

Abstract
Transposable elements (TEs) are parasitic DNA bits capable of mobilization and mutagenesis, typically suppressed by host’s epigenetic silencing. Since the selfish DNA concept, it is appreciated that genomes are also molded by arms-races against natural TE inhabitants. However, our understanding of evolutionary processes shaping TEs adaptive populations is scarce. Here, we review the events of recombination associated to reverse-transcription in LTR retrotransposons, a process shuffling their genetic variants during replicative mobilization. Current evidence may suggest that recombinogenic retrotransposons could beneficially exploit host suppression, where clan behavior facilitates their speciation and diversification. Novel refinements to retrotransposons life-cycle and evolution models thus emerge.
Keywords: epigenetics, recombination, retroelements, reverse-transcription, LTR retrotransposons, transcriptional gene silencing, transposons, transposable elements
“We must not only consider how things are, but how they came to be so.”
Thomas Burnet (1635–1715)
Introduction
Transposable elements (TEs) are selfish intragenomic parasites capable of replicative mobilization, inducing deleterious insertional mutations or potentially altering the regulation of nearby host genes (Weil and Martienssen 2008; Chuong et al. 2017; Gaubert et al. 2017). Classically, two types of TEs have been recognized: class I elements comprise “copy-and-paste” retrotransposons replicating through RNA intermediates, while class II elements comprise excising “cut-and-paste” TEs using DNA intermediates (Wicker et al. 2007). Since their discovery, much has been learned about their structural features, life-cycles, and active mobilization (Sabot and Schulman 2006; Feschotte and Pritham 2007; Wicker et al. 2007; Bennetzen and Wang 2014). Considerable attention has focused on how genomes recognize and epigenetically silence TEs, and how their numerous copies impact host trait variation, phenotypic diversity, and whole genome evolution (Rebollo et al. 2012; Bennetzen and Wang 2014; Fultz et al. 2015; Goodier 2016; Chuong et al. 2017). The dynamics of TEs within genomes has also been studied, for example, by using evolutionary models, in which extant TEs populations are explained by their historical burst-mediated increase in copy number counterbalanced by natural selection against those with harmful effects on the host (Le Rouzic and Capy 2006; Le Rouzic et al. 2007; Barron et al. 2014). However, the adaptive molecular evolution of TEs is much less understood (Feschotte and Pritham 2007).
Here, we analyze a process proposed to be involved in the evolution of particular TEs; specifically, extrachromosomal “reverse-transcription-related” recombination in LTR retrotransposons. We review available experimental data supporting the occurrence of such phenomena, and infer conceivable scenarios in which this type of interelement recombination becomes a driver of retrotransposon diversification and evolution, highlighting its relevance for intragenomic parasitic survival.
The Recombinogenic Nature of Retroelements
Retroelements represent a type of eukaryotic parasitic elements defined by a replicative mode that involves the reverse-transcription of their genomic RNA (gRNA) (Koonin et al. 2015). Retroelements include class I “copy-and-paste” TEs, comprising long-terminal-repeat (LTR) retrotransposons and non-LTR retrotransposons (Wicker et al. 2007). They also include animal retroviruses, which are thought to be related to ancestral forms of LTR retrotransposons (Koonin et al. 2015). Given their evolutionary relationship and life-cycle resemblances, it is plausible that retroviruses and retrotransposons share similar mechanisms to secure molecular variability and evolvability.
In retroviruses such as HIV, most genetic variability arises during the course of animal infection through the host cytidine deaminase mutating viral sequences, whereas virus replicative infidelity seems to play only a minor role (Cuevas et al. 2015). In addition, retroviral quasispecies shuffle their genetic information by means of recombination events, taking place during reverse-transcription (Onafuwa-Nuga and Telesnitsky 2009). In analogy to eukaryotes, a recombinatorial stage is thought to be advantageous for accelerating the exploration of the retroviral sequence space (Burke 1997). Simulations on HIV empirical fitness landscapes indeed underpin the notion that retroelement recombination accelerates adaptation (Moradigaravand et al. 2014).
It was thought that this step of reverse-transcription-related recombination is a common inherent character shared among all retroelements. This view was strongly supported by early studies in the yeast Saccharomyces cerevisiae, demonstrating that artificial Ty LTR retrotransposons recombined in vivo (Boeke et al. 1986; Wilhelm et al. 1999). Furthermore, phylogenetic studies of genome sequences revealed historical interelement recombination in particular LTR retrotransposon families from S. cerevisiae, Drosophila melanogaster, several plants, and mammalian endogenous-retroviruses (Jordan and McDonald 1998; Vicient et al. 2005; Sabot and Schulman 2007; Marco and Marin 2008; Sharma et al. 2008; Du, Tian, Bowen, et al. 2010; Carr et al. 2012; Sharma and Presting 2014; Vargiu et al. 2016). However, for naturally occurring LTR retrotransposons, reverse-transcription-related recombination has only been recently confirmed experimentally for Ty1 from S. cerevisiae and ONSEN/COPIA78 from the model plant Arabidopsis thaliana (Bleykasten-Grosshans et al. 2011; Sanchez et al. 2017). Despite its potential importance, interelement recombination still remains an understudied feature of retrotransposon biology.
Extrachromosomal Recombination during the Life-Cycle of LTR Retrotransposons
The structure and life-cycle of LTR retrotransposons are in principle analogous to retroviruses and have been reviewed elsewhere; for detailed understanding the reader is directed to more comprehensive revisions (Sabot and Schulman 2006; Wicker et al. 2007; Berkhout and Jeang 2013; Grandbastien 2015). However, we will briefly describe their assembly and replicative steps (fig. 1), necessary to grasp the interelement recombination events considered here. LTR retrotransposons are characterized by an internal coding area flanked by two LTRs which contain so-called U3/R/U5 domains, involved in transcriptional regulation (U3 domain harbor trans-activator binding sites, while R/U5 domains contain the transcription-start-site and transcription-termination-site) (fig. 2A). The open-reading frames typically code for a structural GAG and a polyprotein POL that comprises a protease, a reverse-transcriptase/ribonuclease H, and an integrase (figs. 1 and 2A). Their life-cycle starts with transcriptional triggering via the LTR promoter activity (fig. 1A), resulting in gRNA/mRNA translated to functional proteins (fig. 1B). The structural GAG assembles in the cytoplasm of host cells as virus-like particles (fig. 1C), where the enzymes and gRNA are copackaged (fig. 1D). Importantly, analogous to retroviruses, packaging comprises two plus-stranded parental gRNA molecules (fig. 1D) (Feng et al. 2000; Sabot and Schulman 2006; Onafuwa-Nuga and Telesnitsky 2009; Johnson and Telesnitsky 2010). Subsequently, a discontinuous reverse-transcription of gRNA takes place. It involves the priming of a template gRNA (executed by a tRNA recognizing a primer-binding site), followed by cDNA synthesis catalyzed by the reverse-transcriptase (fig. 1E). In addition, two so-called “strong-stop DNA” strand transfers take place (see below). As a result of reverse-transcription, an extrachromosomal DNA (ecDNA) molecule is generated (fig. 1F). Classical life-cycle ends when this ecDNA intermediate translocate to the host nucleus (fig. 1G), and eventually inserts at different host chromosome locations through integrase activity (fig. 1H).
Fig. 1.
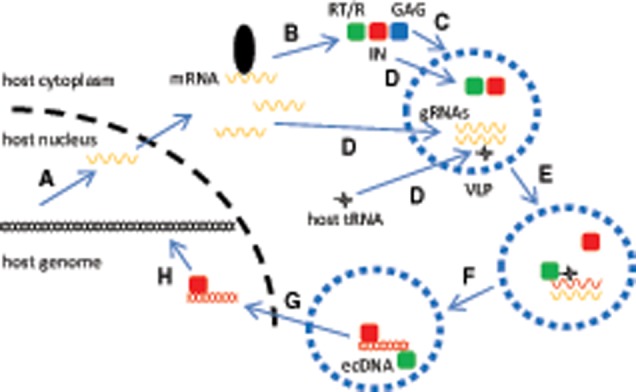
—Simplified classical representation of LTR retrotransposons life-cycle. (A) LTR retrotransposon is activated transcriptionally (black strand, host DNA; yellow strand, TE messenger/genomic RNA [mRNA/gRNA]). (B) mRNA is translated and cleaved into functional proteins, including a reverse-transcriptase/RNaseH (RT/R, green box), an integrase (IN, red box) and a structural GAG (blue box). (C) GAG assembles as virus-like particles (VLP) within the host cytoplasm. (D) Two gRNA molecules are copackaged along with proteins and host tRNA. (E) tRNA anneals the primer-binding-site, and primed RT/R eventually synthetizes a linear double-stranded extrachromosomal DNA (ds-ecDNA, red strand) intermediary, using gRNAs as templates. (F and G) ds-ecDNA associates with the integrase and migrates into the nucleus using yet unidentified mechanisms. (H) ds-ecDNA intermediary eventually inserts into a new location within the host genome, resulting in a new LTR retrotransposon copy.
Fig. 2.

—Schematic representation of discontinuous reverse-transcription and recombination steps. (A) Two members of a LTR retrotransposon clan are transcriptionally activated and the corresponding genomic RNA (gRNA) progenitors (starting and ending in the R regions of 5′- and 3′-LTR, respectively) are copackaged. (B) Reverse-transcriptase/RNaseH activity (RT/R, green) is primed by tRNA annealing the priming-binding-site (pbs) of gRNA1 (orange), and minus-single-strand extrachromosomal cDNA ((-)ss-ecDNA) is synthesized. (C) First strand transfer: strong-stop (-)ss-ecDNA transfers to a second gRNA2 (green) (first recombinogenic step; the hypothesized recombination point is marked with an inverted gray triangle), using sequence homologies in the R region (marked with a dotted box). (D) At the same time that RNAseH activity proceeds (not shown), the (-)ss-ecDNA is extended using gRNAs as alternate templates (second recombinogenic step; a single hypothesized recombination point is marked with an inverted black triangle, but note that more than one event is possible). The color-coded newly synthesized (-)ss-ecDNA molecule is represented as a mosaic of the two progenitor gRNAs. Although RNAseH activity degrades portions of gRNA2 template (not shown), priming of poly-purine-track (ppt) allows nascent plus-single-strand extrachromosomal cDNA ((+)ss-ecDNA) synthesis until the end of the (-)ss-ecDNA molecule used as template. (E) Second strand transfer: strong-stop (+)ss-ecDNA swaps toward the 5′ area of the (-)ss-ecDNA. In addition, RT/R final extensions take place. (F) After final extensions, a mosaic blunt-ended linear extrachromosomal DNA (ds-ecDNA) molecule with two identical LTRs is generated. U3/R/U5, domains within LTRs; GAG, structural protein; POL, polyprotein.
During reverse-transcription, two complex strong-stop DNA strand transfers mentioned previously are required to ultimately generate new identical LTRs within the resulting progeny. These DNA transfers have been exquisitely characterized for retroviruses and to a lesser extent for yeast Ty family retrotransposons (Pochart et al. 1993; Lauermann and Boeke 1997; Wilhelm et al. 1999; Basu 2008; Rausch et al. 2017). The first transfer proceeds after priming and cDNA extension till the end of the first gRNA template (fig. 2B, (-)ss-ecDNA), when this nascent strong-stop minus-single-strand cDNA swaps positions from the 5′-LTR to the 3′-LTR area of transcripts (dotted arrow from fig. 2B to C). This transfer is possible thanks to R domain homologies (fig. 2C, dotted box), and can take place within (intramolecular) or between (intermolecular) parental gRNAs (fig. 2C depicts the latter type) (Wilhelm et al. 1999). In downstream events, the synthesis of a plus-single-strand cDNA initiates from the priming of a poly-purine-track present in the minus-single-strand cDNA now used as template (fig. 2D, ppt), with further cDNA extension toward the end of the intermediate 3′ area (fig. 2D, (+)ss-ecDNA). The second transfer takes place when this nascent strong-stop plus-single-strand cDNA swaps position within the minus-single-strand cDNA template, from the 3′-LTR to the 5′-LTR area (dotted arrow from fig. 2D to E), apparently facilitated by primer-binding-site domain homologies (fig. 2E, dotted box). After final extensions of both minus and plus cDNA edges (fig. 2E, RT/R), the outcome is actually a blunt-ended linear double-stranded extrachromosomal DNA (ds-ecDNA) intermediate with identical LTRs (fig. 2F).
This life-cycle is inherently pseudodiploid, involving two gRNA progenitors that generate a single ecDNA molecule (Onafuwa-Nuga and Telesnitsky 2009). As mentioned earlier for retroviruses, such a pseudosexual scheme may benefit from recombination, which takes place during the discontinuous reverse-transcription stages and results in the shuffling of parental sequences (fig. 2F). Therefore, it becomes apparent when the progeny arises from two dissimilar gRNA molecules. At least two recombinogenic steps may be recognized in this scheme, with the earliest one resulting from the first aforementioned minus-single-strand transfer (fig. 2B and C). Here, an intermolecular swap will reconstitute next-generation LTRs as mosaics, merging the 5′-LTR R/U5 domains from the first primed gRNA template with the U3 domain from the 3′-LTR of the other copacked gRNA (fig. 2C, inverted gray triangle) (Basu 2008). As a consequence, LTR regulatory areas become mixed between progenitor elements. A second recombinogenic step may result from the reverse-transcriptase switching templates between the gRNAs during cDNA extension (fig. 2D, inverted black triangle), a phenomenon analogous to that described as “copy-choice” in RNA virus biology (Poirier and Vignuzzi 2017). Although reverse-transcriptase copy-choice can be understood as transfers of its product during cDNA synthesis (e.g., the minus-single-strand; Basu 2008), here, we will refer to it as reverse-transcriptase switching templates, to avoid confusion with the first DNA strand transfer. Sequence homologies between donor and acceptor molecules are required for the efficient template switches of reverse-transcriptase, which dissociates from one template and anneals to the other during cDNA extension (Onafuwa-Nuga and Telesnitsky 2009; Delviks-Frankenberry et al. 2011).
Importantly, we want to emphasize that reverse-transcription-related recombination takes place extrachromosomally (i.e., presumably within cytoplasmic virus-like particles, away from host chromosomes); unlike recombination of a different sort resulting from host genomic events such as unequal, illegitimate, ectopic, and homologous recombination (Devos et al. 2002; Ma et al. 2004; Sharma et al. 2008; Barron et al. 2014; Bennetzen and Wang 2014).
Naturally Occurring LTR Retrotransposons Display Clan Behavior
Given the shortage of data regarding the adaptive molecular evolution of TEs, LTR retrotransposons have been thought to acquire genetic variability largely through the accumulation of mutations introduced by the error-prone reverse-transcriptase during cDNA synthesis (Eickbush and Jamburuthugoda 2008). However, this view may eventually change in the face of mounting evidence resulting from in vivo observations connected to reverse-transcription-related recombination. Early research in S. cerevisiae used artificial elements to demonstrate that interelement recombination was operative in eukaryotic LTR retrotransposons (Boeke et al. 1986; Wilhelm et al. 1999). But to the best of our knowledge, only two reverse-transcription-related recombination cases among natural inhabitant LTR retrotransposons were caught in the act experimentally, namely for Ty1 and ONSEN/COPIA78 (Bleykasten-Grosshans et al. 2011; Sanchez et al. 2017).
Ty1 and ONSEN/COPIA78 are multimember LTR retrotransposon families with full-length elements, most of which can be unambiguously recognized by a set of sequence polymorphisms in the form of SNPs or indels (Carr et al. 2012; Sanchez et al. 2017). Older members typically present a higher number of discriminative polymorphisms, presumably acquired randomly since the time of their insertion. In some cases, these polymorphisms lead to the interruption of functional coding areas thus rendering partially defective TEs. These defective elements are usually thought to replicate nonautonomously, cis parasitizing their autonomous counterparts by hijacking required life-cycle proteins (Le Rouzic and Capy 2006; Sabot and Schulman 2006).
Notably, as a result of successful transposition bursts, chromosomal copies of newly inserted Ty1 and ONSEN/COPIA78 revealed contributions from both young and older family members. These neoinsertions were sequence mosaics entirely compatible with the occurrence of parental reverse-transcription-related recombination as described for retroviruses. In their LTRs, they showed signatures of inter- or intramolecular cDNA transfers—between distinct parental gRNAs or within particular older elements in which 5′ and 3′ LTRs diverged, respectively. Such mosaic new copies also frequently presented at least one, but usually more, apparent recombination events in-between LTRs as in reverse-transcriptase copy-choice template switches (Bleykasten-Grosshans et al. 2011; Sanchez et al. 2017). These results confirmed that sequence polymorphisms in naturally occurring LTR retrotransposons may be shuffled in a single cycle of replicative transposition; the fact that such events were detected independently in different kingdoms conceivably points toward a general principle of LTR retrotransposon evolution.
Importantly, not all members of Ty1 and ONSEN/COPIA78 families appeared to be involved in recombination events. Hence, we introduce here the novel concept of a retrotransposon “clan,” not only to convey the idea of sequence similarities revealing genealogy (as interpreted by the terms family or subfamily; Wicker et al. 2007) but also to reflect enabled transposition potential with cross-hybridization capabilities. The retrotransposon clan thus comprises family members capable of activation and generation of mosaic progenies through interelement recombination. Since TEs families usually also accommodate derived and inactive historical remnant elements, in most cases it is expected that the clan will represent only the youngest fraction of a family.
Evolutionary Implications of Recombination Associated to Reverse-Transcription
The previous observations revealed that even moderately disrupted LTR retrotransposons may contribute to family progenies, in the form of new seemingly competent full-length copies. This point was not necessarily expected given that old TEs are typically considered inactive, or at best replicating only nonautonomously (Sabot and Schulman 2006). Interestingly, some supposedly nonautonomous Ty1 and ONSEN/COPIA78 members generated both putative nonautonomous and autonomous progenies when engaged in recombination with manifest autonomous members (Bleykasten-Grosshans et al. 2011; Sanchez et al. 2017). Hence, it is possible that both, parasitical competition and recombinogenic complementation, may be operative replicative modes for defective elements of an LTR retrotransposon clan.
The number of nonparental polymorphisms observed in Ty1 and ONSEN/COPIA78 neoinsertions, which could be attributed to errors during transcription or reverse-transcription, reflected a degree of replication infidelity comparable to that observed for retroviruses (Eickbush and Jamburuthugoda 2008; Bleykasten-Grosshans et al. 2011; Sanchez et al. 2017). Nevertheless, nonparental error-related polymorphisms were much less abundant than those polymorphisms acquired from parental sequences via interelement recombination. Therefore, most molecular novelty in newly evolved copies may originate from sequence changes gained at host chromosomal level, apparently by the gradual ageing of parental clan members.
As with retroviruses, recombination of LTR retrotransposons should enable a faster exploration of the sequence space available for molecular evolution (Burke 1997). However, ageing becomes influential insofar older clan members recurrently and significantly contribute to reverse-transcription-related recombination. In other words, interelement recombination involving older members must consistently extend toward the evolutionary scale. Although conceivable, this still remains to be demonstrated. In ONSEN/COPIA78, reverse-transcription-related recombination effectively took place between family members separated by roughly 0.5–1 Myr of divergence (Sanchez et al. 2017), a figure comparable to the estimated half-life of LTR retrotransposons in plant genomes (Pereira 2004; Wicker and Keller 2007; Du, Tian, Hans, et al. 2010; Wicker et al. 2018; Carpentier et al. 2019; Liu et al. 2019). This suggests that the acquisition of polymorphisms may not be harmful for LTR retrotransposons fitness as long as it progresses in a time frame attuned with their population dynamics. It is plausible that the rate at which a thriving clan successfully bursts could be, on an average, higher than the rate at which the random acquisition of mutations in due course deleteriously disturbs its life-cycle. If this condition is met, then the time a clan spends quiescent between successful burst events, even under host’s epigenetic silencing, could be viewed as a variability acquiring stage. It could be said that genetic variation in a population of these recombinogenic TEs becomes a property also derived from their natural ageing. We thus anticipate that the life-cycle of prosperous LTR retrotransposon clans include two phases for gaining genetic variability: a slow phase that involves the “acquisition” of ageing polymorphisms perpetuated by host chromosomes, and a fast phase that “generates” variability from the overall replication infidelity during transposition bursts (fig. 3). Note that both ageing and infidelity polymorphisms may be shuffled by reverse-transcription-related recombination.
Fig. 3.
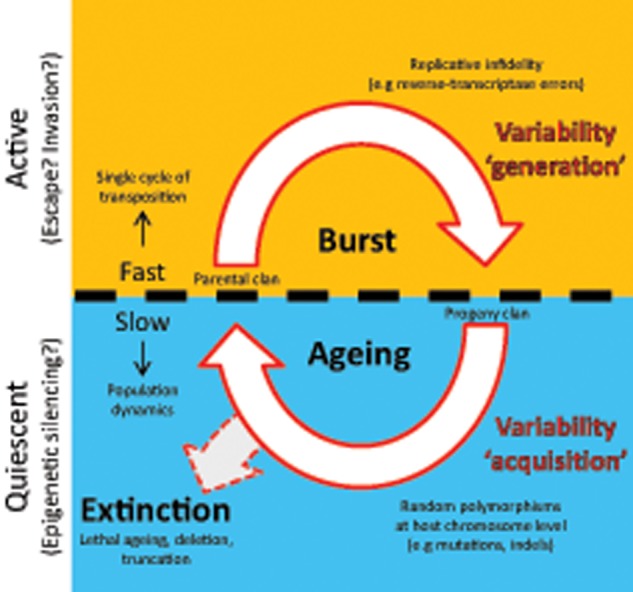
—Diagram of mechanisms for gaining genetic variability during LTR retrotransposons life-cycle. The life-cycle of LTR retrotransposon clans may include two hypothetical phases for gaining genetic variability: a fast phase “generating” variability from the overall replication infidelity during transposition bursts (top, orange background), and a long-lasting quiescent phase that involves the “acquisition” of polymorphisms resulting from ageing (bottom, blue background). In this model, the rate at which a clan bursts is assumed to be higher—on long-term average—than the rate at which the random acquisition of deleterious mutations eventually disturbs its life-cycle. If this condition is not met, a clan may ultimately be driven to extinction due to lethal ageing. Other contingent factors may also be implicated in clan extinction, such as truncation or deletion of its members mediated by host chromosomal recombination or rearrangements. Note that the classical life-cycle from figure 1 and the recombinogenic steps from figure 2 would be contained within the upper fast “generating” variability phase.
At present, it is not clear if host chromosomal recombination events involving TE sequences could considerably contribute to the slow phase, but it is conceivable that processes such as illegitimate recombination may increase the rate of polymorphisms occurring in silenced TEs (Devos et al. 2002; Ma et al. 2004; Sharma et al. 2008; Barron et al. 2014; Bennetzen and Wang 2014). In addition, although current available experimental data appear to suggest that the slow phase is of greater importance, the underlying notion is that the occurrence probability of spontaneous transposition is very low for any given host individual. But in principle, it is certainly possible that the fast phase may become the primary source of variability in clans displaying a relatively high mobilization rate—considered at host population level—thus, drastically decreasing the amount of evolutionary time allocated for the accumulation of chromosomal mutations. In addition, contributions to the fast phase from host cytidine deaminase edits in animal elements, as with retroviruses, cannot be ruled out (Goodier 2016). Altogether, this model (fig. 3) provides an initial mechanistic explanation for the extraordinary genetic variability and speed of molecular evolution displayed by LTR retrotransposons (Grandbastien 2015).
Another interesting empirical observation was that, despite the occurrence of pervasive reverse-transcription-related recombination, still some neoinsertions were not mosaics, presenting sequences indistinguishable from any clan parent (Bleykasten-Grosshans et al. 2011; Sanchez et al. 2017). This is most likely due to the copackaging of identical gRNA molecules, and it could be interpreted as a safe guard strategy against the excessive combinatorial capabilities of the life-cycle. Since many ageing changes may be expected to be functionally unfavorable, this effectively decreases the chances of negative consequences to fitness from reshuffling detrimental mutations. It follows that the rapid exploration of the sequence space enabled by interelement recombination was not fully exploited, ensuring long-term survival of functional sequences from the successful original stock. The limit seemed intrinsically imposed by differential transcriptional activation, since in both Ty1 and ONSEN/COPIA78 the majority of new mosaic and nonmosaic copies derived from the most transcriptionally competent parents (Morillon et al. 2002; Bleykasten-Grosshans et al. 2011; Sanchez et al. 2017). However, current available empirical data cannot rule out a relative bias toward heterodimeric gRNA copackaging.
On the other hand, the mechanisms of reverse-transcription-related recombination ensure that new mosaic copies will not receive all accumulated mutations from a particularly aged but still transcriptionally active parent, thus decreasing the chances of extreme inherent suboptimal performance in the next generations. Both properties could aid LTR retrotransposon clans in maximizing diversity without lethally compromising fitness, avoiding the accumulation of deleterious mutations that may lead to loss of fitness with eventual downward spiral decline in population size.
Limitations to Reverse-Transcription-Related Recombination
Clan behavior as documented within Ty1 and ONSEN/COPIA78 families may imply the existence of recombination barriers; the most obvious candidates being sequence homologies and functional recognition supporting gRNA copackaging, complementation, and propagation (Motomura et al. 2008; Ali et al. 2016). For retroviruses such as HIV-1 and HIV-2, gRNAs cross-packed and further recombined despite relatively low similarity, albeit at very low frequencies (Motomura et al. 2008). This reflects a potential trade-off between retroelements homology/complementation barriers and the frequency of recombination.
Less stringent barriers could conceivably allow much older LTR retrotransposons to often indulge into recombinogenic behavior with younger elements, although this may be further restricted by activation at appropriate time and space in vivo. Differential transcription was mentioned in the previous section, and developmental patterns of retrotransposon activation signals are interesting to briefly consider. It is plausible that the occurrence likelihood of interelement recombination will be highest in the host germline, from where genomic parasites spread vertically. Some reports demonstrated that LTR retrotransposons may inhabit particular eukaryotic cellular niches contributing to the host next generation, sometimes even invading the germline from somatic tissue (Wang et al. 2018; Sanchez et al. 2019). Since animals differentiate the germline early in development, interelement recombination-competent niches will most likely be gametic, zygotic, and early embryonic (Rodriguez-Terrones and Torres-Padilla 2018; Wang et al. 2018). In plants, the germline differentiates in the final steps of their life cycle, expanding these opportunities to vegetative tissues carrying the germline at various discernible developmental stages. However, extrachromosomal recombination events could be inferred for ONSEN/COPIA78 upon activation in whole seedlings mostly composed of vegetative nongermline plant tissue (Sanchez et al. 2017), suggesting that recombination may still occur at any host cell where TEs are activated and competent for reverse-transcription. In summary, cellular niches with enabled interelement recombination potential may differ depending on TEs and hosts lineages. What exact barriers and windows of opportunity curb reverse-transcription-related recombination events among clan LTR retrotransposons remain to be thoroughly investigated.
It is also important to point out that our understanding of interelement recombination phenomena is not only constrained by biological impediments but also by available technology. Classical cloning and Sanger-sequencing were essential for validating episodes of recombination as revealed in the progeny, and will probably remain as the ultimate accurate demonstration of retrotransposon mobilization and genetic shuffling (Bleykasten-Grosshans et al. 2011; Sanchez et al. 2017). However, this evidently required contemporary transposition busts to be caught in the act, a feat currently accessible in only few exemplary cases. Current short-read next-generation-sequencing techniques, combining whole-genome RNA and DNA sequencing, allowed the real-time tracking of TE activity estimating the contribution of individual elements to the next generation while screening for recombinant progeny (Gaubert et al. 2017; Sanchez et al. 2017). This also enabled the direct detection of extrachromosomal recombination events, albeit at low sensitivity due to confounding effects from intrinsic sequencing errors and dilution of extrachromosomal copies under a plentiful genomic DNA background (Sanchez et al. 2017). Future applications of sequence capture technology may overcome this last drawback (e.g., as applied in Quadrana et al. 2016). Note that in the context of next-generation sequencing, PCR-free technologies are required to ascertain with confidence the shuffling of genetic polymorphisms, due to heteroduplex formation during mixed-template polymerization (Thompson et al. 2002). The coming wave of long-read sequencing data will certainly open up unprecedented possibilities to overcome limitations imposed by short-window sequencing (van Dijk et al. 2018), facilitating the finding of novel TEs insertions at low coverage (Debladis et al. 2017). However, its current high error rate is of concern, and may restrict its efficacy to uncovering only interelement recombination between sufficiently dissimilar gRNAs. A sensible application of mixed technologies could undoubtedly expedite the exploration of this field.
Reverse-Transcription-Related Recombination at the Population Level
In sexually reproducing organisms, meiotic recombination enables the shuffling of genetic variants brought together by interbreeding, which is a major tenet of the biological species concept. Interbreeding drives flows of genetic information within a population (de Queiroz 2005). Speciation events lead to reproductive isolation, where in principle this gene flow is no longer possible. Loosely resembling sexual organisms, reverse-transcription-related recombination could drive the evolutionary trajectories of LTR retrotransposons “species.” Extreme examples have been phylogenetically documented as instances of apparent interelement recombination between more- or less-distant TEs families/subfamilies, where gRNAs were probably heterodimeric cross-packaged resulting in new TEs lineages (Jordan and McDonald 1998; Vicient et al. 2005; Sabot and Schulman 2007; Marco and Marin 2008; Sharma et al. 2008; Du, Tian, Bowen, et al. 2010; Carr et al. 2012; Sharma and Presting 2014). Thus, divergence may be initiated with the emergence of active founder variants unable to recombine back with their original clan, isomorphic to genetic isolation (fig. 4). This provides a source for the emergence of novel elements, a nonmutually exclusive alternative for the appearance of new TE inhabitants through host genome invasion mediated by sexual interspecific hybridization or nonsexual horizontal transfer (fig. 4) (Le Rouzic et al. 2007; Schaack et al. 2010; Carr et al. 2012; El Baidouri et al. 2014).
Fig. 4.
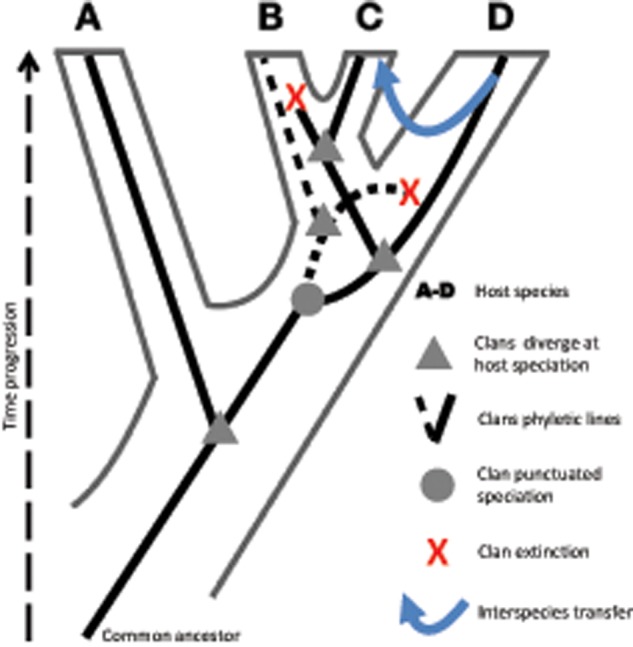
—Schematic population-level evolutionary trajectories influenced by reverse-transcription-related recombination. Four hypothetical host lineages (A–D) are depicted, with the corresponding conjectural evolutionary trajectories of retrotransposon clans. Phyletic transformation may take place along a clan’s evolutionary trajectory (continuous and dotted black lines), speeded up by reverse-transcription-related recombination. Common ancestor clans are shown at host divergence points (gray triangles). Clan punctuated speciation may occur when an active founder variant emerge, which is unable to recombine back with the clan (gray circle). A hypothetical event of interspecies transfer is also shown (blue arrow). Note that phyletic transformation may eventually result in diversification, in this case depicted as the lack of recombination between the original clan line in (C) and the invasion clan line from (D). Clan extinction events are also assumed (red cross). For simplicity, the host speciation event for the clan denoted by the phyletic dotted line toward (C) is not shown.
It is worth mentioning that interelement recombination between very distant or unrelated cross-packaged gRNA must be rare, thus explaining why so far only a handful of phylogenetic studies uncovered these events. Again, a trade-off between retroelements homology/complementation barriers and recombination frequency seems to be revealed. The most common cases of reverse-transcription-related recombination will arise from “conspecific” gRNA copackaging, which will not manifest punctuated historical events with conspicuous discontinuity of parental identity. Nevertheless, recombination between copackaged conspecifics is expected to increase the rate by which clans evolve, thus speeding-up LTR retrotransposon diversification through phyletic (vertical) transformation (fig. 4).
A noteworthy topic is that the advantages gained by reverse-transcription-related recombination in eukaryotic retrotransposons may not be necessarily constrained by the copy number of family elements occurring within the genome of a single host individual. Effective interelement recombination can still be expected between variants occurring in genomes of other host individuals or even between ecotypes. In other words, sexual host populations represent a reservoir of segregating nonidentical LTR retrotransposon copies of the same clan, brought together by host interbreeding and hybridization between subpopulations. It follows that the universe of sequence variability, potentially available for recombinogenic molecular evolution of retrotransposons, will be governed not only by the copy number of clan members within an individual genome but also by host population size, element occurrence frequencies within this population, and host propagation strategy (e.g., inbreeding vs. outbreeding). Therefore, the clan should be recognized as all active recombinogenic elements of a family/subfamily within a host pan-genome, although in practice those inhabitants from different host subpopulations may never effectively recombine. Unfortunately, without experimental evidence, it seems currently unlikely to predict exactly which members of a family may actually represent the whole clan, particularly for those elements acquiring large number of polymorphisms through genetic drift.
These points are compatible with the view of genomes as ecological communities of TEs (Venner et al. 2009). In classical Darwinian thinking, the unit of selection is the individual, but the population is the unit of evolution (Lewontin 1970). In analogy, when considered in the context of ecological communities, we envision that the individual LTR retrotransposon is under selection but the clan drives its evolution.
Topics on Selection of Mosaic Elements
Retroviral quasispecies appear to thrive near the limits of their critical mutation rate (error threshold), maximizing diversity while retaining genomic identity (Tripathi et al. 2012). Here, the stages of gaining genetic variability, recombinogenic shuffling, and selection for proximal functional optimization all occur within a restricted time scale (i.e., in the course of host infection; Onafuwa-Nuga and Telesnitsky 2009). However, lacking an infective phase enabling horizontal spreading, selection postintegration may be vastly stretched chronologically in TEs.
Natural selection over TEs may act at least at three levels: at host population, host individual, and TEs sequence levels (Tenaillon et al. 2010). In the former, host demography and historical contingencies related to survival of host populations must pose a sieve over the persistence of TE lineages (e.g., consider host extinction). At the host individual level, selection would be negative over those individuals carrying deleterious elements, thus selecting against TE insertions that mutated essential genes or otherwise had a negative impact on gene function or regulation (Weil and Martienssen 2008; Tenaillon et al. 2010; Barron et al. 2014). On the other hand, selection would be positive over newly inserted elements that benefit the host (e.g., insertions deregulating genes toward an increase in host fitness; Lanciano and Mirouze 2018).
Finally, natural selection is also expected to operate at individual TE sequence level, which is of critical interest in the case of recombinogenic elements. First, selection must act negatively against discrete element variants unfit for proper selfish maneuvers; for instance, that cannot undergo efficient replicative mobilization (at least at a rate that would compensate for the natural loss of their copy number; Le Rouzic et al. 2007). It could be envisioned that those responsible polymorphisms will be purged from a successful active clan aid by the workings of interelement recombination. Second, it is conceivable that selection will be positive on element sequence variants promoting their survival, such as those carrying mutations that increase the chances of escaping silencing (mounting evidence provide conceivable escaping scenarios in both animal and plant retrotransposons; see, e.g., Wang et al. 2018; Sanchez et al. 2019), or propitiate activation and mobilization (presumably, insofar they are not relatively more deleterious at previous levels). For instance, if a clan member is not recognized by host silencing, reverse-transcription-related recombination may ensure the occurrence of a future offspring clan which will remain free from suppression. In another case, it may be hypothesized that diversifying recombination within a normally recognized and silenced clan could result in “rejuvenated” elements capable of escaping silencing. Although the emphasis of this speculation was placed on vulnerability to host epigenetic machinery, selection could be hypothesized to operate ensuring a certain degree of effective silencing over recombinogenic LTR retrotransposons, which could guarantee the accumulation of ageing polymorphisms during the gaining of genetic variability in a clan’s slow “acquisition” phase (fig. 3).
Concluding Remarks
Extrachromosomal reverse-transcription-related recombination, in conjunction with host intra- or interspecific hybridization and interspecies transfer, is most likely at the heart of retrotransposon evolvability (Schaack et al. 2010; Bleykasten-Grosshans et al. 2011; Carr et al. 2012; El Baidouri et al. 2014; Sanchez et al. 2017). Recombination significance lies not only in permitting clan behavior, increasing the rate of adaptive exploration of the sequence space while purging deleterious mutations but also in the ensuing diversification when it is absent. The potential universality of such mechanisms within retrotransposons becomes more palpable when considering also particular non-LTR retrotransposons, for which interelement recombination has been established not only by phylogenetic analysis but also empirically for cultured animal cells or an artificial element in a protist model (Hayward et al. 1997; Gilbert et al. 2005; Yadav et al. 2012; Sookdeo et al. 2013). Interestingly, chimerization of copies and vertical diversification have also been recognized in some class II “cut-and-paste” TEs (Fischer et al. 2003; Feschotte and Pritham 2007; Novick et al. 2011; Vergilino et al. 2013), for which there is growing evidence of pervasive horizontal transfer (Schaack et al. 2010; Peccoud et al. 2017). Perhaps interelement recombination might be a convergent property of all TEs, proceeding through different underlying molecular mechanisms depending on the TE type or replication strategy.
Based on early observations, a daring proposition was that TEs may be “hidden” from the genome by epigenetic silencing, allowing their accumulation in high copy numbers (Martienssen 1998). We here entertain the conjectural notion that recombinogenic TEs might benefit from host genome identification and targeting, exploiting epigenetic suppression to decrease their clan activation rate. This would in theory aid not only in diminishing detrimental consequences of transposition (Weil and Martienssen 2008) but also in gradually accumulating polymorphisms that could eventually enhance their own molecular evolution through diversifying recombination, improving their adaptability to hosts. Note that this would imply the evolution of self-restrain, which is an already recognized property of retrotransposons (Tucker et al. 2015; Gaubert et al. 2017).
These may represent molecular processes enabling TEs to express a scientifically ill-explored repertoire of survival strategies within the context of intragenomic parasites versus host arms-races. Further empirical studies exploring the viewpoints presented herein may unveil the precise coevolutionary relationship between TEs and their host genomes on a population genomic scale.
Acknowledgments
We would like to thank Adrian Valli for critical reading of the article, and two anonymous reviewers for positive feedback and suggestions.
Literature Cited
- Ali LM, Rizvi TA, Mustafa F.. 2016. Cross- and co-packaging of retroviral RNAs and their consequences. Viruses 8(10):276.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron MG, Fiston-Lavier AS, Petrov DA, Gonzalez J.. 2014. Population genomics of transposable elements in Drosophila. Annu Rev Genet. 48:561–581. [DOI] [PubMed] [Google Scholar]
- Basu VP. 2008. Strand transfer events during HIV-1 reverse transcription. Virus Res. 134(1–2):19–38. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, Wang H.. 2014. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu Rev Plant Biol. 65(1):505–530. [DOI] [PubMed] [Google Scholar]
- Berkhout B, Jeang KT.. 2013. Retrovirus replication strategies. Curr Opin Virol. 3(6):601–603. [DOI] [PubMed] [Google Scholar]
- Bleykasten-Grosshans C, et al. 2011. The Ty1 LTR-retrotransposon population in Saccharomyces cerevisiae genome: dynamics and sequence variations during mobility. FEMS Yeast Res. 11(4):334–344. [DOI] [PubMed] [Google Scholar]
- Boeke JD, Styles CA, Fink GR.. 1986. Saccharomyces cerevisiae SPT3 gene is required for transposition and transpositional recombination of chromosomal Ty elements. Mol Cell Biol. 6(11):3575–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke DS. 1997. Recombination in HIV: an important viral evolutionary strategy. Emerg Infect Dis. 3(3):253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier MC, et al. 2019. Retrotranspositional landscape of Asian rice revealed by 3000 genomes. Nat Commun. 10(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr M, Bensasson D, Bergman CM.. 2012. Evolutionary genomics of transposable elements in Saccharomyces cerevisiae. PLoS One 7(11):e50978.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, Feschotte C.. 2017. Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet. 18(2):71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas JM, Geller R, Garijo R, López-Aldeguer J, Sanjuán R.. 2015. Extremely high mutation rate of HIV-1 in vivo. PLoS Biol. 13(9):e1002251.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Queiroz K. 2005. Ernst Mayr and the modern concept of species. Proc Natl Acad Sci U S A. 102(Suppl 1):6600–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debladis E, Llauro C, Carpentier M-C, Mirouze M, Panaud O.. 2017. Detection of active transposable elements in Arabidopsis thaliana using Oxford nanopore sequencing technology. BMC Genomics 18(1):537.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delviks-Frankenberry K, et al. 2011. Mechanisms and factors that influence high frequency retroviral recombination. Viruses 3(9):1650–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos KM, Brown JK, Bennetzen JL.. 2002. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 12(7):1075–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Tian Z, Bowen NJ. et al. 2010. Bifurcation and enhancement of autonomous-nonautonomous retrotransposon partnership through LTR swapping in soybean. Plant Cell 22(1):48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Tian Z, Hans CS, et al. 2010. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. Plant J. 63(4):584–598. [DOI] [PubMed] [Google Scholar]
- Eickbush TH, Jamburuthugoda VK.. 2008. The diversity of retrotransposons and the properties of their reverse transcriptases. Virus Res. 134(1–2):221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Baidouri M, et al. 2014. Widespread and frequent horizontal transfers of transposable elements in plants. Genome Res. 24(5):831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y-X, Moore SP, Garfinkel DJ, Rein A.. 2000. The genomic RNA in Ty1 virus-like particles is dimeric. J Virol. 74(22):10819.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ.. 2007. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 41(1):331–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SE, Wienholds E, Plasterk RH.. 2003. Continuous exchange of sequence information between dispersed Tc1 transposons in the Caenorhabditis elegans genome. Genetics 164(1):127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fultz D, Choudury SG, Slotkin RK.. 2015. Silencing of active transposable elements in plants. Curr Opin Plant Biol. 27:67–76. [DOI] [PubMed] [Google Scholar]
- Gaubert H, Sanchez DH, Drost HG, Paszkowski J.. 2017. Developmental restriction of retrotransposition activated in Arabidopsis by environmental stress. Genetics 207(2):813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N, Lutz S, Morrish TA, Moran JV.. 2005. Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol Cell Biol. 25(17):7780–7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL. 2016. Restricting retrotransposons: a review. Mob DNA. 7:16.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandbastien MA. 2015. LTR retrotransposons, handy hitchhikers of plant regulation and stress response. Biochim Biophys Acta. 1849(4):403–416. [DOI] [PubMed] [Google Scholar]
- Hayward BE, Zavanelli M, Furano AV.. 1997. Recombination creates novel L1 (LINE-1) elements in Rattus norvegicus. Genetics 146(2):641–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SF, Telesnitsky A.. 2010. Retroviral RNA dimerization and packaging: the what, how, when, where, and why. PLoS Pathog. 6(10):e1001007.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan IK, McDonald JF.. 1998. Evidence for the role of recombination in the regulatory evolution of Saccharomyces cerevisiae Ty elements. J Mol Evol. 47(1):14–20. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Dolja VV, Krupovic M.. 2015. Origins and evolution of viruses of eukaryotes: the ultimate modularity. Virology 479-480:2–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciano S, Mirouze M.. 2018. Transposable elements: all mobile, all different, some stress responsive, some adaptive? Curr Opin Genet Dev. 49:106–114. [DOI] [PubMed] [Google Scholar]
- Lauermann V, Boeke JD.. 1997. Plus-strand strong-stop DNA transfer in yeast Ty retrotransposons. EMBO J. 16(21):6603–6612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rouzic A, Boutin TS, Capy P.. 2007. Long-term evolution of transposable elements. Proc Natl Acad Sci U S A. 104(49):19375–19380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rouzic A, Capy P.. 2006. Population genetics models of competition between transposable element subfamilies. Genetics 174(2):785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin RC. 1970. The units of selection. Annu Rev Ecol Syst. 1(1):1–18. [Google Scholar]
- Liu Y, et al. 2019. Comparative analysis of miniature inverted-repeat transposable elements (MITEs) and long terminal repeat (LTR) retrotransposons in six Citrus species. BMC Plant Biol. 19(1):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Devos KM, Bennetzen JL.. 2004. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 14(5):860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Marin I.. 2008. How Athila retrotransposons survive in the Arabidopsis genome. BMC Genomics 9(1):219.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen R. 1998. Transposons, DNA methylation and gene control. Trends Genet. 14(7):263–264. [DOI] [PubMed] [Google Scholar]
- Moradigaravand D, et al. 2014. Recombination accelerates adaptation on a large-scale empirical fitness landscape in HIV-1. PLoS Genet. 10(6):e1004439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillon A, Benard L, Springer M, Lesage P.. 2002. Differential effects of chromatin and Gcn4 on the 50-fold range of expression among individual yeast Ty1 retrotransposons. Mol Cell Biol. 22(7):2078–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motomura K, Chen J, Hu W-S.. 2008. Genetic recombination between human immunodeficiency virus type 1 (HIV-1) and HIV-2, two distinct human lentiviruses. J Virol. 82(4):1923–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick PA, Smith JD, Floumanhaft M, Ray DA, Boissinot S.. 2011. The evolution and diversity of DNA transposons in the genome of the lizard Anolis carolinensis. Genome Biol Evol. 3:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onafuwa-Nuga A, Telesnitsky A.. 2009. The remarkable frequency of human immunodeficiency virus type 1 genetic recombination. Microbiol Mol Biol Rev. 73(3):451–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peccoud J, Loiseau V, Cordaux R, Gilbert C.. 2017. Massive horizontal transfer of transposable elements in insects. Proc Natl Acad Sci U S A. 114(18):4721–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira V. 2004. Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol. 5(10):R79.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochart P, et al. 1993. Biochemical and electron microscope analyses of the DNA reverse transcripts present in the virus-like particles of the yeast transposon Ty1. Identification of a second origin of Ty1DNA plus strand synthesis. Nucleic Acids Res. 21(15):3513–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier EZ, Vignuzzi M.. 2017. Virus population dynamics during infection. Curr Opin Virol. 23:82–87. [DOI] [PubMed] [Google Scholar]
- Quadrana L, et al. 2016. The Arabidopsis thaliana mobilome and its impact at the species level. Elife 5:e15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch JW, Miller JT, Le Grice SF.. 2017. Reverse transcription in the Saccharomyces cerevisiae long-terminal repeat retrotransposon Ty3. Viruses 9(3):44.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo R, Romanish MT, Mager DL.. 2012. Transposable elements: an abundant and natural source of regulatory sequences for host genes. Annu Rev Genet. 46(1):21–42. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Terrones D, Torres-Padilla M-E.. 2018. Nimble and ready to mingle: transposon outbursts of early development. Trends Genet. 34(10):806–820. [DOI] [PubMed] [Google Scholar]
- Sabot F, Schulman AH.. 2006. Parasitism and the retrotransposon life cycle in plants: a hitchhiker’s guide to the genome. Heredity 97(6):381–388. [DOI] [PubMed] [Google Scholar]
- Sabot F, Schulman AH.. 2007. Template switching can create complex LTR retrotransposon insertions in Triticeae genomes. BMC Genomics 8(1):247.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez DH, Gaubert H, Drost HG, Zabet NR, Paszkowski J.. 2017. High-frequency recombination between members of an LTR retrotransposon family during transposition bursts. Nat Commun. 8(1):1283.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez DH, Gaubert H, Yang W.. 2019. Evidence of developmental escape from transcriptional gene silencing in MESSI retrotransposons. New Phytol. 223(2):950–964. [DOI] [PubMed] [Google Scholar]
- Schaack S, Gilbert C, Feschotte C.. 2010. Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol. 25(9):537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Presting GG.. 2014. Evolution of centromeric retrotransposons in grasses. Genome Biol Evol. 6(6):1335–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Schneider KL, Presting GG.. 2008. Sustained retrotransposition is mediated by nucleotide deletions and interelement recombinations. Proc Natl Acad Sci U S A. 105(40):15470–15474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sookdeo A, Hepp CM, McClure MA, Boissinot S.. 2013. Revisiting the evolution of mouse LINE-1 in the genomic era. Mob DNA. 4(1):3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon MI, Hollister JD, Gaut BS.. 2010. A triptych of the evolution of plant transposable elements. Trends Plant Sci. 15(8):471–478. [DOI] [PubMed] [Google Scholar]
- Thompson JR, Marcelino LA, Polz MF.. 2002. Heteroduplexes in mixed-template amplifications: formation, consequence and elimination by ‘reconditioning PCR’. Nucleic Acids Res. 30(9):2083–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi K, Balagam R, Vishnoi NK, Dixit NM.. 2012. Stochastic simulations suggest that HIV-1 survives close to its error threshold. PLoS Comput Biol. 8(9):e1002684.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker JM, Larango ME, Wachsmuth LP, Kannan N, Garfinkel DJ.. 2015. The Ty1 retrotransposon restriction factor p22 targets gag. PLoS Genet. 11(10):e1005571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C.. 2018. The third revolution in sequencing technology. Trends Genet. 34(9):666–681. [DOI] [PubMed] [Google Scholar]
- Vargiu L, et al. 2016. Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology 13(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venner S, Feschotte C, Biemont C.. 2009. Dynamics of transposable elements: towards a community ecology of the genome. Trends Genet. 25(7):317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergilino R, Elliott TA, Desjardins-Proulx P, Crease TJ, Dufresne F.. 2013. Evolution of a transposon in Daphnia hybrid genomes. Mob DNA. 4(1):7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicient CM, Kalendar R, Schulman AH.. 2005. Variability, recombination, and mosaic evolution of the barley BARE-1 retrotransposon. J Mol Evol. 61(3):275–291. [DOI] [PubMed] [Google Scholar]
- Wang L, Dou K, Moon S, Tan FJ, Zhang ZZ.. 2018. Hijacking oogenesis enables massive propagation of LINE and retroviral transposons. Cell 174(5):1082–1094.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil C, Martienssen R.. 2008. Epigenetic interactions between transposons and genes: lessons from plants. Curr Opin Genet Dev. 18(2):188–192. [DOI] [PubMed] [Google Scholar]
- Wicker T, et al. 2007. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 8(12):973–982. [DOI] [PubMed] [Google Scholar]
- Wicker T, et al. 2018. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 19(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Keller B.. 2007. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 17(7):1072–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M, Boutabout M, Heyman T, Wilhelm FX.. 1999. Reverse transcription of the yeast Ty1 retrotransposon: the mode of first strand transfer is either intermolecular or intramolecular. J Mol Biol. 288(4):505–510. [DOI] [PubMed] [Google Scholar]
- Yadav VP, Mandal PK, Bhattacharya A, Bhattacharya S.. 2012. Recombinant SINEs are formed at high frequency during induced retrotransposition in vivo. Nat Commun. 3(1):854.. [DOI] [PubMed] [Google Scholar]