizes tissue damage (Speiser et al., 2014), although the impact of the attenuated immunity on pathogen control remains unknown. These findings collectively imply that the consequence of PD-1-mediated regulation of immune responses depends on the context of host-microbe interactions, such as microenvironmental cues and inflammatory signals.
In this study, we sought to define the impact of PD-1 signaling on long-term viral control and the contexts that modulate its function in host-pathogen interactions. We found that loss of PD-1 signals during the early stages of chronic viral infection results in long-term viral persistence. Defective antiviral immunity resulted from atrophy of lymphoid tissues and global lymphopenia with marked loss of germinal center (GC) B cells due to excessive T cell responses. Intriguingly, a transient blockade of type I interferons (IFN-Is) not only reversed tissue damage caused by PD-1 blockade but also enhanced viral clearance in combination with a subsequent PD-1 blockade. Thus, functional outcomes of the engagement or blockade of the PD-1 pathway depend on inflammatory conditions established by IFN-Is, and thus, the PD-1 pathway must be engaged not only for host protection against immunopathology but also for durable antiviral immunity.
RESULTS
PD-1 Blockade at the Peak of Viremia Enhances CD8 T Cell Activation with No Immediate Impact on Viral Control
Cd274−/− mice die from immunopathology by infection with the chronic LCMV-c13, but not the acute LCMV-Armstrong (LCMV-Arm) strain (Barber et al., 2006). To determine whether high viral load is sufficient to induce the lethal immunopathology in the absence of PD-1 signaling, we infected C57BL/6 (B6) mice with LCMV-c13 and blocked PD-1 by injecting anti-PD-L1 monoclonal antibody (mAb) on day 8 post-infection (dpi), corresponding to the peak of viremia (Wherry et al., 2003). As a control, we infected B6 mice with LCMV-c13 and treated with anti-PD-L1 at the time of infection, recapitulating lethal immunopathology as shown using Cd274−/− mice (Figure 1A; Barber et al., 2006; Frebel et al., 2012). Depletion of CD8 T cells prior to infection completely rescued the mice from death, although depletion of CD4 T cells had little impact on their survival, confirming CD8 T-cell-dependent immunopathology (Figure S1A; Frebel et al., 2012). Although overriding PD-1-mediated suppression during the initial stages after infection achieved expansion of Ag-specific CD8 T cells on 5 dpi, it had little impact on the establishment of high viremia (Figures S1B–S1D), suggesting a minimal role of PD-1 in controlling virus in this context.
Figure 1. PD-1 Blockade at the Peak of Viremia Enhances CD8 T Cell Activation with No Immediate Impact on Viral Control.

(A) Survival LCMV-c13-infected B6 mice following anti-PD-L1 treatment. n = 25 (control), n = 6 (days 0–8), and n = 30 (days 8–22) pooled from 3 experiments.
(B) Survival of LCMV-c13-infected B6 mice after adoptive transfer of unexhausted LCMV-specific CD8 T cells and treatment with anti-PD-L1 or isotype control. Data are from 3 experiments.
(C) Expression of CD4, CD8, CD44, and LCMV-gp276-specific T cell receptor (TCR) in peripheral blood mononuclear cells (PBMCs) from control and anti-PD-L1-treated mice on 14 dpi with LCMV-c13 infection. Representative plots are shown with mean ± SD from 3 experiments.
(D and E) Expression of CD4, CD8, CD44, PD-1, LAG-3, and LCMV-gp33-specific TCR in PBMCs on 22 dpi. Representative plots (D) and mean ± SD from 2 experiments (E) are shown.
(F) Plasma viral RNA load in LCMV-c13-infected mice on 22 dpi. Data pooled from 5 experiments are shown with median. The statistical difference was tested by the Mann-Whitney U test.
See also Figure S1.
In marked contrast, the majority of mice survived when anti-PD-L1 treatment started on 8 dpi (Figure 1A), indicating that high viral load is insufficient to cause death of the infected mice. To rule out the possibility that the lack of death resulted from resistance of target tissues or reduced Ag presentation, we transferred unexhausted CD8 T cells harvested from LCMV-Arm-infected mice into LCMV-c13-infected mice and subsequently treated the recipient mice with anti-PD-L1. All LCMV-c13-infected recipient mice died quickly after anti-PD-L1 treatment (Figure 1B), indicating that the resistance to lethal pathology of mice treated with anti-PD-L1 on 8–22 dpi is independent of T-cell-extrinsic resistance of the infected animals.
The increased frequency of total and LCMV-specific CD8 T cells in the peripheral blood on 14 dpi in mice with anti-PDL1 treatment started on 8 dpi indicated that virus-specific CD8 T cells are suppressed by PD-1 signaling during this period (Figure 1C). However, the increase in CD8 T cells was no longer observed on 22 dpi with expression of PD-1 and LAG-3 by LCMV-specific CD8 T cells being elevated in anti-PD-L1-treated compared with control antibody-treated or untreated mice (Figures 1D and 1E). PD-1 blockade has been shown to promote pathogen clearance in certain infection models, including LCMV-c13 infection (Barber et al., 2006; Bhadra et al., 2011; Fuller et al., 2013). However, plasma viral titers were unchanged on day 22 dpi (Figure 1F). These results indicate that engagement of the PD-1 pathway during the acute and subacute phases has little immediate impact on control of viral infection.
PD-1 Signaling Protects Host from Sustained Immunopathology in Lymphoid Organs
Although the majority of LCMV-infected mice treated with anti-PD-L1 initiated on 8 dpi survived, we found a significant delay in recovery of body weight, suggesting that PD-1 blockade during this period causes non-lethal immunopathology (Figure S2A). Lymphoid organs are transiently disrupted following LCMV-c13 infection due predominantly to T-cell-mediated tissue damage (Müller et al., 2002; Teijaro et al., 2013; Wilson et al., 2013; Zinkernagel et al., 1999). We thus determined whether excessive loss of the integrity of lymphoid organs, presumably impacting subsequent durable immunity, is a direct consequence of PD-1 blockade. Indeed, spleen sizes and splenocyte numbers in anti-PD-L1-treated mice were significantly reduced compared to controls (Figures 2A and 2B). Histological analyses of spleens on 14 and 22 dpi revealed smaller B cell follicles and disrupted red and white pulp boundaries in anti-PD-L1-treated animals (Figures 2C and S2B). This tissue damage in the spleen was CD8 T cell dependent because it was prevented by depletion of CD8 T cells prior to infection (Figure S2C). Frequencies of LCMV-specific CD8 T cells, including “terminally exhausted” PD-1+ TIM-3+ cells and stem-cell-like PD-1+ TCF-1+ cells (Im et al., 2016; Utzschneider et al., 2016; Wu et al., 2016), were similar in control and treated mice (Figures 2D and 2E). However, absolute numbers of these populations were diminished in proportion to reductions in spleen size and cellularity (Figure 2E).
Figure 2. PD-1 Blockade at the Peak of Viral Load Results in Reduced Spleen Sizes and Pan-lymphopenia.

(A) Gross images of spleens from control and anti-PD-L1-treated mice on 22 dpi.
(B) Splenocyte counts of control and anti-PD-L1-treated mice on 22 dpi. n > 8 mice per group, shown by mean and SD.
(C) H&E staining of spleen sections from control and day 8–22 anti-PD-L1-treated mice on 22 dpi. Scale bars, 200 μm. Representative images of 6 mice per group are shown.
(D) Expression of CD44 and LCMV-gp33-specific TCR of total splenocytes and expression of TCF-1 and TIM-3 by LCMV-gp33-specific CD8 T cells in control and anti-PD-L1-treated mice on 22 dpi.
(E) Statistical analysis of frequencies and absolute numbers of the indicated cell populations pooled from 2 experiments. n = 4–6 mice per group.
(F) Expression of IFN-γ and TNF-α by splenocytes from LCMV-c13-infected control and anti-PD-L1-treated B6 mice following stimulation with the gp276–284 peptide. Representative data from 2 experiments are shown with mean ± SD; n = 4.
(G) Expression of Ki-67 by gp33-specific CD8 T cells.
(H) Frequencies of CD45.2 PD-1+ donor CD8 T cells in PBMCs of CD45.1-recipient mice that received PD-1+ CD8 T cells from LCMV-c13-infected anti-PD-L1-treated B6 mice and subsequently challenged with LCMV-c13. Data are pooled from two experiments.
(I) Expression of Fas and GL7 expression in B220+ splenocytes from control and anti-PD-L1-treated mice. Data from 2 experiments are shown with mean and SD.
(J) Anti-LCMV IgG2c antibody titers on 22 dpi.
See also Figure S2.
It has now become clear that the consequences of loss of PD-1 signaling differ depending on contexts in which the signal is blocked. Although the blockade at later time points in LCMV infection or in the tumor microenvironment enhances CD8 T cell responses, its constitutive loss paradoxically leads to more pronounced cell-intrinsic dysfunction of CD8 T cells (Barber et al., 2006; Odorizzi et al., 2015). To determine whether intrinsic CD8 T cell function is altered by PD-1 blockade during the subacute period, we analyzed cytokine production ex vivo and proliferative capacity of CD8 T cells in vivo by adoptive transfer to congenic mice followed by LCMV-c13 infection. Immediately following blockade of PD-L1, we detected increased frequencies of tumor necrosis factor alpha (TNF-α)+ interferon (IFN)-γ+ co-producing CD8 T cells ex vivo as well as an increased frequency of Ki-67+ LCMV-specific CD8 T cells (Figures 2F and 2G), suggesting PD-1 blockade started on 8 dpi resulted in enhanced activity of Ag-specific CD8 T cells. Additionally, adoptively transferred PD-1+ CD8 T cells from control or anti-PD-L1-treated mice were capable of comparable secondary expansion in the recipient mice (Figure 2H), indicating that Ag-specific CD8T cells preserved polyfunctionality and proliferative capacity, regardless of PD-1 blockade during this period. These results suggest that the decreased overall CD8 T cell response in the treated mice is secondary to tissue damage in lymphoid organs caused by CD8 T cells.
CD8 T cells also directly target Ag-specific B cells in LCMV infection and thus impair antibody responses (Moseman et al., 2016). We hypothesized PD-1 blockade exacerbates CD8 T-cell-dependent cytotoxicity to B cells. Consistently, despite comparable frequencies of CD8 T cells and total B220+ B cells, we observed a significant reduction in the frequencies and numbers of GL7+ Fas+ GC B cells known to be directly infected by LCMV (Moseman et al., 2016) as well as in anti-LCMV IgG2c titers (Figures 2I and 2J). Although PD-1 blockade could also act directly on B cells and cause their death by hyperactivation, we did not detect PD-1 expression on B cells at this stage of infection, making this explanation unlikely (Figure S2D). Moreover, depletion of CD8 T cells prior to infection restored GC B cell numbers and anti-LCMV IgG2c titers (Figures S2E and S2F), indicating that the GC B cell reduction was mediated by Ag-specific killing by uninhibited CD8 T cells. These data collectively indicate that PD-1 signaling during the subacute stage of LCMV infection protects hosts from excess tissue damage, particularly in the lymphoid organs that provide a microenvironment for proper adaptive immune responses. Thus, engagement of the PD-1 pathway may be an essential component of protective immunity to control persistent viral infection targeting lymphoid organs.
Engagement of PD-1/PD-L1 Is Essential for Control of Chronic LCMV Infection
We have shown that PD-1 is necessary to prevent tissue damage with no immediate impact on viral control. We thus hypothesized that insufficient adaptive immunity resulting from the defective lymphoid tissue integrity impairs long-term control of virus. Accordingly, we conducted a longitudinal analysis of viremia using different regimens of PD-1 blockade. As demonstrated previously (Barber et al., 2006), PD-1 blockade late in the infection, initiated on 23 dpi, accelerated resolution of viremia, with viral RNA undetectable by 50 dpi (Figure 3A, blue dots). In sharp contrast, PD-1 blockade initiated on 8 dpi impaired viral control with a majority of infected mice exhibiting viral persistence (Figure 3A, red dots). Thus, PD-1 engagement in the subacute phase of infection, after T cell priming and initial clonal expansion, is required to preserve durable antiviral responses.
Figure 3. Engagement of PD-1 in the Subacute Phase Is Required for Durable Control of LCMV Infection.

(A) Plasma viral RNA load in LCMV-c13-infected B6 mice treated with anti-PD-L1 on 8–22 dpi (red dots, same as Figure 1) or 23–37 dpi (blue dots). Data are pooled from 5 experiments shown with median.
(B–D) Expression of CD4, CD8, CD44, PD-1, LAG3, and LCMV-gp33-specific TCR by splenocytes (B and C) and peripheral blood mononuclear cells (D) on day 37 of LCMV-c13 infection. Representative plots are shown with median ± SD from at least 2 experiments.
To further delineate the effects of a temporary PD-1 blockade on 8–22 dpi, we first examined how function of Ag-specific CD8 T cells was impacted. It has been shown that loss of PD-1 signaling depletes T-bet+ CD8 T cells, resulting in defective antiviral responses (Odorizzi et al., 2015). To this end, similar to analysis on 22 dpi, frequencies of LCMV-specific CD8 T cells, including those of PD-1+ TCF-1+ and T-bet+ CD8 T cells, were comparable between 8–22 dpi treated and control groups in the spleen on 37 dpi (Figures 3B and 3C). However, expression of TIM-3 by Ag-specific CD8 T cells in the spleen was increased in the treated mice (Figures 3B and 3C), suggesting an accumulation of severely exhausted cells due to the changes in the microenvironment. This change of CD8 T cells in the spleen was reflected by more pronounced reduction of both total and LCMV-specific CD8 T cells in the peripheral blood (Figure 3D). Thus, it is likely that the decline in CD8 T cells and diminished anti-LCMV antibody responses led to defective viral control in mice treated with anti-PD-L1 between 8–22 dpi. Together, our results indicate that engagement of the PD-1 pathway in the face of persistent Ag is essential for durable antiviral responses by generating requisite numbers of adaptive immune cells through the maintenance of lymphoid tissue integrity.
IFN-Is Are Required for Anti-PD-L1-Mediated Immunopathology
Our results demonstrate the requirement of PD-1 signaling for protecting the host from tissue damage that compromises antiviral responses. However, immunopathology preferentially occurred during early time points in the infection, potentially due to elevated inflammatory cytokines. Among the cytokines induced upon viral infection, IFN-Is are transiently produced through by plasmacytoid dendritic cells (pDCs) and other myeloid cells upon infection by LCMV-c13 (Macal et al., 2012; Teijaro et al., 2013; Wang et al., 2012; Wilson et al., 2013; Ng et al., 2015). The expression of IFN-Is peaks in the acute stage and would thus be consistent with their roles in enhancing immunopathology following anti-PD-L1 treatment in the acute phase (Baccala et al., 2014; Oldstone et al., 2018). To first test whether IFN-Is were required for lethal immunopathology in LCMV-c13-infected mice resulting from PD-1 blockade, we combined administration of blocking mAbs against interferon alpha/beta receptor 1 (IFNAR) immediately before infection (day −1) and PD-L1 on 0–8 dpi. In striking contrast to mice treated with anti-PD-L1 alone, almost all infected mice were rescued from death by the co-treatment (Figure 4A), indicating that IFN-Is enhance effector function of Ag-specific CD8 T cells.
Figure 4. Type I Interferon Receptor Blockade Rescues Immunopathology Caused by Loss of PD-1 Signaling in LCMV-Infected Mice.

(A) Survival of LCMV-c13-infected B6 mice treated with anti-IFNAR 1 day prior to infection, anti-PD-L1 (days 0, 2, 4, 6, and 8), or both. Data pooled from 3 experiments with n > 6 mice per group are shown.
(B and C) Frequencies of total CD8 T cells, LCMV-gp33-specific CD8 T cells, and PD-1+ cells in indicated PBMC populations on 8 dpi in LCMV-c13-infected B6 mice with indicated treatment. Representative plots (B) with mean ± SD from 2 experiments (C) are shown.
(D) Plasma LCMV viral RNA load in LCMV-c13-infected B6 mice with indicated treatment from 2 experiments shown with median.
(E) Macroscopic images of spleen and splenocyte counts of LCMV-c-13-infected B6 mice with indicated treatment as in (A) on 22 dpi. Data from two experiments are shown.
See also Figures S3 and S4.
We next examined how loss of IFNAR signaling impacted the CD8 T cell response to LCMV-c13 infection. We found reduced frequencies of total and LCMV-specific CD8 T cells in peripheral blood on 8 dpi in anti-IFNAR-treated mice (Figure 4B). This diminished CD8 T cell expansion was independent of natural killer (NK)-cell-dependent cytotoxicity, as CD8 T cell frequencies remained unchanged with depletion of NK cells (Crouse et al., 2014; Madera et al., 2016; Xu et al., 2014; Figure S3A). In Ifnar1−/− mice, splenocyte counts were increased by two-fold, consistent with diminished CD8 T-cell-mediated tissue damage, although total and LCMV-specific CD8 T cell numbers were reduced (Figures S3B–S3E). Among LCMV-specific CD8 T cells, we observed skewed differentiation into TCF-1+ memory-like CD8 T cells over TIM-3+ effector/exhausted cells (Figures S3B–S3E), consistent with a previous study (Wu et al., 2016). These results suggest that differentiation of activated CD8 T cells into effectors is suppressed in the absence of IFNAR signaling. In addition, LCMV-specific CD8 T cells deprived of IFNAR signaling expressed substantially higher PD-1 compared to control CD8 T cells (Figure 4B) and therefore may be more sensitive to PD-L1-mediated suppression. Consistently, additional blockade of PD-1 signaling rescued the reduction in LCMV-specific CD8 T cells without impacting total CD8 T cell frequencies (Figures 4B and 4C).
We next sought to establish which cellular compartments IFN-Is target to cause immunopathology. Bone marrow (BM) chimeras reconstituted with Ifnar1−/− hematopoietic cells died following LCMV-c13 infection and anti-PD-L1 treatment with similar kinetics to control chimeric mice reconstituted with wild-type (WT) BM cells (Figure S4A). Although Ifnar1−/− host mice reconstituted with Ifnar1−/− hematopoietic cells were resistant to PD-1-blockade-mediated death as expected, Ifnar1−/− hosts reconstituted with WT BM cells also succumbed to PD-1 blockade following LCMV-c13 infection (Figure S4B). These results collectively indicate that IFN-Is program enhanced CD8 T cell effector responses by acting on both non-hematopoietic and hematopoietic cells and in part by restricting induction of PD-1 in activated CD8 T cells to desensitize them to PD-L1-mediated suppression.
To determine whether early PD-1 blockade can promote control of infection in the absence of excess tissue damage, we tracked antiviral responses following anti-IFNAR and anti-PD-L1 treatment. As shown previously (Teijaro et al., 2013; Wilson et al., 2013), anti-IFNAR alone on day −1 prior to infection accelerated resolution of viremia compared to control (Figure 4D). The viral control was further enhanced by combination of anti-IFNAR on day −1 with anti-PD-L1 on 8 dpi. In contrast, anti-IFNAR administration on 7 dpi, at which time IFN-Is are already undetectable in serum (Macal et al., 2012; Teijaro et al., 2013; Wang et al., 2012; Wilson et al., 2013), had no effect (Figure 4D). Consistently, atrophy of the spleen caused by anti-PD-L1 was completely rescued in mice co-treated with anti-IFNAR and anti-PD-L1 (Figure 4E). These results suggest IFN-Is as a target to tune the outcomes of immune checkpoint blockade.
DISCUSSION
Our data demonstrate a critical, temporal function of PD-1 in the regulation of host immunity against persistent infections. PD-1 is a major inhibitory receptor that causes attenuated CD8 T cell responses to some infections and tumors. Blockade of the PD-1/PD-L1 interaction to restore function of exhausted CD8 T cells was originally demonstrated using a mouse chronic LCMV infection model and has now been applied broadly as a cancer immunotherapy in humans. However, the biological significance of PD-1-mediated suppression on long-term control of persistent viral infection is understudied. We now demonstrate that PD-1-mediated restriction of T cell responses is beneficial for control of chronic infection.
Immunopathology in the LCMV-c13 infection model is a consequence of abundant and broad expression of viral Ag across different cell types. LCMV is a non-cytopathic virus, and the pathogenesis of LCMV infection is predominantly host T cell response dependent, as is the case with some viral infections in humans, such as hepatitis B virus (HBV), HCV, and HIV infections. Context-dependent PD-1-mediated protection through restriction of immunopathology may be beneficial to establish durable immunity against those infections, although the benefit of restraining host T cell responses may not be applicable to all viral infections. Constitutive absence of PD-1 signal causes severe exhaustion of CD8 T cells in a cell-intrinsic manner (Odorizzi et al., 2015). In contrast, our results show the requirement for PD-1 to preserve CD8 T cell responses in a T-cell-extrinsic manner and thus reveal the additional role of immune regulation in durable antiviral immunity. Collectively, restriction of CD8 T cell function by PD-1 or acquisition of the exhausted state is a host protective mechanism in response to persistent Ag, for which long-term durable immunity is prioritized at the expense of immediate immune responses.
Our results also show that IFN-Is enhance immunopathology in the setting of PD-1/PD-L1 deficiency. Sequential blockade of IFNAR and PD-1 signaling synergistically improved control of LCMV infection, further supporting our conclusion that preventing initial immunopathology is critical for overall antiviral responses, even if it temporarily compromises viral control. Although IFNAR is broadly expressed in both hematopoietic and non-hematopoietic compartments, its expression in either compartment was sufficient to cause lethal immunopathology in LCMV-c13-infected mice depleted of PD-1 signaling. IFNAR signaling thus likely contributes to maximizing CD8 T cell responses through multiple mechanisms, including effector differentiation and modulation of PD-1 expression by CD8 T cells. Although blockade of IFNAR signaling enhances control of persistent LCMV infection (Teijaro et al., 2013; Wilson et al., 2013), the therapeutic effects appear to be mediated by regulation of CD8 T cells and preservation of host immune functions, similar to PD-1-mediated suppression, rather than direct enhancement of T cell function. This conclusion is supported by protection of LCMV-specific B cells by IFNAR blockade (Fallet et al., 2016; Moseman et al., 2016). The IFN-I pathway could be a potential target for modulation of immunotherapies against infections and cancer beyond its direct effect on the virus or transformed cells.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Takeshi Egawa (egawat@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Male C57BL/6N and B6-CD45.1 mice were purchased from Charles River Laboratory. Ifnar1−/− were obtained from Michael S. Diamond (Washington University). All mice were housed in a specific pathogen-free facility at Washington University in St. Louis, and were used at 8 to 10 weeks of age, unless stated otherwise. We used both males and females except that only male mice were used as recipient mice for bone marrow chimeras to minimize the risk of graft rejection due to the male antigen. All experiments were performed according to a protocol approved by Washington University’s Animal Studies Committee.
METHOD DETAILS
Mouse Infection and Viral Load Quantification
Stocks of LCMV were made by propagating virus in BHK (baby hamster kidney) cells, followed by titering of culture supernatants by focus forming assay on Vero (African green monkey kidney) cells. For LCMV infection, mice were infected with 2 × 105 plaque-forming units (PFU) of LCMV-Arm via the intraperitoneal route or 2 × 106 (PFU) of LCMV-c13 by intravenous injection. For the quantification of plasma viral load, RNA was extracted from 10 μL of plasma using Trizol (Life Technologies). Before RNA extraction a spike-in of exogenous control mRNA encoding clover was added to the plasma samples as previously described. The amounts of the LCMV gp transcript relative to that of ‘spiked- in’ clover RNA were determined by real-time qRT-PCR using qScript cDNA synthesis and Luminaris HiColor Green Master Mix as previously described (Chou et al., 2016; McCausland and Crotty, 2008; Raju et al., 2018).
Antibody treatments
200 μg of anti-PD-L1 (29F.1.G2, BioXcell) was injected every day or every other day for treatments initiated on day 0, otherwise administration was every three days for a full two weeks of treatment. anti-IFNAR (MAR1, Leinco) was intraperitoneally injected at the indicated time points after infection at a dose of 1 mg. For depletion of CD4 T cells, 200 μg of anti-CD4 (GK1.5, Leinco) was injected on day −1 and +1 of infection. For depletion of CD8 T cells, 400 μg of anti-CD8 (YTS-169, Leinco) was injected on a day before infection. For NK cell depletion, 100 μg of anti-NK1.1 (PK136, Leinco) was injected on a day before infection.
Histology
Organs were fixed in 4% PFA overnight, then dehydrated in 70% ethanol. Samples were subject paraffin embedding and H&E staining using standard methods performed by the Developmental Biology Histology Core at Washington University in St. Louis. Images were collected using an EVOS FL Auto Imaging System (Thermofisher).
Cell Preparation, Staining, and Flow Cytometry
Single-cell suspensions were prepared by manual disruption of spleens with frosted glass slides. Absolute live cell counts were determined by Trypan-blue exclusion using Vi-Cell (Beckman Coulter). Tetramer staining was performed using iTag-PE LCMV gp33–44 and gp276–284 (MBL international) for 1 hour at RT followed by surface staining was performed at 4°C for 30 minutes. For intracellular cytokine staining, splenocytes were cultured in RPMI-1640 supplemented with 10% fetal bovine serum in the presence of 1 ug/ml of LCMV-gp peptide (Genscript) and 5 ug/ml of Brefeldin A (Biolegend) for 4 hours. Cells were stained for surface makers and then subject to LIVE/DEAD Aqua staining (Thermofisher) for 30 minutes at 4°C before being fixed with 4% PFA for 10 minutes at RT. Cells were then washed twice with 0.03% saponin in 2% FBS/PBS before being stained with the indicated antibodies in 0.3% Saponin in 2% FBS/PBS for 20 min at 4°C. Staining for transcription factors was performed using the Foxp3 staining kit (eBioscience) according to the manufacturer’s instructions. Stained samples were analyzed with FACS LSR Fortessa or X20 (BD) and data processing with FlowJo Software (FlowJo. LLC).
Anti-LCMV antibody ELISA
Anti-LCMV antibody ELISA was performed as described previously (Raju et al., 2018). Briefly, Nunc Polysorp plates were coated with 10ug/mL sonicated cell lysate from LCMV-infected BHK-21 cells as capture antigen or uninfected BHK-21 cell lysate overnight followed by UV irradiation (300 mJ in Stratalinker 1800; Stratagene). Plasma antibody was detected with biotinylated anti-mouse IgG2c antibody (1077–08; Southern Biotech), followed by Streptavin-HRP (BD), and TMB substrate (Dako). Endpoint titers were calculated by a sigmoidal-dose response curve using Graphpad Prism software.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using Prism (Graphpad Software). P-values were calculated with an unpaired two-tailed Student’s t test or the Mann-Whitney U-test for two-group comparisons and by one-way ANOVA for multi-group comparisons with the Tukey post hoc test. *p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
DATA AND CODE AVAILABILITY
This study did not generate code or new datasets.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-B220-FITC | Biolegend | CAT#103206; RRID:AB_312991 |
| Anti-B220-PB | Biolegend | CAT#103227; RRID:AB_492876 |
| Anti-CD4-BV605 | Biolegend | CAT#100451; RRID:AB_2564591 |
| Anti-CD44-AF700 | Biolegend | CAT#103026; RRID:AB_493713 |
| Anti-CD8a-BUV395 | BD | CAT#563786; RRID:AB_2732919 |
| Anti-CD8a-FITC | Biolegend | CAT#100706; RRID:AB_312745 |
| Anti-CD8b-PerCP-Cy5.5 | Biolegend | CAT#126610; RRID:AB_2260149 |
| Anti-EOMES-PerCP-e710 | Invitrogen | CAT#46–4875–82; RRID:AB_10597455 |
| Anti-Fas-PE-Cy7 | BD | CAT#557653; RRID:AB_396768 |
| Anti-IFNg-PE | Biolegend | CAT#505808; RRID:AB_315402 |
| Anti-Ki-67-APC | BD | CAT#561126; RRID:AB_10611874 |
| Anti-LAG-3-BV421 | Biolegend | CAT#125221; RRID:AB_2572080 |
| Anti-mouse IgG2c-biotin | SOUTHERN BIOTECHNOLOGY | CAT#1077–08; RRID:AB_2794453 |
| Anti-PD1-PE-Cy7 | Biolegend | CAT#135216; RRID:AB_10689635 |
| anti-Rabbit-IgG-A488 | Invitrogen | CAT#A27034; RRID:AB_2536097 |
| Anti-T-bet-APC | Biolegend | CAT#644814; RRID:AB_10901173 |
| Anti-TCF-1 | CST | CAT#2203S; RRID:AB_2199302 |
| Anti-TIM3-BV421 | Biolegend | CAT#119723; RRID:AB_2616908 |
| Anti-TNFa-APC | Biolegend | CAT#506308; RRID:AB_315429 |
| GL7-A647 | Biolegend | CAT#144606; RRID:AB_2562185 |
| Streptavidin-HRP | BD | CAT#554066 |
| Anti-CD4 (for In vivo administration) | Leinco | CAT#C1333; RRID:AB_2737452 |
| Anti-CD8 (for In vivo administration) | Leinco | CAT#C2442 |
| Anti-IFNAR (for In vivo administration) | Leinco | CAT#I-401; RRID:AB_2491621 |
| Anti-NK1.1 (for In vivo administration) | Leinco | CAT#N123; RRID:AB_2737553 |
| Anti-PD-L1 (for In vivo administration) | BioXcell | CAT#BE0101; RRID:AB_10949073 |
| Isotype control (for In vivo administration) | BioxCell | CAT#BE0090; RRID:AB_1107780 |
| Bacterial and Virus Strains | ||
| LCMV-Armstrong | Marco Colonna’s Lab | N/A |
| LCMV-clone 13 | Marco Colonna’s Lab | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Brefeldin A | Biolegend | CAT#420601 |
| H2-Db (LCMV gp33–41) tetramer | MBL International | CAT#TB-5002–1 |
| H2-Db(LCMV gp276–284) tetramer | MBL International | CAT#TB-5009–1 |
| Fetal Bovine Serum | Thermofisher | CAT#10437028 |
| LCMV gp276–284 peptide | Genscript | CAT#RP19983 |
| LCMV gp33–41 peptide | Genscript | CAT#RP20257 |
| Linear Acrylamide | Ambion | CAT#AM9520 |
| LIVE/DEAD Aqua | Invitrogen | CAT#L34966 |
| RPMI-1640 | Thermofisher | CAT#SH3009601 |
| TMB Substrate Dako | Dako | CAT#T03026 |
| Critical Commercial Assays | ||
| Foxp3/transcription factor staining buffer | Thermofisher | CAT#00–5523–00 |
| Luminaris Color HiGreen qPCR Master Mix | Thermofisher | CAT#K0394 |
| Negative Selection CD8 Kit | Biolegend | CAT#480008 |
| qScript cDNA SuperMix | Quantabio | CAT#95048–100 |
| Experimental Models: Cell Lines | ||
| BHK cells | Marco Colonna’s Lab | N/A |
| Vero Cells | Marco Colonna’s Lab | N/A |
| Experimental Models: Organisms/Strains | ||
| B6-CD45.1 | Charles River | CAT#564 |
| C57BL/6NCr | Charles River | CAT#556 |
| Ifnar1−/− | Michael Diamond’s Lab | N/A |
| Oligonucleotides | ||
| GFP FWD primer: GACAACCACTACCTGAGCCA | Raju et al., 2018 | N/A, Sigma Custom Synthesis |
| GFP REV primer: GTCCATGCCATGTGTAATCC | Raju et al., 2018 | N/A, Sigma Custom Synthesis |
| LCMV gp FWD primer: CATTCACCTGGACTTTGTCAGACTC | McCausland and Crotty, 2008 | N/A, Sigma Custom Synthesis |
| LCMV gp REV primer: GCAACTGCTGTGTTCCCGAAAC | McCausland and Crotty, 2008 | N/A, Sigma Custom Synthesis |
| Software and Algorithms | ||
| Prism, version 8 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Flowjo, version 10 | FlowJo, LLC | https://www.flowjo.com/ |
Highlights.
Host response to PD-1 blockade is timing dependent in LCMV-c13 infection
PD-1 blockade in the subacute period leads to persistent viremia
The impaired antiviral response results from lymphoid tissue damage by CD8 T cells
Blocking IFNAR rescues LCMV-infected mice from immunopathology by PD-1 blockade
ACKNOWLEDGMENTS
We thank M. Colonna and M. Cella for LCMV stocks; M.S. Diamond for Ifnar1−/− mice; J. Boon, C. Fujii, and M. Holmgren for technical support; and C.-S. Hsieh, E.M. Oltz, and E. Tonc for discussion and critical reading of the manuscript. This study was supported by United States National Institutes of Health (NIH) grants R01AI130152-01A1 and R03AI139875-01 (to T.E.), T32HL007317 (to S.R.), and T32GM007200 (to S.R. and D.J.V.). T.E. is a Scholar of the Leukemia and Lymphoma Society (https://www.lls.org, United States). The ORCID for T.E. is 0000-0001-7489-1051.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.092.
REFERENCES
- Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, Sharpe AH, Freeman GJ, Irving BA, and Ahmed R (2018). Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl. Acad. Sci. USA 115, 4749–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccala R, Welch MJ, Gonzalez-Quintial R, Walsh KB, Teijaro JR, Nguyen A, Ng CT, Sullivan BM, Zarpellon A, Ruggeri ZM, et al. (2014). Type I interferon is a therapeutic target for virus-induced lethal vascular damage. Proc. Natl. Acad. Sci. USA 111, 8925–8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, and Ahmed R (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687. [DOI] [PubMed] [Google Scholar]
- Bhadra R, Gigley JP, Weiss LM, and Khan IA (2011). Control of Toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1-PDL-1 blockade. Proc. Natl. Acad. Sci. USA 108, 9196–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, and Flies DB (2013). Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol 13, 227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C, Verbaro DJ, Tonc E, Holmgren M, Cella M, Colonna M, Bhattacharya D, and Egawa T (2016). The transcription factor AP4 mediates resolution of chronic viral infection through amplification of germinal center B cell responses. Immunity 45, 570–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, Von Laer D, Kalinke U, Vivier E, Jonjic S, and Oxenius A (2014). Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity 40, 961–973. [DOI] [PubMed] [Google Scholar]
- Doherty PC, and Zinkernagel RM (1974). T-cell-mediated immunopathology in viral infections. Transplant. Rev. 19, 89–120. [DOI] [PubMed] [Google Scholar]
- Fallet B, Narr K, Ertuna YI, Remy M, Sommerstein R, Cornille K, Kreutzfeldt M, Page N, Zimmer G, Geier F, et al. (2016). Interferon-driven deletion of antiviral B cells at the onset of chronic infection. Sci. Immunol 1, eaah6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, Wagner CA, Loffing-Cueni D, Kurrer M, Ludewig B, and Oxenius A (2012). Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J. Exp. Med 209, 2485–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MJ, Callendret B, Zhu B, Freeman GJ, Hasselschwert DL, Satterfield W, Sharpe AH, Dustin LB, Rice CM, Grakoui A, et al. (2013). Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death-1 (PD-1). Proc. Natl. Acad. Sci. USA 110, 15001–15006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham AL, Allen JE, and Read AF (2005). Evolutionary causes and consequences of immunopathology. Ann. Rev. Ecol. Evol. Syst 36, 373–397. [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu L-F, and Rudensky AY (2012). Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol 30, 531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kägi D, Ledermann B, Bürki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, and Hengartner H (1994). Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369, 31–37. [DOI] [PubMed] [Google Scholar]
- Lee J, Ahn E, Kissick HT, and Ahmed R (2015). Reinvigorating exhausted T cells by blockade of the PD-1 pathway. For. Immunopathol. Dis. Therap 6, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macal M, Lewis GM, Kunz S, Flavell R, Harker JA, and Zúñiga EI (2012). Plasmacytoid dendritic cells are productively infected and activated through TLR-7 early after arenavirus infection. Cell Host Microbe 11, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madera S, Rapp M, Firth MA, Beilke JN, Lanier LL, and Sun JC (2016). Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J. Exp. Med 213, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M, Concepcion RJ, and Ahmed R (1994). CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol 68, 8056–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCausland MM, and Crotty S (2008). Quantitative PCR technique for detecting lymphocytic choriomeningitis virus in vivo. J. Virol. Methods 147, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KW, de Waal Malefyt R, Coffman RL, and O’Garra A (2001). Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol 19, 683–765. [DOI] [PubMed] [Google Scholar]
- Moseman EA, Wu T, de la Torre JC, Schwartzberg PL, and McGavern DB (2016). Type I interferon suppresses virus-specific B cell responses by modulating CD8+ T cell differentiation. Sci. Immunol 1, eaah3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskophidis D, Lechner F, Pircher H, and Zinkernagel RM (1993). Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 362, 758–761. [DOI] [PubMed] [Google Scholar]
- Müller S, Hunziker L, Enzler S, Bühler-Jungo M, Di Santo JP, Zinkernagel RM, and Mueller C (2002). Role of an intact splenic microarchitecture in early lymphocytic choriomeningitis virus production. J. Virol 76, 2375–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S, Sheehan KCF, Schreiber RD, and Oldstone MBA (2015). Blockade of interferon beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe 17, 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorizzi PM, Pauken KE, Paley MA, Sharpe A, and Wherry EJ (2015). Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 212, 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldstone MBA, Ware BC, Horton LE, Welch MJ, Aiolfi R, Zarpellon A, Ruggeri ZM, and Sullivan BM (2018). Lymphocytic choriomeningitis virus clone 13 infection causes either persistence or acute death dependent on IFN-1, cytotoxic T lymphocytes (CTLs), and host genetics. Proc. Natl. Acad. Sci. USA 115, E7814–E7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju S, Kometani K, Kurosaki T, Shaw AS, and Egawa T (2018). The adaptor molecule CD2AP in CD4 T cells modulates differentiation of follicular helper T cells during chronic LCMV infection. PLoS Pathog. 14, e1007053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse BT, and Sehrawat S (2010). Immunity and immunopathology to viruses: what decides the outcome? Nat. Rev. Immunol 10, 514–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE (2001). Antiviral actions of interferons. Clin. Microbiol. Rev 14, 778–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speiser DE, Utzschneider DT, Oberle SG, Münz C, Romero P, and Zehn D (2014). T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat. Rev. Immunol 14, 768–774. [DOI] [PubMed] [Google Scholar]
- Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KCF, Welch M, Schreiber RD, de la Torre JC, and Oldstone MBA (2013). Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427. [DOI] [PubMed] [Google Scholar]
- Wang Y, Swiecki M, Cella M, Alber G, Schreiber RD, Gilfillan S, and Colonna M (2012). Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 11, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, and Ahmed R (2003). Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol 77, 4911–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, and Brooks DG (2013). Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, et al. (2016). The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol 1, eaai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HC, Grusdat M, Pandyra AA, Polz R, Huang J, Sharma P, Deenen R, Köhrer K, Rahbar R, Diefenbach A, et al. (2014). Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 40, 949–960. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM (2005). Immunology and immunity against infection: general rules. J. Comput. Appl. Math 184, 4–9. [Google Scholar]
- Zinkernagel RM, Planz O, Ehl S, Battegay M, Odermatt B, Klenerman P, and Hengartner H (1999). General and specific immunosuppression caused by antiviral T-cell responses. Immunol. Rev 168, 305–315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate code or new datasets.