

Abstract
Hematoma expansion (HE), defined as a greater than 33% increase in intracerebral hemorrhage (ICH) volume within the first 24 hours, results in significant neurological deficits, and enhancement of ICH-induced primary and secondary brain injury.
An escalation in the use of oral anticoagulants has led to a surge in the incidences of oral anticoagulation-associated ICH (OAT-ICH), which has been associated with a greater risk for HE and worse functional outcomes following ICH. The oral anticoagulants in use include vitamin K antagonists, and direct thrombin and factor Xa inhibitors. Fibrinolytic agents are also frequently administered. These all act via differing mechanisms and thus have varying degrees of impact on HE and ICH outcome.
Additionally, antiplatelet medications have also been increasingly prescribed, and result in increased bleeding risks and worse outcomes after ICH. Aspirin, thienopyridines, and GPIIb/IIIa receptor blockers are some of the most common agents in use clinically, and also have different effects on ICH and hemorrhage growth, based on their mechanisms of action. Recent studies have found that reduced platelet activity may be more effective in predicting ICH risk, hemorrhage expansion, and outcomes, than antiplatelet agents, and activating platelets may thus be a novel target for ICH therapy.
This review explores how dysfunctions or alterations in the coagulation and platelet cascades can lead to, and/or exacerbate, hematoma expansion following intracerebral hemorrhage, and describe the mechanisms behind these effects and the drugs that induce them. We also discuss potential future therapy aimed at increasing platelet activity after ICH.
Keywords: Hematoma expansion, intracerebral hemorrhage, anticoagulant, antiplatelet, oral-anticoagulant-associated ICH, C-type lectin-like receptor 2, platelet activation
Graphical Abstract
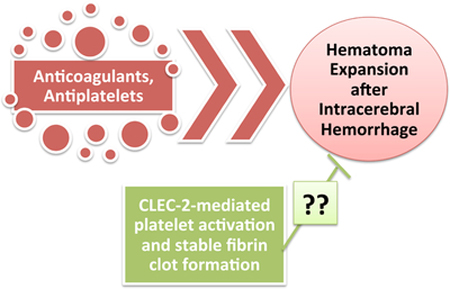
1. Introduction
Hematoma expansion (HE) is a detrimental outcome of spontaneous intracerebral hemorrhage (ICH), resulting in significant neurological deficits, and enhancement of ICH-induced brain injury [1, 2]. This phenomenon is defined as a >33% growth in the initial hemorrhage volume over the first 24 hours after ICH, which contributes to a midline shift and acceleration of neurological deterioration; seventy-three percent of patients experience some degree of expansion and approximately one-third of them expand more than 33% [3–7]. Primary brain injury after ICH is due to mechanical damage from the hematoma, which is also one of the main causes of secondary brain injury, as iron and heme from lysed erythrocytes create a highly oxidative and cytotoxic environment, damaging brain tissue [8–11]. Primary brain injury involves an increase in pressure and damage to the surrounding tissue, which leads to edema and early neurological deficits, while secondary brain injury involves thrombin activation, red blood cell lysis, cell death, migration of inflammatory agents to the site of the hematoma, and possible metabolic changes [12–15]. These all lead to significant deterioration after ICH; thus, it is essential to understand and effectively target the formation and growth of the hematoma.
ICH is the second most common form of stroke, accounting for 15–20% of all stroke subtypes [16], and results from the rupture of small blood vessels within the basal ganglia, thalamus, pons, or cerebellum. Intracerebral hemorrhaging can lead to a sudden increase in intracranial pressure, resulting in headaches, nausea, vomiting, unconsciousness, and death [1]. The increase in intracranial pressure and concomitant edema can lead to reduced venule blood flow and brain ischemia [17]. As such, though it does not account for the majority of stroke occurrences, these fatal outcomes make ICH the most mortal and least treatable subtype, being associated with a 30–50% mortality rate at 30 days [18–20]. After the first year, more than 75% of patients are deceased or severely injured [21] and only 20% of survivors regain normal function [18].
Primary ICH is mainly caused by long-standing hypertension, which causes the formation of microaneurysms at the bifurfaction of intracerebral arterioles that can rupture; however, it may also be caused by cerebral amyloid angiopathy, arteriovenous malformation, ischemic stroke with hemorrhagic transformation, or a dysfunction in the platelet or coagulation cascade, usually due to the use of anticoagulant or antiplatelet medications [22, 23].
2. Hematoma Expansion
The initial hematoma volume and location are strong predictors of 30-day mortality and functional outcome [24, 25]. Deep and cerebellar hemorrhages were found to be associated with a higher mortality than lobar hemorrhages. Flaherty and colleagues found 30-day mortality rates according to location to be as follows: 44% for deep, 46% for lobar, 60% for brainstem, and 34% for cerebellar [25]. However, the hemorrhage volume was reported to be the most important predictor of 30-day outcome: for deep hemorrhages, the 30-day mortality was 93%, 64%, and 23% for ICH volumes of more than 60cm3, 30–60cm3, and less than 30cm3, respectively, showing a strong correlation [24]. In fact, each 10% increase in expansion results in a 5% increased risk of mortality [5].
Risk factors identified for hematoma expansion include blood pressure, and vascular and coagulation disorders [26, 27]. Coagulopathies are a major cause of secondary ICH, and it is therefore essential to understand how the anticoagulant drugs being used can increase its incidence.
3. The Coagulation Cascade
Coagulation involves a series of zymogen activation reactions in which a zymogen (precursor protein) is converted into an active protease by cleavage of one or more peptide bonds at each stage of the process. The final protease to be generated is thrombin (factor IIa), which is the active form of prothrombin (factor II),by the prothrombin activator (factor X). Coagulation occurs by two pathways: intrinsic and extrinsic, both of which result in factor Xa (FXa) activation, leading to a common pathway by which thrombin is activated, leading to fibrin formation from fibrinogen (factor I). The extrinsic pathway is initiated by substances outside of the blood, from damaged tissue cells (tissue factor – factor III) and is for the initial formation of thrombin, while the intrinsic pathway is initiated by blood coming into contact with collagen from the vessel wall, activating factor XII, and is for sustaining thrombin production. The intrinsic pathway therefore results in the formation of much larger amounts of thrombin, and thus larger clots. The protease zymogens involved in coagulation include factors II, IX, X, and prekalikrein. Factors V and VII are non-proteolytic cofactors. The coagulation cascade is outlined in Figure 1.
Figure 1. The coagulation cascade and anticoagulant targets.

Coagulation occurs via two pathways: intrinsic and extrinsic. It is a series of reactions in which precursor proteins are converted to their active forms, eventually resulting in the formation of fibrin from fibrinogen (common pathway). Fibrin cross-links to form a clot. Warfarin and other vitamin K antagonists inhibit the vitamin K-induced conversion of prothrombin to thrombin (and other vitamin K-dependent factors). Apixaban, rivaroxaban, and other direct factor Xa inhibitors inhibit the action of factor Xa in activating prothrombin. Ximelagatran, dabigatran, and other direct thrombin inhibitors, block the action of thrombin in converting fibrinogen to fibrin. Recombinant activated factor VII (rFVIIa) mimics the activity of FVIIa in activating factor X.
F (I through XIII) = Factor (I through XII); F(I through XIII)a = activated Factor (I through XIII); TF = Tissue Factor
3.1. Coagulation and Hematoma Expansion
Choudri et al. [28], found that targeting the intrinsic pathway via competitive inhibition of factor IX was not associated with increased ICH, though it improved outcomes following cerebral infarction by reducing fibrin and platelet accumulation. Similar findings were reported in another study, which targeted factor XII instead of factor IX as part of the intrinsic pathway [29]. Furthermore, several clinical trials have provided strong evidence for the use of anticoagulants (mainly warfarin) in significantly reducing the risk of ischemic stroke in patients with atrial fibrillation (AF), the most common type of cardiac arrhythmia [30, 31]. AF patients have an increased risk of ischemic strokes. The incidences of AF have risen exponentially with the shift towards an older population, thus resulting in the increased use of anticoagulant treatments.
This escalation in the use of oral anticoagulants has led to a surge in the incidences of oral anticoagulation-associated ICH (OAT-ICH) [25, 32]. Moreover, the use of these drugs for the prevention of thrombotic and thromboembolic disease is expected to continue to rise, further increasing the rates of this type of ICH. As a matter of fact, the rate of OAT-ICH is about 7–10 fold higher than in the untreated population [33], and is increasing to the point where the incidence is almost as high as that of subarachnoid hemorrhages [34]. Flaherty and colleagues also found that OAT-ICH patients had more comorbidities than ICH patients without coagulopathy, and OAT-ICH was more common in whites than in blacks, coinciding with the fact that whites have a greater incidence of AF (and thereby, possible warfarin use) than blacks, though blacks have been shown to be more prone to ICH than whites overall (possibly due to higher hypertension rates) [25]. Additionally, patients on warfarin exhibit a larger initial hemorrhage volume upon presentation and more frequently have hematoma expansion [35]. Currently, there are contradicting reports on the locations of ICH that are more likely to result from the use of anticoagulants. Flaherty et al. as well as Kase and colleagues, report that OAT-ICH predominantly occurs in the cerebellar region, while Maas et al. and Fredriksson et al. found that OAT-ICH cases more commonly present as lobar hemorrhages [25, 36–38]. As a result, it is not clear whether ICH resulting from anticoagulopathy is more prone to occur in a certain brain region versus another. However, mortality rates do not change based on the location of hemorrhages formed secondary to anticoagulant use.
Generally, OAT-ICH incidences have been clinically shown to have larger hematoma volumes and worse functional outcomes, when compared to spontaneous ICH with normal coagulation status [32, 39, 40]. In a study of coagulopathy in progressive hypertensive ICH, it was found that patients who had an enlarged hematoma had significantly lower plasma levels of coagulation parameters on admission (fibrinopeptide A (FPA): 4.0±0.6 ng/ml vs. 17.2±2.8ng/ml in the group that had no hematoma volume expansion; and thrombin-antithrombin III complex (TAT): 7.4±2.8ng/ml vs. 21.9±3.1ng/ ml in the unchanged hematoma group [27]. These results suggest that there is insufficient coagulation (thrombin) activity in these patients, resulting in enlargement of the hematoma, and continuous bleeding. Thrombin is a serine protease that plays a vital role in the coagulation cascade (Fig 1). Immediately upon initiation of hemorrhaging (in both adults and in our own neonatal studies), thrombin is released to stop the bleeding [41, 42]. Very small doses of thrombin can be neuroprotective. However, direct infusion of a high dose results in edema formation, inflammation, blood-brain barrier disruption, and death [12, 43, 44]. Thrombin also plays a role in hydrocephalus formation after intraventricular hemorrhage, which is associated with ventricular wall damage and BBB disruption, effects that can be reversed by a protease activated receptor (PAR)-1 (a thrombin receptor) antagonist [45]. Furthermore, Cheng et al. [46] found that thrombin induced significant intracerebral hemorrhages via activation of PAR-1. Additionally, TAT levels are increased in ICH animals compared to controls, and are even higher in the hematoma fluid than the plasma of ICH animals [47]. These findings highlight a role for thrombin, and its activation or inhibition, in ICH, and possibly, in hematoma expansion.
4. Mechanisms of Anticoagulant/Antithrombotic Drugs
Of course, different anticoagulant drugs have been found to have differing effects, with some being much more effective at reducing the risk of ischemic stroke from AF than others, and some being much more likely to result in bleeding, and thereby ICH induction, than others. The mechanisms by which they inhibit clotting may play a significant role in this, and understanding them could greatly aid in regulating both their therapeutic and adverse effects. Thus, we will do an overview of the mechanisms by which the various anticoagulant (and later, antiplatelet) drugs act, and their relation to hemorrhaging and hematoma expansion.
4.1. Warfarin (Vitamin K Antagonists)
Vitamin K antagonists (VKAs) are widely used in the treatment of cardioembolic stroke and AF [48], but pose a risk for ICH [49]. Further, VKA-associated ICH has a worse prognosis than spontaneous ICH [50, 51]. In randomized clinical trials, the risk of major bleeding from VKA administration was up to 19.3% [51, 52]. Several studies have examined the effects of warfarin, a VKA and the most widely used oral anticoagulant, on hematoma expansion and ICH severity [4, 40, 53–56], and have found that prior warfarin use is correlated with a greater risk of hematoma expansion, and therefore worse neurological deficits. It has also been reported that warfarin increases the volume and risk of intraventricular hemorrhages (IVH) in deep and lobar ICHs, and this may be how anticoagulation worsens outcomes following ICH, as IVH can be an independent predictor of poor outcome after ICH [57, 58].
Warfarin and other VKAs work by antagonizing the effect of Vitamin K in promoting the synthesis of coagulation factors; it noncompetitively inhibits epoxide reductase, the enzyme that converts the vitamin epoxide (2,3-epoxide) to active Vitamin K [59, 60]. This blocks the clotting cascade, as Vitamin K is an essential cofactor in the postribosomal modification of the liver protein precursors that lead to biologically active prothrombin (factor II) and the other Vitamin K-dependent plasma clotting factors (factors VII, IX, and X) [51, 59]. Several reports have shown that patients have varied responses to warfarin treatment, due to its very narrow therapeutic index, and its tendency to cause bleeding. Against this background, Li and companions [60] did a review of the literature on the genetics of warfarin-related anticoagulation and found that single-nucleotide polymorhisms in the Vitamin K Epoxide Reductase Complex (VKORC1) affected a patient’s response to, and their required dosage of, warfarin treatment, resulting in mixed effects. It was also found that polymorphisms in the Cytochrome P450 (CYP450) 2C9 enzyme, which influences warfarin metabolism, contribute to these varied responses to warfarin [61]. Patients who carry the CYP 2C9*2 and CYP 2C9*3 alleles, or the VKORC1 A haplotype tend to require less warfarin due to slower metabolism, or to decreased expression of messenger RNAs which produce the proteins necessary for VKORC1 synthesis, respectively [62–65]. Based on this data, genotyping prior to administering warfarin could potentially be one way to reduce the incidences of ICH secondary to warfarin usage [66]. However, this has not been a recommended option since it has been reported to add only about one day to the patient’s lifespan compared to the standard dosing, and is a very costly undertaking [67]. Prothrombin complex concentrates (PCCs), which rapidly replace the vitamin K-dependent clotting factors (and is one of the most effective treatments), fresh frozen plasma (FFP), recombinant activated factor VII (rFVIIa), or actual Vitamin K have been the most common treatments for reversing the bleeding effects of warfarin and other VKAs in ICH patients [37, 54, 68, 69]. However, these are not always effective. Interestingly, it was found in a recent study that although warfarin pretreatment in an ICH model increased hematoma volumes, it reduced cell death and matrix metalloproteinase (MMP)-9 activity [55]. These results highlight the need for more studies to fully understand the effects and mechanisms of warfarin (and other VKAs) in HE after ICH.
4.2. Direct Thrombin and Factor Xa inhibitors
Due to the extensive adverse effects of warfarin on bleeding, recent experimental and clinical studies have sought to find other anticoagulants that may be just as, or even more efficacious than warfarin, but with a much higher benefit to risk ratio [70]. Numerous clinical trials report that the newer anticoagulants (direct thrombin and factor Xa inhibitors) reduce the risk of ICH by half, compared with warfarin [71]. We describe a few of these newer anticoagulants and their mechanisms of action below.
4.2.1. Ximelagatran
Ximelagatran, a direct thrombin inhibitor that, upon absorption, is rapidly hrdrolyzed to melagatran, its active form, and inhibits both thrombin activity and generation, was one of the first direct thrombin inhibitors to be tested [72]. Thrombin is required for the conversion of fibrinogen to fibrin (Fig 1); thus, inhibition of thrombin decreases clot formation. Melagatran also inhibits thrombin’s activation of the protease-activated receptors on platelets, thus affecting platelet activity [73], as well as thrombin bound to thrombomodulin, thereby inhibiting protein C activation [74].
Melagatran has low oral bioavailability; hence its prodrug, ximelagatran, which is 170 times more lipophilic and has sufficient oral bioavailability, is administered as the antithrombotic agent [75]. The mean elimination half-life is 3 hours for melagatran, and its pharmacokinetic profile remains stable over time, regardless of patient age, sex, weight, etc [76]. Furthermore, there is a very low risk of drug interactions with melagatran because it does not use the hepatic P450 system for metabolism [77]. Factor VIII inhibitor bypassing activity (FEIBA), a coagulation factor concentrate, was able to reverse the melagatran-induced prolonged bleeding time, indicating that melagatran treatment could possibly be readily regulated.
Unlike warfarin, which has a very narrow therapeutic index, it was reported that melagatran can be given across a wide range of doses without any significant increase in risk of bleeding [78]. Coinciding with this, clinical trials found that the risk of bleeding after ximelagatran treatment was significantly reduced compared to warfarin [79–81]. Conversely, however, a recent study in ICH rats reported that melagatran increased ICH volume and prothrombin time (PT), and at higher doses, there was 100% mortality, due to severe ICH [82]. The prolonged PT is in agreement with the study by Gustafsson et al. [72], which found that melagatran prolonged clotting time, thrombin time (TT), activated partial thromboplastin time (APTT), and PT. The change in these parameters suggests that melagatran could increase the risk of ICH and HE, and should therefore be more thoroughly studied in ICH.
4.2.2. Dabigatran
Dabigatran undergoes rapid transformation from its prodrug dabigatran etexilate (DE) by esterases in the gastrointestinal system [83], and inhibits thrombin directly by binding to both its free and clot-bound forms. DE was recently approved for stroke prevention in AF and was found to be as efficacious as warfarin in reducing stroke, but with less risk of hemorrhage at its lower dose [84, 85]. However, DE resulted in prolonged tail bleeding and clotting times, as well as increased hematoma expansion in a mouse ICH model [86]. Factor VIIa was unable to reverse the effects of dabigatran on hematoma expansion [86]. On the contrary, Lauer et al. found that dabigatran, but not warfarin, had no effect on hematoma volumes when given as a pretreatment to ICH animals [87]. Increased age and concomitant antiplatelet (aspirin) use were identified as risk factors for ICH secondary to dabigatran therapy for AF [88].
Dabigatran, unlike warfarin, is not metabolized by the cytochrome p450 (CYP450) system, and thus has fewer drug interactions [89]. However, a major concern regarding DE and other direct thrombin and factor Xa inhibitors is that here is currently no optimal reversal mechanism for them, though several studies suggest that administration of PCCs effectively antagonizes the bleeding effect of these inhibitors [86]. Additionally, dabigatran treatment results in significant adverse effects on the gastrointestinal tract [70].
4.2.3. Apixaban and Rivaroxaban
Apixaban is a reversible, selective, direct inhibitor of factor Xa (the cofactor responsible for the conversion of prothrombin to thrombin). It inhibits both the free factor Xa and Xa that is bound in the prothrombinase complex. Since it is selective for factor Xa, apixaban inhibits thrombin without affecting factors Xlla, XIa, IXa, or VIIa [90, 91].
Rivaroxaban inhibits FXa, and was found to increase hematoma expansion in a mouse ICH model and prolong PT. The increase in hematoma volume, but not the prolonged PT, could be reversed by PCC, FFP, and factor VIIa [92].
Apixaban and rivaroxaban are both metabolized by the CYP450 system (specifically, CYP3A4) and thus are more likely to have drug-drug interactions. Furthermore, the bioavailability of rivaroxaban decreases with fasting [93]. This is important as it may dictate the effective doses of these drugs, and could therefore potentially limit the risk of hemorrhage from apixaban or rivaroxaban treatment. Unlike warfarin, however, apixaban and rivaroxaban do not interfere with the tissue factor (TF)-factor VII complex formation; they directly inhibit factor X. Also, the upstream vitamin K-dependent factors inhibited by warfarin that are necessary for the initiation and amplification of thrombin production are unhindered in apixaban or rivaroxaban treatment; thus, with these inhibitors, the amount of thrombin generated by the coagulation cascade is more than warfarin, and could be the mechanism by which they reduce the risk of ICH [71]. Edoxaban is another factor Xa inhibitor being tested and it was found to increase ICH volumes and prolong PTT in a rat ICH model [82]. Nevertheless, it also reduces the risk of hemorrhaging, in comparison to warfarin [94].
5. Mechanisms of Fibrinolytic Drugs
Fibrinolysis (Figure 2) is the cleavage of fibrin into fibrin degradation products. Fibrin serves as the cofactor for the tissue plasminogen activator (tPA)-mediated activation of plasminogen to plasmin. Plasminogen activator inhibitor-1(PAI-1) is the inhibitor of tPA; when its levels are low (or there is an abundance of tPA), clots are more effectively cleared, and bleeding increases.
Figure 2. Fibrinolysis and the action of fibrinolytic agents.

Fibrinolysis is the breakdown of fibrin to fibrin degradation products. This reduces clot formation and increases bleeding. The body maintains a balance between coagulation and fibrinolysis to prevent excessive clotting or bleeding. Recombinant tissue plasminogen activator (rtPA) mimics the action of naturally occurring tPA in activating plasminogen to plasmin, which catalyzes fibrin breakdown, increasing bleeding.
FXIIIa = Activated Factor XIII
5.1. Recombinant Tissue Plasminogen Activator (rtPA)
The intravenous use of rtPA as a thrombolytic agent is the only approved therapy in the treatment of ischemic stroke. Before administration of tPA, however, ICH has to be ruled out by imaging, as it is one of the most significant risks of this kind of treatment. Both clinical and experimental studies have reported a significant number of ICH incidences after tPA treatment for ischemic stroke, with a 10-fold increase in mortality related to symptomatic hemorrhage [95, 96]. Studies also report that patients who develop an ICH secondary to thrombolytic therapy are usually older, more hypertensive, and have higher National Institutes of Health Stroke Scale (NIHSS) scores prior to treatment [97]. Conversely, Berrouschot and colleagues found that age did not alter the incidence of ICH after tPA administration, though older patients did have worse outcomes [98]. Hyperglycemic stroke patients (typically already associated with worse outcomes) who are given tPA also have a higher risk of hemorrhagic transformation and worse outcome [99–102]. Of course, the number of patients saved by tPA has to be balanced against those that have a worse outcome (hemorrhaging and death); it was reported that though mortality related to hemorrhages was increased in tPA-treated groups, overall mortality was reduced due to the reduction in non-hemorrhagic deaths [96].
Because of its fibrin specificity, though tPA has a really short half-life, it can remain in fibrin rich clots for one or more days, which is what causes it to have an effect on hemorrhaging [103]. Montagne et al. found that recombinant tPA increased hematoma expansion after ICH, and resulted in significant deficits [104]. Yet, it is interesting that several studies have found that administration of tPA did not lead to any significant changes in hematoma growth [105, 106], but the tPA-treated group in the study by Marinkovic et al. did have greater mortality and neurological deficits than other groups.
The fibrinolytic effect of tPA is mediated by plasminogen activation and subsequent fibrinolysis. tPA cleaves plasminogen, so it becomes active plasmin, the major function of which is to dissolve fibrin-based clots, creating fibrin degradation products from the splitting of fibrin and fibrinogen (Fig 2). Hence, the overall effect of tPA is to remove clots and increase blood flow to the affected region. Against this background, rtPA is also being administered to ICH patients by an intrahematomal catheter for hematoma lysis as a possible therapy, and clinical trials are ongoing to determine the best dosing [107]. However, tPA also has several other non-fibrinolytic functions, which may create further deficits in tPA-treated patients [108]. tPA interacts with matrix metalloproteinases (MMPs), particulary MMP-9, thus enhancing the disruption of the blood-brain barrier [109, 110] and this is thought to be the mechanism by which it induces hemorrhagic transformation of ischemic strokes [111]. This interaction is lipoprotein receptor (LRP)-mediated [112]. These findings correlate with clinical findings that MMP-9 levels were increased in tPA-treated patients that became hemorrhagic, and it could therefore be used as a predictive marker of ICH due to thrombolysis [113]. tPA has also been shown to be involved in neurotoxicity [114, 115] and this is reported to involve N-methyl D-aspartate (NMDA) activation [104]. The NMDAR antagonist, memantine, was found to reduce hematoma expansion following experimental ICH via reduction of endogenous tPA levels and, consequently, reduction in MMP-9 expression and activity [116]. This suggests that inhibition of tPA activity may a beneficial target for reduction of hematoma expansion after ICH.
5.2. Urokinase
Urokinase is another fibrinolytic agent, with similar actions to tPA, activating plasminogen to plasmin, and inducing fibrinolysis. Thus, urokinase therapy for ischemic stroke could result in increased hemorrhaging/hematoma expansion. Administration of urokinase significantly prolonged PT, APTT, and TT in comparison to control and untreated stroke patients [117]. PT is a reflection of the extrinsic coagulation pathway, while APTT reflects the intrinsic pathway; thus, urokinase affected both coagulation pathways. Warfarin prolongs PT, affecting the extrinsic pathway, while dabigatran increases APTT, affecting the intrinsic pathway [87]; prolonged TT indicates the presence of anticoagulants. These results imply that urokinase could enhance hematoma expansion after ICH.
5.3. Other fibrinolytic drugs
Other fibrinolytic and thrombolytic agents have been employed in experimental studies and clinical trials. This includes streptokinase, which works by forming a complex with plasminogen, in turn causing plasmin activity (the action of naturally occurring tPA in the fibrinolytic cascade) [103]. Because streptokinase increases systemic levels of plasmin so it is much greater than that which can be effectively inhibited by antiplasmin, thus allowing plasmin to inhibit other coagulation factors (V, VII, XII) and von Willebrand factor, it is categorized as non-fibrin-specific, as is urokinase. Streptokinase has a longer half-life than tPA and, as a result, reocclusion is less common after ischemic stroke with streptokinase treatment. However, non-fibrin-specific fibrinolytic agents are less desired as they have reduced efficiency at clot resolution. Other agents being tested include pro-urokinase, desmoteplase, reteplase, etc [103]. These all have similar mechanisms of action.
6. Platelet Function and Hematoma Expansion
Platelets are highly specialized to react to vessel injury, and trigger a series of signaling pathways that result in the formation of a shear-resistant hemostatic plug, sealing the wound and limiting excessive bleeding [118]. Platelet activation and formation of the plug involves several membrane receptors; the rapid onset of glycoprotein (GP) Ib-V-IX and von WIllebrand factor (vWF) interaction is one of the first steps [119]. The activated platelets then release adenosine diphosphate (ADP) from their dense granules, which activates surrounding platelets via the G-protein coupled receptors, P2Y1 and P2Y12 [120]. Though a weak platelet agonist itself, ADP amplifies the platelet response induced by stronger agonists (collagen and vWF) [121]. The P2Y1 receptor mediates cytoplasmic calcium mobilization, which plays a major role in platelet activation and stabilization. This is then amplified by the P2Y12 receptor, which also induces the secretion of dense granules by stimulating agonists such as thromboxane A2 (TxA2) and thrombin [122]. Thromboxane A2 stimulates local activation of plasma coagulation factors, generating a fibrin clot that reinforces the platelet aggregate. Thrombin, one of the most potent platelet activators, acts via four receptors: protease activated receptor-1 (PAR-1), PAR-4, glycoprotein (GP) Ibα, and GP V [123]. Platelet activation is illustrated in Figure 3.
Figure 3. Platelet activation and antiplatelet mechanisms.

Platelets can be activated by thrombin, collagen, and vWF, among other proteins. Activated platelets release ADP and TxA2 from their dense granules. This eventually leads to the release of fibrin, which results in a stable clot formation. Aspirin inhibits the action of COX-1 in producing TxA2; Clopidogrel, ticlopidine, and other thienopyridines, inhibit the ADP receptor P2Y12; GPIIb/IIIa blockers inhibit platelet aggregation by blocking the activity of this recptor that is vital in the final pathway of platelet activation. The novel platelet receptor, CLEC-2, can be activated by its endogenous ligand, podoplanin, or the snake venom protein, rhodocytin, leading to platelet activation and stable clot formation.
vWF = von Willebrand Factor; COX-1 = cyclooxygenase-1; TxA2 = thromboxane A2; ADP = adenosine diphosphate; GPIIb/IIIa = glycoprotein IIb/IIIa; CLEC-2 = C-type lectin-like receptor 2; Src = proto-oncogene tyrosine-protein kinase Src (sarcoma); PLCγ2 = phospholipase C gamma-2
Platelets also play a role in vascular integrity by releasing factors such as sphingosine-1-phosphate (S1P), which is important for maintaining blood-brain barrier integrity by interacting with its G-protein coupled receptors on endothelial cells [124, 125]. Additionally, platelets within the clot contain contractile proteins that pull the edges of the wound together, preventing further hemorrhage. This could be significant for targeting hematoma expansion.
Antiplatelet medications, such as aspirin, have been more and more frequently prescribed to the aging population at risk for myocardial infarctions, unstable anginas, and transient ischemic attacks, and have been found to reduce the risk of these vascular events, though to a lesser extent than warfarin [126]. The combination of aspirin with thienopyridines, ADP receptor inhibitors, has become the mainstay of treatment for the prevention of arterothrombotic events [127].
Currently, one-third of ICH cases occur in patients that are taking antiplatelet drugs [120, 128], and previous studies have found that reduced platelet activity is associated with early ICH growth and worse functional outcomes [129–132], suggesting an important role for platelet function in ICH and hemorrhage expansion. In fact, several reports indicate that administration of antiplatelet medications can increase hematoma expansion and mortality [133, 134]. However, there are studies that have found no increase in hematoma expansion with antiplatelet treatment [120, 135, 136]; though one of these studies did find that platelet inhibition increased tail bleeding times [120].
7. Mechanisms of Antiplatelet Drugs
Inhibition of platelet aggregation prevents them from secreting thrombin, which consequently inhibits the conversion of fibrinogen to fibrin; hence, there may be excessive bleeding and inability to form a stable clot. Platelet inhibition can occur via interference with the release of substances from their dense granules, inhibition of aggregation, or hindrance of adhesion to the vessel wall. The naturally occurring platelet inhibitor, prostacyclin (PGI2), inhibits the adherence of platelets to the injured endothelium and inhibits their aggregation. This it does through the production of adenylyl cyclase via its G-protein coupled receptor [137]. Prostacyclin production in the endothelium is stimulated by thrombin, which is also released from activated platelets. Descriptions of different antiplatelet drug mechanisms are given below.
7.1. Aspirin
Low dose acetylsalicylic acid (aspirin) is used widely in the prevention of primary and secondary cardiovascular diseases, as it is an anti-aggregatory agent [138], and is increasingly prescribed to the aging US population. In fact, the use of aspirin is as high as 61% [139]. McLeod et al. found that doses of acetylsalicylic acid, ranging from 50–3900mg was able to significantly inhibit platelet function and increase bleeding times compared to controls in healthy subjects [140]. This was also shown in a recent study by Chylova et al. which reported that doses of aspirin between 100 and 500mg significantly reduced platelet aggregation in healthy volunteers and ischemic stroke patients [141]. Aspirin interferes with the synthesis of thromboxane A2, an inducer of platelet aggregation, by inhibiting cyclooxygenase-1 (COX-1), the enzyme responsible for producing the thromboxane A2 precursor, arachidonic acid [120, 142]. COX-1 also catalyzes the conversion of arachidonic acid to prostaglandins; thus, aspirin inhibits these as well [143–145]. It does this by covalently acetylating a serine residue near the active site of cyclooxygenase [146]. Platelets are unable to synthesize new proteins; hence, the action of aspirin on platelet cyclooxygenase lasts for the lifetime of the platelets (7 to 10 days). Consequently, repeated dosing of aspirin, which applies to the majority of patients to whom it is prescribed, has a cumulative effect on platelet activity. This has been shown to increase hemorrhagic complications and the occurrence of cerebral microbleeds [147]. Complete inactivation of cyclooxygenase is achieved when 160 mg of aspirin is taken daily, and it has been found to be maximally effective as an antithrombotic agent at doses of 160–320mg/ day [148].
Randomized clinical trials have reported that aspirin is not as effective as warfarin in reducing the risk of ischemic stroke [126, 149]. However, warfarin results in significantly greater risk of bleeding and intracranial hemorrhaging. As such, it has been suggested that patients who only have minimal risk of stroke from atrial fibrillation be placed on aspirin therapy instead of warfarin, to reduce the risk of further stroke (ICH). Some suggest that aspirin should rarely ever be used to treat atrial fibrillation as well-managed warfarin therapy has little bleeding risk [150]. However, aspirin use has been reported to increase the risk of ICH recurrence in lobar ICHs [151], and was also found to be a predictor for the volume of this type of ICH and to increase said volume [152].
7.2. Thienopyridines
Thienopyridines are a class of selective, irreversible ADP receptor (P2Y12) inhibitors that are widely used as antiplatelet agents, and include ticlopidine, clopidogrel, and prasugrel. Clopidogrel is the main thienopyridine that is administered, but prasugrel has been found to be more efficacious at reducing thrombotic events, but with an increased risk of bleeding [153].
7.2.1. Ticlopidine
Ticlopidine is a thienopyridine that inhibits platelet function by interacting with the platelet receptor glycoprotein IIb/IIIa, thus inhibiting the binding of fibrinogen to activated platelets. The fibrinogen binding site on GPIIb/IIIa receptor is opened upon platelet activation [123]. This reduces the rigidity of the clot. As a result, ticlopidine increases bleeding time, with its maximal effect observed following several days of therapy, and platelet function is hindered for several days after treatment discontinuation [154]. As a consequence, patients on ticlopidine may be at greater risk for HE following ICH. Ticlopidine is currently prescribed for the prevention of thrombosis in cerebrovascular and coronary artery disease, and is recommended for patients unable to tolerate aspirin. It is metabolized by the hepatic CYP450 enzymes, resulting in the formation of the active metabolite that irreversibly inhibits the P2Y12 receptor [155]. Ticlopidine was the first thienopyridine to be introduced to the clinic, but due to its unfavorable side effects, was replaced by clopidogrel [156].
7.2.2. Clopidogrel
Clopidogrel is the most widely prescribed ADP receptor inhibitor (antiplatelet) for the reduction of ischemia and thrombosis in cardiovascular disease [122, 157]. It inhibits the ADP-induced platelet activation by interfering with platelet secretion via inhibition of the P2Y12 receptor on platelets [120]. Clopidogrel is metabolized by the hepatic CYP450 system by several different cytochromes, mainly CYP2C19, and by an esterase paraoxonase-1 (PON1) [158, 159]. Data have shown that variations in these two genes (CYP2C19 and PON1) result in reduced response to clopidogrel therapy by decreasing the formation of the active metabolite of the drug [122]. The PON1Q192 variant was shown to have a less efficient conversion of the pro-drug, 2-oxo-clopidogrel, to the active metabolite, than the PON1R192 variant of the gene [159]. Additionally, healthy individuals with the CYP2C19*2 loss-of-function allele, in particularl, have a distinct reduction in platelet responsiveness to clopidogrel [160], and this genetic variation is believed to be largely involved in clopidogrel resistance. CYP2C19*3 is also a loss-of-function allele that affects platelet response to clopidogrel. On the other hand, CYP2C19*17 is a gain of function allele that has been associated with ultra-rapid metabolization of and increased response to clopidogrel (and thus reduced platelet aggregation) [161]. These data indicate that, similar to warfarin, genotyping in order to determine clopidogrel resistance in patients may be necessary before administering the treatment. Acordingly, it has been suggested that genotyping and ex-vivo platelet function testing be a strategy for identifying patients who are more likely to respond to clopidogrel therapy [162–164]. Alternative treatments have also been suggested, such as using other, more direct acting drugs, including prasugrel, cangrelor, elinogrel, and ticagrelor [137, 165]. From this data, we can induce that individuals with a CYP2C19*17 allele may be more prone to the development of an ICH secondary to clopidogrel therapy than individuals with the CYP2C19*2 or CYP2C19*3 variants, or even the CYP2C19*1 allele, which is the normal function variant. Hence, genotyping may indeed be an effective way to manage clopidogrel therapy and identify high-risk patients for ICH. Interestingly, patients who have reduced response to inhibition of ADP-induced platelet aggregation have a more heightened response to PAR-1 (thrombin)-mediated activation and that this may contribute to the variability in response to treatment by clopidogrel and other ADP inhibitors [123]. It was therefore suggested that blocking alternative platelet activation pathways (such as thrombin-induced activation through PAR-1) might be beneficial in the patients who display a reduced response. However, this may be more detrimental as it relates to the occurrence of an ICH. Thus, further studies are needed to further understand and target platelet activation while limiting the risk of ICH. Other P2Y12 inhibitors, such as prasugrel, have been associated with a greater reduction in ischemic events than clopidogrel; however, they produce a much greater risk of bleeding [137]. Hence, the use of these drugs should be limited in patients at higher risk for ICH.
7.2.3. GPIIb/IIIa blockers
Glycoprotein (GP) IIb/IIIa is one of the most dominant platelet surface receptors, ranging between 60,000–80,000 copies per platelet [166]; it plays a major role in platelet activation, being the receptor in the final common pathway of platelet aggregation, and is therefore a relevant target for stroke therapies. GPIIb/IIIa inhibitors are currently the most potent regulators of platelet activity. GPIIb/IIIa mainly binds fibrinogen, but can also bind vWF, fibronectin, or vitronectin, to mediate platelet aggregation under shear stress. There are currently three approved inhibitors of GPIIb/IIIa; these are: abciximab, eptifibatide, and tirofiban [166]. These all inhibit the binding of fibrinogen, via different mechanisms: abciximab is a humanized form of a murine monoclonal antibody against GPIIb/IIIa, eptifibatide is a heptapeptide that mimics the arginine-glycine-aspartic (RGD) acid sequence on GPIIb/IIIa, and tirofiban is a non-peptide antagonist that mimics the RGD-containing loop of the disintegrin echistatin [142]. Lamifiban is another GPIIb/IIIa anatagonist that was being tested clinically, but was not approved.
Blockade of GPIIb/IIIa in a mouse ischemic model resulted in a significantly high number of ICH incidences and increased mortality in a dose-dependent manner [167]. It also increased tail bleeding times to more than 20 minutes. Furthermore, a phase III clinical trial with the GPIIb/IIIa specific antagonist, abciximab, was discontinued due to the increased rate of ICH occurrences that resulted from the treatment [168]. Thus, GPIIb/IIIa inhibitors are recommended only for very high risk thrombotic cases, and P2Y12 receptor inhibitors are suggested to be given instead [166]. Additionally, studies have found that administration of GPIIb/IIIa blockers secondary to P2Y12 receptor inhibitors did not add any beneficial effect, but may increase the risk of bleeding [142]. On the contrary, there have been studies that report no significant increase in hemorrhage occurrences with GPIIb/IIIa inhibition combined with a fibrinolytic agent; though a lower dose of the GPIIb/IIIa blocker was given in the combination therapy [169–171]. More studies are needed to define the role of GPIIb/IIIa and other platelet receptors in the progression of intracerebral hemorrhages.
8. Current Management and Future Directions
Current management of ICH includes monitoring hemodynamic parameters, control of intracranial pressure, and hematoma evacuation. There are several clinical trials aimed at preventing hematoma expansion also ongoing: aggressive blood pressure reduction, treatment with rFVIIa, and surgical intervention for superficial hematomas without intraventricular extension [19,26, 172–174]. rFVIIa dose-dependently reduced hematoma expansion and improved mortality in ICH patients, compared to those receiving placebo [19]. However, there was an increase in the number of thromboembolic events in the rFVIIa-treated groups compared to placebo. Furthermore, in a larger clinical trial, rFVIIa reduced hematoma volumes, but did not improve survival or functional outcome following ICH [172]. Hence, we see that activating the coagulation cascade after ICH is likely to result in thromboembolism, while inhibiting coagulation after an embolic stroke involves the risk of ICH. Thus, there is a balance that must be attained, as well as, we have to weigh the risk to benefit ratio. ICH is the most fatal stroke subtype, and it is therefore urgent that effective therapies be developed for its treatment.
ICHs that occur due to coagulopathy have a different pathophysiology and are more likely to result in increased bleeding and hematoma growth. To this extent, Cervera et al. [51], and Aguilar and Freeman [175], provide extensive reviews on treatment options for ICH with coagulopathy. Briefly, the most common treatments include: administration of Vitamin K (to reverse vitamin K antagonists such as warfarin), FFP (widely available, and most commonly used, next to vitamin K), rFVIIa, and PCCs. In a study done by Huttner and colleagues, the incidence and growth of the hematoma was significantly lower in patients receiving PCCs, compared with FFP and vitamin K; and was suggested to be associated with a more rapid international normalized ratio (INR) reversal by PCCs [176]. This coincides with the finding by Curtze et al. that a higher baseline INR was correlated to increased mortality and larger hematomas on admission in ICH secondary to warfarin use [177]. PCCs lead to the rapid replacement of vitamin K-dependent factors, and could therefore be the means by which they reduce growth of the hematoma.
Several antiplatelet agents are currently in use for acute coronary syndrome, myocardial infarction, and other cardioembolic diseases, and all pose risks of bleeding, particularly the GPIIb/IIIa receptor blockers, though to a lesser extent than the anticoagulants and antifibrinolytics. Thus, for the benefit of ICH, platelet targeting may be the more effective and safer route. Naidech et al. found that early platelet transfusion (<12 hours) resulted in smaller hemorrhage size and independence at 3 months in ICH patients, via increased platelet activity [178]. Additionally, reduced platelet activity, and not necessarily the use of antiplateplet medicatiions, was associated with increased occurrence of intraventricular hemorrhage, a greater ICH score, and increased mortality in ICH patients [179]. Thus, this group identified platelet activity as being more predictive of hematoma expansion and ICH outcomes than the use of antiplatelets in ICH patients [180]. Along this line, desmopressin, which leads to the release of vWF, an important platelet activator, was able to increase platelet activity after ICH and limit growth of the hematoma [181]. Against this background, increasing platelet activity (by targeting platelet receptors and other activators) may be a novel treatment strategy for ICH, and warrants further studies.
8.1. CLEC-2: Possible target for hematoma reduction and improved ICH outcomes?
A new platelet transmembrane receptor, the C-type lectin-like receptor 2 (CLEC-2), has recently been discovered, and may be a new target for reduction of hematoma expansion after ICH. CLEC-2 has been identified in the last decade as the receptor for rhodcytin, a very potent platelet activating protein isolated from the Malayan pit viper Calloselasma rhodostoma [182]. CLEC-2 is also activated upon the binding of podoplanin, its endogenous ligand [124, 125, 183]. This receptor signals through a single cytoplasmic YXXL motif, known as a hemi-immunoreceptor tyrosine-based activation motif (hemilTAM); it is tyrosine phosphorylated upon binding of its ligand [184]. This then promotes interaction with Syk, via the two Src-homology domain 2 (SH2) domains on Syk [182, 185]. Ligand binding of CLEC-2 also triggers the tyrosine phosphorylation of Syk, PLCγ2, PI3K, Vav3, LAT, SLP-76, and Btk [185–187].
Because CLEC-2 mediates such potent platelet activation, and its loss has been associated with protection from occlusive thrombus formation, it has been suggested as a therapeutic target in thromboembolic events [119, 184]. However, this could mean bleeding complications. We suggest that activating platelet CLEC-2 could be a novel mechanism for reducing hematoma expansion by inducing rapid platelet aggregation. Furthermore, CLEC-2 has been shown to play an important role in the formation of a stable thrombus, and loss of CLEC-2 resulted in prolonged bleeding times [119]. On the other hand, activation of CLEC-2 significantly reduces bleeding times [188]. The responsiveness of CLEC-2 to its ligands, particularly rhodocytin, indicates that small doses could produce the desired activation, and could therefore mean little or no risk of adverse thromboembolic events, as was the case with the use of procoagulants such as rFVIIa for hematoma reduction. Moreover, the fact that antiplatelet medications are not as efficacious in improving ischemic stroke outcomes as anticoagulants could suggest that activating platelets would have very little effect on thromboembolism. Furthermore, following the loss of the endogenous ligand, podoplanin, it was shown that components of the endothelial adherens junction such as β-catenin were decreased [124]. In addition, inhibition of the podoplanin-CLEC-2 interaction resulted in a loss of high endothelial venule integrity [124], suggesting that this interaction plays a critical role in maintaining vascular integrity, which is important for favorable outcomes following ICH. Taken together, these findings suggest that CLEC-2 may be an important target for hematoma reduction following ICH, and that its activation may be able to reduce the growth of the hemorrhage.
Conclusions
Anticoagulants and antiplatelets have been increasingly prescribed to patients experiencing ischemic strokes, myocardial infarctions, and other cardiovascular ischemic events, but have consistently shown an increase in bleeding as an adverse side effect. Anticoagulants pose a more significant risk than antiplatelets, but are more beneficial for thromboembolic stroke reduction, and are thus more commonly administered. This means an increase in the number of oral anticoagulant-associated ICHs. However, platelets play an important role in clot stabilization, and reduced platelet activity has been shown to worsen ICH prognoses. Targeting platelet activation after ICH may be the direction to move in, thus reducing hematoma expansion and its effects on outcome by increasing clot stabilization and preventing further growth. We propose that more in-depth studies of the mechanisms of the activity of the novel platelet receptor, CLEC-2, in ICH, and its potential role in reducing hematoma expansion, are a worthy pursuit.
Abbreviations:
- HE
Hematoma expansion
- ICH
Intracerebral hemorrhage
- AF
Atrial fibrillation
- OAT-ICH
Oral anticoagulant-associated intracerebral hemorrhage
- PAR
protease activated receptor
- VKAs
vitamin K antagonists
- VKORC
Vitamin K Epoxide Reductase Complex
- CYP450
cytochrome p450
- PCCs
Prothrombin complex concentrates
- FFP
fresh frozen plasma
- rFVIIa
recombinant activated factor VII
- PT
prothrombin time
- APTT
activated partial thromboplastin time
- TT
thrombin time
- rtPA
recombinant tissue plasminogen activator
- tPA
tissue plasminogen activator
- GP
glycoprotein
- vWF
von WIllebrand factor
- ADP
adenosine diphosphate
- PON-1
esterase paraoxonase-1
- CLEC-2
C-type lectin-like receptor 2
Footnotes
Conflicts of Interest:
The authors report no conflicts of interest.
References
- 1.Elijovich L, Patel PV, Hemphill JC 3rd, Intracerebral hemorrhage. Semin Neurol. 2008;28(5):657–67. [DOI] [PubMed] [Google Scholar]
- 2.Emiru T, Bershad EM, Zantek ND, et al. Intracerebral hemorrhage: a review of coagulation function. Clinical and applied thrombosis/hemostasis : official journal of the International Academy of Clinical and Applied Thrombosis/Hemostasis. 2013;19(6):652–62. [DOI] [PubMed] [Google Scholar]
- 3.Brott T, Broderick J, Kothari R, et al. Early Hemorrhage Growth in Patients With Intracerebral Hemorrhage. Stroke. 1997;28(1):1–5. [DOI] [PubMed] [Google Scholar]
- 4.Flibotte JJ, Hagan N, O’Donnell J, Greenberg SM, Rosand J. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology. 2004;63(6):1059–64. [DOI] [PubMed] [Google Scholar]
- 5.Davis SM, Broderick J, Hennerici M, et al. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology. 2006;66(8):1175–81. [DOI] [PubMed] [Google Scholar]
- 6.Dowlatshahi D, Demchuk AM, Flaherty ML, et al. Defining hematoma expansion in intracerebral hemorrhage: relationship with patient outcomes. Neurology. 2011;76(14):1238–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brouwers HB, Goldstein JN. Therapeutic strategies in acute intracerebral hemorrhage. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2012;9(1):87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Mori T, Sumii T, Lo EH. Hemoglobin-Induced Cytotoxicity in Rat Cerebral Cortical Neurons: Caspase Activation and Oxidative Stress. Stroke. 2002;33(7):1882–8. [DOI] [PubMed] [Google Scholar]
- 9.Wu J, Hua Y, Keep RF, Schallert T, Hoff JT, Xi G. Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain research. 2002;953(1–2):45–52. [DOI] [PubMed] [Google Scholar]
- 10.Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42(6):1781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong XY, Wang J, Qian ZM, Yang QW . Iron and intracerebral hemorrhage: from mechanism to translation. Translational stroke research. 2014;5(4):429–41. [DOI] [PubMed] [Google Scholar]
- 12.Xi GH, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurology. 2006;5(1):53–63. [DOI] [PubMed] [Google Scholar]
- 13.Babu R, Bagley JH, Di C, Friedman AH, Adamson C. Thrombin and hemin as central factors in the mechanisms of intracerebral hemorrhage-induced secondary brain injury and as potential targets for intervention. Neurosurg Focus. 2012;32(4):E8. [DOI] [PubMed] [Google Scholar]
- 14.Zhao X, Sun G, Zhang H, et al. Polymorphonuclear neutrophil in brain parenchyma after experimental intracerebral hemorrhage. Translational stroke research. 2014;5(5):554–61. [DOI] [PubMed] [Google Scholar]
- 15.Chen S, Yang Q, Chen G, Zhang JH. An update on inflammation in the acute phase of intracerebral hemorrhage. Translational stroke research. 2015;6(1):4–8. [DOI] [PubMed] [Google Scholar]
- 16.Nieswandt B, Pleines I, Bender M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J Thromb Haemost. 2011;9 Suppl 1:92–104. [DOI] [PubMed] [Google Scholar]
- 17.Zhang JH. Vascular neural network in subarachnoid hemorrhage. Translational stroke research. 2014;5(4):423–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Counsell C, Sandercock P. Use of Anticoagulants in Patients with Acute Ischemic Stroke. Stroke. 1995;26(3):522-. [PubMed] [Google Scholar]
- 19.Mayer SA, Brun NC, Begtrup K, et al. Recombinant activated factor VII for acute intracerebral hemorrhage. The New England journal of medicine. 2005;352(8):777–85. [DOI] [PubMed] [Google Scholar]
- 20.Qureshi AI, Mohammad YM, Yahia AM, et al. A prospective multicenter study to evaluate the feasibility and safety of aggressive antihypertensive treatment in patients with acute intracerebral hemorrhage. J Intensive Care Med. 2005;20(1):34–42. [DOI] [PubMed] [Google Scholar]
- 21.van Asch CJ, Oudendijk JF, Rinkel GJ, Klijn CJ. Early intracerebral hematoma expansion after aneurysmal rupture. Stroke. 2010;41(11):2592–5. [DOI] [PubMed] [Google Scholar]
- 22.Sahni R, Weinberger J. Management of intracerebral hemorrhage. Vasc Health Risk Manag. 2007;3(5):701–9. [PMC free article] [PubMed] [Google Scholar]
- 23.Rutledge WC, Ko NU, Lawton MT, Kim H. Hemorrhage rates and risk factors in the natural history course of brain arteriovenous malformations. Translational stroke research. 2014;5(5):538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Broderick JP, Brott TG, Duldner JE, Tomsick T, Huster G. Volume of intracerebral hemorrhage. A powerful and easy-to-use predictor of 30-day mortality. Stroke. 1993;24(7):987–93. [DOI] [PubMed] [Google Scholar]
- 25.Flaherty ML, Haverbusch M, Sekar P, et al. Location and outcome of anticoagulant-associated intracerebral hemorrhage. Neurocrit Care. 2006;5(3):197–201. [DOI] [PubMed] [Google Scholar]
- 26.Brouwers HB, Greenberg SM. Hematoma expansion following acute intracerebral hemorrhage. Cerebrovasc Dis. 2013;35(3):195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takahashi H, Urano T, Nagai N, Takada Y, Takada A. Progressive expansion of hypertensive intracerebral hemorrhage by coagulopathy. American Journal of Hematology. 1998;59(2):110–4. [DOI] [PubMed] [Google Scholar]
- 28.Choudhri TF, Hoh BL, Prestigiacomo CJ, et al. Targeted Inhibition of Intrinsic Coagulation Limits Cerebral Injury in Stroke without Increasing Intracerebral Hemorrhage. Journal of Experimental Medicine. 1999;190(1):91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleinschnitz C, Stoll G, Bendszus M, et al. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006;203(3):513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The effect of low-dose warfarin on the risk of stroke in patients with nonrheumatic atrial fibrillation. The Boston Area Anticoagulation Trial for Atrial Fibrillation Investigators. The New England journal of medicine. 1990;323(22):1505–11. [DOI] [PubMed] [Google Scholar]
- 31.Risk Factors for Stroke and Efficacy of Antithrombotic Therapy in Atrial Fibrillation. Archives of Internal Medicine. 1994;154(13):1449. [PubMed] [Google Scholar]
- 32.Sjalander A, Engstrom G, Berntorp E, Svensson P. Risk of haemorrhagic stroke in patients with oral anticoagulation compared with the general population. Journal of Internal Medicine. 2003;254(5):434–8. [DOI] [PubMed] [Google Scholar]
- 33.Steiner T, Rosand J, Diringer M. Intracerebral hemorrhage associated with oral anticoagulant therapy: current practices and unresolved questions. Stroke. 2006;37(1):256–62. [DOI] [PubMed] [Google Scholar]
- 34.Flaherty ML, Kissela B, Woo D, et al. The increasing incidence of anticoagulant-associated intracerebral hemorrhage. Neurology. 2007;68(2):116–21. [DOI] [PubMed] [Google Scholar]
- 35.Kuwashiro T, Yasaka M, Itabashi R, et al. Enlargement of acute intracerebral hematomas in patients on long-term warfarin treatment. Cerebrovasc Dis. 2010;29(5):446–53. [DOI] [PubMed] [Google Scholar]
- 36.Kase CS, Robinson RK, Stein RW, et al. Anticoagulant-related intracerebral hemorrhage. Neurology. 1985;35(7):943–8. [DOI] [PubMed] [Google Scholar]
- 37.Fredriksson K, Norrving B, Stromblad LG. Emergency reversal of anticoagulation after intracerebral hemorrhage. Stroke. 1992;23(7):972–7. [DOI] [PubMed] [Google Scholar]
- 38.Maas MB, Rosenberg NF, Kosteva AR, Prabhakaran S, Naidech AM. Coagulopathy disproportionately predisposes to lobar intracerebral hemorrhage. Neurocrit Care. 2013;18(2):166–9. [DOI] [PubMed] [Google Scholar]
- 39.Cucchiara B, Messe S, Sansing L, Kasner S, Lyden P, Investigators C. Hematoma growth in oral anticoagulant related intracerebral hemorrhage. Stroke. 2008;39(11):2993–6. [DOI] [PubMed] [Google Scholar]
- 40.Flaherty ML, Tao H, Haverbusch M, et al. Warfarin use leads to larger intracerebral hematomas. Neurology. 2008;71(14):1084–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hua Y, Keep RF, Hoff JT, Xi G. Brain injury after intracerebral hemorrhage: the role of thrombin and iron. Stroke. 2007;38(2 Suppl):759–62. [DOI] [PubMed] [Google Scholar]
- 42.Lekic T, Manaenko A, Rolland W, et al. Rodent neonatal germinal matrix hemorrhage mimics the human brain injury, neurological consequences, and post-hemorrhagic hydrocephalus. Exp Neurol. 2012;236(1):69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? Journal of Neurochemistry. 2002;84(1):3–9. [DOI] [PubMed] [Google Scholar]
- 44.Kevin RL, Nobuyuki K, Seoung K, Oren S, Julian TH. Mechanisms of edema formation after intracerebral hemorrhage: effects of thrombin on cerebral blood flow, blood-brain barrier permeability, and cell survival in a rat model. J Neurosurg. 1997;86(2):272–8. [DOI] [PubMed] [Google Scholar]
- 45.Gao F, Liu F, Chen Z, Hua Y, Keep RF, Xi G. Hydrocephalus after intraventricular hemorrhage: the role of thrombin. J Cereb Blood Flow Metab. 2014;34(3):489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng Y, Xi G, Jin H, Keep RF, Feng J, Hua Y. Thrombin-induced cerebral hemorrhage: role of protease-activated receptor-1. Translational stroke research. 2014;5(4):472–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu CH, Yang RL, Huang SY, et al. Analysis of thrombin-antithrombin complex contents in plasma and hematoma fluid of hypertensive intracerebral hemorrhage patients after clot removal. Eur J Neurol. 2011;18(8):1060–6. [DOI] [PubMed] [Google Scholar]
- 48.Alberts MJ, Eikelboom JW, Hankey GJ. Antithrombotic therapy for stroke prevention in non-valvular atrial fibrillation. Lancet Neurol. 2012;11(12):1066–81. [DOI] [PubMed] [Google Scholar]
- 49.Flaherty ML. Anticoagulant-associated intracerebral hemorrhage. Semin Neurol. 2010;30(5):565–72. [DOI] [PubMed] [Google Scholar]
- 50.Suarez-Pinilla M, Fernandez-Rodriguez A, Benavente-Fernandez L, Calleja-Puerta S. Vitamin K antagonist-associated intracerebral hemorrhage: lessons from a devastating disease in the dawn of the new oral anticoagulants. J Stroke Cerebrovasc Dis. 2014;23(4):732–42. [DOI] [PubMed] [Google Scholar]
- 51.Cervera A, Amaro S, Chamorro A. Oral anticoagulant-associated intracerebral hemorrhage. J Neurol. 2012;259(2):212–24. [DOI] [PubMed] [Google Scholar]
- 52.Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126(3 Suppl):287S–310S. [DOI] [PubMed] [Google Scholar]
- 53.Rosand J, Eckman MH, Knudsen KA, Singer DE, Greenberg SM. The effect of warfarin and intensity of anticoagulation on outcome of intracerebral hemorrhage. Arch Intern Med. 2004;164(8):880–4. [DOI] [PubMed] [Google Scholar]
- 54.Foerch C, Arai K, Van Cott EM, van Leyen K, Lo EH. Rapid reversal of anticoagulation reduces hemorrhage volume in a mouse model of warfarin-associated intracerebral hemorrhage. J Cereb Blood Flow Metab. 2009;29(5):1015–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schlunk F, Schulz E, Lauer A, et al. Warfarin Pretreatment Reduces Cell Death and MMP-9 Activity in Experimental Intracerebral Hemorrhage. Translational stroke research. 2014. [DOI] [PubMed] [Google Scholar]
- 56.Yaghi S, Dibu J, Achi E, Patel A, Samant R, Hinduja A. Hematoma expansion in spontaneous intracerebral hemorrhage: predictors and outcome. Int J Neurosci. 2014. [DOI] [PubMed] [Google Scholar]
- 57.Biffi A, Battey TW, Ayres AM, et al. Warfarin-related intraventricular hemorrhage: imaging and outcome. Neurology. 2011;77(20):1840–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Q, Zhang J, Guo J, et al. Chronic Hydrocephalus and Perihematomal Tissue Injury Developed in a Rat Model of Intracerebral Hemorrhage with Ventricular Extension. Translational stroke research. 2014. [DOI] [PubMed] [Google Scholar]
- 59.Whitlon DS, Sadowski JA, Suttie JW. Mechanism of Coumarin Action - Significance of Vitamin-K Epoxide Reductase Inhibition. Biochemistry. 1978;17(8):1371–7. [DOI] [PubMed] [Google Scholar]
- 60.Li J, Wang S, Barone J, Malone B. Warfarin pharmacogenomics. P T. 2009;34(8):422–7. [PMC free article] [PubMed] [Google Scholar]
- 61.Aithal GP, Day CP, Kesteven PJL, Daly AK. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. The Lancet. 1999;353(9154):717–9. [DOI] [PubMed] [Google Scholar]
- 62.Rieder MJ, Reiner AP, Gage BF, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. The New England journal of medicine. 2005;352(22):2285–93. [DOI] [PubMed] [Google Scholar]
- 63.Rettie AE, Tai G. The pharmocogenomics of warfarin: closing in on personalized medicine. Mol Interv. 2006;6(4):223–7. [DOI] [PubMed] [Google Scholar]
- 64.Zhu Y, Shennan M, Reynolds KK, et al. Estimation of warfarin maintenance dose based on VKORC1 (−1639 G>A) and CYP2C9 genotypes. Clin Chem. 2007;53(7):1199–205. [DOI] [PubMed] [Google Scholar]
- 65.Limdi NA, Veenstra DL. Warfarin pharmacogenetics. Pharmacotherapy. 2008;28(9):1084–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.International Warfarin Pharmacogenetics C, Klein TE, Altman RB, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. The New England journal of medicine. 2009;360(8):753–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eckman MH. Cost-Effectiveness of Using Pharmacogenetic Information in Warfarin Dosing for Patients With Nonvalvular Atrial Fibrillation. Annals of Internal Medicine. 2009;150(2):73. [DOI] [PubMed] [Google Scholar]
- 68.Jaffer AK. Managing anticoagulant related coagulopathy. J Thromb Thrombolysis. 2008;25(1):85–90. [DOI] [PubMed] [Google Scholar]
- 69.Grobler C, Callum J, McCluskey SA. Reversal of vitamin K antagonists prior to urgent surgery. Can J Anaesth. 2010;57(5):458–67. [DOI] [PubMed] [Google Scholar]
- 70.Ogbonna KC, Jeffery SM. Risk versus benefit of non-vitamin K dependent anticoagulants compared to warfarin for the management of atrial fibrillation in the elderly. Drugs Aging. 2013;30(7):513–25. [DOI] [PubMed] [Google Scholar]
- 71.Hankey GJ. Intracranial hemorrhage and novel anticoagulants for atrial fibrillation: what have we learned? Curr Cardiol Rep. 2014;16(5):480. [DOI] [PubMed] [Google Scholar]
- 72.Gustafsson D, Antonsson T, Bylund R, et al. Effects of melagatran, a new low-molecular-weight thrombin inhibitor, on thrombin and fibrinolytic enzymes. Thromb Haemost. 1998;79(1):110–8. [PubMed] [Google Scholar]
- 73.Nylander S, Mattsson C. Thrombin-induced platelet activation and its inhibition by anticoagulants with different modes of action. Blood Coagul Fibrinolysis. 2003;14(2):159–67. [DOI] [PubMed] [Google Scholar]
- 74.Mattsson C, Menschik-Lundin A, Nylander S, Gyzander E, Deinum J. Effect of Different Types of Thrombin Inhibitors on Thrombin/Thrombomodulin Modulated Activation of Protein C In Vitro. Thrombosis Research. 2001;104(6):475–86. [DOI] [PubMed] [Google Scholar]
- 75.Gustafsson D, Nystrom J, Carlsson S, et al. The direct thrombin inhibitor melagatran and its oral prodrug H 376/95: intestinal absorption properties, biochemical and pharmacodynamic effects. Thromb Res. 2001;101(3):171–81. [DOI] [PubMed] [Google Scholar]
- 76.Akins PT, Feldman HA, Zoble RG, et al. Secondary stroke prevention with ximelagatran versus warfarin in patients with atrial fibrillation: pooled analysis of SPORTIF III and V clinical trials. Stroke. 2007;38(3):874–80. [DOI] [PubMed] [Google Scholar]
- 77.Bredberg E, Andersson TB, Frison L, et al. Ximelagatran, an oral direct thrombin inhibitor, has a low potential for cytochrome P450-mediated drug-drug interactions. Clin Pharmacokinet. 2003;42(8):765–77. [DOI] [PubMed] [Google Scholar]
- 78.Gustafsson D, Elg M. The pharmacodynamics and pharmacokinetics of the oral direct thrombin inhibitor ximelagatran and its active metabolite melagatran: a mini-review. Thrombosis Research. 2003;109:S9–S15. [DOI] [PubMed] [Google Scholar]
- 79.Albers GW, Investigators S. Stroke prevention in atrial fibrillation: pooled analysis of SPORTIF III and V trials. Am J Manag Care. 2004;10(14 Suppl):S462–9; discussion S9–73. [PubMed] [Google Scholar]
- 80.Albers GW, Diener HC, Frison L, et al. Ximelagatran vs warfarin for stroke prevention in patients with nonvalvular atrial fibrillation: a randomized trial. JAMA. 2005;293(6):690–8. [DOI] [PubMed] [Google Scholar]
- 81.Diener HC, Executive Steering Committee of the S, III, Investigators V. Stroke prevention using the oral direct thrombin inhibitor ximelagatran in patients with non-valvular atrial fibrillation. Pooled analysis from the SPORTIF III and V studies. Cerebrovasc Dis. 2006;21(4):279–93. [DOI] [PubMed] [Google Scholar]
- 82.Shirasaki Y, Morishima Y, Shibano T. Comparison of the effect of edoxaban, a direct factor Xa inhibitor, with a direct thrombin inhibitor, melagatran, and heparin on intracerebral hemorrhage induced by collagenase in rats. Thromb Res. 2014;133(4):622–8. [DOI] [PubMed] [Google Scholar]
- 83.Yates S, Sarode R. Novel thrombin and factor Xa inhibitors: challenges to reversal of their anticoagulation effects. Curr Opin Hematol. 2013;20(6):552–7. [DOI] [PubMed] [Google Scholar]
- 84.Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. The New England journal of medicine. 2009;361(12):1139–51. [DOI] [PubMed] [Google Scholar]
- 85.Wallentin L, Yusuf S, Ezekowitz MD, et al. Efficacy and safety of dabigatran compared with warfarin at different levels of international normalised ratio control for stroke prevention in atrial fibrillation: an analysis of the RE-LY trial. The Lancet. 2010;376(9745):975–83. [DOI] [PubMed] [Google Scholar]
- 86.Zhou W, Schwarting S, Illanes S, et al. Hemostatic therapy in experimental intracerebral hemorrhage associated with the direct thrombin inhibitor dabigatran. Stroke. 2011;42(12):3594–9. [DOI] [PubMed] [Google Scholar]
- 87.Lauer A, Cianchetti FA, Van Cott EM, et al. Anticoagulation with the oral direct thrombin inhibitor dabigatran does not enlarge hematoma volume in experimental intracerebral hemorrhage. Circulation. 2011;124(15):1654–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hart RG, Diener HC, Yang S, et al. Intracranial hemorrhage in atrial fibrillation patients during anticoagulation with warfarin or dabigatran: the RE-LY trial. Stroke. 2012;43(6):1511–7. [DOI] [PubMed] [Google Scholar]
- 89.Ebner T, Wagner K, Wienen W. Dabigatran acylglucuronide, the major human metabolite of dabigatran: in vitro formation, stability, and pharmacological activity. Drug Metab Dispos. 2010;38(9):1567–75. [DOI] [PubMed] [Google Scholar]
- 90.Roser-Jones C, Becker RC. Apixaban: an emerging oral factor Xa inhibitor. J Thromb Thrombolysis. 2010;29(1):141–6. [DOI] [PubMed] [Google Scholar]
- 91.Turpie AG. New oral anticoagulants in atrial fibrillation. Eur Heart J. 2008;29(2):155–65. [DOI] [PubMed] [Google Scholar]
- 92.Zhou W, Zorn M, Nawroth P, et al. Hemostatic therapy in experimental intracerebral hemorrhage associated with rivaroxaban. Stroke. 2013;44(3):771–8. [DOI] [PubMed] [Google Scholar]
- 93.Gong IY, Kim RB. Importance of pharmacokinetic profile and variability as determinants of dose and response to dabigatran, rivaroxaban, and apixaban. Can J Cardiol. 2013;29(7 Suppl):S24–33. [DOI] [PubMed] [Google Scholar]
- 94.Morishima Y, Honda Y, Kamisato C, et al. Comparison of antithrombotic and haemorrhagic effects of edoxaban, an oral direct factor Xa inhibitor, with warfarin and enoxaparin in rats. Thromb Res. 2012;130(3):514–9. [DOI] [PubMed] [Google Scholar]
- 95.Sloan MA, Sila CA, Mahaffey KW, et al. Prediction of 30-Day Mortality Among Patients With Thrombolysis-Related Intracranial Hemorrhage. Circulation. 1998;98(14):1376–82. [DOI] [PubMed] [Google Scholar]
- 96.Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. The New England journal of medicine. 1995;333(24):1581–7. [DOI] [PubMed] [Google Scholar]
- 97.Rao NM, Levine SR, Gornbein JA, Saver JL. Defining clinically relevant cerebral hemorrhage after thrombolytic therapy for stroke: analysis of the National Institute of Neurological Disorders and Stroke tissue-type plasminogen activator trials. Stroke. 2014;45(9):2728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Berrouschot J, Rother J, Glahn J, Kucinski T, Fiehler J, Thomalla G. Outcome and severe hemorrhagic complications of intravenous thrombolysis with tissue plasminogen activator in very old (> or =80 years) stroke patients. Stroke. 2005;36(11):2421–5. [DOI] [PubMed] [Google Scholar]
- 99.Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA. Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by Increasing superoxide production. Ann Neurol. 2011;70(4):583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hafez S, Coucha M, Bruno A, Fagan SC, Ergul A. Hyperglycemia, acute ischemic stroke, and thrombolytic therapy. Translational stroke research. 2014;5(4):442–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mandava P, Martini SR, Munoz M, et al. Hyperglycemia worsens outcome after rt-PA primarily in the large-vessel occlusive stroke subtype. Translational stroke research. 2014;5(4):519–25. [DOI] [PubMed] [Google Scholar]
- 102.Gonzalez-Moreno EI, Camara-Lemarroy CR, Gonzalez-Gonzalez JG, Gongora-Rivera F. Glycemic variability and acute ischemic stroke: the missing link? Translational stroke research. 2014;5(6):638–46. [DOI] [PubMed] [Google Scholar]
- 103.Murray V, Norrving B, Sandercock PA, Terent A, Wardlaw JM, Wester P. The molecular basis of thrombolysis and its clinical application in stroke. J Intern Med. 2010;267(2):191–208. [DOI] [PubMed] [Google Scholar]
- 104.Montagne A, Hebert M, Jullienne A, et al. Memantine improves safety of thrombolysis for stroke. Stroke. 2012;43(10):2774–81. [DOI] [PubMed] [Google Scholar]
- 105.Marinkovic I, Mattila OS, Strbian D, et al. Evolution of intracerebral hemorrhage after intravenous tPA: reversal of harmful effects with mast cell stabilization. J Cereb Blood Flow Metab. 2014;34(1):176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Foerch C, Rosidi NL, Schlunk F, et al. Intravenous tPA therapy does not worsen acute intracerebral hemorrhage in mice. PloS one. 2013;8(2):e54203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Malinova V, Stockhammer F, Atangana EN, Mielke D, Rohde V. Catheter placement for lysis of spontaneous intracerebral hematomas: is a navigated stylet better than pointer-guided frameless stereotaxy for intrahematomal catheter positioning? Translational stroke research. 2014;5(3):407–14. [DOI] [PubMed] [Google Scholar]
- 108.Wang X, Tsuji K, Lee SR, et al. Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke. 2004;35(11 Suppl 1):2726–30. [DOI] [PubMed] [Google Scholar]
- 109.Lapchak PA, Chapman DF, Zivin JA, Hsu CY. Metalloproteinase Inhibition Reduces Thrombolytic (Tissue Plasminogen Activator)-Induced Hemorrhage After Thromboembolic Stroke Editorial Comment. Stroke. 2000;31(12):3034–40. [DOI] [PubMed] [Google Scholar]
- 110.Sumii T, Lo EH. Involvement of Matrix Metalloproteinase in Thrombolysis-Associated Hemorrhagic Transformation After Embolic Focal Ischemia in Rats. Stroke. 2002;33(3):831–6. [DOI] [PubMed] [Google Scholar]
- 111.Castellanos M, Leira R, Serena J, et al. Plasma Metalloproteinase-9 Concentration Predicts Hemorrhagic Transformation in Acute Ischemic Stroke * Editorial Comment. Stroke. 2002;34(1):40–6. [PubMed] [Google Scholar]
- 112.Wang X, Lee SR, Arai K, et al. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med. 2003;9(10):1313–7. [DOI] [PubMed] [Google Scholar]
- 113.Montaner J Matrix Metalloproteinase-9 Pretreatment Level Predicts Intracranial Hemorrhagic Complications After Thrombolysis in Human Stroke. Circulation. 2003;107(4):598–603. [DOI] [PubMed] [Google Scholar]
- 114.Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Tissue plasminogen activator (tPA) increase neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nature Medicine. 1998;4(2):228–31. [DOI] [PubMed] [Google Scholar]
- 115.Nagai N, Yamamoto S, Tsuboi T, et al. Tissue-type plasminogen activator is involved in the process of neuronal death induced by oxygen-glucose deprivation in culture. J Cereb Blood Flow Metab. 2001;21(6):631–4. [DOI] [PubMed] [Google Scholar]
- 116.Lee ST, Chu K, Jung KH, et al. Memantine reduces hematoma expansion in experimental intracerebral hemorrhage, resulting in functional improvement. J Cereb Blood Flow Metab. 2006;26(4):536–44. [DOI] [PubMed] [Google Scholar]
- 117.Ji X, Meng R, Zhou J, Ling F, Jia J. Dynamic change of coagulation and anticoagulation markers of patients with acute cerebral infarction during intravenous urokinase thrombolysis. Neurological research. 2006;28(1):46–9. [DOI] [PubMed] [Google Scholar]
- 118.Boulaftali Y, Hess PR, Getz TM, et al. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest. 2013;123(2):908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.May F, Hagedorn I, Pleines I, et al. CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood. 2009;114(16):3464–72. [DOI] [PubMed] [Google Scholar]
- 120.Lauer A, Schlunk F, Van Cott EM, Steinmetz H, Lo EH, Foerch C. Antiplatelet pretreatment does not increase hematoma volume in experimental intracerebral hemorrhage. J Cereb Blood Flow Metab. 2011;31(8):1736–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Alexopoulos D p2y12 receptor inhibitors in acute coronary syndromes: from the research laboratory to the clinic and vice versa. Cardiology. 2014;127(4):211–9. [DOI] [PubMed] [Google Scholar]
- 122.Yin T, Miyata T. Pharmacogenomics of clopidogrel: evidence and perspectives. Thromb Res. 2011;128(4):307–16. [DOI] [PubMed] [Google Scholar]
- 123.Gremmel T, Kopp CW, Seidinger D, Koppensteiner R, Steiner S, Panzer S. Preserved thrombin-inducible platelet activation in thienopyridine-treated patients. Eur J Clin Invest. 2013;43(7):689–97. [DOI] [PubMed] [Google Scholar]
- 124.Herzog BH, Fu J, Wilson SJ, et al. Podoplanin maintains high endothelial venule integrity by interacting with platelet CLEC-2. Nature. 2013;502(7469):105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Navarro-Nunez L, Langan SA, Nash GB, Watson SP. The physiological and pathophysiological roles of platelet CLEC-2. Thromb Haemost. 2013;109(6):991–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Moher M, Lancaster T. Who needs antiplatelet therapy? Br J Gen Pract. 1996;46(407):367–70. [PMC free article] [PubMed] [Google Scholar]
- 127.Bhatt DL, Fox KA, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. The New England journal of medicine. 2006;354(16):1706–17. [DOI] [PubMed] [Google Scholar]
- 128.Stead LG, Jain A, Bellolio MF, et al. Effect of anticoagulant and antiplatelet therapy in patients with spontaneous intra-cerebral hemorrhage: Does medication use predict worse outcome? Clin Neurol Neurosurg. 2010;112(4):275–81. [DOI] [PubMed] [Google Scholar]
- 129.Roquer J, Rodriguez Campello A, Gomis M, Ois A, Puente V, Munteis E. Previous antiplatelet therapy is an independent predictor of 30-day mortality after spontaneous supratentorial intracerebral hemorrhage. J Neurol. 2005;252(4):412–6. [DOI] [PubMed] [Google Scholar]
- 130.Naidech AM, Jovanovic B, Liebling S, et al. Reduced platelet activity is associated with early clot growth and worse 3-month outcome after intracerebral hemorrhage. Stroke. 2009;40(7):2398–401. [DOI] [PubMed] [Google Scholar]
- 131.Creutzfeldt CJ, Weinstein JR, Longstreth WT Jr., Becker KJ, McPharlin TO, Tirschwell DL. Prior antiplatelet therapy, platelet infusion therapy, and outcome after intracerebral hemorrhage. J Stroke Cerebrovasc Dis. 2009;18(3):221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yildiz OK, Arsava EM, Akpinar E, Topcuoglu MA. Previous antiplatelet use is associated with hematoma expansion in patients with spontaneous intracerebral hemorrhage. J Stroke Cerebrovasc Dis. 2012;21(8):760–6. [DOI] [PubMed] [Google Scholar]
- 133.Broderick JP, Diringer MN, Hill MD, et al. Determinants of intracerebral hemorrhage growth: an exploratory analysis. Stroke. 2007;38(3):1072–5. [DOI] [PubMed] [Google Scholar]
- 134.Thompson BB, Bejot Y, Caso V, et al. Prior antiplatelet therapy and outcome following intracerebral hemorrhage: a systematic review. Neurology. 2010;75(15):1333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sansing LH, Messe SR, Cucchiara BL, et al. Prior antiplatelet use does not affect hemorrhage growth or outcome after ICH. Neurology. 2009;72(16):1397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Moussouttas M, Malhotra R, Fernandez L, et al. Role of antiplatelet agents in hematoma expansion during the acute period of intracerebral hemorrhage. Neurocrit Care. 2010;12(1):24–9. [DOI] [PubMed] [Google Scholar]
- 137.Cattaneo M New P2Y(12) inhibitors. Circulation. 2010;121(1):171–9. [DOI] [PubMed] [Google Scholar]
- 138.Bode-Böger S, Böger R, Schubert M, Frölich J. Effects of very low dose and enteric-coated acetylsalicylic acid on prostacyclin and thromboxane formation and on bleeding time in healthy subjects. Eur J Clin Pharmacol. 1998;54(9–10):707–14. [DOI] [PubMed] [Google Scholar]
- 139.Ajani UA, Ford ES, Greenland KJ, Giles WH, Mokdad AH. Aspirin use among US adults - Behavioral risk factor surveillance system. American Journal of Preventive Medicine. 2006;30(1):74–7. [DOI] [PubMed] [Google Scholar]
- 140.McLeod L, Roberts M, Cossum P, Vial J. The effects of different doses of some acetylsalicylic acid formulations on platelet function and bleeding times in healthy subjects. Scand J Haematol. 1986;36(4):379–84. [DOI] [PubMed] [Google Scholar]
- 141.Chylova M, Motovska Z, Osmancik P, Prochazka B, Kalvach P. The effect of different doses and different routes of acetylsalicylic Acid administration on platelet aggregation in healthy volunteers and ischemic stroke patients. Translational stroke research. 2015;6(2):160–5. [DOI] [PubMed] [Google Scholar]
- 142.Starnes HB, Patel AA, Stouffer GA. Optimal use of platelet glycoprotein IIb/IIIa receptor antagonists in patients undergoing percutaneous coronary interventions. Drugs. 2011;71(15):2009–30. [DOI] [PubMed] [Google Scholar]
- 143.Vane JR. Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-Like Drugs. Nature-New Biology. 1971;231(25):232-&. [DOI] [PubMed] [Google Scholar]
- 144.Vane JR, Botting RM. The mechanism of action of aspirin. Thrombosis Research. 2003;110(5–6):255–8. [DOI] [PubMed] [Google Scholar]
- 145.Smith JB, Willis AL. Aspirin selectively inhibits prostaglandin production in human platelets. Nat New Biol. 1971;231(25):235–7. [DOI] [PubMed] [Google Scholar]
- 146.Botting R Vane’s discovery of the mechanism of action of aspirin changed our understanding of its clinical pharmacology. Pharmacol Rep. 2010;62(3):518–25. [DOI] [PubMed] [Google Scholar]
- 147.Ge L, Niu G, Han X, et al. Aspirin treatment increases the risk of cerebral microbleeds. Can J Neurol Sci. 2011;38(6):863–8. [DOI] [PubMed] [Google Scholar]
- 148.Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Lancet. 1988;3(8607):349–60. [PubMed] [Google Scholar]
- 149.Adjusted-dose warfarin versus low-intensity, fixed-dose warfarin plus aspirin for high-risk patients with atrial fibrillation: Stroke Prevention in Atrial Fibrillation III randomised clinical trial. The Lancet. 1996;348(9028):633–8. [PubMed] [Google Scholar]
- 150.Sabir IN, Matthews GD, Huang CL. Antithrombotic therapy in atrial fibrillation: aspirin is rarely the right choice. Postgrad Med J. 2013;89(1052):346–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Biffi A, Halpin A, Towfighi A, et al. Aspirin and recurrent intracerebral hemorrhage in cerebral amyloid angiopathy. Neurology. 2010;75(8):693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Falcone GJ, Biffi A, Brouwers HB, et al. Predictors of hematoma volume in deep and lobar supratentorial intracerebral hemorrhage. JAMA Neurol. 2013;70(8):988–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. The New England journal of medicine. 2007;357(20):2001–15. [DOI] [PubMed] [Google Scholar]
- 154.Quinn MJ, Fitzgerald DJ. Ticlopidine and Clopidogrel. Circulation. 1999;100(15):1667–72. [DOI] [PubMed] [Google Scholar]
- 155.Cattaneo M ADP receptors: Inhibitory strategies for antiplatelet therapy. Drug News & Perspectives. 2006;19(5):253–9. [DOI] [PubMed] [Google Scholar]
- 156.Kaul U, Mansoor AH. Platelet adenosine diphosphate receptor antagonists: ticlopidine to ticagrelor — a long continuing journey. Indian Heart Journal. 2012;64(1):54–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sabatine MS, Mega JL. Pharmacogenomics of antiplatelet drugs. Hematology Am Soc Hematol Educ Program. 2014;2014(1):343–7. [DOI] [PubMed] [Google Scholar]
- 158.Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. The New England journal of medicine. 2009;360(4):354–62. [DOI] [PubMed] [Google Scholar]
- 159.Bouman HJ, Schomig E, van Werkum JW, et al. Paraoxonase-1 is a major determinant of clopidogrel efficacy. Nat Med. 2011;17(1):110–6. [DOI] [PubMed] [Google Scholar]
- 160.Hulot JS, Bura A, Villard E, et al. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood. 2006;108(7):2244–7. [DOI] [PubMed] [Google Scholar]
- 161.Sibbing D, Koch W, Gebhard D, et al. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation. 2010;121(4):512–8. [DOI] [PubMed] [Google Scholar]
- 162.Gurbel PA, Tantry US, Shuldiner AR, Kereiakes DJ. Genotyping: one piece of the puzzle to personalize antiplatelet therapy. J Am Coll Cardiol. 2010;56(2):112–6. [DOI] [PubMed] [Google Scholar]
- 163.Shanker J, Yuri Gasparyan AD. Kitas G,V. Kakkar V Platelet Function and Antiplatelet Therapy in Cardiovascular Disease: Implications of Genetic Polymorphisms. Current Vascular Pharmacology. 2011;9(4):479–89. [DOI] [PubMed] [Google Scholar]
- 164.Oikonomou E, Papageorgiou N, Papaioannou S, et al. Genetic testing and antiplatelet treatment: Still way to go? Int J Cardiol. 2015;187:63–5. [DOI] [PubMed] [Google Scholar]
- 165.Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF / AHA / SCAI Guideline for Percutaneous Coronary Intervention: executive summary: a report of the American College of Cardiology Foundation/ American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Circulation. 2011;124(23):2574–609. [DOI] [PubMed] [Google Scholar]
- 166.Armstrong PC, Peter K. GPIIb/IIIa inhibitors: from bench to bedside and back to bench again. Thromb Haemost. 2012;107(5):808–14. [DOI] [PubMed] [Google Scholar]
- 167.Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. 2007;115(17):2323–30. [DOI] [PubMed] [Google Scholar]
- 168.Adams HP Jr., Effron MB, Torner J, et al. Emergency administration of abciximab for treatment of patients with acute ischemic stroke: results of an international phase III trial: Abciximab in Emergency Treatment of Stroke Trial (AbESTT-II). Stroke. 2008;39(1):87–99. [DOI] [PubMed] [Google Scholar]
- 169.Savonitto S Risk of intracranial haemorrhage with combined fibrinolytic and glycoprotein IIb/IIIa inhibitor therapy in acute myocardial infarction Dichotomous response as a function of age in the GUSTO V trial. European Heart Journal. 2003;24(20):1807–14. [DOI] [PubMed] [Google Scholar]
- 170.Ihn YK, Sung JH, Kim B-S. Intravenous Glycoprotein IIb/IIIa Inhibitor (Tirofiban) Followed by L ow-Dose Intra-Arterial Urokinase and Mechanical Thrombolysis for the Treatment of Acute Stroke. Neuroradiology Journal. 2011;24(6):907–13. [DOI] [PubMed] [Google Scholar]
- 171.Kwon JH, Shin SH, Weon YC, Hwang JC, Baik SK. Intra-arterial adjuvant tirofiban after unsuccessful intra-arterial thrombolysis of acute ischemic stroke: preliminary experience in 16 patients. Neuroradiology. 2011;53(10):779–85. [DOI] [PubMed] [Google Scholar]
- 172.Mayer SA, Brun NC, Begtrup K, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. The New England journal of medicine. 2008;358(20):2127–37. [DOI] [PubMed] [Google Scholar]
- 173.Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. The Lancet. 2009;373(9675):1632–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Pandey AS, Xi G. Intracerebral hemorrhage: a multimodality approach to improving outcome. Translational stroke research. 2014;5(3):313–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Aguilar MI, Freeman WD. Treatment of coagulopathy in intracranial hemorrhage. Curr Treat Options Neurol. 2010;12(2):113–28. [DOI] [PubMed] [Google Scholar]
- 176.Huttner HB, Schellinger PD, Hartmann M, et al. Hematoma growth and outcome in treated neurocritical care patients with intracerebral hemorrhage related to oral anticoagulant therapy: comparison of acute treatment strategies using vitamin K, fresh frozen plasma, and prothrombin complex concentrates. Stroke. 2006;37(6):1465–70. [DOI] [PubMed] [Google Scholar]
- 177.Curtze S, Strbian D, Meretoja A, et al. Higher baseline international normalized ratio value correlates with higher mortality in intracerebral hemorrhage during warfarin use. Eur J Neurol. 2014;21(4):616–22. [DOI] [PubMed] [Google Scholar]
- 178.Naidech AM, Liebling SM, Rosenberg NF, et al. Early platelet transfusion improves platelet activity and may improve outcomes after intracerebral hemorrhage. Neurocrit Care. 2012;16(1):82–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Naidech AM, Bernstein RA, Levasseur K, et al. Platelet activity and outcome after intracerebral hemorrhage. Ann Neurol. 2009;65(3):352–6. [DOI] [PubMed] [Google Scholar]
- 180.Naidech AM, Bassin SL, Bernstein RA, et al. Reduced platelet activity is more common than reported anti-platelet medication use in patients with intracerebral hemorrhage. Neurocrit Care. 2009;11(3):307–10. [DOI] [PubMed] [Google Scholar]
- 181.Naidech AM, Maas MB, Levasseur-Franklin KE, et al. Desmopressin improves platelet activity in acute intracerebral hemorrhage. Stroke. 2014;45(8):2451–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Suzuki-Inoue K, Fuller GL, Garcia A, et al. A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood. 2006;107(2):542–9. [DOI] [PubMed] [Google Scholar]
- 183.Suzuki-Inoue K, Kato Y, Inoue O, et al. Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J Biol Chem. 2007;282(36):25993–6001. [DOI] [PubMed] [Google Scholar]
- 184.O’Callaghan CA. Thrombomodulation via CLEC-2 targeting. Curr Opin Pharmacol. 2009;9(2):90–5. [DOI] [PubMed] [Google Scholar]
- 185.Severin S, Pollitt AY, Navarro-Nunez L, et al. Syk-dependent Phosphorylation of CLEC-2: A NOVEL MECHANISM OF HEM-IMMUNORECEPTOR TYROSINE-BASED ACTIVATION MOTIF SIGNALING. Journal of Biological Chemistry. 2010;286(6):4107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Fuller GL, Williams JA, Tomlinson MG, et al. The C-type lectin receptors CLEC-2 and Dectin-1, but not DC-SIGN, signal via a novel YXXL-dependent signaling cascade. J Biol Chem. 2007;282(17):12397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pollitt AY, Grygielska B, Leblond B, Desire L, Eble JA, Watson SP. Phosphorylation of CLEC-2 is dependent on lipid rafts, actin polymerization, secondary mediators, and Rac. Blood. 2010;115(14):2938–46. [DOI] [PubMed] [Google Scholar]
- 188.Manne BK, Getz TM, Hughes CE, et al. Fucoidan is a novel platelet agonist for the C-type lectin-like receptor 2 (CLEC-2). J Biol Chem. 2013;288(11):7717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]