

Abstract
Common and rare variants of the CACNA1C voltage-gated calcium channel gene have been associated with autism and other neurodevelopmental disorders including schizophrenia, bipolar disorder and ADHD. However, little is known about how CACNA1C variants affect cellular processes to alter neurodevelopment. The Timothy syndrome mutation is a rare de novo gain-of-function variant in CACNA1C that causes autism with high penetrance, providing a powerful avenue into investigating the role of CACNA1C variants in neurodevelopmental disorders. Here, we use egl-19, the C. elegans homolog of CACNA1C, to investigate the role of voltage-gated calcium channels in autism. We show that an egl-19(gof) mutation that is equivalent to the Timothy syndrome mutation can alter axon targeting and affect behavior in C. elegans. We find that wildtype egl-19 negatively regulates axon termination. The egl-19(gof) mutation represses axon termination to cause axon targeting defects that lead to the misplacement of electrical synapses and alterations in habituation to light touch. Moreover, genetic interactions indicate that the egl-19(gof) mutation functions with genes that promote selective autophagy to cause defects in axon termination and behavior. These results reveal a novel genetic mechanism whereby a de novo mutation in CACNA1C can drive alterations in circuit formation and behavior.
Author summary
Autism is a disorder that affects neuronal development, leading to alterations in cognition and behavior. Imaging studies have revealed alterations in axonal connectivity as a key feature of autism. However, the underlying perturbations in cell biology that drive these alterations remain largely unknown. To address this issue, we have taken advantage of the Timothy syndrome mutation, a variant in a voltage-gated calcium channel that has the unusual property of causing autism with high penetrance. We identify a role for wild-type voltage-gated calcium channels in regulating axon targeting in C. elegans. Moreover, we find that two different versions of the Timothy syndrome mutation disrupt axon targeting. Our results suggest that the Timothy syndrome mutations disrupt axon targeting and behavior by interacting with genes that promote selective autophagy, the process through which cellular components are selected for degradation. These results reveal a mechanism through which variants in voltage-gated calcium channels can cause the disruptions in axonal connectivity that underlie autism.
Introduction
Variants in the CACNA1C voltage-gated calcium channel (VGCC) gene are common risk factors for autism and other neurodevelopmental disorders including schizophrenia, bipolar disorder and attention deficit hyperactivity disorder (ADHD). For example, genome wide association studies (GWAS) have associated common variants in CACNA1C to autism [1, 2]. Moreover, statistical analysis of whole genome sequencing data indicates that rare variants in CACNA1C are also associated with autism [3–10]. Whereas the evidence is strongest for CACNA1C, variants in other VGCC subunit genes are also associated with autism [2, 8, 11, 12]. Despite these insights from statistical analysis, little is currently known about how variants in VGCC genes affect cellular processes to disrupt neurodevelopment.
A major impediment to understanding how autism-associated variants affect cellular processes is that most variants have a small effect size. Because each variant only has a small effect, it is thought that multiple variants engage in genetic interactions that give rise to the neurodevelopmental defects underlying autism [13, 14]. Therefore, a key goal in understanding the biological basis for autism is to understand genetic interactions between autism-associated variants. However, currently little is known about how autism-associated variants interact with each other. Moreover, in most cases the cellular mechanisms perturbed by each variant are also unknown.
Morphological abnormalities in axon development are associated with autism and other neurodevelopmental disorders [15–19]. Most mechanistic studies of autism-associated genes have focused on dendrite and synapse structure. However, imaging studies have suggested that alterations in axon targeting are a key feature of autism. For example, diffusion tensor imaging has revealed alterations in the Inferior Longitudinal Fasciculus in autistic individuals relative to healthy controls [15–19]. Moreover, functional MRI has revealed alterations in long range connectivity that can predict autism in individuals before the onset of symptoms [20–22]. These observations suggest that alterations in axon targeting are likely to underlie autism. However, little is currently known about how autism-associated variants can alter axon targeting.
In this work, we use the Timothy syndrome mutation as a platform to discover how autism-associated variants interact with each other to alter cellular processes and disrupt axon development. The Timothy syndrome mutation is actually a class of three rare de novo mutations in CACNA1C that encode either a G402R, G402S or G406R mutation in the CACNA1C protein. [10, 23]. Although the Timothy syndrome mutation is rare with large effect, common variants with small effect in CACNA1C are also associated with autism [1, 2]. As the mechanisms by which CACNA1C affects axon development are unknown, studies of the Timothy Syndrome variant may uncover genetic mechanisms that also apply to the more common CACNA1C risk variants.
Here, we investigate the role of egl-19, the C. elegans homolog of CACNA1C, in axon targeting. Like CACNA1C, EGL-19 is the pore-forming subunit for L-type voltage-gated calcium channels. We identify two egl-19 gain-of-function mutations that are equivalent to the Timothy syndrome G402R and G406R mutations. We find that each of these egl-19 gain-of-function mutations cause overextension of the PLM axon, leading to the misplacement of electrical synapses. Moreover, we find that egl-19 gain-of-function mutations interact genetically with a homolog of the autism-associated WDFY3 selective autophagy gene to disrupt axon development and alter behavior.
Results
A VGCC mutation that causes autism in humans and disrupts axon termination in C. elegans
Both common and rare variants in the human CACNA1C gene have been associated with autism and other neurodevelopmental disorders [1, 2, 8, 9, 12, 24–26]. The Timothy syndrome mutations, G402R, G402S and G406R in human CACNA1C, are of particular interest because each causes autism with high penetrance [10, 27, 28]. To establish a model for investigating how the Timothy syndrome mutations affect neurodevelopment, we searched for an equivalent mutation in egl-19, the C. elegans orthologue of CACNA1C. We found that the egl-19(n2368) mutation (hereafter referred to as egl-19(gof)) encodes a G365R variant of EGL-19 that is equivalent to the G402R Timothy syndrome variant in human CACNA1C (Fig 1A). In both human CACNA1C and C. elegans EGL-19, this Timothy syndrome mutation is a gain-of-function variant: it disrupts slow inactivation of the voltage-gated channel, thereby increasing calcium permeation [10].
Fig 1. Mutations equivalent to the G402R and G406R Timothy syndrome mutations cause PLM axon termination defects.
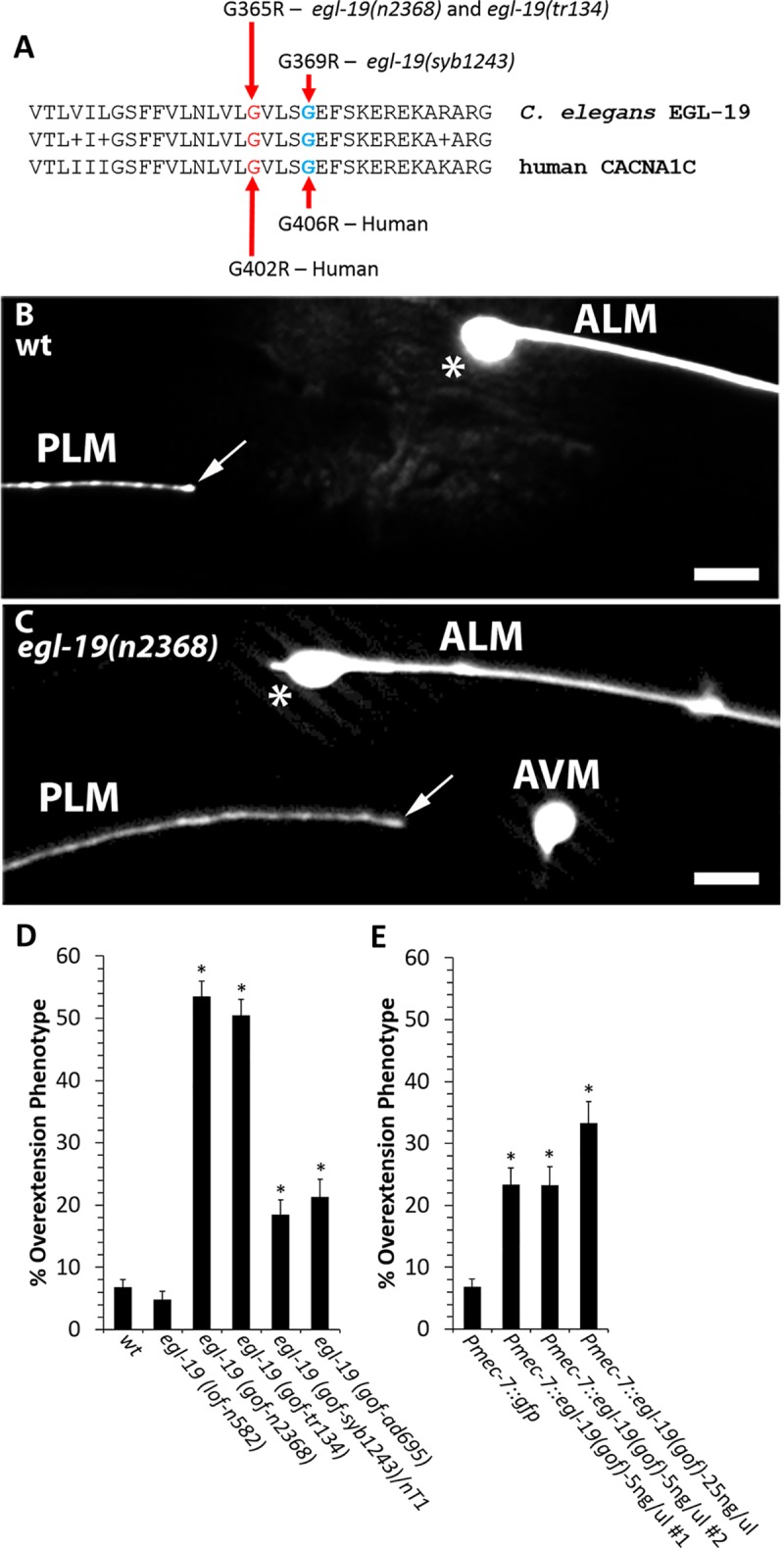
(A) The egl-19(n2368) and egl-19(tr134) mutations are equivalent to the Timothy Syndrome mutation in CACNA1C that causes autism in humans. The egl-19(syb1243) mutation is equivalent to the CACNA1C G406R mutation that causes Timothy Syndrome in humans. (B) Example of normal axon termination in a L4 stage wild-type PLM neuron, where the axon terminates posterior to the ALM cell body. (C) Example of axon termination defect in a L4 stage egl-19(gof) mutant, where the axon terminates anterior to the ALM cell body. Axons are visualized with the muIs32 transgene that encodes Pmec-7::gfp. Arrows point to the tip of the PLM axon. Asterisk marks the ALM cell body. Scales bar are 10um. (D) Gain-of-function mutations in egl-19 cause axon termination defects. The egl-19(lof) mutation does not affect axon termination. (E) Transgenic expression of EGL-19(GOF) specifically within touch receptor neurons causes axon termination defects. The Pmec-7::egl-19(gof) transgenes use the mec-7 promoter to drive expression of an egl-19 cDNA that includes a mutation identical to the egl-19(n2368) mutation. For Pmec-7::egl-19(gof)-5 ng/ul, 2 independent transgenic strains were analyzed. For Pmec-7::egl-19(gof)-25 ng/ul, 1 transgenic strain was analyzed. Between 200 and 400 axons were observed in L4 stage hermaphrodites per genotype. Asterisks indicate statistically significant difference, Z-test for proportions (***p<0.0001). Error bars represent the standard error of the proportion.
To determine how the Timothy syndrome mutation affects neurodevelopment, we observed the PLM touch receptor neuron in egl-19(gof) mutants. The PLM cell body is located in the tail, with an axon extending along the lateral body wall. For this experiment we used two independently isolated egl-19(gof) alleles, egl-19(n2368) and egl-19(tr134). Both of these alleles produce a G365R mutation in EGL-19, which is equivalent to the G402R Timothy syndrome mutation in humans (Fig 1A) [29, 30]. In wild-type animals, nearly all of the PLM axons terminate posterior to the ALM cell body (Fig 1B and 1D). However, in the egl-19(n2368) and egl-19(tr134) G365R gain-of-function mutants, around 52% of the PLM axons terminate anterior to the ALM cell body (Fig 1C and 1D). By contrast, egl-19(lof) mutants exhibit normal PLM axon termination (Fig 1D), with nearly all PLM axons terminating posterior to the ALM cell body. We also tested the egl-19(ad695) gain-of function mutation, which has previously been characterized as a weaker gain-of-function relative to egl-19(n2368) [30]. Consistent with these prior observations, we found that the egl-19(ad695) gain-of-function mutation causes axon termination defects with a lower penetrance relative to egl-19(n2368) and egl-19(tr134) (Fig 1D).
In humans, Timothy syndrome is also caused by a G406R mutation in CACNA1C [10]. Therefore, we tested egl-19(syb1243), a mutation that produces a G369R mutation in EGL-19, which is equivalent to G406R in human CACNA1C (Fig 1A). We found that the egl-19(syb1243) mutation is homozygous lethal, with no maternal rescue. However, we observed the PLM axon in egl-19(syb1243) heterozygotes and found that around 18% of the PLM axons had axon termination defects (Fig 1D). Together, these observations indicate that mutations equivalent to the Timothy syndrome mutations can cause defects in axon targeting.
To determine if the Timothy syndrome mutation functions cell autonomously to disrupt axon termination, we used Pmec-7::egl-19(gof) transgenes to express the EGL-19 G365R mutant protein specifically within touch receptor neurons (PLM, ALM, AVM and PVM) (Fig 1E). We tested one transgene that was created by injecting Pmec-7::egl-19(gof) at a concentration of 5 ng/ul and another at 25 ng/ul. We found that both of these transgenes caused axon termination defects. These results suggest that EGL-19(GOF) functions cell autonomously in the PLM neuron to disrupt axon termination.
Wild-type EGL-19 and other VGCC subunits negatively regulate axon termination
To determine if and how wild-type EGL-19 regulates axon termination, we conducted genetic analysis with mutations in the genes that encode the RPM-1 (PAM, Highwire) signaling pathway. RPM-1 is an E3 ubiquitin ligase that promotes axon termination by ubiquitinating DLK-1 (MAP3K12), thereby marking it for proteasomal degradation [31–33]. This function of RPM-1 is mediated through an interaction between RPM-1 and a SCF (Skp/Cullin/F-box) complex that includes the FSN-1 (FBXO45) F-box protein. In addition, RPM-1 also promotes axon termination by functioning with GLO-4 (RCBTB1), a guanine nucleotide exchange factor (GEF) for the GLO-1 (RAB38) Rab GTPase [34]. GLO-4 functions with RPM-1, but in parallel to FSN-1, to promote axon termination.
Since the egl-19(gof) mutation disrupts axon termination, it is possible that wild-type EGL-19 also negatively regulates axon termination. Alternatively, it is possible that egl-19(gof) acts in a neomorphic role not normally controlled by wild-type EGL-19. To determine the function of wild-type EGL-19, we conducted genetic analysis with a loss-of-function allele of egl-19. For this experiment, we used a null allele of the fsn-1 that causes axon termination defects [34, 35]. In fsn-1(null) mutants, 53% of the PLM axons are overextended (Fig 2A). However, in fsn-1(null); egl-19(lof) double mutants, the phenotype is suppressed to only 33% of PLM axons overextended. These observations suggest that wild-type EGL-19 acts to negatively regulate axon termination.
Fig 2. VGCC genes negatively regulate axon termination.

(A) Loss-of-function mutations in VGCC genes suppress axon termination defects caused by the fsn-1(null) mutation. The egl-19 gene encodes the pore-forming subunit of the L-type VGCC. The unc-2 gene encodes the pore-forming subunit of the P/Q-type VGCC and the unc-36 gene encodes the Alpha2-Delta3 subunit that works with both the L-type and P/Q-type VGCCs. (B) Mutations in VGCC genes do not suppress axon termination defects caused by loss-of-function mutations in glo-4 or rpm-1. (C) Loss of VGCC function can partially suppress axon termination defects caused by the fsn-1(null); glo-4(null) double mutant background. Asterisks indicate statistically significant difference, Z-test for proportions (*p<0.005, ***p<0.0001). Between 200 and 400 axons were observed in L4 stage hermaphrodites per genotype using the muIs32 transgene. Error bars represent the standard error of the proportion. For Pmec-7::unc-36::rfp, 2 independent transgenic strains were analyzed. Alleles: fsn-1(null) is fsn-1(gk429), egl-19(lof) is egl-19(n582), unc-2(null) is unc-2(e55), unc-36(null) is unc-36(e251), glo-4(null) is glo-4(ok362), rpm-1(lof) is rpm-1(ok364).
EGL-19 is the pore forming subunit of the L-type VGCC. To further explore the role of voltage-gated calcium channels, we tested loss-of-function mutations in other genes that encode VGCC components. The unc-2 gene encodes the pore-forming subunit of the P/Q-type VGCC [36]. The human homolog of unc-2, CACNA1A, has also been associated with autism [1, 37, 38]. We found that a null allele of unc-2 could also suppress axon termination defects caused by a fsn-1(null) mutation (Fig 2A). We also tested a null mutation in unc-36, which encodes the alpha2-delta3 subunit that works with both the EGL-19 and UNC-2 pore-forming subunits to modulate voltage dependence, activation kinetics, and calcium conductance [39–41]. The human homolog of UNC-36, CACNA2D3 has also been associated with autism [6, 8, 42]. We found that a null allele of unc-36 could also suppress axon termination defects caused by a fsn-1(null) mutation (Fig 2A). Together, these observations suggest that both L-type and P/Q-type voltage-gated calcium channels can negatively regulate axon termination. We note that the different levels of suppression observed between unc-36(null), unc-2(null) and egl-19(lof) mutations are likely the result of differing roles played by each VGCC subunit or could reflect different strengths of the alleles that we used.
To determine if VGCCs function cell-autonomously to regulate axon termination, we constructed a Pmec-7::unc-36::rfp transgene, which uses the mec-7 promoter to drive expression of UNC-36::RFP within the touch receptor neurons. If UNC-36 functions cell autonomously, we expect that the Pmec-7::unc-36::rfp transgene will reverse the suppression of axon termination defects observed in fsn-1(null);unc-36(null) double mutants relative to fsn-1(null) single mutants. Indeed, we found that fsn-1(null);unc-36(null) double mutants with the Pmec-7::unc-36::rfp transgene had a higher penetrance of axon termination defects relative to fsn-1(null);unc-36(null) double mutants without the Pmec-7::unc-36::rfp transgene (Fig 2A). These observations suggest that UNC-36 functions cell-autonomously to negatively regulate axon termination.
VGCC regulation of axon termination is specific to the FSN-1 pathway
FSN-1 functions in parallel to GLO-4 to promote PLM axon termination [34, 43]. Although both pathways promote axon termination, they do so through distinct molecular mechanisms. Whereas FSN-1 is an F-box protein that regulates a MAP Kinase cascade [35], GLO-4 is a guanine nucleotide exchange factor for the GLO-1 Rab GTPase [43]. To determine if VGCCs regulate the GLO-4 pathway, we constructed glo-4(null);unc-2(null) and glo-4(null);unc-36(null) double mutants. We found that neither loss of unc-2 nor loss of unc-36 function suppresses the axon termination defects caused by loss of glo-4 function, suggesting that VGCCs do not regulate the GLO-4 pathway (Fig 2B). To further explore the role of VGCCs within the context of the parallel FSN-1 and GLO-4 pathways, we constructed an fsn-1(null);glo-4(null);unc-36(null) triple mutant and a fsn-1(null);glo-4(null) double mutant (Fig 2C). Consistent with prior studies, we found that fsn-1(null);glo-4(null) double mutants had termination defects in 86% of PLM axons [34]. In the triple mutant, loss of unc-36 function reduced this penetrance to 46%, which is similar to the glo-4 single mutants. Together, these observations suggest that VGCCs can negatively regulate axon termination in response to the FSN-1 pathway, but not GLO-4 pathway.
The RPM-1 ubiquitin ligase functions with the FSN-1 F-box protein to negatively regulate downstream proteins [34, 35]. Loss-of-function mutations in these downstream proteins suppress the phenotype of loss-of-function mutations in FSN-1 and RPM-1. For example, FSN-1 and RPM-1 function together to negatively regulate the DLK-1 MAP Kinase [31]. Loss of DLK-1 function suppresses the phenotype that is caused by either loss of RPM-1 function or loss of FSN-1 function. Since loss of VGCC function can suppress the axon termination phenotype caused by loss of FSN-1 function, we considered the possibility that FSN-1 might function with RPM-1 to negatively regulate VGCCs. If this is true, loss of VGCC function should suppress the axon termination defect caused by loss of RPM-1 function. However, we found that axon termination defects caused by loss of RPM-1 function could not be suppressed by loss of function mutations in unc-36, unc-2 or egl-19 (Fig 2B). These observations suggest that VGCCs are not downstream targets of RPM-1 and FSN-1.
The egl-19(gof) mutation alters PLM axon connectivity
To determine if the egl-19(gof) mutation affects connectivity of the PLM axon, we examined its chemical and electrical synapses. In wild-type animals, the PLM axon extends a synaptic branch that forms a cluster of chemical synapses onto axons in the ventral nerve cord [32]. We used a mec-7::rfp transgene [44] to visualize the PLM synaptic branch and found that it appears normal in egl-19(gof) mutants (Fig 3A and 3B). We also used a mec-7::gfp::rab-3 transgene [45] to visualize synaptic vesicles and found that these also appear normal in egl-19(gof) mutants (Fig 3A and 3B). We measured the length of the synaptic vesicle clusters in wildtype and egl-19(gof) mutants and found no significant difference (t-test, p>0.05): egl-19(gof) = 4.7±0.11μm (n = 50); wildtype = 5.09±0.18μm (n = 50). Moreover, consistent with prior findings [34], we found that about 15% of fsn-1 null mutants were missing the PLM branch (Fig 3C). However, this missing branch phenotype was not suppressed in fsn-1(null); egl-19(lof) double mutants (Fig 3C). These observations suggest that the EGL-19(GOF) mutation does not affect the PLM synaptic branch or its chemical synapses. Moreover, wildtype EGL-19 does not affect the PLM branch. However, we cannot rule out the possibility that the synaptic branch and chemical synapses are affected in more subtle ways.
Fig 3. The egl-19(gof) Timothy syndrome mutation causes mislocalization of PLM electrical synapses.

(A) Example of normal PLM chemical synapse in a wild-type animal. The PLM axon and branch are labeled in red. (B) Example of chemical synapse in a egl-19(gof) mutant. In A and B, the axon and its synaptic branch are shown in red as visualized with the jsls973 transgene that encodes Pmec-7::rfp. The chemical synapse is shown in green as visualized by the jsls821 transgene encodes Pmec-7::gfp::rab-3. Arrowheads mark the synaptic vesicles, which appear yellow due to overlap with the red signal. (C) The PLM synaptic branch is unaffected by changes in egl-19 function. The egl-19(gof) mutation does not cause defects in the PLM synaptic branch. The egl-19(lof) mutation does not suppress the PLM synaptic branch defect caused by the fsn-1(null) mutation. Between 200 and 400 synaptic branches were observed in L4 stage hermaphrodites per genotype using the muIs32 transgene. Error bars represent the standard error of the proportion. (D) Example of zone 2 electrical synapses at the tip of the PLM axon in wildtype animals. (E) Example of zone 2 electrical synapses at the tip of the PLM axon in egl-19(gof) mutants. The egl-19(gof) mutation causes mislocalization of PLM zone 2 electrical synapses. The zone 2 electrical synapses should be localized posterior to the ALM cell body. However, in egl-19(gof) mutants, the zone 2 electrical synapses are aberrantly localized anterior to the ALM cell body. Electrical synapses were visualized with the yadIs12 transgene that encodes Pmec-4::gfp::unc-9 [46]. Arrow marks electrical synapses at the tip of the PLM axon. Asterisk marks the ALM cell body. Scalebars are 10um. Alleles: egl-19(gof) is egl-19(n2368), fsn-1(null) is fsn-1(gk429), egl-19(lof) is egl-19(n582).
We next asked if the egl-19(gof) mutation affects electrical synapses. In wild-type PLM axons, electrical synapses are clustered in two distinct zones [46]. Zone 1 electrical synapses are located close to the PLM cell body, whereas zone 2 electrical synapses are located at the PLM axon tip, which is posterior to the ALM cell body (Fig 3D). Since the egl-19(gof) mutation causes overextension of the PLM axon, it is possible that it could also cause misplacement of zone 2 electrical synapses to a location anterior to the ALM cell body. Alternatively, it is possible that egl-19(gof) causes axon overextension, but leaves the zone 2 synapses in their normal location posterior to the ALM cell body. To differentiate between these two possibilities, we used a mec-7::unc-9::gfp transgene [46] to visualize the UNC-9 Innexin, a marker for electrical synapses [46]. We found that the egl-19(gof) mutation caused misplacement of the zone 2 electrical synapses to a point anterior to the ALM cell body (Fig 3E). Misplacement of the electrical synapse occurred in 52.9±7.0% of egl-19(gof) PLM axons, but only in 7.1±3.4% of wildtype PLM axons (n = 50 for both genotypes, significantly different by z-test for proportions, p<0.0001). Therefore, the PLM axon overextension caused by egl-19(gof) is also associated with misplacement of zone 2 electrical synapses.
The egl-19(gof) mutation interacts with selective autophagy genes to disrupt axon termination
As part of an ongoing effort to test autism-associated genes for roles in axon development, we identified a genetic interaction between egl-19(gof) and wdfy-3, a homolog of the autism-associated WDFY3 selective autophagy gene. For this experiment, we used the wdfy-3(ok912) deletion allele, hereafter called wdfy-3(lof). This allele is likely to be a null or strong loss of function because it creates a frameshift that disrupts the FYVE domain, WD repeat domain and nearly all of the beach domain. We found that the PLM axon was normal in wdfy-3(lof) single mutants. In egl-19(gof); wdfy-3(lof) double mutants, the axon termination defects observed in egl-19(gof) single mutants were almost completely suppressed (Fig 4A). Moreover, PLM axon termination defects caused by transgenic expression of EGL-19(GOF) in touch receptor neurons were also suppressed by wdfy-3(lof). The PLM neurons are likely to express WDFY-3 because RNAseq on purified touch receptor neurons identified wdfy-3 mRNA transcripts [47]. The wdfy-3 gene is an orthologue of the human autism-associated WDFY3 gene that encodes a protein required for cargo selection during selective autophagy [48, 49]. Therefore, these observations establish a genetic pathway between two autism-associated genes that regulates axon termination.
Fig 4. EGL-19(GOF) functions with selective autophagy to cause axon termination defects.

(A) Axon termination defects caused by the egl-19(gof) mutation are suppressed by loss-of-function mutations in wdfy-3(lof), epg-7(null) and cup-5(lof). (B) Axon termination defects caused by the fsn-1(null) mutation are suppressed by the wdfy-3(lof) mutation. However, axon termination defects caused by rpm-1(lof) mutation are not suppressed by the wdfy-3(lof) mutation. Axons are visualized with the muIs32 transgene that encodes Pmec-7::gfp. Asterisks indicate statistically significant difference, Z-test for proportions (*p<0.005, ***p<0.0001). Error bars represent the standard error of the proportion. n = 200–400 axons per genotype. Alleles: egl-19(gof) is egl-19(n2368), wdfy-3(lof) is wdfy-3(ok912), epg-7(null) is epg-7(tm2508), cup-5(lof) is cup-5(ar465), rpm-1(lof) is rpm-1(ok364), fsn-1(null) is fsn-1(gk429).
To further explore a potential interaction between selective autophagy and EGL-19(GOF), we constructed double mutants between egl-19(gof) and mutations in two other genes that are expected to disrupt selective autophagy: epg-7 (RB1CC1, FIP200) and cup-5 (Mucolipin-3). The epg-7 gene encodes an additional component required for selection of cargo for autophagy [50], and the cup-5 gene encodes a scaffold protein that promotes lysosome biogenesis [51, 52]. We found that axon termination defects caused by the egl-19(gof) mutation could be suppressed by either a likely null mutation in epg-7 or a hypomorphic mutation in cup-5 (Fig 4A). These observations suggest that the egl-19(gof) mutation causes axon termination through a mechanism that requires selective autophagy.
WDFY-3 negatively regulates PLM axon termination
Having found that wdfy-3 can negatively regulate axon termination in the egl-19(gof) mutant, we wanted to ask if wdfy-3 could also regulate axon termination independently of the gain-of-function egl-19 mutation. For this experiment, we used a fsn-1(null) mutation to induce axon termination defects. We found that loss of wdfy-3 function completely suppresses the axon termination defects caused by the fsn-1(null) mutation (Fig 4B). Like the VGCC loss-of-function mutants, we also found that the wdfy-3(lof) mutant does not suppress axon termination defects caused by rpm-1(lof). Together, these observations suggest that WDFY-3, like VGCCs, can regulate axon termination signaling downstream of FSN-1, but is not a downstream target of FSN-1 and RPM-1.
The egl-19(gof) mutation interacts with the wdfy-3 selective autophagy gene to regulate habituation to light touch
The PLM neuron is a mechanosensory neuron that is responsible for sensing light touch in the posterior of C. elegans [53]. When light touch is applied to the tail, the animal responds by moving forward [54–56]. However, after repeated touches, the animal habituates and becomes less likely to respond to each touch. Since we observed that the egl-19(gof) mutation alters the morphology of the PLM neuron, we wanted to determine if this mutation also alters the response to light touch.
We conducted a touch assay to determine if the egl-19(gof) mutation affects the response to light touch. Each animal was subjected to ten eyelash touches alternating between the head and tail (Fig 5). For the initial touch, there was no significant difference in the response rate between egl-19(gof) mutants and wild-type animals. However, for each subsequent touch, the egl-19(gof) mutants had a significantly lower response rate relative to wild-type animals. These observations suggest that the egl-19(gof) mutation enhances habituation to light touch.
Fig 5. The wdfy-3 gene functions with the egl-19(gof) mutation to alter habituation to light touch.

Animals were subjected to eyelash touches alternating between the head and tail and the response rate was recorded after each touch. (A) Response rate for anterior touches. (B) Response rate for posterior touches. For both anterior and posterior touches habituation was significantly increased in egl-19(gof) mutants relative to wild-type. This change in habituation in egl-19(gof) mutants was suppressed by wdfy-3(lof). Asterisks indicate statistically significant difference compared to wild type worms, Z-test for proportions (*p<0.0001). Error bars represent the standard error of the proportion. For each genotype, the assay was repeated 3 times by 3 different observers who were blind to the genotype. Each experiment included 20 worms for a total n of 60 for each genotype. Alleles: egl-19(gof) is egl-19(n2368), wdfy-3(lof) is wdfy-3(ok912).
Because we found that the egl-19(gof) mutation interacts with wdfy-3 to disrupt axon termination, we next asked if this genetic interaction can also affect habituation to light touch. If the egl-19(gof) mutation functions with wdfy-3 to alter habituation, we expect that loss of wdfy-3 function will reduce the effect of the egl-19(gof) mutation on habituation. Indeed, we found that in egl-19(gof);wdfy-3(lof) mutants, the habituation to light touch was not significantly different relative to wild-type animals. Together, these observations suggest that the egl-19(gof) mutation acts through wdfy-3 to affect both axon termination and habituation to light touch.
Discussion
The Timothy syndrome mutation in CACNA1C has the unusual property of being causative for autism with high penetrance, providing an opportunity to discover the downstream cellular processes that are perturbed to cause autism. However, the cellular processes that interact with this mutation to give rise to autism have remained unknown. To address this question, we created a disease model in C. elegans that utilizes a mutation equivalent to the Timothy syndrome mutation in humans. Our results reveal that selective autophagy genes interact with the Timothy syndrome mutation to disrupt axon termination and alter behavior. Because common variants of CACNA1C are associated with autism, it is likely that this mechanism will be broadly applicable to autism in humans.
An understanding of genetic interactions between variants is key to understanding autism
Only a small fraction of autism cases are thought to be caused by a single variant. Rather, most cases of autism are thought to be caused by genetic interactions between variants [9, 14, 57]. For example, likely gene-disrupting (LGD) mutations have been associated with 15–20% of autism cases. In addition, autistic individuals carry more missense mutations in autism-associated genes relative to healthy controls [9, 58].
However, both LGD and missense mutations are rare and therefore almost always heterozygous. Therefore, in most cases, it is thought that each of these mutations have little or no effect on their own. Thus, it is likely that the disorder arises from genetic interactions between autism-associated variants. Indeed, statistical analysis of sequencing data suggests that autism arises from the combined action of multiple variants [9, 14, 59].
Although the heritability of autism has been estimated at 83% [60], the complexity of the genetic interactions that give rise to autism make it difficult to predict and diagnose autism from whole genome sequencing data. In fact, with current knowledge, no genetic cause can be found from whole genome sequencing data for most cases of autism. The solution to this challenge could come from genetic analysis. For example, in most cases, single heterozygous null mutations have no phenotype. However, many cases exist to show that an animal carrying two heterozygous null mutations can exhibit a phenotype when each of the mutated genes function in the same genetic pathway [61, 62]. Thus, if an individual is heterozygous for two LGD variants in each of two autism-associated genes that function in a pathway, this individual would carry a higher risk for autism. Therefore, knowledge of the pathways that link autism-associated genes will help promote our understanding of the genetic basis of autism.
Selective autophagy functions with EGL-19(GOF) to alter axon development and behavior
A key finding of our study is the identification of a genetic interaction between the homologs of two autism-associated genes, egl-19 and wdfy-3. Our genetic analysis indicates that wdfy-3 and other selective autophagy genes are required for the egl-19(gof) mutation to disrupt axon termination. Moreover, we also find that wdfy-3 can negatively regulate axon termination independently of the egl-19(gof) mutation. These genetic interactions could be explained by a few different molecular models. First, it is possible that EGL-19 functions in a pathway with WDFY-3 to negatively regulate axon termination. Alternatively, it is possible that EGL-19 and WDFY-3 function in two separate parallel pathways that can both negatively regulate axon termination. We favor the former model, because both egl-19(lof) and wdfy-3(lof) are able to suppress axon termination defects caused by loss of fsn-1 function, but not by loss of rpm-1 function. Moreover, the strength of the suppression of the egl-19(gof) phenotype by the wdfy-3(null) mutation is most consistent with the idea that EGL-19 and WDFY-3 function together in a pathway. If EGL-19 and WDFY-3 do function together in a pathway, it is possible that the Timothy syndrome mutation induces excessive selective autophagy that causes a disruption of axon termination. As an alternative, it is also possible that selective autophagy could function upstream of the EGL-19(GOF) mutant protein. In this scenario, selective autophagy could promote the function of the EGL-19(GOF) protein by affecting its turnover, stability or localization.
The genetic interaction between egl-19 and wdfy-3 also regulates habituation to light touch. It is possible that this genetic interaction affects behavior by functioning in the developing nervous system to regulate connectivity. Alternatively, it is possible that the genetic interaction between egl-19 and wdfy-3 functions in the mature nervous system to regulate neural function. Although our data cannot distinguish between these two possibilities, recent work on mice favor the former possibility [63]. Conditional knockout of CACNA1C in forebrain neurons during development results in anxiety in adult mice, whereas knockout of CACNA1C during adulthood does not. These observations lend support to the possibility that CACNA1C acts during development to alter circuit formation, which in turn affects behavior in the adult.
The interaction between the Timothy syndrome mutation and wdfy-3 provides biological evidence for a role of selective autophagy in autism. A major challenge in autism genetics is to confirm and characterize the roles of autism candidate genes. For example, WDFY3 is a candidate gene for autism because whole genome sequencing has found that 3 out of 6707 sequenced autism genomes contain a heterozygous de novo likely-gene-disrupting mutation in WDFY3 [6, 8, 42, 64]. However, despite this association, it is not possible to determine if WDFY3 variants contribute to the cause of autism. Our results place genes that promote selective autophagy in a pathway with a mutation that is causative for autism in humans, thereby providing the first biological evidence for a role of selective autophagy in autism.
Selective autophagy is also required for normal axon development. Aside from its function with EGL-19(GOF) in inducing axon defects, our results suggest that WDFY-3 also functions independently of EGL-19(GOF) to regulate axon development. Consistent with this idea, loss of WDFY3 causes the disorganization and loss of many commissural axon tracts in mice [65]. Loss of WDFY3 in mice also attenuates the response to guidance cues in vitro, suggesting that selective autophagy could regulate the response to guidance cues. Despite these insights, the mechanism through which selective autophagy regulates axon targeting is currently unknown. Based on our genetic analysis, we propose a mechanism whereby selective autophagy functions with voltage-gated calcium channels to regulate the response to axon targeting cues.
Selective autophagy and bulk autophagy may have distinct functions in neurodevelopment. Whereas our results suggest a mechanism for selective autophagy in the negative regulation of axon termination, prior studies have reported a role for bulk autophagy in promoting synapse development and inhibiting axon growth. In cultured mouse neurons, knockdown of an autophagy gene promotes axon growth, whereas induction of autophagy inhibits axon growth [66]. In Drosophila, autophagy promotes development of the neuromuscular junction [67]. In C. elegans, autophagosomes form at synaptic sites and are required for presynaptic assembly [68, 69]. This role for autophagy in synaptogenesis is specific to bulk autophagy, since mutations in selective autophagy genes do not affect synaptogenesis [69]. Interestingly, our results suggest that selective autophagy may regulate axon termination without affecting synaptogenesis.
Selective autophagy could function with VGCCs to regulate other aspects of autism-related pathology. Whereas our study focuses on the role of the Timothy syndrome mutation in misregulating axon termination and behavior, previous work has found that the Timothy syndrome mutation can promote activity-dependent dendrite retraction in cultured mouse neurons and can inhibit the elaboration of mouse dendrites in vivo [70]. The downstream cellular mechanisms for this effect on dendrites are not yet known, but it is possible that selective autophagy could also be involved in this process. Alternatively, it is possible that the Timothy syndrome mutation functions through distinct mechanisms to affect dendrite development and axon development.
Potential role for VGCCs in regulating the RPM-1 pathway
Our results identify specific genetic interactions between VGCC genes and RPM-1 pathway genes. These genetic interactions indicate that loss of VGCC function suppresses axon termination events that are caused by loss of fsn-1 function, but not loss of glo-4 function. Moreover, we find that loss of VGCC function does not suppress defects caused by loss of rpm-1 function, but can partially suppress defects caused by the double loss of fsn-1 and glo-4. Taken together, these observations suggest that VGCCs specifically regulate axon termination signaling downstream of FSN-1, but are not themselves downstream targets of RPM-1 and FSN-1.
The genetic interactions between the VGCC genes and RPM-1 pathway genes could be explained by a model where VGCCs negatively regulate an unknown protein that functions with RPM-1 to enhance signaling events that promote axon termination downstream of FSN-1, but not GLO-4 (S1 Fig). In fact, prior work has found that FSN-1 binds to RPM-1 and promotes axon termination by negatively regulating the DLK-1 MAP kinase signaling pathway [31, 35]. Moreover, the PPM-1 phosphatase also binds to RPM-1 and promotes axon termination by negatively regulating DLK-1 MAPK signaling [71]. Thus, it is possible that EGL-19 might repress axon termination by negatively regulating PPM-1, or another protein that plays a similar role (S2 Fig).
Role for VGCC-mediated calcium transients in axon growth
Our study focuses on the genetic mechanisms that mediate the role of VGCC genes in axon termination, but does not address how alterations in calcium permeation might be involved in this process. Interestingly, a recent study of cultured prenatal mouse neurons has revealed that VGCCs function during axon outgrowth to produce calcium transients that have very different properties compared to those produced during synaptic transmission [72]. These transients have been named Spontaneous Regenerative Calcium Transients (SRCaTs) and are mediated by CaV1.2, which includes the homolog of EGl-19, CACNA1C. Unlike its function in adult neurons, CaV1.2 appears to open near resting potential, suggesting that the CaV1.2 channel may open spontaneously in developing axons. Knockout of CACNA1C in these cultured neurons causes a decrease in axon growth. Thus, CaV1.2 functions in axons to regulate axon growth, using a mechanism that is very different than how it functions in synaptic transmission. The role of SRCaTs in regulating axon growth are unknown. However, our results suggest the possibility that these SRCaTs may regulate signaling downstream of FSN-1 (FBXO45), utilizing a mechanism that involves selective autophagy.
Potential role for common CACNA1C variants in affecting selective autophagy and axon development in autism
We propose that the effect of CACNA1C variation in altering axon development is not limited to the Timothy syndrome mutation, but rather extends to the other autism-associated CACNA1C variants. This idea is supported by our genetic analysis suggesting that the effect of the egl-19(gof) Timothy syndrome mutation on axon termination is not neomorphic, but rather reflects an increase in the normal function of egl-19. Therefore, other gain-of-function and loss-of-function variants in CACNA1C could contribute to autism by altering axon development. Consistent with this idea, statistical analysis has identified some candidate variants in VGCC genes that are likely to be gain-of-function and others that are likely to be loss-of-function [6, 10, 11, 42, 73]. Therefore, we speculate that both under-activation and over-activation of the signaling pathways that promote axon termination could contribute to autism.
The Timothy syndrome mutation is a very rare de novo mutation, and is therefore only responsible for a very tiny fraction of autism cases. However, several common variants in CACNA1C have also been associated with autism [1, 2, 74]. For example, the A genotype at the rs1006737 locus in CACNA1C confers risk for autism and is present in about 33% of the human population. This A genotype at rs1006737 is located within a large intron and is thought to cause CACNA1C gain-of-function because neurons with the risk genotype have higher levels of CACNA1C mRNA and increased L-type calcium currents relative to neurons with the non-risk genotype [75]. Therefore, this risk variant may be associated with a gain-of-function of CACNA1C that could disrupt axon development in a way analogous to the Timothy syndrome mutation. However, the small effect size of the rs1006737 locus suggests that this is a relatively weak CACNA1C gain-of-function.
Although common alleles have a small effect size relative to the Timothy syndrome mutation, they could interact with risk variants in other genes that function in a genetic pathway with CACNA1C. For example, the rs1006737 risk variant could provide a weak gain-of-function in CACNA1C that does not cause autism on its own. However, the rs1006737 risk variant could synergize with a gain-of-function risk variant in WDFY3 to contribute to autism. Alternatively, a weak loss of function in CACNA1C could synergize with a weak loss-of-function in WDFY3 to give rise to autism.
Methods
C. elegans genetics
C. elegans strains were cultured and maintained on nematode growth medium (NGM)-agar plates using standard methods at 20°C (Brenner, 1974). The following alleles were used in this study: wild-type N2, rpm-1(ok364), glo-4(ok362), fsn-1(gk429), unc-2(e55), unc-36(e251), egl-19(n2368), egl-19(n582), egl-19(syb1243), cup-5(ar465), epg-7(tm2508), wdfy-3(ok912). Unless otherwise noted, double and triple mutants were constructed following standard procedures, and were confirmed by the associated phenotypes and by PCR/sequence genotyping.
Transgenic fluorescent markers
The muIs32 transgene was obtained from the CGC and encodes Pmec-7::gfp + lin-15(+) [76] and was used to observe the PLM axon. The jsls973 and jsls821 transgenes were obtained from Michael Nonet. The jsls973 transgene encodes Pmec-7::rfp [45] and was used to observe the PLM axon. The jsls821 transgene encodes Pmec-7::gfp::rab-3 [44] and was used to observe the localization of chemical synapses in the PLM axon. The yadIs12 transgene was obtained from Dong Yan and encodes Pmec-4::GFP::unc-9 [46] and was used to observe electrical synapses in the PLM axon. The egl-19(syb1243) mutation was obtained from SunyBiotech. The cueEx17 and cueEx18 transgenes were created by injecting Pmec-7::unc-36::rfp at 5 ng/ul + Pstr-1::gfp at 50 ng/ul. The cueEx19 and cueEx20 transgenes were created by injecting Pmec-7::egl-19(gof) at 5 ng/ul + Podr-1::rfp at 50 ng/ul. The cueEx21 transgene was created by injecting Pmec-7::egl-19(gof) at 25 ng/ul + Podr-1::rfp at 50 ng/ul.
Analysis of phenotypes
For analysis of axon termination phenotypes, animals were mounted on a 5% agarose pad and observed with a 40x objective. For PLM axon termination, an axon was scored as defective if it grew anterior to the ALM cell body. PLM neurons were visualized with the muIs32 transgene which encodes Pmec-7::gfp and is expressed in all mechanosensory neurons.
For analysis of the PLM chemical synapses, a Pmec-7::gfp::rab-3 transgene that expresses the RAB-3 synaptic vesicle marker in the touch receptor neurons was used to visualize synaptic vesicle clusters [44]. The size of each synaptic cluster was measured as previously described [77]. For analysis of PLM electrical synapses, a Pmec-4::gfp::unc-9 transgene was used to express the UNC-9 innexin fused to GFP in the touch receptor neurons [46].
For analysis of mechanosensation, we adopted an eyelash touch assay [78]. We assayed gentle touch responses by touching the lateral side of animals with an eyebrow hair. Each animal was subjected to five touches alternating between the anterior and posterior ends and scored by the number of responses elicited. Assays were performed blind to genotype. Three independent samples of 20 animals each were collected by three independent observers and reported as mean percentage scores.
Ethics statement
Because C. elegans are invertebrate animals they do not require review by an ethics committee.
Supporting information
This hypothetical model could explain the genetic interactions observed between VGCC genes and genes that encode members of the RPM-1 pathway. In this model, VGCCs function with WDFY-3 to negatively regulate an unknown protein (labeled as X). Protein X functions with RPM-1 to promote signaling events downstream of FSN-1 that promote axon termination. Loss of VGCC function causes an increase in protein X function. The additional protein X function works with RPM-1 to promote axon termination mechanism B, thereby compensating for loss of FSN-1 function. Loss of VGCCs does not suppress loss of RPM-1 function because the function of protein X requires RPM-1.
(TIF)
It is possible that PPM-1, or another protein with a similar role, could be protein X (see S1 Fig). Both FSN-1 and PPM-1 promote axon termination by functioning with RPM-1 to negatively regulate the DLK-1 MAP kinase pathway. Thus, it is possible that VGCCs and WDFY-3 could negatively regulate PPM-1, or a protein with a similar role. The extra PPM-1 function could enhance negative regulation of the DLK-1 pathway, thereby compensating for loss of FSN-1 function.
(TIF)
Acknowledgments
We thank Shohei Mitani, Michael Nonet, Dong Yan, Peter Roy and the Caenorhabditis Genetics Center for strains and reagents. We also thank Claire de la Cova for discussion of the manuscript.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
Research reported in this publication was supported by the National Institute Of Mental Health of the National Institutes of Health (NIH) under Award Number R01MH119157 to CCQ. Additional funding was provided by the National Institute of Neurological Disorders and Stroke of the NIH under Award Number R03NS101524 to CCQ. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional funding came from a Research Growth Initiative grant #101X356 from the University of Wisconsin-Milwaukee to CCQ, and a Shaw Scientist Award from the Greater Milwaukee Foundation to CCQ. The Caenorhabditis Genetics Center was funded by NIH P40 OD010440. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Li J, Zhao L, You Y, Lu T, Jia M, Yu H, et al. Schizophrenia Related Variants in CACNA1C also Confer Risk of Autism. PLoS One. 2015;10(7):e0133247 10.1371/journal.pone.0133247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu AT, Dai X, Martinez-Agosto JA, Cantor RM. Support for calcium channel gene defects in autism spectrum disorders. Mol Autism. 2012;3(1):18 10.1186/2040-2392-3-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvarez-Mora MI, Calvo Escalona R, Puig Navarro O, Madrigal I, Quintela I, Amigo J, et al. Comprehensive molecular testing in patients with high functioning autism spectrum disorder. Mutat Res. 2016;784–785:46–52. 10.1016/j.mrfmmm.2015.12.006 . [DOI] [PubMed] [Google Scholar]
- 4.Brett M, McPherson J, Zang ZJ, Lai A, Tan ES, Ng I, et al. Massively parallel sequencing of patients with intellectual disability, congenital anomalies and/or autism spectrum disorders with a targeted gene panel. PLoS One. 2014;9(4):e93409 10.1371/journal.pone.0093409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D'Gama AM, Pochareddy S, Li M, Jamuar SS, Reiff RE, Lam AN, et al. Targeted DNA Sequencing from Autism Spectrum Disorder Brains Implicates Multiple Genetic Mechanisms. Neuron. 2015;88(5):910–7. 10.1016/j.neuron.2015.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–21. 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2013;93(2):249–63. 10.1016/j.ajhg.2013.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.RK CY, Merico D, Bookman M, J LH, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20(4):602–11. 10.1038/nn.4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaaf CP, Sabo A, Sakai Y, Crosby J, Muzny D, Hawes A, et al. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum Mol Genet. 2011;20(17):3366–75. 10.1093/hmg/ddr243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31. 10.1016/j.cell.2004.09.011 . [DOI] [PubMed] [Google Scholar]
- 11.Breitenkamp AF, Matthes J, Nass RD, Sinzig J, Lehmkuhl G, Nurnberg P, et al. Rare mutations of CACNB2 found in autism spectrum disease-affected families alter calcium channel function. PLoS One. 2014;9(4):e95579 10.1371/journal.pone.0095579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strom SP, Stone JL, Ten Bosch JR, Merriman B, Cantor RM, Geschwind DH, et al. High-density SNP association study of the 17q21 chromosomal region linked to autism identifies CACNA1G as a novel candidate gene. Mol Psychiatry. 2010;15(10):996–1005. 10.1038/mp.2009.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson EB, St Pourcain B, Anttila V, Kosmicki JA, Bulik-Sullivan B, Grove J, et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat Genet. 2016;48(5):552–5. 10.1038/ng.3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turner TN, Coe BP, Dickel DE, Hoekzema K, Nelson BJ, Zody MC, et al. Genomic Patterns of De Novo Mutation in Simplex Autism. Cell. 2017;171(3):710–22 e12. 10.1016/j.cell.2017.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farias GG, Guardia CM, De Pace R, Britt DJ, Bonifacino JS. BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc Natl Acad Sci U S A. 2017;114(14):E2955–E64. 10.1073/pnas.1616363114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koldewyn K, Yendiki A, Weigelt S, Gweon H, Julian J, Richardson H, et al. Differences in the right inferior longitudinal fasciculus but no general disruption of white matter tracts in children with autism spectrum disorder. Proc Natl Acad Sci U S A. 2014;111(5):1981–6. 10.1073/pnas.1324037111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lazar M, Miles LM, Babb JS, Donaldson JB. Axonal deficits in young adults with High Functioning Autism and their impact on processing speed. Neuroimage Clin. 2014;4:417–25. 10.1016/j.nicl.2014.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Travers BG, Adluru N, Ennis C, Tromp do PM, Destiche D, Doran S, et al. Diffusion tensor imaging in autism spectrum disorder: a review. Autism Res. 2012;5(5):289–313. 10.1002/aur.1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolff JJ, Gu H, Gerig G, Elison JT, Styner M, Gouttard S, et al. Differences in white matter fiber tract development present from 6 to 24 months in infants with autism. Am J Psychiatry. 2012;169(6):589–600. 10.1176/appi.ajp.2011.11091447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Just MA, Cherkassky VL, Keller TA, Kana RK, Minshew NJ. Functional and anatomical cortical underconnectivity in autism: evidence from an FMRI study of an executive function task and corpus callosum morphometry. Cereb Cortex. 2007;17(4):951–61. 10.1093/cercor/bhl006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Just MA, Cherkassky VL, Keller TA, Minshew NJ. Cortical activation and synchronization during sentence comprehension in high-functioning autism: evidence of underconnectivity. Brain. 2004;127(Pt 8):1811–21. 10.1093/brain/awh199 . [DOI] [PubMed] [Google Scholar]
- 22.Schipul SE, Keller TA, Just MA. Inter-regional brain communication and its disturbance in autism. Front Syst Neurosci. 2011;5:10 10.3389/fnsys.2011.00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bader PL, Faizi M, Kim LH, Owen SF, Tadross MR, Alfa RW, et al. Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proc Natl Acad Sci U S A. 2011;108(37):15432–7. 10.1073/pnas.1112667108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Mubarak B, Abouelhoda M, Omar A, AlDhalaan H, Aldosari M, Nester M, et al. Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: a trio study from Saudi families. Sci Rep. 2017;7(1):5679 10.1038/s41598-017-06033-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stessman HA, Xiong B, Coe BP, Wang T, Hoekzema K, Fenckova M, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet. 2017;49(4):515–26. 10.1038/ng.3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21(2):185–91. 10.1038/nm.3792 . [DOI] [PubMed] [Google Scholar]
- 27.Diep V, Seaver LH. Long QT syndrome with craniofacial, digital, and neurologic features: Is it useful to distinguish between Timothy syndrome types 1 and 2? Am J Med Genet A. 2015;167A(11):2780–5. 10.1002/ajmg.a.37258 . [DOI] [PubMed] [Google Scholar]
- 28.Napolitano C, Antzelevitch C. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac voltage-dependent L-type calcium channel. Circ Res. 2011;108(5):607–18. 10.1161/CIRCRESAHA.110.224279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwok TC, Hui K, Kostelecki W, Ricker N, Selman G, Feng ZP, et al. A genetic screen for dihydropyridine (DHP)-resistant worms reveals new residues required for DHP-blockage of mammalian calcium channels. PLoS Genet. 2008;4(5):e1000067 Epub 2008/05/10. 10.1371/journal.pgen.1000067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee RY, Lobel L, Hengartner M, Horvitz HR, Avery L. Mutations in the alpha1 subunit of an L-type voltage-activated Ca2+ channel cause myotonia in Caenorhabditis elegans. EMBO J. 1997;16(20):6066–76. Epub 1997/10/08. 10.1093/emboj/16.20.6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, et al. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell. 2005;120(3):407–20. 10.1016/j.cell.2004.12.017 . [DOI] [PubMed] [Google Scholar]
- 32.Schaefer AM, Hadwiger GD, Nonet ML. rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron. 2000;26(2):345–56. 10.1016/s0896-6273(00)81168-x . [DOI] [PubMed] [Google Scholar]
- 33.Zhen M, Huang X, Bamber B, Jin Y. Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron. 2000;26(2):331–43. 10.1016/s0896-6273(00)81167-8 . [DOI] [PubMed] [Google Scholar]
- 34.Grill B, Bienvenut WV, Brown HM, Ackley BD, Quadroni M, Jin Y. C. elegans RPM-1 regulates axon termination and synaptogenesis through the Rab GEF GLO-4 and the Rab GTPase GLO-1. Neuron. 2007;55(4):587–601. 10.1016/j.neuron.2007.07.009 . [DOI] [PubMed] [Google Scholar]
- 35.Liao EH, Hung W, Abrams B, Zhen M. An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature. 2004;430(6997):345–50. 10.1038/nature02647 . [DOI] [PubMed] [Google Scholar]
- 36.Schafer WR, Kenyon CJ. A calcium-channel homologue required for adaptation to dopamine and serotonin in Caenorhabditis elegans. Nature. 1995;375(6526):73–8. 10.1038/375073a0 . [DOI] [PubMed] [Google Scholar]
- 37.Damaj L, Lupien-Meilleur A, Lortie A, Riou E, Ospina LH, Gagnon L, et al. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet. 2015;23(11):1505–12. 10.1038/ejhg.2015.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lelieveld SH, Reijnders MR, Pfundt R, Yntema HG, Kamsteeg EJ, de Vries P, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19(9):1194–6. 10.1038/nn.4352 . [DOI] [PubMed] [Google Scholar]
- 39.Frokjaer-Jensen C, Kindt KS, Kerr RA, Suzuki H, Melnik-Martinez K, Gerstbreih B, et al. Effects of voltage-gated calcium channel subunit genes on calcium influx in cultured C. elegans mechanosensory neurons. J Neurobiol. 2006;66(10):1125–39. 10.1002/neu.20261 . [DOI] [PubMed] [Google Scholar]
- 40.Saheki Y, Bargmann CI. Presynaptic CaV2 calcium channel traffic requires CALF-1 and the alpha(2)delta subunit UNC-36. Nat Neurosci. 2009;12(10):1257–65. 10.1038/nn.2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laine V, Frokjaer-Jensen C, Couchoux H, Jospin M. The alpha1 subunit EGL-19, the alpha2/delta subunit UNC-36, and the beta subunit CCB-1 underlie voltage-dependent calcium currents in Caenorhabditis elegans striated muscle. J Biol Chem. 2011;286(42):36180–7. Epub 2011/09/01. 10.1074/jbc.M111.256149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–15. 10.1038/nature13772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hermann GJ, Schroeder LK, Hieb CA, Kershner AM, Rabbitts BM, Fonarev P, et al. Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Mol Biol Cell. 2005;16(7):3273–88. 10.1091/mbc.E05-01-0060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bounoutas A, Zheng Q, Nonet ML, Chalfie M. mec-15 encodes an F-box protein required for touch receptor neuron mechanosensation, synapse formation and development. Genetics. 2009;183(2):607–17, 1SI-4SI. Epub 2009/08/05. 10.1534/genetics.109.105726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marcette JD, Chen JJ, Nonet ML. The Caenorhabditis elegans microtubule minus-end binding homolog PTRN-1 stabilizes synapses and neurites. Elife. 2014;3:e01637 Epub 2014/02/27. 10.7554/eLife.01637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meng L, Chen CH, Yan D. Regulation of Gap Junction Dynamics by UNC-44/ankyrin and UNC-33/CRMP through VAB-8 in C. elegans Neurons. PLoS Genet. 2016;12(3):e1005948 10.1371/journal.pgen.1005948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaletsky R, Lakhina V, Arey R, Williams A, Landis J, Ashraf J, et al. The C. elegans adult neuronal IIS/FOXO transcriptome reveals adult phenotype regulators. Nature. 2016;529(7584):92–6. Epub 2015/12/18. 10.1038/nature16483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy. 2010;6(3):330–44. 10.4161/auto.6.3.11226 . [DOI] [PubMed] [Google Scholar]
- 49.Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell. 2010;38(2):265–79. 10.1016/j.molcel.2010.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin L, Yang P, Huang X, Zhang H, Lu Q, Zhang H. The scaffold protein EPG-7 links cargo-receptor complexes with the autophagic assembly machinery. J Cell Biol. 2013;201(1):113–29. 10.1083/jcb.201209098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hersh BM, Hartwieg E, Horvitz HR. The Caenorhabditis elegans mucolipin-like gene cup-5 is essential for viability and regulates lysosomes in multiple cell types. Proc Natl Acad Sci U S A. 2002;99(7):4355–60. 10.1073/pnas.062065399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Treusch S, Knuth S, Slaugenhaupt SA, Goldin E, Grant BD, Fares H. Caenorhabditis elegans functional orthologue of human protein h-mucolipin-1 is required for lysosome biogenesis. Proc Natl Acad Sci U S A. 2004;101(13):4483–8. 10.1073/pnas.0400709101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chalfie M, Sulston JE, White JG, Southgate E, Thomson JN, Brenner S. The neural circuit for touch sensitivity in Caenorhabditis elegans. J Neurosci. 1985;5(4):956–64. 10.1523/JNEUROSCI.05-04-00956.1985 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hobert O, Moerman DG, Clark KA, Beckerle MC, Ruvkun G. A conserved LIM protein that affects muscular adherens junction integrity and mechanosensory function in Caenorhabditis elegans. J Cell Biol. 1999;144(1):45–57. 10.1083/jcb.144.1.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rankin CH, Beck CD, Chiba CM. Caenorhabditis elegans: a new model system for the study of learning and memory. Behav Brain Res. 1990;37(1):89–92. 10.1016/0166-4328(90)90074-o . [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, Chalfie M. MTD-1, a touch-cell-specific membrane protein with a subtle effect on touch sensitivity. Mech Dev. 2002;119(1):3–7. 10.1016/s0925-4773(02)00293-9 . [DOI] [PubMed] [Google Scholar]
- 57.Grice SJ, Liu JL, Webber C. Synergistic interactions between Drosophila orthologues of genes spanned by de novo human CNVs support multiple-hit models of autism. PLoS Genet. 2015;11(3):e1004998 10.1371/journal.pgen.1004998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Geisheker MR, Heymann G, Wang T, Coe BP, Turner TN, Stessman HAF, et al. Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nat Neurosci. 2017;20(8):1043–51. 10.1038/nn.4589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen S, Fragoza R, Klei L, Liu Y, Wang J, Roeder K, et al. An interactome perturbation framework prioritizes damaging missense mutations for developmental disorders. Nat Genet. 2018;50(7):1032–40. 10.1038/s41588-018-0130-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A. The Heritability of Autism Spectrum Disorder. JAMA. 2017;318(12):1182–4. 10.1001/jama.2017.12141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yook KJ, Proulx SR, Jorgensen EM. Rules of nonallelic noncomplementation at the synapse in Caenorhabditis elegans. Genetics. 2001;158(1):209–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu Y, Quinn CC. MIG-10 functions with ABI-1 to mediate the UNC-6 and SLT-1 axon guidance signaling pathways. PLoS Genet. 2012;8(11):e1003054 Epub 2012/12/05. 10.1371/journal.pgen.1003054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dedic N, Pohlmann ML, Richter JS, Mehta D, Czamara D, Metzger MW, et al. Cross-disorder risk gene CACNA1C differentially modulates susceptibility to psychiatric disorders during development and adulthood. Mol Psychiatry. 2018;23(3):533–43. 10.1038/mp.2017.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang T, Guo H, Xiong B, Stessman HA, Wu H, Coe BP, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun. 2016;7:13316 10.1038/ncomms13316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dragich JM, Kuwajima T, Hirose-Ikeda M, Yoon MS, Eenjes E, Bosco JR, et al. Autophagy linked FYVE (Alfy/WDFY3) is required for establishing neuronal connectivity in the mammalian brain. Elife. 2016;5 10.7554/eLife.14810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ban BK, Jun MH, Ryu HH, Jang DJ, Ahmad ST, Lee JA. Autophagy negatively regulates early axon growth in cortical neurons. Mol Cell Biol. 2013;33(19):3907–19. 10.1128/MCB.00627-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shen W, Ganetzky B. Autophagy promotes synapse development in Drosophila. J Cell Biol. 2009;187(1):71–9. 10.1083/jcb.200907109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hill SE, Kauffman KJ, Krout M, Richmond JE, Melia TJ, Colon-Ramos DA. Maturation and Clearance of Autophagosomes in Neurons Depends on a Specific Cysteine Protease Isoform, ATG-4.2. Dev Cell. 2019. 10.1016/j.devcel.2019.02.013 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stavoe AK, Hill SE, Hall DH, Colon-Ramos DA. KIF1A/UNC-104 Transports ATG-9 to Regulate Neurodevelopment and Autophagy at Synapses. Dev Cell. 2016;38(2):171–85. 10.1016/j.devcel.2016.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krey JF, Pasca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16(2):201–9. 10.1038/nn.3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tulgren ED, Baker ST, Rapp L, Gurney AM, Grill B. PPM-1, a PP2Calpha/beta phosphatase, regulates axon termination and synapse formation in Caenorhabditis elegans. Genetics. 2011;189(4):1297–307. Epub 2011/10/05. 10.1534/genetics.111.134791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kamijo S, Ishii Y, Horigane SI, Suzuki K, Ohkura M, Nakai J, et al. A Critical Neurodevelopmental Role for L-Type Voltage-Gated Calcium Channels in Neurite Extension and Radial Migration. J Neurosci. 2018;38(24):5551–66. Epub 2018/05/19. 10.1523/JNEUROSCI.2357-17.2018 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Limpitikul WB, Dick IE, Ben-Johny M, Yue DT. An autism-associated mutation in CaV1.3 channels has opposing effects on voltage- and Ca(2+)-dependent regulation. Sci Rep. 2016;6:27235 10.1038/srep27235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cross-Disorder Group of the Psychiatric Genomics C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371–9. 10.1016/S0140-6736(12)62129-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yoshimizu T, Pan JQ, Mungenast AE, Madison JM, Su S, Ketterman J, et al. Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Mol Psychiatry. 2015;20(2):284 10.1038/mp.2014.181 . [DOI] [PubMed] [Google Scholar]
- 76.Ch'ng Q, Williams L, Lie YS, Sym M, Whangbo J, Kenyon C. Identification of genes that regulate a left-right asymmetric neuronal migration in Caenorhabditis elegans. Genetics. 2003;164(4):1355–67. Epub 2003/08/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu Y, Quinn CC. Transition between synaptic branch formation and synaptogenesis is regulated by the lin-4 microRNA. Dev Biol. 2016;420(1):60–6. Epub 2016/10/25. 10.1016/j.ydbio.2016.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chalfie M, Hart AC, Rankin CH, Goodman MB. Assaying mechanosensation. WormBook. 2014. 10.1895/wormbook.1.172.1, http://www.wormbook.org [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This hypothetical model could explain the genetic interactions observed between VGCC genes and genes that encode members of the RPM-1 pathway. In this model, VGCCs function with WDFY-3 to negatively regulate an unknown protein (labeled as X). Protein X functions with RPM-1 to promote signaling events downstream of FSN-1 that promote axon termination. Loss of VGCC function causes an increase in protein X function. The additional protein X function works with RPM-1 to promote axon termination mechanism B, thereby compensating for loss of FSN-1 function. Loss of VGCCs does not suppress loss of RPM-1 function because the function of protein X requires RPM-1.
(TIF)
It is possible that PPM-1, or another protein with a similar role, could be protein X (see S1 Fig). Both FSN-1 and PPM-1 promote axon termination by functioning with RPM-1 to negatively regulate the DLK-1 MAP kinase pathway. Thus, it is possible that VGCCs and WDFY-3 could negatively regulate PPM-1, or a protein with a similar role. The extra PPM-1 function could enhance negative regulation of the DLK-1 pathway, thereby compensating for loss of FSN-1 function.
(TIF)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.