

ABSTRACT
BACKGROUND:
Pulmonary hypertension (PH) is an idiopathic or secondary disorder associated with many systemic illnesses. Long-term survival in PH depends on the severity and functional class. Several new drugs are now available to treat PH, but their impact on clinical outcome and survival are not well established.
OBJECTIVES:
Evaluate severity parameters and the impact of current recommended therapy on survival in PH.
DESIGN:
Cross-sectional.
SETTINGS:
Tertiary care center.
PATIENTS AND METHODS:
The study included adult patients who had undergone right heart catheterization since 2012 and were diagnosed with pulmonary hypertension. Survival was recorded after 6 years, at the end of the study. Nine severity variables for PH were assessed including right ventricular size by echocardiogram and pulmonary artery diameter (PA diameter) and the ratio of pulmonary artery diameter to ascending aorta diameter (PA/Ao ratio) by CT.
MAIN OUTCOME MEASURES:
Evaluation of severity parameters.
SAMPLE SIZE:
30 patients.
RESULTS:
Twenty-five patients were positive for 8/9 severity parameters. Eight of 30 (26.6%) patients died. In nonsurvivors, right ventricular size was increased by 25% (P=.427), pulmonary vascular resistance increased by 29.4% in nonsurvivors (P=.302), the 6-minute walk distance decreased by 21% (P=.875), median brain natriuretic peptide increased by 96% (P=.890), median GGT and alkaline phosphatase were 3 times higher in nonsurvivors (P=.893 and P=.047, respectively) and PA/Ao was nonsignificantly decreased in nonsurvivors (P=.373), Survival was decreased by a median of 2.3 years in nonsurvivors.
CONCLUSION:
Our study identified a subgroup of PH patients with NYHA functional class III and above with worsening severity indicators who were labeled as a high-risk group. These patients showed continuous deterioration in their clinical status despite escalation of therapy with current guidelines. We recommend these high-risk group patients be referred for early lung transplantation.
LIMITATIONS:
Low sample size and only a single center. Needs confirmation with a larger multicenter trial.
CONFLICT OF INTEREST:
None.

INTRODUCTION
Pulmonary hypertension (PH) may present as a primary idiopathic disorder or as a secondary disorder associated with many systemic illnesses.1 Current guidelines define PH as an increase in mean pulmonary arterial pressure (mPAP) >25 mm Hg at rest as assessed by right heart catheterization (RHC).1 The clinical classification of PH defines five groups according to clinical presentation, pathological findings, hemodynamic characteristics and treatment strategy. Comparative epidemiological data on the prevalence of the different groups of PH are not widely available. A group of risk factors have been described including genetic mutations (e.g. bone morphogenetic protein receptor 2, eukaryotic translation initiation factor 2 alpha kinase 4 and potassium channel subfamily K member 3), drugs and medical conditions (e.g. heart failure, lung diseases and chronic thromboembolic pulmonary hypertension). RHC is the standard diagnostic tool, but a diagnostic algorithm (including echocardiography, radiological assessments and blood tests) are recommended before proceeding to RHC. Risk stratification of patients with PH includes several parameters (including signs of right heart failure, a history of syncope, NT (N-terminal) proBNP (brain natriuretic peptide) plasma levels, New York Heart Association (NYHA) classification and the 6-minute walk distance. The NYHA classification provides a simple way of classifying the extent of PH, based on their limitations during physical activity from I-IV, class IV being the worst.1,2
Factors that may influence long-term survival in PH are not well established despite progress in diagnosis and management.3-10 Radiological findings of enlarged pulmonary artery size on computed tomography (CT) of the chest may correlate with the presence of PH but does not predict severity.12-16
In patients with advanced disease NYHA class III and above the impact of escalating therapy with current guidelines is not well established, several new drugs have recently been introduced with claims of significant benefit with a huge cost to the health care system, NYHA III and above, increased proBNP and dilated right ventricle with increased right ventricular pressure have been reported as indicators of poor prognosis. But impact of current therapy in reversing the parameters or improving clinical status not well established.11 In this study, we evaluated nine severity parameters of PH and the impact of current therapy on these severity parameters and the impact on long-term survival in a high-risk group.
PATIENTS AND METHODS
The patients were registered in 2012 and treated at King Faisal Specialist Hospital and Research Centre (KFSHRC), a tertiary care hospital in Riyadh. The enrollment included adult patient (>18 years old) who underwent RHC and were diagnosed with PH. There were no exclusion criteria. Postregistration follow up lasted until 2018, after 6 years. All of the five PH subgoups were represented among the patients.
All patients received treatment for PH that included sildenafil, bosentan or macitentan, systemic or inhaled prostanoids and anticoagulation, following the standard guidelines for management of PH.1 Right-sided cardiac catheterization was used to measure mean pulmonary artery systolic pressure (mPAP, mm Hg), systolic pulmonary artery pressure (sPAP, mm Hg), mean pulmonary vascular resistance (PVR, Wood units) and left ventricular ejection fraction (LVEF, %). Right ventricular size was measured by echocardiogram. Pulmonary artery diameter (PA diameter) and the ratio of pulmonary artery diameter to ascending aorta diameter (PA/Ao ratio) was measured by CT of the chest (Figure 1). Physical assessment included the 6-minute walk distance (6MWD). Laboratory measurements included Pro-BNP and liver function tests.
Figure 1.
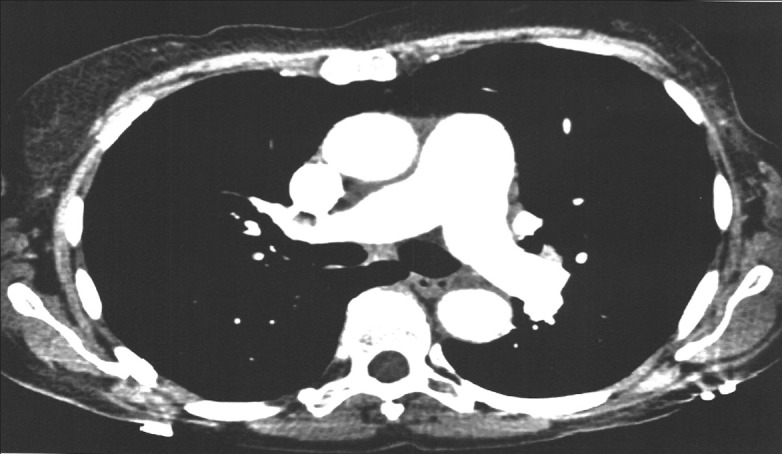
Measurement of the main pulmonary artery and ascending aorta at the level of bifurcation.
The main pulmonary artery (PA) size is typically taken at the level of the bifurcation of the main pulmonary artery perpendicular to the vessel wall. The aortic dimension of the ascending aorta is taken at the same level to calculate the PA to the aortic diameter (PA/Ao) ratio. The diameter is determined using the internal diameter in the contrast-enhanced image.
Survival was calculated from the time of registry until either death of the patient or the end of the study. The impact of escalating treatment with recommended current guidelines on survival was recorded.
For statistical data analysis we used SAS version 9.4 (SAS institute Inc. Cary, NC, USA). Descriptive statistics for the continuous variables are reported median and interquartile range (with the exception of age, which was normally distributed) and categorical variables are summarized as frequencies and percentages. Severity variables were correlated with mPAP by the Pearson test. Normality was tested using the Shapiro-Wilks test. The nonparametric Mann-Whitney U test or the t test was used as appropriate to compare variables between those who lived and died. The level of statistical significance was set at P<.05.
RESULTS
The mean (SD) age of the 30 patients was 40.4 (12.3) years. Males (n=12) were older (n-12, mean [SD], 44.2 (12.4) years) than females (n=18, mean [SD], 37.9 (11.9) years) (Figure 2). Most of the patients had advanced disease (8 of 9 positive parameters). A patient with advanced disease had high risk mortality parameters, including mean pulmonary artery pressure >35 mmHg, functional class III-IV. A positive parameter is any parameter above normal range. The 8 patients (26.6%) who died were labelled the high-risk group.
Figure 2.
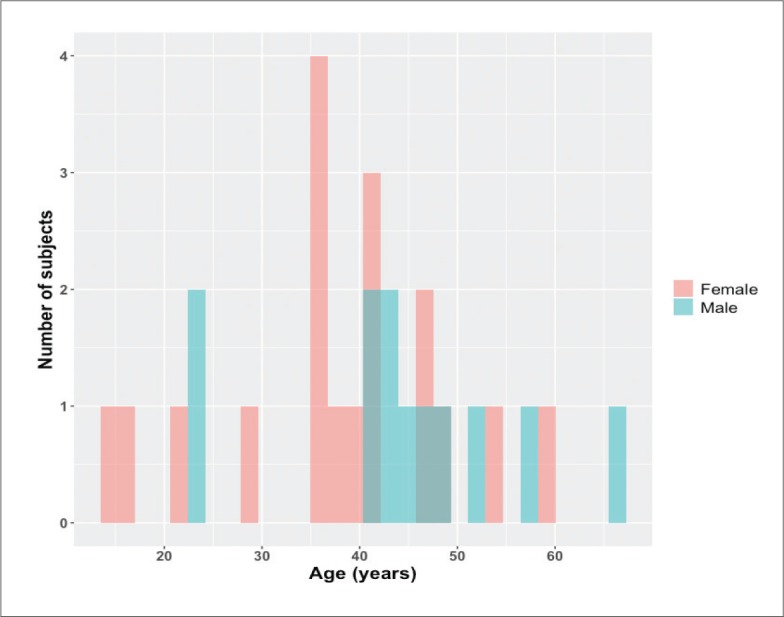
Age distribution of the study population (n=30).
Patients were classified into five groups by type of lung disease (Table 1). Patients with pulmonary arterial hypertension (PAH) and chronic thromboembolic pulmonary hypertension of groups I and IV were on combination therapy, while other patients were initially treated with one agent (either sildenafil or macitentan), but did not tolerate therapy.
Table 1.
Distribution of patients by five pulmonary hypertension groups (n=30).
| Pulmonary arterial hypertension | 5 (16.6) |
| Idiopathic pulmonary artery hypertension | 3 (1.0) |
| Congenital heart disease (ASD) | 1 (3.3) |
| Pulmonary capillary hemangiomatosis | 1 (3.3) |
| Pulmonary hypertension due to left heart disease | 0 (0) |
| Pulmonary hypertension due to lung diseases and/or hypoxia | 18 (60.0) |
| COPD (emphysema) | 1 (3.3) |
| Bronchiectasis | 4 (13.3) |
| Interstitial lung fibrosis | 13 (43.3) |
| Chronic thromboembolic pulmonary hypertension | 1 (3.3) |
| Pulmonary hypertension with unclear and/or multifactorial mechanisms | 6 (20.0) |
| Systemic disorders (Sarcoidosis) | 4 (13.3) |
| Hematological disorders (Beta thalassemia and sickle cell disease) | 2 (6.7) |
Data are number (percentage). ASD: atrial septal defect, COPD: chronic obstructive pulmonary disease. Based on guidelines of European Respiratory Society and American Thoracic Society.
During the follow up the nonsurvivors showed significant worsening of 6 parameters despite escalating treatment with current therapeutic guidelines. Although differences between survivors and nonsurvivors were not statistically significant, the differences indicated a worsening condition despite escalating treatment according to current guidelines (Figure 3). The PA/Ao ratio was higher in survivors but the difference was not statistically significant. In 22 patients in whom it was measured, LVEF was >55 in 16 survivors and in 4 who died. In nonsurvivors, right ventricular size was increased by 25% (P=.427). The 6-minute walk distance was reduced by 21% (299.0 meters in survivors patients vs 247 meters in nonsurvivors, P=.875). Differences in the 6-minute walk test were not statistically significant (Figure 4). Figure 5 shows differences in laboratory values. While proBNP was 96% higher in nonsurvivors, the difference was not statistically significant (P=.890). The median value for alkaline phosphatase was three times higher in the patients nonsurvivors (Figure 5, P=.047). Median survival was 2.3 years less in nonsurvivors compared with survivors despite escalation of therapeutic measures. Interstitial lung diseases were present in 13 patients (Table 2).
Figure 3.

Cardiovascular factors in survivors (n=22) and nonsurvivors (n=8) (median, interquartile range, maximum and minimum).
Figure 4.

Six-minute walk test in survivors (n=22) and nonsurvivors (n=8) (median, interquartile range, maximum and minimum).
Figure 5.

Laboratory measures in survivors (n=22) and nonsurvivors (n=8) (median, interquartile range, maximum and minimum).
Table 2.
Interstitial lung diseases among 13 patients.
| Idiopathic pulmonary fibrosis | 6 (20) |
| Interstitial lung fibrosis 2nd to dermatomyositis | 1 (3.3) |
| Interstitial lung fibrosis 2nd to scleroderma | 1 (3.3) |
| Interstitial lung fibrosis 2nd to systemic lupus erythematosus | 3 (10) |
| Interstitial lung fibrosis 2nd Sjogren syndrome | 1 (3.3) |
| Interstitial lung fibrosis 2nd Mixed connective tissue disease | 1 (3.3) |
Data are number (%)
DISCUSSION
Due to the rapid development of new treatment strategies for PH, it has become difficult to assess long-term survival in these patients. Claims of performance and survival benefits are minimal with a huge cost to the healthcare system and the individual patients.16-19 Timely interventions like lung transplant may be delayed because of the false security of potential benefits from new treatment strategies.20,21 In our study, we found indications of worsening in patients with NYHA functional class III and above that might be confirmed by a larger sample. Average survival in the high-risk subgroup (non-survivors) was less than 2.3 years. The high-risk patients required frequent hospitalizations and escalating treatment interventions but did not derive significant benefit in terms of longer survival.
Most of the therapeutic interventions seek to improve the 6MWD, which may show some variation or positive response but not have an impact on patient survival.22-26 CT chest findings of pulmonary artery size are not helpful in assessing severity of PH, but an increase in the PA/Ao ratio has a positive correlation with the severity of the disease and poor correlation with survival.12-16 Increased PA/Po ratio was probably due to a decrease in left ventricular end-diastolic volume and stroke volume with increasing PAP that lead to the decrease in the aortic diameter hence causing an increase in PA/Ao ratio, suggesting an increased severity of the illness. The patients who died also had an increased IVC diameter and an increased left main bronchus diameter of the left main PA branch diameter ratio.
Patients in the high-risk group did not show any significant change in clinical status or reversibility in their parameters despite escalating the standard therapy, as mentioned above. We have a newly developed lung transplant program and most of our patients with PH are referred out of Saudi Arabia for lung transplantation because organ availability is sparse. The delay in referral for lung transplantation in the high-risk group and a false expectation of possible positive outcome with newer costly drugs for pulmonary hypertension may deny them a chance of a timely lung transplantation. In our opinion, high-risk patients should be referred for lung transplantation when a deteriorating trend in severity parameters is noticed. Escalating medical therapy and waiting for a possible beneficial outcome carries a huge cost to the healthcare system and the patient, while survival is unpredictable.
The limitations of our study were the low number of patients and the lack of a control group. The results needs to be further substantiated in a larger multicenter study. However, our study identified a subgroup of PH patients with NYHA functional class III and above with worsening severity indicators who were identified as being at high risk. These patients showed continuous deterioration in their clinical status and poor survival despite escalation of therapy per current guidelines. We recommend that patients in this high-risk group be referred for an early lung transplantation.
Funding Statement
None.
REFERENCES
- 1.Galiè N, McLaughlin VV, Rubin LJ, Simonneau G.. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur Respir J. 2019;53(1):1802148. Published 2019 Jan 24. doi: 10.1183/13993003.02148-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Criteria Committee, New York Heart Association, Inc. Diseases of the Heart and Blood Vessels. Nomenclature and Criteria for diagnosis, 6th edition Boston, Little, Brown and Co. 1964, p 114. [Google Scholar]
- 3.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006; 173(9):1023-30. [DOI] [PubMed] [Google Scholar]
- 4.Pugh ME, Sivarajan L, Wang L, Robbins IM, Newman JH, Hemnes AR.. Causes of pulmonary hypertension in the elderly. Chest. 2014; 146(1):159–166. doi: 10.1378/chest.13-1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.York M, Farber HW.. Pulmonary Hypertension: Screening and Evaluation in Scleroderma. Current Opinion in Rheumatology. 2011; 23 (6): 536–544. doi: 10.1097/BOR.0b013e32834ba6a7. [DOI] [PubMed] [Google Scholar]
- 6.Mittoo S, Fell CD.. Pulmonary Manifestations of Systemic Lupus Erythematosus. Semin Respir Crit Care Med. 2014; 35(2):249-54. doi: 10.1055/s-0034-1371537 [DOI] [PubMed] [Google Scholar]
- 7.Hoeper MM, Mayer E, Simonneau G, Rubin LJ.. Chronic Thromboembolic Pulmonary Hypertension. Circulation. 2006; 113 (16): 2011–2020. doi: 10.1161/CIRCULATIONAHA.105.602565. ISSN 0009-7322. PMID . [DOI] [PubMed] [Google Scholar]
- 8.Hayes MM, Vedamurthy A, George G, Dweik R, Klings ES, Machado RF,et al. Pulmonary hypertension in sickle cell disease. Ann Am Thorac Soc. 2014;11(9):1488–1489. doi: 10.1513/AnnalsATS.201408-405CME [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minai OA, Chaouat A, Adnot S.. Pulmonary hypertension in COPD : Epidemiology, significance, and management: pulmonary vascular disease: the global perspective. Chest. 2010; 137 (6_suppl): 39S–51S. doi: 10.1378/chest.10-0087. ISSN 0012-3692. [DOI] [PubMed] [Google Scholar]
- 10.Balachandran JS, Masa JF, Mokhlesi B.. Obesity Hypoventilation Syndrome Epidemiology and Diagnosis. Sleep medicine clinics. 2014; 9 (3): 341–347. doi: 10.1016/j.jsmc.2014.05.007. ISSN 1556-407X. PMC 4210766. PMID . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howard LS. Prognostic factors in pulmonary arterial hypertension: assessing the course of the disease, European Respiratory Review 2011; 20: 236-242; DOI: 10.1183/09059180.00006711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu M, Ma Z, Guo X, Zhang H, Yang Y, Wang C.. Computed tomographic pulmonary angiography in the assessment of severity of chronic thromboembolic pulmonary hypertension and right ventricular dysfunction. Eur J Radiol. 2011;80: 462–9. [DOI] [PubMed] [Google Scholar]
- 13.Liu M, Ma Z, Guo X, Chen X, Yang Y, Wang C.. Cardiovascular parameters of computed tomographic pulmonary angiography to assess pulmonary vascular resistance in patients with chronic thromboembolic pulmonary hypertension. Int J Cardiol. 2013;164:295–300. [DOI] [PubMed] [Google Scholar]
- 14.Corson N, Armato SG, Labby ZE, Straus C, Starkey A, Gomberg-Maitland M.. CT-based pulmonary artery measurements for the assessment of pulmonary hypertension. Acad Radiol. 2014; 21(4):523–530. doi: 10.1016/j.acra.2013.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terpenning S, Deng M, Hong-Zohlman SN, Lin CT, Kligerman SJ, Jeudy J, et al. CT measurement of central pulmonary arteries to diagnose pulmonary hypertension (PHTN): more reliable than valid?. Clin Imaging. 2016;40(4):821-7. doi: 10.1016/j.clinimag.2016.02.024. [DOI] [PubMed] [Google Scholar]
- 16.Kam J, Pi J, Doraiswamy V, Elnahar Y, Abdul-Jawad S, DeBari V, et al. CT scanning in the evaluation of pulmonary hypertension. Lung. 2013; 191(4):321-6. [DOI] [PubMed] [Google Scholar]
- 17.Dornia C, Lange TJ, Behrens G, Stiefel J, Müller-Wille R, Poschenrieder F, et al. Multidetector computed tomography for detection and characterization of pulmonary hypertension in consideration of WHO classification. J Comput Assist Tomogr. 2012; 36:175–80. [DOI] [PubMed] [Google Scholar]
- 18.Liu C, Chen J, Gao Y, Deng B, Liu K.. Endothelin receptor antagonists for pulmonary arterial hypertension. Cochrane Database Syst Rev. 2013;2: CD004434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryerson CJ, Nayar S, Swiston JR, Sin DD.. Pharmacotherapy in pulmonary arterial hypertension: a systematic review and meta-analysis. Respir Res. 2010;11:12. doi: 10.1186/1465-9921-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He B, Zhang F, Li X, Tang C,Lin G,Du J, et al. Meta-analysis of randomized controlled trials on treatment of pulmonary arterial hypertension. Circ J. 2010;74:1458–1464. doi: 10.1253/circj.CJ-09-0971. [DOI] [PubMed] [Google Scholar]
- 21.Mardi GM, Cherylanne GK, Watson S, Frantz R, Park M, Frost A, et al. Survival in pulmonary arterial hypertension patients awaiting lung transplantation. The Journal of Heart and Lung Transplantation. 2013; 32: 12. [DOI] [PubMed] [Google Scholar]
- 22.Dandel M, Lehmkuhl HB, Mulahasanovic S, Weng Y, Kemper D, Grauhan O, et al. Survival of patients with idiopathic pulmonary arterial hypertension after listing for transplantation: impact of iloprost and bosentan treatment. J. Heart Lung Transplant.2007; 26(9): 898–906. [DOI] [PubMed] [Google Scholar]
- 23.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010; 122: 156–163. [DOI] [PubMed] [Google Scholar]
- 24.Savarese G, Paolillo S, Costanzo P, D'Amore C, Cecere M, Losco T, et al. Do changes of 6-minute walk distance predict clinical events in patients with pulmonary arterial hypertension?: a meta-analysis of 22 randomized trials. J Am Coll Cardiol 2012; 60:1192–1201. [DOI] [PubMed] [Google Scholar]
- 25.FDA Advisory Committee Briefing Document Cardiovascular and Renal Drugs Advisory Committee. Use of ?PVRI for dosing recommendations of adult-approved drugs in pediatric PAH patients. www.fda.gov/downloads/AdvisoryCommittees/Commit-teesMeeting/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/UCM220250.pdf Date last updated: July 29, 2010Date last accessed: July 21, 2011
- 26.Lee WT, Peacock AJ, Johnson MK.. The role of percent predicted 6-min walk distance in pulmonary arterial hypertension. Eur Respir J 2010; 36: 1294–1301. [DOI] [PubMed] [Google Scholar]