

Abstract
Controlling which particular members of a large protein family are targeted by a drug is key to achieving a desired therapeutic response. In this study, we report a rational data-driven strategy for achieving restricted polypharmacology in the design of anti-tumor agents selectively targeting the TYRO3, AXL and MERTK (TAM) family tyrosine kinases. Our computational approach, based on the concept of FRAgments in Structural Environments (FRASE), distills relevant chemical information from structural and chemogenomic databases to assemble a three-dimensional inhibitor structure directly in the protein pocket. Target engagement by the inhibitors designed led to disruption of oncogenic phenotypes as demonstrated in enzymatic assays and in a panel of cancer cell lines, including acute lymphoblastic and myeloid leukemia (ALL/AML) and non-small cell lung cancer (NSCLC). Structural rationale underlying the approach was corroborated by x-ray crystallography. The lead compound demonstrated potent target inhibition in a pharmacodynamic study in leukemic mice.
Graphical Abstract
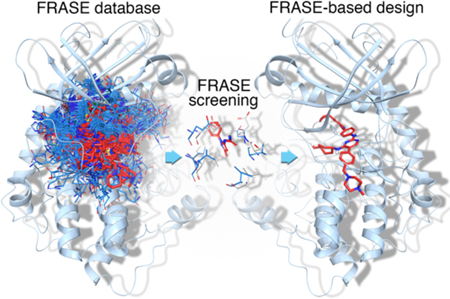
Introduction
Polypharmacology: embracing the inevitable
Large families of structurally and often functionally related proteins, such as GPCR [1,2], protein kinases [3,4], and more recently HDAC [5] or bromodomains [6], are the most important source of drug targets. Due to the high intra-family similarity, especially within smaller subfamilies, a single small-molecule drug, in general, binds multiple family members, presenting both a challenge and an opportunity for drug discovery [7]. Beyond the basic consideration that promiscuous binding to multiple targets is a major cause of adverse effects [8] and general toxicity [9], it has also been shown that excessive selectivity for a single target may be a fatal safety concern [10], while binding several related targets could be a prerequisite for more efficacious drug action [11]. Hence, to achieve a desired therapeutic response, a drug discoverer should ideally be able to control the polypharmacology of a drug candidate; that is, to disable its promiscuous binding to unwanted targets, while enhancing potency against therapeutically relevant targets. However, controlling which particular members of a large protein family would be inhibited by a drug is an extremely challenging endeavor. Here we describe a data-driven approach to design small-molecule drugs with fine-tuned selectivity profiles in the context of highly homologous protein targets. In our approach, 3D structures of bioactive ligands are directly “assembled” from smaller building blocks in the protein binding pocket.
From virtual screener to virtual chemist
Previous computational efforts to design multi-target drugs focused on extending bioinformatics models or scaling up cheminformatics techniques. In particular, various flavors of binding-pocket similarity have been used to make predictions based on the principle “similar targets bind similar ligands” [12–14]. Alternatively, broad panels of computational structure-activity relationship (SAR) models were used to predict targets for ligand libraries [15–18]. Most practical applications focused on the two largest drug-target families: G-protein coupled receptors (GPCRs) [19] and protein kinases [20]. Although highly diverse in terms of underlying principles and assumptions, all previously reported approaches share one common function: they act as a “virtual screener”, that is, receive a ligand structure as the input and report its activity against a given target as the output. “Virtual screeners” are highly efficient when applied to a small panel of well-studied targets with large SAR datasets. However, they are suboptimal for dealing with larger panels of predominantly novel targets: first, because there are no SAR data to develop predictive SAR models, and, second, a “virtual screener” still needs a human chemist to feed it with newly designed compounds. Yet, traditional SAR-based medicinal chemistry is difficult to scale up to multi-target discovery. Intuitively, the design of chemically novel ligands for understudied targets may benefit from (i) thousands of experimental structures of protein-ligand complexes, (ii) hundreds of thousands of biological activities for those ligands in public databases, and (iii) an intricate network of evolutionary relationships within large protein families. Tools are needed to jointly exploit disparate pieces of information from heterogeneous sources. The approach to polypharmacological ligand design, that we introduce, is based on the concept of a FRAgment in Structural Environment (FRASE). A single FRASE extracted from a high-affinity ligand explicitly encapsulates 3D structural information and, implicitly, SAR and sequence alignment data. Several FRASEs extracted from different protein-ligand complexes can be readily combined into a novel ligand for an orphan protein target. Hence, FRASE-based design represents a first step in the evolution of computer-aided drug from “virtual screener” to “virtual chemist”.
Targeting tumor-survival promoters TYRO3, AXL and MERTK
Here we demonstrate the potential of the FRASE-based approach by using it to design ligands that can interfere with the function of the TYRO3, AXL and MERTK (TAM) family of receptor tyrosine kinases [21]. These receptors play a physiologic role in dampening inflammatory responses, a role that is subverted in cancer-associated myeloid cells in the immunosuppressive tumor microenvironment [22,23]. TAM kinases, in particular MERTK, have been shown to upregulate expression of the T cell immune checkpoint molecule PD-L1 and to promote phosphatidylserine-dependent efferocytosis and AKT-mediated chemoresistance in tumor cells [22,24]. In addition, TAM kinases are ectopically expressed in many human cancers, including both hematologic malignancies and solid tumors, where they function to promote tumor cell survival, chemoresistance and motility [23]. Consequently, targeted inhibition of TAM family members could reverse the immunosuppressive microenvironment and directly target tumor cells in a wide range of cancers.
The therapeutic value of TAM inhibitors may be largely dependent on their respective polypharmacologic profiles, both within the TAM family and with respect to other protein kinases. Therefore, it is important to design chemical probes selective either for a single TAM-family member or for any combination thereof. However, controlling the intra-family selectivity is challenging because TAM kinases were among the latest to evolve and, hence, they have highly similar kinase-domain structures and almost identical ATP pockets, providing almost no opportunity for developing intra-family selectivity. Here, we made use of FRASE-based design to develop inhibitors selective for specific TAM-family members and hence explore their potential as anti-cancer agents.
Results
FRASE-based design
For a long time, ligand-and structure-based strategies in computer-aided drug design evolved separately, one toward embracing bigger data for more predictive and comprehensive activity models and the other toward faster and more accurate prediction of ligand-protein interactions [25,26]. However, despite significant progress, both strategies conserve their essential limitations. Ligand-based approaches cannot predict activities for “orphan” targets and structure-based techniques still have a long way to go before it might be possible to profile hundreds of ligands against hundreds of targets with a suitable accuracy in a reasonable time. FRASEs are structural descriptors that are intended to merge the chemical and protein structure spaces to overcome both of the above limitations. FRASE-based design enables processing of thousands of 3D protein-ligand complexes and tens of thousands of bioactivity data points to find the most appropriate building blocks for new active ligands. Moreover, the suggested building blocks are shown as 3D protein-bound poses and can be easily shuffled to yield synthetically tractable combinations with diverse family-wide polypharmacology profiles. A typical FRASE (Fig. 1A) includes a chemically sound fragment of a 3D protein-bound ligand pose, e.g., a cycle and/or functional group, as well as the nearby protein fragments (in this study, full residue structures). There are two primary assumptions behind the FRASE-based design: (a) a ligand has high affinity to a given protein due to an “endorsement” by every fragment it consists of; (b) a fragment is “good” due to its favorable interactions with its immediate protein environment. By combining (a) and (b) we obtain: (c) if a fragment is “good” in one protein it will also be “good” in any other protein that features an identical structural environment.
Figure 1: FRASE-based approach to “assemble” inhibitors selective for specific TAM-family members.

A. Example of a typical FRASE: the constituent fragments are rendered as thick sticks (gray for the protein portion of the FRASE, blue for the ligand portion);
B. Simplified representation of the FRASE-based design: (left to right) A target kinase (gray cartoon) with projected reference FRASEs from the database (magenta and blue sticks); Evaluation of how individual components of a reference protein environment (magenta sticks) match the nearby target residues (gray sticks)(the numbers represent per-residue Tscore values, for a total Tscore of 0.79); Ligand fragments from the matching FRASEs (blue sticks) that can be used for design.
C. Adenine (cyan) and back-pockets (pink) in a kinase enzyme;
D. Aligned sequences of residues lining the back-pocket show 44 pairwise differences;
E. The only amino-acid position (rendered as ball-and-sticks) showing variance in the adenine pocket of TAM kinases and the x-ray structure for the template inhibitor 1 in complex with MERTK.
Conceptually, design of a new ligand for a given kinase involves (i) identification of structural environments (stored in the FRASE database) that match those in the binding pocket of the kinase of interest, and (ii) inspect locations and orientations of the ligand fragments belonging to the matching FRASEs (Fig. 1B) and combine them into synthetically tractable compounds. The algorithmic implementation of step (i) is straightforward. First, it must be noted that all kinase structures (either those used to create the FRASE database or those for which ligands are to be identified) are aligned in 3D space thus enabling an instantaneous projection of all FRASEs on the target structure (Fig. 1B). Hence, the i-th protein residue belonging to a structural environment from the FRASE database (reference environment) can be directly compared to the closest (i’-th) residue in the target protein and the match between them can be quantified as a function of their chemical similarity and proximity in 3D space, , where dii is the distance between the residue centers and Tii is the Tanimoto[27] similarity between them. The total score for a reference environment is an average over all residues in the reference protein environment, Tscore = 〈Tscorei〉 (Tscore would be equal to one if all reference residues are chemically identical to and spatially aligned with their target counterparts). Fig. 1B exemplifies evaluation of a single reference environment with Tscorei shown for each individual residue. The total Tscore value in this example is 0.79 which is high enough and would warrant the selection of the respective ligand fragment as a potential building block for design. The database search for matching FRASEs can be significantly restrained by setting a particular location for the FRASE’s ligand fragment. This would allow one to build a new ligand sequentially, by finding the next ligand fragment close to the attachment site of the previous one (which is the way ligands were designed in this study). The final selection of ligand fragments is closely associated with step (ii), that is, assembling fragments into synthetically tractable compounds. More than one factor is taken into consideration. First, Tscore is used to narrow down the choice. As in any screening process, the quantitative endpoint only serves to select a manageable number of hits that can be evaluated using alternative, more time-and resource-demanding criteria (a few thousand FRASEs are usually selected based on Tscore). There is no a clearly defined, optimal Tscore threshold. Certainly, it should not be set significantly lower than 0.5, but most of the times the threshold is determined by how many FRASE hits one can process. The next step is triage of the initial fragment hits. One notable aspect of ligand fragments in the FRASE database is their significant redundancy due to the fact that a fragment can be cut out of a ligand in multiple overlapping ways (this redundancy was intentionally allowed to maximize the sensitivity of the database search). We remove this redundancy by clustering on chemical similarity of the ligand fragments and the location of their geometrical centers. Finally, binding poses of the remaining fragments (irrespectively of their Tscores) are visually inspected and their relevance is assessed based on the synthetic tractability, availability of reagents, as well as the opportunity of connecting the selected fragment (to other fragments or the pre-existing structural template) by a common minimal linker, such as a single bond, ether, amide or urea. Sometimes a fragment can be slightly modified to facilitate its fusion with the structural template.
FRASE database, the key prerequisite for the design, integrates the information from 3D structures of protein-ligand complexes, ligand structures and their inhibitory potencies. First, a cross-search in the Uniprot[28], PDB[29] and PDBbind[30] databases was performed to yield 2,800 3D structures of high-affinity complexes (involving 2,100 unique ligands and 230 kinases). Next, each ligand in every protein-ligand complex was broken into fragments (cycles and acyclic functional groups). Finally, each ligand fragment from the previous step is combined into a single structure with the nearby protein fragments (here, whole residues were used as protein fragments). A total of ~230,000 FRASEs were identified. We observed that, on average, a single structural environment can be found in ~30 kinases, that is, ~6% of the kinome. This appears to be an acceptable balance between uniqueness and redundancy. Indeed, if a structural environment occurs in every kinase it would not be useful for designing selective inhibitors, while if it occurs in only one kinase it would only be useful for designing an inhibitor for this particular kinase (and none other). The selectivity profile of a FRASE-based inhibitor would be an intersection of kinase lists that each individual FRASE is “good” for. Technical details on the development of the database and related tools are given in the Methods section.
Kinase back-pocket offers structural variance for selective TAM-member inhibitors
Our strategy consists of taking a type I TAM inhibitor and expanding it to new protein pockets featuring more significant structural differences in the vicinity of the ligand, thus providing a better opportunity to tune the inhibitor selectivity for any given TAM kinase. Structural analysis of all TAM members suggests that the “back-pocket” (Fig. 1C), located between the c-helix and the adjacent antiparallel b-sheet, displays significant structural diversity and hence provides multiple opportunities for enhancing inhibitors’ selectivity. The three TAM sequences of C-helices (the rear-wall of the pocket) and adjacent antiparallel b-sheets (the ceiling) display 44 pairwise-differing residues (Fig. 1D), whose side chains may either directly interact with the ligand or induce changes in relative orientations of these structural modules. This structural diversity in the back-pocket is in sharp contrast with the high conservation of the adenine pocket, to which typical type I kinase inhibitors, such as our pyridine-pyrimidine 1 [31] bind. In this pocket, there is only one amino-acid position showing variation across the TAM-family: MERTK:Ile650 vs. AXL:Met599 vs. TYRO3:Ala581 (Fig. 1E). In all three cases, the differing residues do not have close contacts or make high-energy interactions with the ligand. Hence, we hypothesized that the back-pocket (Fig. 1C, E) would be a more suitable target for pyridine-pyrimidine expansion. A sequential FRASE-based strategy was applied to design TAM ligands interacting with the back-pocket. Inhibitor 1 was used as a structural template with its 2-amino nitrogen on pyrimidine ring as an attachment site for a new fragment. Then, the position of each further fragment was constrained by the proximity of the previous one and lying on an imaginary line pointing toward the back-pocket.
Opening the back-door and reaching the back-pocket
A first step in expanding the type I inhibitor 1 towards the back-pocket was to find a spacer that would “deliver” a functional group of choice to the back-pocket while avoiding bad contacts with the gatekeeper residue (MERTK:Leu671 / AXL:Leu621 / TYRO3:Leu603). To this end, we have extracted from our FRASE database those FRASEs that would possess the required spacer properties and match the respective structural environments of TAM family members (1,786 FRASEs from 73 unique PDB structures representing 30 kinases). The triage process yielded 203 non-redundant fragments from which several most minimalistic fragments were retained as relevant for the design (Fig. 2A). They were extracted from the following structures: AKT2 (PDB: 3D0E; Tscore: 0.74), CHK1 (4FST/0.53) and MELK (4UMP/0.52, 4UMT/0.57, 4UMU/0.56) kinases. The respective TAM inhibitor 2 was readily inferred, synthesized and tested in an enzymatic microcapillary electrophoresis (MCE) assay [32] (Fig. 2A). It had low nanomolar IC50 values against MERTK and TYRO3 and its potency against AXL was approximately one order of magnitude weaker (Fig. 2A and Supplementary Table S1 for a full account of IC50 values in this study). The new spacer is expected to efficiently deliver any functional group to the back pocket; any substitution at the alkyne-bound methyl would be past the gatekeeper residue. The next step was to identify a fragment that would penetrate into the back-pocket. One of the relevant FRASEs, donated by CHK1 (4FST), features an alkyne-aryl fragment that aligns with the aryl-alkyne group of 2. We also searched for fragments and features located at the entry to the back-pocket and identified 151 such fragments (from 9 unique PDB structures). The most frequent feature at this location was a hydrogen bond (H-bond) acceptor pointing toward the large lobe (Fig. 2B, top), which suggested using a 2-pyridine ring as an aromatic scaffold within the back-pocket. The respective structural environments come from 6 PDB structures from 4 unique kinases (VEGFR2, c-Met, and p38a) with Tscores ranging from 0.50 to 0.84. Combining this 2-pyridine ring with 2 resulted in 3 (Fig. 2A). Although it demonstrated reduced potencies against all TAM members (Fig. 2A), inhibitor 3 provides a promising template to explore the back-pocket SAR.
Figure 2: Steps toward potent TAM back-pocket binders.

A, Growing the aryl-alkyne-aryl spacer: template compound 1 (green), aryl-alkyne and aryl FRASEs (magenta), and TAM/FLT3 profiles of inhibitors (IC50 values given in nM) with a partial (2) and full (3) spacers. B, Filling the back-pocket by aromatic groups: template compound (green), source FRASEs (magenta) and sample inhibitors 4–6 not showing sufficient potency and selectivity. C, Filling the back-pocket with smaller aliphatic groups: template compound (green), source FRASEs (magenta) and sample inhibitors 7, 8 showing high potency and promising selectivity. D, Co-crystal structure of 7 in complex with MERTK and its major interactions with the adenine pocket (Pro672, Phe673 and Glu595) and back-pocket (Lys619).
Small chemical modifications in the back-pocket alter intra-family selectivity
To explore the chemical space to fill the back-pocket, we used two one-point queries, with the radius of 2 Å, that mostly cover its volume. One of the queries yielded 1,837 fragment hits (119 non-redundant, 30 unique PDB, 7 kinases) and the other, 155 hits (21 non-redundant, 11 unique PDB, 8 kinases). The fragments from both queries can be split into two broad categories: rigid, flat aromatic groups and more flexible and bulky aliphatic ones (a few fragments featured both aromatic and aliphatic moieties). Our first step was to investigate the impact of aromatic groups that could be readily attached to template 3 (such groups were observed in several p38a structures at the previous step of design). In the course of further visual analysis, a set of aromatic fillings was retained to inspire our ligand design. These fragments were “donated” by 8 diverse kinases including BRAF1 (PDB: 3NNW, Tscore: 0.98), c-MET (3EFK/0.54, 2RFN/0.53), FAK2 (3FZT, 0.53), HCK (2C0O/0.69), JNK2 (3NPC/0.99), JNK3 (3E92/0.92, 3D7Z/0.89, 3DA6/0.65), p38a (2BAJ/0.93, 3BYS/0.91, 3HV3/0.87, 3NNV/0.87, 3BYU/0.84, 3HV5/0.84, 3HV4/0.84, 3GCU/0.84, 3GCV/0.83, 1W82/0.83, 2OFV/0.80, 3NNX/0.78, 3HV6/0.78, 3HV7/0.72, 3GCQ/0.80, 1KV2/0.69, 3NNU/0.68), and PLK (3DB6/0.70). A series of such aromatic substituents were attached to 3 through a minimal linker (see examples in Fig. 2B). The respective inhibitors 4–6 have slightly improved potencies against TAM members showing, respectively, pan-TAM (4), MERTK/TYRO3/FLT3 (5) and MERTK/FLT3 (6) profiles. Unfortunately, the potency improvement achieved for 4–6 is not sufficient to warrant further progression of these compounds toward in vivo application. Hence, we explored flexible, aliphatic back-pocket-filling fragments located in potentially more diverse structural environments. They originate from 5 kinases, including BRAF1 (PDB: 3NNW, Tscore: 0.98), FAK2 (3FZT, 0.53), JNK2 (3DS6/0.66), p38a (3F82/0.84, 3BYS/0.91, 3NNV/0.87, 3CTQ/0.85, 3BYU/0.84, 3E93/0.80, 2OFV/0.80, 3NNX/0.78, 2OG8/0.71, 3P7A/0.69, 1W83/0.69, 3P7C/0.67, 3GCQ/0.80, 3P7B/0.75, 3P79/0.75, 1WBS/0.71, 3NNU/0.68), and TIE2 (2OO8, 0.72). Incorporation of these aliphatic fragments in the template compound was less straightforward synthetically than in the previous steps of the design. We eventually developed a large series of modified inhibitors whose “decoration” in the back-pocket closely matches the respective FRASEs. Two of them, inhibitors 7 and 8, are exemplified in Fig. 2C and show significant improvement in potency as well as a higher diversity of selectivity profiles compared to 3–6. In particular, compound 7 (UNC3437A) is primarily selective for MERTK with slightly lower potency against FLT3; 8 is clearly AXL-biased, a remarkable result since none of our previously published inhibitors were selective for AXL; and 9 is primarily a TYRO3-biased inhibitor with slightly lower MERTK potency. The sensitivity of the TAM/FLT3 polypharmacology profile to small chemical changes demonstrates the high potential of FRASE-based design to provide a rational method of optimization. To corroborate the structural rationale underlying the approach we solved the co-crystal structure of MERTK in complex with inhibitor 7 (Fig. 3d and Supplementary Table S2). The structure confirmed that indeed, the pyridine-dioxolane group is bound to the back pocket extended toward the small lobe, consistent with the respective FRASE locations. On the other hand, the pyridine-pyrimidine core binding to the adenine pocket featured the same interactions with hinge residues Pro672, Phe673, and Glu595 or Asp678 as the template pyridine-pyrimidine compound 1. Overall, the x-ray structure confirmed the assumptions underlying the design of the novel series and, more generally, the utility of the FRASE-based approach for modulating TAM/FLT3 selectivity profiles.
Figure 3: Kinome selectivity profiles.

MIB-MS kinome profiles for inhibitor 10 (see structure and TAM/FLT3 IC50 values) and the initial template 1. Each kinase features an orange (inhibitor 1) and a blue (inhibitor 10) bars, with bar heights corresponding to log2 of the inhibitor/control ratio. Thresholds of −1 and 1 (shown in dotted lines) specify significant enrichment or decrease of kinases on the beads with respect to control. The brackets with labels on the top of the charts indicate the respective kinase family.
Kinome profiling and in vivo pharmacokinetics
We selected a pan-TAM inhibitor 10 (UNC4042A) (Fig. 3) as a lead compound for more comprehensive biological characterization. At this initial step of the series evaluation, a probe with a broad TAM potency spectrum was intentionally chosen to test whether the in vitro inhibitory potency translates into target engagement in a broad range of cellular and in vivo assays.
We next sought to know whether, and how, extending type I inhibitors toward the back-pocket would affect the inhibitors’ kinome-wide polypharmacology. To this end, we compared the selectivity profile of inhibitor 10 to that of inhibitor 1 (Fig. 1), the initial chemical template for FRASE-based design. Multiplexed inhibitor bead kinome enrichment coupled to quantitative mass spectrometry analysis (MIB-MS) was used to determine specificity of the compounds in an unbiased manner. In MIB-MS, mixtures of Sepharose beads with covalently immobilized, linker-adapted kinase inhibitors are used to capture a large portion of the kinome present in the analyzed cell lysate [33]. The identity of the kinases captured is then established by means of MS analysis. In a competition MIB-MS assay, an inhibitor is preincubated with the lysate prior to the analysis, so that the inhibitor-bound kinases would not be captured by the beads. The kinases captured with and without the inhibitor present in lysate can then be compared to determine which kinases were down-or upregulated by the inhibitor.
In this study, NOMO-1 human AML cells were treated with DMSO, 1 or 10 (both at 100 nM) for one hour and then, kinases were enriched using MIBs and analyzed by LC-MS/MS. A set of 222 kinases were quantified across the samples using a label-free approach. Inhibitor 1 showed significantly lower kinome selectivity compared to 10 (Fig. 3); 62 kinases showed lower enrichment on the beads upon treatment with 1 compared to DMSO control, whereas 27 kinases were decreased upon treatment with 10. Of these 27 kinases, 8 were significantly decreased (p < 0.05 and log2 ratio 10/control ≥ 2). In addition to higher selectivity, the kinases decreased by 10 are not a mere subset of the 62 kinases decreased by 1, suggesting a different mode of action. Detailed MIB-MS data, including results for inhibitors 1 and 10 at 1 μM, are given in Supplementary Datafile “FRASE-Proteomics.xlsx”.
We also assessed whether the lead compound is suitable for in vivo studies. To this end, pharmacokinetic (PK) studies in mice have been performed with compound 10 administered via intravenous (i.v.), intraperitoneal (i.p.) and oral (p.o.) routes. As can be seen from the evaluation of drug concentration in plasma over time, 10 has a PK profile suitable for in vivo application in murine models (Supplementary Fig. S1, Cmax = 435 ng/mL; AUCpo = 1661 ng*h/mL and T1/2 = 4.97 h via i.p. route). The weak oral bioavailability of compound 10 precludes the p.o. route. However, oral administration is not crucial for animal studies and can be addressed by further chemical optimization. Details of the in vivo PK study are described in the Supplementary Information.
Target engagement in cellular and animal models
To confirm potent on-target inhibition and tumor suppression activity mediated by the lead compound 10, we evaluated the effects of treatment with 10 in solid tumor and hematologic malignancy models. First, we determined target engagement by monitoring the level of active, phosphorylated MERTK and AXL in cancer cells as a function of inhibitor concentration. As expected, 10 inhibited both MERTK (IC50 = 28 nM, 95% confidence interval = 16–52 nM) and AXL (IC50 = 32 nM, 95% confidence interval = 18–57 nM) at nanomolar concentrations (Fig. 4A and Supplementary Fig. S2A,B). Near complete inhibition of pMERTK and pAXL was observed in cells treated with higher concentrations of 10. This assay provides a potency level reference for pharmacologic effects of 10 that might be observed in cellular tumor models and be attributable to MERTK or AXL inhibition.
Figure 4: Target engagement in cellular and animal tumor models.

Compound 10 inhibits MERTK and AXL phosphorylation, decreases colony forming potential, and induces cell death in tumor cell lines. A, 697 ALL cells (pMERTK assay) and A549 NSCLC cells (pAXL assay) were cultured with 10 or vehicle for 1 hour and then treated with pervanadate phosphatase inhibitor to stabilize phosphorylated proteins. MERTK and AXL were immunoprecipitated from cell lysates and phosphorylated and total MERTK or AXL proteins were detected by immunoblot. Phosphorylated and total proteins were quantitated by densitometry. B, NSCLC (A549 and COLO-699) cells were cultured in soft agar overlaid with medium containing 10 or vehicle only for 14 days. Media and 10 were refreshed every 3 days. C, Kasumi-1 and NOMO-1 AML cells were cultured with 10 or vehicle for 72 hours, then cells were stained with PoPro-1-iodide (PoPro) and propidium iodide (PI) dyes and apoptotic (PoPro+, PI-) and dead (PI+) cells were detected by flow cytometry. Mean values and standard errors from 3–4 independent experiments are shown. Statistically significant differences relative to vehicle were determined using 1-way ANOVA (**p<0.01, ***p<0.001). D, Compound 10 inhibits MERTK phosphorylation in bone marrow leukemia cells in vivo. NSG mice were transplanted with 697 human leukemia cells and leukemic mice were treated with a single dose of 60 mg/kg 10 or saline vehicle (VEH) by intraperitoneal injection. Bone marrow cells were isolated from femurs 2 hours later and incubated with pervanadate phosphatase inhibitor for 10 minutes to stabilize phosphoproteins. Phosphorylated and total human MERTK proteins were quantitated by densitometry. The ratio of phosphorylated and total MERTK proteins was determined for each sample and values relative to the mean for vehicle-treated mice in each experiment were determined. Mean values and standard errors derived from 2 independent experiments are shown (** p = 0.0026, student’s unpaired t test).
Consistent with previous data demonstrating oncogenic roles for MERTK in AML cells and for both MERTK and AXL in NSCLC cells [34,35], treatment with 10 mediated potent inhibition of colony formation in soft agar cultures of both AML and NSCLC cell lines (Fig. 4B and Supplementary Fig. S2C). More specifically, both OCI-AML5 AML cells and COLO-699 NSCLC cells were very sensitive to 10, with concentrations as low as 50 nM mediating a 70% reduction in colony number and complete abrogation of colony formation in response to treatment with 300 nM 10. The NOMO-1 AML and A549 NSCLC cell lines were sensitive to 10 as well, although with reduced potency compared to the other cell lines. In these cases, a 300 nM concentration was required to affect a significant 60–75% reduction in colony number, perhaps indicating a requirement for more complete target inhibition. In addition, treatment with 300 nM 10 was sufficient to induce tumor cell death in 67% and 41% of Kasumi-1 and NOMO-1 AML cells, respectively (Fig. 4C).
Consistent with the favorable PK profile for 10 in mice, treatment with 10 also inhibited MERTK phosphorylation in vivo. Immune compromised NOD/SCID/IL2Rgamma−/− (NSG) mice were inoculated intravenously with human B-cell ALL cells to generate orthotopic xenografts and mice with advanced leukemia were treated with a single 60 mg/kg dose of 10 administered by i.p. injection. Bone marrow cells were collected two hours post-treatment and phosphorylated and total human MERTK proteins were detected by immunoblot. MERTK phosphoprotein was significantly decreased in leukemia cells collected from mice treated with 10 relative to mice treated with vehicle (0.23 ± 0.116 versus 1.00 ± 0.031, p=0.0026) (Fig. 4D and Supplementary Fig. S2D). These pharmacodynamic data confirm inhibition of MERTK mediated by 10 in vivo and demonstrate the utility of 10 for translational studies utilizing murine models.
Discussion and Conclusions
FRASE-based series of in vivo antitumor agents
The enzymatic, structural, cellular, and in vivo data obtained in this study suggest that the pyridine-pyrimidine-alkyne series represent useful in vivo chemical probes with readily adjustable intra-TAM/FLT3 selectivity profiles and potential to significantly affect their kinome-wide polypharmacology profiles. This is exemplified by the compound 10, which shows an improved kinome selectivity profile in cells compared to the initial chemical template 1, cell-based activity consistent with the presumed pharmacology of the target, abrogation of oncogenic phenotypes and induction of cytotoxicity in cell-based assays, chemical stability, and an SAR profile suggestive of a specific interaction with the target [36]. We have shown that small chemical changes, suggested by FRASE analysis, induce significantly different selectivity profiles within the TAM family. Hence, probes with varying TAM and kinome-wide selectivity can be readily derived using additional FRASEs. Development of a more comprehensive probe set would enable a more specific analysis of the role of the TAM tyrosine kinase family and individual members in their physiologic and pathologic functions, including prevention of chronic inflammatory responses and auto-immunity. In particular, availability of more selective kinase inhibitors with specific TAM profiles may facilitate a better understanding of the extent to which inhibition of individual or all TAM kinases may relieve the deleterious immunosuppressive action of tumor associated macrophages and myeloid derived suppressor cells. Selective and potent inhibition would also reverse the overexpressed TAM kinase action to drive tumor cell survival and chemoresistance. Remarkably, these probes have been designed with the help of a “virtual medicinal chemist”, a cheminformatics technique that is able to “assemble” new bioactive compounds based on available 3D structures and chemogenomic data.
Interaction-Activity Relationships
We have chosen the polypharmacology of TAM kinase inhibitors as an opportune test case, given the potential importance of TAM inhibitors for cancer therapy [23]. While the focus of this study was on protein kinases, we expect the FRASE approach to be applicable to any other protein family, provided there is a sufficient amount of 3D and SAR data, and even across protein families. Indeed, FRASE could potentially serve as an informational key for data integration across multiple protein families, so that structures and data abundant in one family, such as protein kinases, would benefit protein classes with sparse structure-activity information. Such extrapolation of SAR beyond a given protein, or even a protein family, should be possible due to the malleable redundancy of FRASE descriptors. That is, the size of ligand and protein fragments constituting a single FRASE can be set small enough for similar structural environments to occur in multiple structurally diverse proteins. Hence, FRASE-based design might overcome some fundamental limitations of SAR analysis and structure-based design. The traditional SAR analysis is target-blind: no structural or sequence similarity between the target proteins is being considered. The activity (or the target) is identified by a label and if the labels in two SAR data sets are different, the sets cannot be cross-linked irrespective of how similar the respective target proteins are. New compounds are prioritized for synthesis based on a formal or informal examination of chemical similarity between active and inactive compounds. At the same time, structure-based design is historically SAR-blind. That is, a new compound is prioritized for synthesis based on a computed or an ad hoc assessment of its binding affinity to the protein of interest. There is no formal algorithm that allows direct involvement of available SAR data in this decision. FRASEs, par excellence, jointly exploit explicit representations of both ligand and protein structures, thus converting conventional SAR into Interaction-Activity Relationships to guide construction and prioritization of new compounds for synthesis. Here, we have shown that even unweighted FRASEs, i.e., those whose “goodness” was inferred from a mere occurrence in a high-affinity complex, can be effectively used as building blocks for biologically active compounds. Future studies will focus on development and application of algorithms to identify FRASEs that are more likely than others to be “good” and to quantify the degree of “goodness”. This can potentially be achieved through a combination of machine learning techniques with a proven efficiency in chemoinformatics [37,38] and specialized affinity assessment techniques developed for structure-based design [39–41].
Methods Summary
Methods and any associated references are available in the online version of the paper.
Supplementary Material
Acknowledgements
This work was supported by the University Cancer Research Fund (UCRF), Federal Funds from the National Cancer Institute, National Institute of Health, under Contract No. HHSN261200800001E, and a philanthropic gift to the CICBDD. We thank Dr. Brenda Temple for her help in depositing the X-ray crystallographic structure to the Protein Data Bank (PDB). This research is based in part upon work conducted using the UNC Proteomics Core Facility, which is supported by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center.
Footnotes
The authors declare the following competing financial interest(s): D.D., D.K.G., H.S.E., S.V.F., X.W., and D.K. have stock in Meryx, Inc.
Supporting Information
Hitlists including fragment structures, environment information, source pdb ids and Tscores are provided as supplementary files. The full FRASE database used in this study was made available to public as a shared archive at: https://drive.google.com/open?id=1xkIIdMDz_XMZdBBVikmivr8RVRoX9Q6X.
References
- 1.Sriram K, Insel PA. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol Pharmacol. 2018. April 3;93(4):251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017. October 27;16(12):829–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen P, Alessi DR. Kinase Drug Discovery – What’s Next in the Field? ACS Chem Biol. 2013. January 18;8(1):96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov Today. 2016. January 1;21(1):5–10. [DOI] [PubMed] [Google Scholar]
- 5.Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med. 2016. October 3;6(10):a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Theodoulou NH, Tomkinson NC, Prinjha RK, Humphreys PG. Clinical progress and pharmacology of small molecule bromodomain inhibitors. Curr Opin Chem Biol. 2016. August 1;33:58–66. [DOI] [PubMed] [Google Scholar]
- 7.Barnash KD, James LI, Frye S V. Target class drug discovery. Nat Chem Biol. 2017. September 19;13(10):1053–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edwards IR, Aronson JK. Adverse drug reactions: definitions, diagnosis, and management. Lancet. 2000. October 7;356(9237):1255–9. [DOI] [PubMed] [Google Scholar]
- 9.Smith GF. Designing Drugs to Avoid Toxicity. Prog Med Chem. 2011. January 1;50:1–47. [DOI] [PubMed] [Google Scholar]
- 10.Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006. January 4;116(1):4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004;3(4):353–9. [DOI] [PubMed] [Google Scholar]
- 12.Zavodszky MI, Rohatgi A, Van Voorst JR, Yan H, Kuhn LA. Scoring ligand similarity in structure-based virtual screening. J Mol Recognit. 2009/February/24 2009;22(4):280–92. [DOI] [PubMed] [Google Scholar]
- 13.Vulpetti A, Kalliokoski T, Milletti F. Chemogenomics in drug discovery: computational methods based on the comparison of binding sites. Future Med Chem. 2012. October 22;4(15):1971–9. [DOI] [PubMed] [Google Scholar]
- 14.Glinca S, Klebe G. Cavities Tell More than Sequences: Exploring Functional Relationships of Proteases via Binding Pockets. J Chem Inf Model. 2013. August 26;53(8):2082–92. [DOI] [PubMed] [Google Scholar]
- 15.Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, Whaley R, Glennon RA, Hert J, Thomas KLH, Edwards DD, Shoichet BK, Roth BL. Predicting new molecular targets for known drugs. Nature. 2009;462(7270):175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, Lavan P, Weber E, Doak AK, Cote S, Shoichet BK, Urban L. Large-scale prediction and testing of drug activity on side-effect targets. Nature. 2012;486(7403):361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng F, Zhou Y, Li J, Li W, Liu G, Tang Y. Prediction of chemical–protein interactions: multitarget-QSAR versus computational chemogenomic methods. Mol Biosyst. 2012. July 31;8(9):2373. [DOI] [PubMed] [Google Scholar]
- 18.Martin E, Mukherjee P, Sullivan D, Jansen J. Profile-QSAR: A Novel meta-QSAR Method that Combines Activities across the Kinase Family To Accurately Predict Affinity, Selectivity, and Cellular Activity. J Chem Inf Model. 2011. August 22;51(8):1942–56. [DOI] [PubMed] [Google Scholar]
- 19.Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang X-P, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FRC, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, Hopkins AL. Automated design of ligands to polypharmacological profiles. Nature. 2012;492(7428):215–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erickson JA, Mader MM, Watson IA, Webster YW, Higgs RE, Bell MA, Vieth M. Structure-guided expansion of kinase fragment libraries driven by support vector machine models. Biochim Biophys Acta-Proteins Proteomics. 2010. March 1;1804(3):642–52. [DOI] [PubMed] [Google Scholar]
- 21.Linger RMA, Keating AK, Earp HS, Graham DK. TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer In: George FVW, George K, editors. Advances in Cancer Research. Academic Press; 2008. p. 35–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasikara C, Kumar S, Kimani S, Tsou W-I, Geng K, Davra V, Sriram G, Devoe C, Nguyen K-QN, Antes A, Krantz A, Rymarczyk G, Wilczynski A, Empig C, Freimark B, Gray M, Schlunegger K, Hutchins J, Kotenko SV., Birge RB. Phosphatidylserine Sensing by TAM Receptors Regulates AKT-Dependent Chemoresistance and PD-L1 Expression. Mol Cancer Res. 2017. June;15(6):753–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14(12):769–85. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen K-QN, Tsou W-I, Calarese DA, Kimani SG, Singh S, Hsieh S, Liu Y, Lu B, Wu Y, Garforth SJ, Almo SC, Kotenko S V, Birge RB. Overexpression of MERTK receptor tyrosine kinase in epithelial cancer cells drives efferocytosis in a gain-of-function capacity. J Biol Chem. 2014. September 12;289(37):25737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, Dearden J, Gramatica P, Martin YC, Todeschini R, Consonni V, Kuz’Min VE, Cramer R, Benigni R, Yang C, Rathman J, Terfloth L, Gasteiger J, Richard A, Tropsha A. QSAR modeling: Where have you been? Where are you going to? J Med Chem. 2014;57(12):4977–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Wu Y, Deng Y, Kim B, Pierce L, Krilov G, Lupyan D, Robinson S, Dahlgren MK, Greenwood J, Romero DL, Masse C, Knight JL, Steinbrecher T, Beuming T, Damm W, Harder E, Sherman W, Brewer M, Wester R, Murcko M, Frye L, Farid R, Lin T, Mobley DL, Jorgensen WL, Berne BJ, Friesner RA, Abel R. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J Am Chem Soc. 2015;137(7):2695–703. [DOI] [PubMed] [Google Scholar]
- 27.Nikolova N, Jaworska J. Approaches to Measure Chemical Similarity– a Review. QSAR Comb Sci. 2003. December 1;22(910):1006–26. [Google Scholar]
- 28.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32(suppl 1):D115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 1999/12/11. 2000;28(1):235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang R, Fang X, Lu Y, Wang S. The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J Med Chem. 2004;47(12):2977–80. [DOI] [PubMed] [Google Scholar]
- 31.Zhang W, Zhang D, Stashko MA, DeRyckere D, Hunter D, Kireev D, Miley MJ, Cummings C, Lee M, Norris-Drouin J, Stewart WM, Sather S, Zhou Y, Kirkpatrick G, Machius M, Janzen WP, Earp HS, Graham DK, Frye S V., Wang X Pseudo-cyclization through intramolecular hydrogen bond enables discovery of pyridine substituted pyrimidines as new mer kinase inhibitors. J Med Chem. 2013;56(23):9683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang W, DeRyckere D, Hunter D, Liu J, Stashko MA, Minson KA, Cummings CT, Lee M, Glaros TG, Newton DL, Sather S, Zhang D, Kireev D, Janzen WP, Earp HS, Graham DK, Frye S V, Wang X. UNC2025, a potent and orally bioavailable MER/FLT3 dual inhibitor. J Med Chem. 2014/July/30 2014;57(16):7031–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, Usary J, Kuan PF, Smalley DM, Major B, He X, Hoadley KA, Zhou B, Sharpless NE, Perou CM, Kim WY, Gomez SM, Chen X, Jin J, Frye S V, Earp HS, Graves LM, Johnson GL. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012/April/17 2012;149(2):307–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee-Sherick AB, Eisenman KM, Sather S, McGranahan A, Armistead PM, McGary CS, Hunsucker SA, Schlegel J, Martinson H, Cannon C, Keating AK, Earp HS, Liang X, DeRyckere D, Graham DK. Aberrant Mer receptor tyrosine kinase expression contributes to leukemogenesis in acute myeloid leukemia. Oncogene. 2013. November 11;32(46):5359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Linger RMA, Cohen RA, Cummings CT, Sather S, Migdall-Wilson J, Middleton DHG, Lu X, Barón AE, Franklin WA, Merrick DT. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene. 2013;32(29):3420–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frye S V The art of the chemical probe. Nat Chem Biol. 2010/February/16 2009;6(3):159–61. [DOI] [PubMed] [Google Scholar]
- 37.Varnek A, Baskin I. Machine Learning Methods for Property Prediction in Chemoinformatics: Quo Vadis? J Chem Inf Model. 2012;52(6):1413–37. [DOI] [PubMed] [Google Scholar]
- 38.Mitchell JBO. Machine learning methods in chemoinformatics. Wiley Interdiscip Rev Comput Mol Sci. 2014;4(5):468–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindstroem A, Edvinsson L, Johansson A, Andersson CD, Andersson IE, Raubacher F, Linusson A. Postprocessing of Docked Protein-Ligand Complexes Using Implicit Solvation Models. J Chem Inf Model. 2011;51(2):267–82. [DOI] [PubMed] [Google Scholar]
- 40.Jain AN. Scoring functions for protein-ligand docking. Curr Protein Pept Sci. 2006/November/01 2006;7(5):407–20. [DOI] [PubMed] [Google Scholar]
- 41.Mobley DL, Gilson MK. Predicting Binding Free Energies: Frontiers and Benchmarks. Annu Rev Biophys. 2017. May 22;46(1):531–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.