


Abstract
Cockayne syndrome is an accelerated aging disorder, caused by mutations in the CSA or CSB genes. In CSB-deficient cells, poly (ADP ribose) polymerase (PARP) is persistently activated by unrepaired DNA damage and consumes and depletes cellular nicotinamide adenine dinucleotide, which leads to mitochondrial dysfunction. Here, the distribution of poly (ADP ribose) (PAR) was determined in CSB-deficient cells using ADPr-ChAP (ADP ribose-chromatin affinity purification), and the results show striking enrichment of PAR at transcription start sites, depletion of heterochromatin and downregulation of H3K9me3-specific methyltransferases SUV39H1 and SETDB1. Induced-expression of SETDB1 in CSB-deficient cells downregulated PAR and normalized mitochondrial function. The results suggest that defects in CSB are strongly associated with loss of heterochromatin, downregulation of SETDB1, increased PAR in highly-transcribed regions, and mitochondrial dysfunction.
INTRODUCTION
Cockayne syndrome (CS) is a rare genetic disorder caused by mutations in the ERCC8 (CSA) or ERCC6 (CSB) genes (1). CSA and CSB are thought to play important roles in DNA repair (2) and transcriptional regulation (3). CS is characterized by early-onset neurodegeneration, vision and hearing loss, as well as impaired mitophagy and accumulation of damaged mitochondria. Thus, CS is considered a ‘progeria’, in which features of normal aging manifest in early life, ultimately leading to premature death.
Although CS is caused by mutations in CSA or CSB, the two proteins share no homology and are structurally and functionally unrelated. CSB participates in transcription, DNA base excision repair (BER) of oxidative DNA lesions, as well as in transcription-coupled nucleotide excision repair (TC-NER) of UV-induced DNA damage (reviewed in Boetefuer et al. (3)), while CSA participates in transcription restart after RNA polymerase II stalled transcription (4). CSB is a member of the SWI2/SNF2 family of DNA-dependent adenosine triphosphatases (ATPases), which act as chromatin remodelers during transcription (5). In the presence of ATP, CSB binds to DNA and shortens the DNA contour length by actively wrapping or unwrapping DNA (6). This activity affects the affinity of DNA binding proteins, including p53 (7), TFAM (8) and CTCF (9) to their cognate binding sites on DNA. Moreover, NAP1-like histone chaperones facilitate the ATP-dependent chromatin remodeling activity of CSB for efficient transcription-coupled DNA repair (10). The data suggest that CSB influences or regulates epigenetic signaling and that this may be important in understanding CS pathology, including features of premature aging. Epigenetic changes, widely accepted as a marker of aging (11), are also diagnostic of premature aging disorders including Hutchinson-Gilford progeria (HGPS) (12) and Werner Syndrome (WS) (13), but they have not been linked to CS.
Epigenetic alterations are non-genomic changes that influence gene expression and alter the structural organization of chromatin. Heterochromatin is tightly packed in a ‘closed’ conformation, less accessible to transcription machinery, while euchromatin is loosely packed, enriched in actively transcribed chromatin in an ‘open’ conformation. The methylation status of core histones strongly influences chromatin conformation: for example, trimethylation of Lys9 of H3 (H3K9me3) promotes and is a marker of heterochromatinization. Thus, enrichment for H3K9me3 can be considered correlative with heterochromatin states.
The ‘heterochromatin loss model of aging’ suggests that the gradual loss of heterochromatin-induced gene silencing leads to aging-related changes in gene expression (14). Many studies report the loss of epigenetic markers including H3K9me3, H3K27me3 and the HP1 protein during normal aging (15) and in HGPS and WS (12,13), while in some species, overexpression of these factors appears to extend lifespan (16,17). Heterochromatin loss with aging may also reduce transcriptional precision and increase genomic instability (15).
Poly (ADP ribose) polymerases (PARPs) play fundamental roles in multiple DNA damage recognition and repair pathways (18). Like many other DNA repair proteins, CSB is recruited to sites of DNA damage by PARylated proteins that are generated by PARP, after which CSB displaces PARP and DNA repair can proceed (19). PARylation occurs on H3K9me3 and H3K27me3 (20), which are enriched in heterochromatin. In CSB-deficient cells, activated PARP remains bound to DNA damage, the DNA lesions are not repaired, PAR accumulates and nicotinamide adenine dinucleotide (NAD) is depleted. These cellular phenotypes are also associated with aging and mitochondrial dysfunction (19,21).
While PARylation has been shown to be increased in CS, its intracellular distribution has not been characterized. The recently developed chromatin affinity purification technique utilized in this study, ADPr-ChAP (ADP ribose-chromatin affinity purification), can assess the genomic distribution of PAR, which would otherwise be difficult to assess using conventional chromatin immunoprecipitation (ChIP). ADPr-ChAP data indicate that oxidative damage-induced PAR mostly occurs in heterochromatic regions in human cells (20). In combination with conventional ChIP, ADPr-ChAP analyses can be used to compare the distribution of H3K9me3 and PAR.
Here, we report reduced H3K9me3 throughout the genome of CSB-deficient cells as well as increased PAR at transcription start sites (TSS) after oxidative stress and decreased expression of the major human H3K9me3 methyltransferases, SUV39H1 and SETDB1. Reduced H3K9me3 and enrichment of PAR in TSS regions were normalized by overexpression of SETDB1. The mitochondrial stress response and mitochondrial dysfunction in CSB-deficient cells (22) were also complemented by expression of SETDB1. These results suggest that decreased heterochromatin in CSB-deficient cells is associated with increased PAR accumulation in TSS regions and perturbations in mitochondrial function.
MATERIALS AND METHODS
Cell culture and stable cell line construction
SV40-transformed CS3BE and CS1AN cells stably transfected with either CSA, CSB (WT CSA, CSB) or empty vectors were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% pen-strep and 400 μg/ml geneticin and grown in 20% O2/5% CO2 at 37°C. Doxycycline-inducible CSA and CSB knockdown HeLa cells were generated by infecting lentivirus with Edit-R™ Inducible Lentiviral Cas9 Nuclease that targets CSA and CSB (Dharmacon, Lafayette, CO, USA). Dox-inducible GFP-tagged lentiviral SUV39H1 or SETDB1 constructs were designed and ordered from VectorBuilder Inc. (Santa Clara, CA, USA). Lentivirus infected CS1AN cells were selected with 400 μg/ml geneticin, 1 μg/ml hygromycin and 1 μg/ml puromycin for 2 weeks and maintained in tet-free media (Atlanta Biologicals, Flowery Branch, GA, USA, S10350H). Cells were treated with concentrations of doxycycline from 10 to 100 ng/ml for 72 h to knockdown CSA/B or to induce SETDB1/SUV39H1. All cell lines used in this study are checked and confirmed that they are free with mycoplasma contamination, using LookOut® Mycoplasma qPCR Detection Kit (Sigma-Aldrich, St. Louis, MO, USA).
Immunofluorescence staining
Human primary fibroblasts were cultured for 48 h (Corning, New York, NY, USA, #354236) on 4-well chamber slides (ThermoFisher scientific, Waltham, MA, USA, #62407–294) in DMEM containing 10% FBS, 2 mM L-glutamine, 1% Penicillin-Streptomycin antibiotics at 37°C in 5% CO2. Slides with cells were washed twice with phosphate-buffered saline (PBS) and fixed in a 4% paraformaldehyde-PBS solution for 20 min followed by permeabilization with 0.1% Triton-X for 10 min. Normal Goat serum (10%) was used to block the cells followed by anti-CSB (Abcam, Cambridge, MA, ab96089) and anti-SETDB1 (Abcam, ab12317) staining overnight at 4°C. Cells were further stained with secondary antibodies (ThermoFisher scientific, CSB: AF647, SETDB1:AF488) for 1 h at room temperature. For nuclear staining, slides were incubated with DAPI for 5 min and mounted with coverslips by using ProLong Diamond Antifade (ThermoFisher scientific, P36961). Confocal images (z-series) were captured with Zeiss LSM 880 inverted laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany) using a 40 × /1.3 Plan-NeoFluar oil-immersion objective at 1 μm z-step intervals with lateral pixel dimensions of 0.22 μm. Captured z-series were imported and analyzed using FIJI-ImageJ.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed as described (23), with slight modifications. Briefly, CSB cells were cross-linked in a solution of 1% formaldehyde in PBS for 10 min at room temperature. The cross-linking reaction was stopped by adding glycine to a final concentration of 0.125 M. The cells were harvested and washed three times with cold PBS, and cytosolic fractions were eliminated with buffer A (5 mM PIPES (pH 8.0), 85 mM KCl, 0.5% NP-40, protease inhibitor cocktail (GenDEPOT, Katy, TX, USA)). Nuclear pellets were resuspended in buffer B (100 mM Tris–Cl (pH 8.1), 1% sodium dodecyl sulfate (SDS), 10 mM ethylenediaminetetraacetic acid (EDTA), protease inhibitor cocktail) and the chromatin was sheared with S-450 sonicator (Branson, Danbury, CT, USA). Sonication condition was optimized by analyzing purified DNA samples with BIOANALYZER (Agilent, DNA1000 Kit). The prepared chromatin fraction (500 μg total sheared DNA per sample) was diluted 1/10 in IP buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–Cl (pH 8.1), 167 mM NaCl and a protease inhibitor cocktail) and incubated with antibodies, overnight at 4°C. Samples were incubated for 2–4 h at 4°C with protein A or G beads. Then the beads were washed with TSE150 (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–Cl (pH 8.1), 150 mM NaCl), TSE500 (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–Cl (pH 8.1), 500 mM NaCl), Buffer III (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-Cl (pH 8.1)) and two times with TE (pH 8.0) for 10 min in each solution. Bead-bound chromatin was eluted with elution buffer (1% SDS, 0.1 M NaHCO3 (pH 8.0)) for 1–2 h at 65°C. The supernatant containing the chromatin, without the beads, was isolated and incubated overnight at 65°C with 200 mM NaCl to reverse cross-linking. Five hundred microliters of the sample were incubated at 50°C after adding 10 μl of 0.5 M EDTA, 20 μl of 1 M Tris (pH 6.5) and 4 μl of Proteinase K (20 mg ml−1) and then purified with phenol/chloroform/isoamyl alcohol. Nucleic acids were precipitated by centrifugation for 30 min at 4°C after mixing the sample with 1 μl of glycogen solution (20 mg ml−1), 20 μl 5 M NaCl and 500 μl isopropanol. Purified nucleic acid pellets were washed with 70% ethanol, dried and dissolved in nuclease-free water.
ADPr-Chromatin affinity purification (ADPr-ChAP)
ADPr-ChAP was performed identically with ChIP, except for the fixing condition (4%, at 4°C, without quenching) and using WWE affinity resin (Tulip Biolabs, Lansdale, PA, USA), instead of antibody and protein A/G beads, according to previously described (24).
ADPr-ChAP and ChIP sequencing
Cells were ChIPed with Anti-trimethyl-Histone H3 (Lys9) antibody (07–442, Millipore, Burlington, MA, USA) and ChAPed with WWE affinity resin. DNA fragments were ligated to a pair of adaptors for sequencing on an Illumina Hiseq-2000. The ligation products were size-fractionated to obtain 200–300-bp fragments on a 2% agarose gel and PCR-amplified for 18 cycles. Each library was diluted to 8 pM for 76 cycles of single-read sequencing on the Illumina Hiseq-2000 following the manufacturer's recommended protocol.
ADPr-ChAP and ChIP-sequencing data analysis
The sequencing data were uploaded to the Galaxy web platform (25), and we used the public server at usegalaxy.org to analyze the data. Sequencing reads were mapped against the human genome (GRCh37/hg19) using Bowtie for Illumina (26) with default parameters. The SAM format outputs were sorted by genomic coordinates and uniquely mapped reliable reads were used in subsequent steps. SAM files were preprocessed using SAMtools (27). We used the MACS tool (28) to select regions that were enriched for H3K9me3 and PAR. We applied the default settings and found significant regions (P-value ≤ 10−5), using non-IP chromatin as a control to eliminate unspecific signals. Using Nebula (29), PAR peaks were annotated for genomic distribution partitioning. Distance of H3K9me3 peak were calculated using GREATv3.0.0 (30). Sequencing reads were aligned on reference coordinates using seqMINER v1.3.3 (31). rDNA analysis was performed following previously described methods (32). Unique reads from each data were mapped onto human rDNA reference sequence (NCBI accession: HUS13369) using BWA. bamCompare tool was used to normalize and subtract reads from each group. Integrative Genomics Viewer (33) was used to visualize the signal.
Quantitative real-time PCR (qPCR) and conventional PCR analysis of relative mRNA levels and ChIP products
Total RNA was extracted with TriZol® (ThermoFisher scientific) and reverse-transcribed (iScript™ cDNA Synthesis Kit, Bio-Rad). mRNA and antibody-bound chromatin levels by ChIP assay were quantified by real-time qPCR with the DyNAmo HS SYBR Green qPCR Kit (F-410L, ThermoFisher scientific) on the iQ5 and CFX Connect Real-time PCR Detection System (Bio-Rad) and then normalized to actin or 1% input chromatin using the 2−ΔΔCT calculation method. The sequences of the primers are listed in Supplementary Table S2.
Cellular oxygen consumption
Oxygen consumption rate (OCR) measurements were performed using the Seahorse XFe96 analyzer (Agilent Technologies, Lexington, MA, USA). 5e4 cells were seeded into a Seahorse tissue culture plate overnight and the following morning, media was changed to freshly prepared unbuffered XF assay media at pH 7.4 (Agilent Technologies), supplemented with 25 mM glucose (SigmaAldrich), 1 mM sodiumpyruvate and 1 mM L-glutamate (ThermoFisher scientific). Cells were incubated for 1 h at 37°C in ambient O2 and CO2 concentration before measurements were initiated. Respiration was measured in four blocks of 3 × 3 min. First, the rate of basal respiration was measured with 3× mix (3 min) wait (1 min) measure (3 min) cycles. Next, oligomycin (Cf: 2.5 μM) (Sigma-Aldrich) was injected into injection port A to inhibit complex V and assess proton leak mediated respiration. Then, FCCP (SigmaAldrich), a proton ionophore (Cf: 2uM) was added to elicit the maximal uncoupled respiration rate Next, complex III inhibitor antimycin A (Cf: 2 μM) plus complex I inhibitor rotenone (Cf: 200 nM) (SigmaAldrich) was added to assess non-mitochondrial respiration. At last, artificial electron donor N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD; Cf 1 μM) TMDP plus ascorbate (10 μM) was injected to assess cytochrome oxidase (complex IV) function. Immediately after the measurements, cells were lysed with RIPA buffer and protein content was measured (BCA, ThermoFisher scientific). Rates of respiration for each well was then normalized to the corresponding protein concentration.
Quantification and statistical analysis
Data are presented as means ± standard deviations and P-values were calculated using the student's t-test calculator (http://www.physics.csbsju.edu/stats/t-test.html), unless otherwise stated. A value of P < 0.05 was considered to be statistically significant. All data are representative of at least three independent experiments. For Figure 7A and B, representative images are maximum intensity z-projection of all the slices from z-stacks. Quantification of mean fluorescence intensity of z-stacks of cells from different sex and aged groups, or different aged groups to measure staining intensity. For each cell line from an individual, a total of 19 and 13 cells were analyzed for CSB and SETDB1, respectively. Captured z-series were imported and analyzed using FIJI-ImageJ.
Figure 7.

SETDB1 expression decreases with chronological aging. Immunofluorescence (IF) analysis of normal human fibroblast from healthy female (ages: 20, 40 and 60) and male (age: 20, 40 and 60) donors. IF was performed with (A) anti-CSB (red) and (B) anti-SETDB1 (green). Cell nuclei were stained with DAPI. Representative images are maximum intensity z-projection of all the slices from z-stacks. Quantification of mean fluorescence intensity of z-stacks of cells from different sex and aged groups, or different aged groups to measure staining intensity. For each cell line from an individual, a total of 19 and 13 cells were analyzed for CSB and SETDB1, respectively. Error bars represents SEM. (C) Brain lysates of young, middle and old mice were probed for the indicated proteins on western blot. Blot density was measured and normalized to GAPDH (H3K9me3 was normalized to H3). Scale bar, 30 μm. Error bars represent SD. NS, not significant; *, <0.05; **, <0.01.
RESULTS
CSB-deficiency is associated with nucleosome decondensation
Heterochromatin loss, or the decondensation of nucleosomes, leads to enlarged nuclei. Heterochromatin loss and enlarged nuclei are observed in aged cells and cells from HGPS and WS patients (12,13). In this study, we compared CS1AN (a fibroblast cell line from a CS patient who carries a truncation mutation in CSB) to CS1AN cells expressing wildtype CSB (CSB-corrected), as well as CS3BE (a fibroblast cell line from a CS patient who carries a missense mutation in CSA) to CS3BE cells expressing wildtype CSA (CSA-corrected). For the present study, CS1AN cell are referred to as ‘CSB-deficient’ cells and CS3BE cells are referred to as ‘CSA-deficient’ cells. Nuclear staining revealed increased nuclear size and decreased nuclear DNA staining intensity in the CS1AN cells (Figure 1A–C). Based on corrected total cellular fluorescence, which reflects the total amount of DNA, we tentatively conclude that CS3BE cells are aneuploid (Figure 1B).
Figure 1.

CSB-deficiency leads to nucleosome decondensation. (A) DAPI staining of CSA-deficient CS3BE (CSA−) and CSB-deficient CS1AN (CSB−) cells with their respective corrected cells, which stably expressing wild-type CS proteins (CSA+ and CSB+). (B and C) Quantification of DAPI signal from A. CTCF, corrected total cell fluorescence. Error bars represent mean ± SEM. ***, <0.001; NS, not significant. (D) CS (CSA− and CSB−) cells and their respective corrected (CSA+ and CSB+) cells were lysed and probed for the indicated proteins on western blot. (E) Doxycycline-inducible CSA and CSB knockdown cells (iKD) were probed for indicated proteins on western blot after 72 h of doxycycline treatment.
As noted above, change in nuclear size and nuclear DNA content suggest changes in heterochromatin content. H3K9me3 and H3K27me3 are well-characterized markers of heterochromatin, while H3K36me3 is a useful marker of euchromatin. Heterochromatin markers H3K9me3, H3K27me3 and H3K36me3 levels were similar between CSA-deficient and CSA-corrected cells, but H3K9me3 was much lower in CSB-deficient cells than in CSB-corrected cells (Figure 1D). This suggests that CSB-deficient cells have reduced heterochromatin. CSA- and CSB-deficient cells also had more phosphorylated H2A.X (pH2A.X) than the matched corrected controls, indicating that the cells contained higher steady-state DNA damage, in agreement with previous reports (34,35). CSB-deficient cells had more pH2A.X, and likely have more DNA damage than CSA-deficient cells (Figure 1D, pH2A.X Long exposure).
To evaluate the relationship between H3K9me3 downregulation and CSB-deficiency, we generated doxycycline-inducible CSA or CSB knockdowns (iKD), in HeLa and U2OS cells. CSB knockdown resulted in H3K9me3 reduction in both HeLa and U2OS cells, suggesting that decreased H3K9me3 is a consequence of decreased CSB (Figure 1E).
CSB-deficient cells have increased PARylation after oxidative damage
CS primary fibroblasts and induced pluripotent stem cells from CS patients are sensitive to oxidative stress (36,37) and have increased PARylation (19,38) even in the absence of external stress. Here, PAR was quantified after oxidative stress in CSA- and CSB-deficient and their respective corrected cells. Exposure to hydrogen peroxide (H2O2) consistently increased PAR in both pairs of cells. However, H2O2 led to higher PAR in CSB-deficient cells than in corrected control cells, while there were no differences between CSA-deficient and corrected cells (Figure 2A). Similar results were obtained in cells treated with oxidant menadione that elevates levels of cellular peroxide and superoxide radicals (Figure 2B). These results suggest that CSB-deficiency leads to higher PAR and more PARylation than CSA-deficiency. Oxidative stress-induced PARylation is highly correlated with heterochromatin markers (20), and so H3K9me3 and PAR were quantified in three genomic regions with distinct chromatin compositions (20) to examine how this correlation was affected by the loss of heterochromatin after CSB loss. The TSS of the GAPDH house keeping gene is constantly transcribed and thus represents a region with very little heterochromatin. In contrast, the promoter of the dynamically regulated IL1B gene represents a denser heterochromatin region, while the alpha satellite repeats (αSAT) represent a constitutively heterochromatinized region.
Figure 2.

CSB-deficient cells have more PARylation sensitivity to oxidative damage. (A) CS (CSA− and CSB−) cells and their respective corrected (CSA+ and CSB+) cells were mock or 5 mM H2O2 treated for 30 min. Nuclear fractions were prepared from formaldehyde cross-linked cells and probed for the indicated proteins on western blot. (B) CSB-deficient (CSB−) and CSB-corrected (CSB+) cells were treated with mock or 30 min of 5 mM H2O2 or 1 h of 100 μM Menadione. Nuclear fractions were probed for the indicated proteins on western blot. (C) H3K9me3 levels in CSB-deficient (CSB−) and CSB-corrected (CSB+) cells were analyzed with ChIP-qPCR on indicated genomic locus. (D) CSB-deficient (CSB−) and corrected (CSB+) cells were treated with 2.5 mM H2O2 for 30 min and PAR levels were analyzed by ADPr-ChAP (ADP ribose-chromatin affinity purification), followed by qPCR on indicated genomic locus. Error bars represent SD. *, <0.05; **, <0.01; ***, <0.001.
As expected, ChIP-qPCR showed low H3K9me3 in the GAPDH TSS, intermediate H3K9me3 in the IL1B promoter, and high H3K9me3 in αSAT heterochromatin (Figure 2C). In CSB-deficient cells, H3K9me3 was significantly lower in IL1B and αSAT than in isogenic corrected cells. There was no significant reduction in the GAPDH region; however, the signal was low and close to the limit of detection/limit of sensitivity for this method. ADPr-ChAP showed highest PAR in αSAT heterochromatin, intermediate at the IL1B promoter and low PAR in GAPDH (Figure 2D). CSB-deficient cells showed more pronounced increase in PAR than isogenic corrected cells after H2O2. The increase in PAR in H2O2-treated CSB-deficient cells was significant in GAPDH and IL1B, but not in αSAT heterochromatin. In order to verify that these three regions were representative of the whole genome, we designed primers for three additional regions per each respective chromatin density and evaluated their H3K9me3 and PAR level. Consistently, H3K9me3 was downregulated all across the regions in CSB-deficient cells (Supplementary Figure S1a). However, H2O2-induced PAR accumulation was significantly increased only on gene coding regions, but not on constitutively heterochromatinized regions of CSB-deficient cells (Supplementary Figure S1b), validating the representativity of the GAPDH, IL1B and αSAT loci. Thus, PAR increased preferentially in the vicinity of gene coding regions rather than in the highly condensed-chromatin regions in CSB-deficient cells after exposure to oxidative stress.
CSB-deficient cells have increased PARylation around TSS regions
In order to explore genomic PAR distribution in more detail, genome wide ADPr-ChAP sequencing was performed in H2O2-treated CSB-deficient and corrected cells. Prior CSB ChIP-sequencing data revealed that CSB was significantly enriched at promoters and enhancers (39), therefore we divided the genome into sub-partitions: TSS, promoters, enhancers and intragenic regions to categorize PAR distribution throughout the genome (Figure 3A). While PAR distribution in CSB-deficient and corrected cells appeared similar (Figure 3B), closer inspection of peak enrichments showed a marked increase in PAR at TSS and flanking regions in CSB-deficient cells (Figure 3C).
Figure 3.

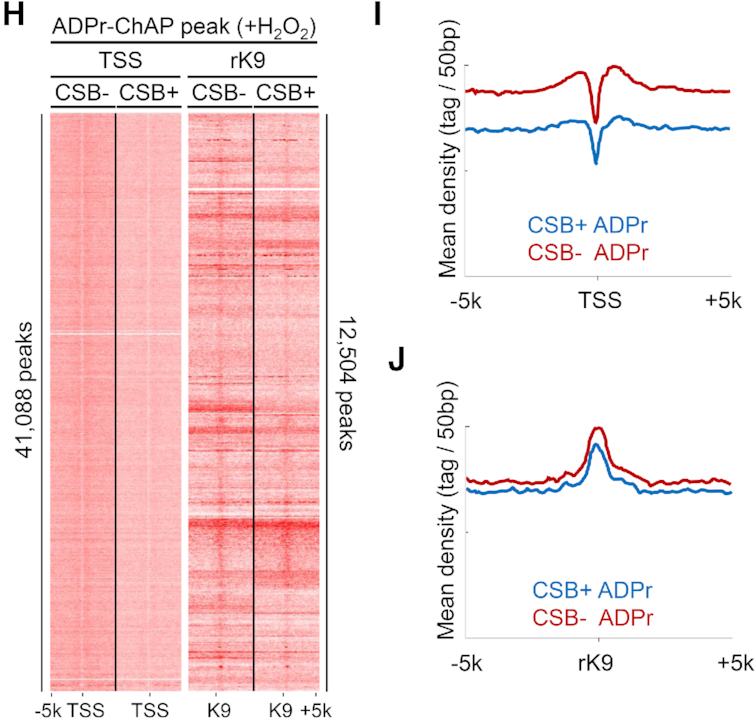
CSB-deficient cells have increased PARylation around TSS regions. (A) Schematic diagram of genomic distribution partitioning. TSS, transcription start site; TTS, transcription termination site. (B) Genomic annotation analysis of ADPr-ChAP sequencing peaks. (C) Annotation of genes with enriched ADPr-ChAP sequencing peaks (CSB-deficient cells to CSB-corrected cells). (D) H3K9me3-ChIP sequencing reads of CSB-deficient (CSB−) and CSB-corrected (CSB+) cells were aligned with seqMINER (Ver 1.3.3) on hg19 refgene TSS and reference H3K9me3 (rK9) peaks as reference coordinates. (E and F) Merged dataset profiles of aligned sequencing reads from D. (G) Distribution distance of rK9 was calculated using Genomic Regions Enrichment of Annotations Tool (GREAT). (H) ADPr-ChAP sequencing reads of H2O2-treated CSB-deficient (CSB−) and CSB-corrected (CSB+) cells were aligned on TSS and rK9 peaks. (I and J) Merged dataset profiles of aligned sequencing reads from H.
Next, we quantified H3K9me3 at TSS in CSB-deficient cells using ChIP-sequencing. To maintain an unbiased approach, we performed H3K9me3-ChIP sequencing from CSB-corrected cells and used this H3K9me3 peak data as reference coordinates (rK9) to align independent H3K9me3 ChIP-sequencing data from CSB-deficient and corrected cells. For further validation, our ChIP-sequencing data were also aligned to TSS from the widely used hg19 refseq dataset to compare H3K9me3 distribution. Both the TSS and rK9 alignments revealed dramatically decreased H3K9me3 in CSB-deficient cells, representing heterochromatin loss at TSS (Figure 3D and E), as well as in constitutively heterochromatinized regions in CSB-deficient cells (Figure 3D and F). To account for possible overlaps between the TSS and rK9 reference coordinates used for alignments, the rK9 peak distribution distance from TSS was calculated. The majority of rK9 peaks were 50–500 kb from TSS, suggesting low likelihood of overlap between the two reference coordinates. This independently confirms the validity of the alignments (Figure 3G).
H3K9me3-ChIP sequencing data from CSB-deficient and CSB-corrected cells were also validated by aligning onto the ENCODE H3K9me3 ChIP-sequencing data from normal human epidermal keratinocytes (GSM1003528). This analysis showed striking H3K9me3-peak overlap (Supplementary Figure S3a and b), validating our analysis and reliability of our H3K9me3-ChIP sequencing data and the deduced genomic distribution of heterochromatin.
H2O2 treatment-induced PAR distribution was also compared in CSB-deficient and corrected cells. Although the distribution patterns of H2O2-induced PAR and H3K9me3 overlapped dramatically in both CSB-deficient and isogenic corrected cells (Figure 3H and J; Supplementary Figure S3c and d), the PAR signal intensity was higher in CSB-deficient than in corrected cells at TSS sites (Figure 3H, TSS alignment, I and Supplementary Figure S2a), while no significant differences were detected at rK9 sites (Figure 3H, rK9 alignment, J and Supplementary Figure S2b). H3K9me3 signal intensities between H2O2-treated CSB-deficient and corrected cells were similar when aligned on TSS (Supplementary Figure S4a–c) and rK9 peaks (Supplementary Figure S4a, d and e), as well as on the ENCODE H3K9me3 peaks (Supplementary Figure S3e and f). In fact, the H3K9me3 signal intensity in corrected cells seemed to be lower than that in CSB-deficient cells when treated with H2O2. This may be a result of the ability of CSB-corrected cells to react to oxidative stress by downregulating heterochromatin marks to enable DNA repair enzyme access (Supplementary Figures S3e-f and 4), whereas a CSB-deficient cell would be impaired in this chromatin remodeling ability. This finding supports the ‘access, repair, restore’ model of DNA repair, which proposes that after the initial phase of DNA damage, chromatin becomes more accessible to repair factors to enable efficient DNA repair (40).
We previously reported altered rDNA transcription in CSB-deficient cells (38). Here, rDNA analysis showed decreased H3K9me3 and H2O2-induced PAR in rRNA coding regions (18S, 5.8S and 28S) in CSB-deficient and corrected cells (Supplementary Figure S5, ΔPAR and -ΔH3K9me3). This data supports our hypothesis that decreased H3K9me3 is associated with increased PAR in transcribed gene regions.
CSB-deficient cells have reduced H3K9me3 methyltransferase expression
It is possible that a decrease in H3K9me3 reflects downregulation of H3K9me3 methyltransferases (HMTases) SUV39H1, SUV39H2 or SETDB1, altered expression of H3K9me3 interacting proteins CBX3, CBX5, upregulation of H3K9me3 demethylases KDM4A, KDM4B, KDM4C, KDM4D and MINA (Figure 4; Supplementary Figures S6 and 7), or a combination of these factors. CSA-deficient and corrected cells showed no differences in the levels of the methyltransferase proteins (Figure 4A and C). In contrast, mRNA and protein expression levels of SUV39H1 and SETDB1, were significantly lower in CSB-deficient cells than in corrected cells (Figure 4B and C).
Figure 4.

CSB-deficient cells have reduced H3K9me3 methyltransferase expression. (A and B) Real-time qPCR mRNA expression analysis of indicated genes from CSA/B-deficient (CSA− and CSB−) cells and their respective corrected (CSA+ and CSB+) cells (normalized with GAPDH). (C) Whole cell extracts of CSA/B-deficient (CSA− and CSB−) cells and their respective corrected (CSA+ and CSB+) cells were probed for the indicated proteins on western blot. Sample loading quantities of CSB-deficient cells were normalized with GAPDH. (D) Venn diagram of ATF3 ChIP-sequencing peak distributed gene sets of indicated datasets. (E) ATF3 ChIP-sequencing data analysis from Epanchintsev et al. (41). Data representing ATF3 ChIP-sequencing peak distribution after 24 h with 12 J/m2 of UV-C irradiation to CS1AN (CSB−) or wild-type CSB expressing CS1AN (CSB+). NS, not significant; **, <0.01; ***, <0.001.
CSB-deficiency may downregulate SETDB1 by lack of ATF3 degradation
It has been reported that UV arrests transcription in CSA- or CSB-deficient cells due to the constitutive presence of ATF3 (activating transcription factor 3) (41). CSB is known to facilitate MDM2-mediated ubiquitination and degradation of ATF3 by the proteasome (41). Thus, we hypothesized high levels of activated ATF3 could repress expression of SUV39H1 and SETDB1 in CSB-deficient cells. In silico analysis of the promoters of these genes showed putative ATF3 binding sites (Supplementary Document S1). Analysis of ATF3 ChIP-sequencing data from the publicly available ENCODE transcription factor database (42) showed direct binding of ATF3 to the promoter of SETDB1 (Figure 4D and Supplementary Table S1). The ATF3 ChIP-sequencing GEO dataset (GSE87562) from Epanchintsev et al. (41) also showed ATF3 binding to the promoters of SETDB1and SUV39H1in CSB-deficient cells. In line with our hypothesis, the GEO dataset showed significantly reduced ATF3 binding to either promoter regions in CSB-corrected cells (Figure 4E), suggesting that SETDB1 expression may be regulated by CSB through ATF3. Thus, we propose that ATF3 represses SETDB1, leading to heterochromatin loss in CSB-deficient cells.
CSB-deficient cells have altered histone expression
Core histone levels have been shown to decrease with age in studies of chronological aging and cellular senescence (15). So, we quantified expression of H3 isoforms in CSB-deficient and control cells. Although the mRNA expression levels of H3 isoforms such as H3.1 and H3.2 were not changed (Supplementary Figure S8a and b), the mRNA level of the H3.3 isoform decreased and correlated with decreased total H3 protein expression in CSB-deficient cells (Supplementary Figure S8c). These results suggest that CSB regulates H3 expression and epigenetic changes.
H3K9me3 recovery by SETDB1 reduces PAR production by oxidative stress
The following experiments tested whether expression of SETDB1 or SUV39H1 alters PAR deposition in CSB-deficient cells. For this purpose, stable cell lines were constructed that express doxycycline-inducible SETDB1 or SUV39H1. Western blot analysis revealed that doxycycline treatment induced the methyltransferases and rescued the decrease of H3K9me3 (Figure 5A and B). H2O2-induced PAR was lower in SETDB1-expressing cells, but did not change in SUV39H1-expressing cells (Figure 5C and E). Consistent with these results, qPCR analysis of ADPr-ChAP samples also showed that induced-expression of SETDB1 reduced H2O2-induced PAR while SUV39H1 had no significant effects (Figure 5D and F). These results confirm above results indicating a relationship between chromatin PARylation, H3K9me3 and chromatin structure/heterochromatinization.
Figure 5.

H3K9me3 recovery by SETDB1 reduces PAR production by oxidative stress. Tet-inducible SETDB1 (A) or SUV39H1 (B) expressing CSB-deficient (CSB−) cells were treated with 10 or 50 ng/ml of doxycycline for 72 h. Whole cell lysates were probed for the indicated proteins on western blot. Sample loading quantities were normalized with either total H3 or GAPDH. Tet-inducible SETDB1 (C) or SUV39H1 (E) cells were treated with DMSO or 50 ng doxycycline for 72 h and treated with 2.5 mM H2O2 for 30 min before ADPr-ChAP sample preparation. Chromatin samples prepared for ADPr-ChAP were probed for nuclear PAR analysis. (D and F) ADPr-ChAP were performed from chromatin preparations from C and E and analyzed for real-time qPCR on indicated genomic locus. Error bars represent SD. *, <0.05.
SETDB1 induction restores mitochondrial homeostasis in CS fibroblasts
Mitochondrial respiration and membrane potential are increased in CSB-deficient cells (22) and PAR suppression by PARP1 inhibition enhances mitochondrial content and function in normal mouse and human CS cells (19,43). We therefore tested whether the expression of SETDB1 is sufficient to correct mitochondrial alterations in CSB-deficient cells. Doxycycline treatment induced SETDB1 expression (Figure 6A) and H3K9me3 was greatly upregulated at TSS in CSB-deficient cells (Figure 6B and Supplementary Figure S9a). H2O2-induced PAR accumulation at TSS was also rescued by SETDB1 restoration (Supplementary Figure S9b). The level of PAR in non-H2O2 treated cells was difficult to detect with conventional western blots and ADPr-ChAP qPCR analysis of GAPDH, IL1B and αSAT loci (Supplementary Figure S9b and c). However, ADPr-ChAP sequencing alignments revealed markedly decreased PAR on TSS regions (Figure 6B).
Figure 6.

SETDB1-induction restores mitochondrial homeostasis in CS fibroblasts. (A) Doxycycline-induced HMTase expression was probed on western blot (CSB+; CSB-corrected cells, SUV; SUV39H1-inducible CSB-deficient cells, SET; SETDB1-inducible CSB-deficient cells). (B) H3K9me3-ChIP and ADPr-ChAP sequencing reads of SETDB1-inducible CSB-deficient (CSB- rtTA SETDB1) cells were aligned on TSS. (C and D) Merged dataset profiles of aligned sequencing reads from (B). Seahorse extracellular flux analysis representative trace (E) and quantification of basal respiration (F), proton leak (G), maximal respiration (H), complex IV function (I), spare capacity (J), ATP-linked respiration (K) and non-mitochondrial respiration (L) in CSB-corrected (CSB+) and SUV39H1/SETDB1-inducible CSB-deficient cells. (n = 5; data are represented as mean ± SEM) *P< 0.05, **P< 0.01, ***P< 0.001 One-way ANOVA.
CSB-deficient cells displayed an increased OCR measured via Seahorse extracellular flux analysis (Figure 6E-L, CSB+ versus CSB- SUV39H1 and CSB- SETDB1). SETDB1 induction in CSB-deficient cells restored mitochondrial oxygen consumption to rates observed in CSB-corrected cells (Figure 6E), including proton leak (Figure 6G), maximal respiration (Figure 6H) and complex IV function (Figure 6I), with a trend toward the normalization of spare respiratory capacity (Figure 6J). Thus, increases in mitochondrial respiration in CSB-deficient cells are reversed by SETDB1 induction, but not by SUV31H1 induction. We propose a model where H3K9me3 recovery by SETDB1 expression suppresses chromatin alterations (PAR, H3K9me3 and histone expression) in CSB-deficient cells thereby improving mitochondrial function (Figure 8).
Figure 8.
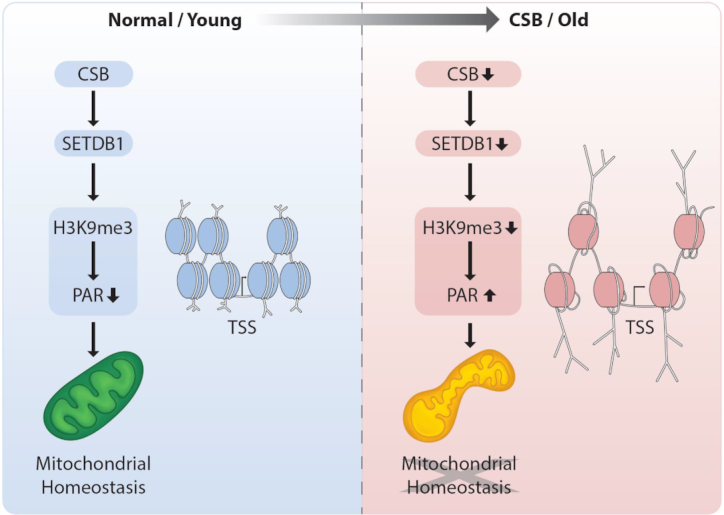
Schematic diagram of CSB-SETDB1 axis. In CS, as well as chronological aging, decreased CSB causes decreased SETDB1 expression, which could lead to reduced H3K9me3 on TSS regions, triggering accumulation of PAR and mitochondrial abnormalities.
SETDB1 expression decreases by chronological aging
At last, we evaluated the validity of our proposed model in relation to chronological aging. CSB and SETDB1 protein levels in fibroblasts from healthy donors from age 20, 40 and 60 years old were analyzed. Both CSB and SETDB1 protein levels decreased significantly with age (Figure 7A and B; Supplementary Figure S10). Furthermore, protein levels of CSB and SETDB1 decreased with age in brain tissue extracts from mice of different ages (Figure 7C). This indicates that heterochromatin loss resulting from the SETDB1 downregulation caused by a CSB deficiency may contribute to heterochromatin loss during normal aging as well.
DISCUSSION
In the present study, we are the first to report heterochromatin loss in CSB-deficient cells. Cellular genomes are continuously damaged by reactive oxygen species (ROS) from aerobic processes. In vitro studies have shown that histones organize DNA into higher order chromatin structures to protect DNA from ROS-induced damage, which is produced by aerobic respiration (44), as well as from radiation-induced damage (45–47). The importance of heterochromatin in protecting DNA is also supported by studies in mice where partial reprogramming of cells to pluripotency from a progeria model mouse (HGPS) increased the levels of the heterochromatin marker H3K9me3, decreased the DNA damage marker pH2AX, and ultimately decreased physiological symptoms of aging and increased lifespans of the mice (48). Thus, the ability of cells to maintain heterochromatin states allows for protection against genomic instability and promotes precise gene transcription.
Here we find that CSB-deficient cells have more pronounced increases in pH2A.X than CSA-deficient cells when compared to their respective corrected counterparts (Figure 1D). A possible explanation is that CSB-deficient cells have a more severe loss of heterochromatin than CSA-deficient cells, making their genomes more susceptible to ROS damage. Indeed, after H2O2 treatment, CSB-deficient cells sustained more chromatin PAR than corrected control cells while CSA-deficient cells had a similar extent of PAR accumulation as their corrected counterparts. This may be one of the reasons for the slight differences in the pathological severities between CSA and CSB complementation groups (49,50), where CSB usually has more severe clinical features than CSA.
CSB has been known as an ATP-dependent chromatin remodeler (51) that can slide histones along DNA to recruit DNA repair enzymes after damage (10). However, in CSB-deficient cells after oxidative stress, more heterochromatin marks are deposited implying more densely packed DNA and less accessibility for DNA repair enzymes (Supplementary Figure S4). These findings likely contribute to the DNA repair deficiency in CS cells.
PARP1 is a major player in multiple DNA damage response pathways. It is activated by many forms of DNA damage including H2O2 and it catalyzes NAD to PAR and nicotinamide upon binding to the DNA damage site. Although PARP1 accounts for about 75–95% of cellular PARylation activity after DNA damage (52), it is possible that PARP2 and PARP3 participate in the DNA damage response, as these two enzymes are also known to play important roles in the repair of DNA strand breaks and are known to be catalytically activated through interaction with damaged DNA (53,54). Consistent with Bartolomei et al. (20), a significant overlap of H3K9me3 and PAR peaks were found in both CSB-deficient and corrected cells after H2O2 treatment (Figure 3H and J; Supplementary Figure S2b). PAR is conjugated to histones and histone modifications, such as H3K9me3, as well as to other proteins recruited to the site of damage and thus believed to participate in the regulation of the DNA damage response. Since the TSS regions of CSB-deficient cells had less H3K9me3 (Figure 3D), one might have expected less PAR, but in fact we find more PAR accumulation after H2O2 at TSS regions (Figure 3H). Thus, CSB-deficient cells seem to accumulate more PAR per single H3K9me3 on TSS when compared to corrected cells.
We can suggest a few scenarios that may explain the increased PAR/H3K9me3 ratio on TSS in CSB-deficient cells compared to corrected cells: (i) despite having less of its substrate H3K9me3, PARP1 persistently builds longer PAR branches on the few H3K9me3 marks on TSS because of the persistent DNA damage caused by decreased heterochromatin structure, which provides less protection from oxidative stress in CSB-deficient cells. (ii) As CSB participates in BER and TC-NER (55), both of which predominantly occur in open chromatin regions (56), CSB-deficient cells likely have more unrepaired DNA damage caused by impaired BER and TC-NER at TSS regions. (iii) G-quadruplexes are an alternative structure formed between guanines in G-rich DNA sequences, mostly in promoters and 5′UTRs of highly transcribed human genes (57). G-quadruplex structures activate PARP1 and lead to accelerated aging in C. elegans, which can be resolved by CSB (38). Since chromatin relaxation increases G-quadruplex structures in regulatory regions, it is possible that heterochromatin loss in TSS regions makes them more prone to PARylation (iv). While PARylation occurs preferentially on substrates such as histones that are enriched with H3K9me3 and H3K27me3 in the rest of the genome, we suggest that PARylation may also occur on substrates other than H3K9me3 at TSS. This idea agrees with a study from Bartolomei et al. (20) noting that TSS regions were an exception to the correlation between high levels of PAR and chromatin density. One or more of these scenarios may explain the high levels of PAR accumulation in TSS of CSB-deficient cells, and further research is required to elucidate the precise mechanism.
To identify the molecular mechanism underlying loss of H3K9me3 in CSB-deficient cells, we screened the expression of multiple methyltransferases and found that SETDB1 and SUV39H1 were specifically downregulated in CSB-deficient cells (Figure 4B and C). Although both SETDB1 and SUV39H1 methylate H3K9, it is likely that these two HMTases have distinct affinities relating to their substrate's genomic distribution. SETDB1 is more responsible for euchromatin and facultative heterochromatin and is particularly involved in dynamic transcription repression (58), while SUV39H1 binding is enriched at constitutive heterochromatin. Our data support that SETDB1 induction is more effective at H3K9me3 restoration in gene coding regions (Supplementary Figure S9a, GAPDH and IL1B) rather than in constitutive heterochromatin regions (Supplementary Figure S9a, αSAT).
In addition to decreased SETDB1 expression, we also found decreased mRNA expression of H3.3 in CSB-deficient cells (Supplementary Figure S8). It was reported that histone isoform H3.3 influences animal lifespan by regulating the pro-longevity transcriptional program (17) and that it plays a critical role during PARP1 derived non-homologous end-joining (59). More interestingly, H3.3 is known to be involved in transcription recovery after UV (60), one of the cardinal features that is defective in CS. So, we speculate that decreased H3.3 expression in CSB-deficient cells not only correlates with the overall low chromatin density but also with transcription and DNA repair dysfunction.
SETDB1 (also known as ESET or KMT1E) is a member of SET domain-containing methyltransferase superfamily, which is one of the major H3K9me3 histone methyltransferases. SETDB1 overexpression is found in various human cancers, including lungs carcinoma (61,62), breast cancer (63,64), colon cancer (65) and liver cancer (66,67) and is reported to increase with cancer progression and metastasis across many cancers. Although speculative, one possible explanation as to why CS patients are not prone to cancer despite their DNA repair deficiency (68) may be related to our findings that CSB-deficient cells have decreased SETDB1 expression.
SUV39H1 is downregulated in old human primary mesenchymal stem cells and in WRN depleted mouse mesenchymal stem cells (13), and was shown to regulate genomic H3K9me3 density in human mesenchymal stem cells. However, restoring SETDB1 rescued H3K9me3 levels in CSB and dramatically suppressed H2O2-induced PAR accumulation in CSB-deficient cells, whereas SUV39H1 did not (Figure 5C–F).
Mitochondrial abnormalities are an important feature of CS (69), are a hallmark of chronological aging, and of a wide range of age-related diseases such as Alzheimer's disease (70,71). Inhibiting PARP1 enhances mitochondrial health (19,43,72). Thus, we tested whether normalization of SETDB1 expression and heterochromatinization of DNA could rescue the mitochondrial dysfunction in CSB-deficient cells (Figure 6E–L) and found that it could. Of note, others have reported that mitochondrial ROS increases with Setdb1 silencing in mouse hematopoietic stem cells (73). Induced-expression of SETDB1, but not of SUV39H1, specifically improved mitochondrial respiration, demonstrating the modulation of chromatin can improve mitochondrial function.
Epigenetic alterations are well established markers of aging, and thus we tested whether SETDB1 and CSB protein expression declines with age in normal fibroblast from healthy individuals and in mouse brain tissue (Figure 7 and Supplementary Figure S10). Both proteins showed decreased expression along with H3K9me3 marks. These results suggest that CSB and SETDB1 may contribute to heterochromatin loss with age. In conclusion, we provide evidence for heterochromatin loss in CSB patient cells and following CSB knockdown. These results are consistent with what occurs in chronological aging and in the progeria syndromes HGPS and WS. After CSB loss, decreased SETDB1 expression results in H3K9me3 loss facilitating PAR accumulation at TSS regions, which then contributes to mitochondrial dysfunction (Figure 8). Importantly, we demonstrate for the first time, that SETDB1 re-expression rescues heterochromatin and PAR alterations and we find improved mitochondrial functionality even though the cells still lack CSB. Additionally, we provided evidence for a novel molecular mechanism that may lead to age-related heterochromatin loss. We also speculate that recovery of lost heterochromatin by re-expression of methyltransferases may improve mitochondrial health in other premature aging models and perhaps in normal aging. These observations provide new insights into CS pathology and implicate chromatin remodeling as a therapeutic target in CS.
DATA AVAILABILITY
The accession number for the raw and processed ChIP sequencing and ADPr-ChAP sequencing data reported in this paper is GEO: GSE133176.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Hyundong Song for providing mouse brain tissue extracts.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program, National Institute on Aging. Funding for open access charge: National Institute on Aging, Intramural Program.
Conflict of interest statement. None declared.
REFERENCES
- 1. Karikkineth A.C., Scheibye-Knudsen M., Fivenson E., Croteau D.L., Bohr V.A.. Cockayne syndrome: clinical features, model systems and pathways. Ageing Res. Rev. 2017; 33:3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hafsi W., Badri T.. StatPearls. 2018; Treasure Island (FL). [Google Scholar]
- 3. Boetefuer E.L., Lake R.J., Fan H.Y.. Mechanistic insights into the regulation of transcription and transcription-coupled DNA repair by Cockayne syndrome protein B. Nucleic Acids Res. 2018; 46:7471–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McKay B.C., Chen F., Clarke S.T., Wiggin H.E., Harley L.M., Ljungman M.. UV light-induced degradation of RNA polymerase II is dependent on the Cockayne's syndrome A and B proteins but not p53 or MLH1. Mutat. Res. 2001; 485:93–105. [DOI] [PubMed] [Google Scholar]
- 5. Citterio E., Van Den Boom V., Schnitzler G., Kanaar R., Bonte E., Kingston R.E., Hoeijmakers J.H., Vermeulen W.. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell Biol. 2000; 20:7643–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beerens N., Hoeijmakers J.H., Kanaar R., Vermeulen W., Wyman C.. The CSB protein actively wraps DNA. J. Biol. Chem. 2005; 280:4722–4729. [DOI] [PubMed] [Google Scholar]
- 7. Lake R.J., Basheer A., Fan H.Y.. Reciprocally regulated chromatin association of Cockayne syndrome protein B and p53 protein. J. Biol. Chem. 2011; 286:34951–34958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berquist B.R., Canugovi C., Sykora P., Wilson D.M. 3rd, Bohr V.A.. Human Cockayne syndrome B protein reciprocally communicates with mitochondrial proteins and promotes transcriptional elongation. Nucleic Acids Res. 2012; 40:8392–8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lake R.J., Boetefuer E.L., Won K.J., Fan H.Y.. The CSB chromatin remodeler and CTCF architectural protein cooperate in response to oxidative stress. Nucleic Acids Res. 2016; 44:2125–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cho I., Tsai P.F., Lake R.J., Basheer A., Fan H.Y.. ATP-dependent chromatin remodeling by Cockayne syndrome protein B and NAP1-like histone chaperones is required for efficient transcription-coupled DNA repair. PLos Genet. 2013; 9:e1003407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Horvath S., Raj K.. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018; 19:371–384. [DOI] [PubMed] [Google Scholar]
- 12. Scaffidi P., Misteli T.. Lamin A-dependent nuclear defects in human aging. Science. 2006; 312:1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang W., Li J., Suzuki K., Qu J., Wang P., Zhou J., Liu X., Ren R., Xu X., Ocampo A. et al.. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015; 348:1160–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Villeponteau B. The heterochromatin loss model of aging. Exp. Gerontol. 1997; 32:383–394. [DOI] [PubMed] [Google Scholar]
- 15. Benayoun B.A., Pollina E.A., Brunet A.. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015; 16:593–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feser J., Truong D., Das C., Carson J.J., Kieft J., Harkness T., Tyler J.K.. Elevated histone expression promotes life span extension. Mol. Cell. 2010; 39:724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Piazzesi A., Papic D., Bertan F., Salomoni P., Nicotera P., Bano D.. Replication-independent histone variant H3.3 controls animal lifespan through the regulation of pro-longevity transcriptional programs. Cell Rep. 2016; 17:987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luo X., Kraus W.L.. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012; 26:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scheibye-Knudsen M., Mitchell S.J., Fang E.F., Iyama T., Ward T., Wang J., Dunn C.A., Singh N., Veith S., Hasan-Olive M.M. et al.. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014; 20:840–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bartolomei G., Leutert M., Manzo M., Baubec T., Hottiger M.O.. Analysis of chromatin ADP-ribosylation at the genome-wide level and at specific loci by ADPr-ChAP. Mol. Cell. 2016; 61:474–485. [DOI] [PubMed] [Google Scholar]
- 21. Fang E.F., Lautrup S., Hou Y., Demarest T.G., Croteau D.L., Mattson M.P., Bohr V.A.. NAD(+) in aging: molecular mechanisms and translational implications. Trends Mol. Med. 2017; 23:899–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scheibye-Knudsen M., Ramamoorthy M., Sykora P., Maynard S., Lin P.C., Minor R.K., Wilson D.M. 3rd, Cooper M., Spencer R., de Cabo R. et al.. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 2012; 209:855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee J.H., Kang B.H., Jang H., Kim T.W., Choi J., Kwak S., Han J., Cho E.J., Youn H.D.. AKT phosphorylates H3-threonine 45 to facilitate termination of gene transcription in response to DNA damage. Nucleic Acids Res. 2015; 43:4505–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bisceglie L., Bartolomei G., Hottiger M.O.. ADP-ribose-specific chromatin-affinity purification for investigating genome-wide or locus-specific chromatin ADP-ribosylation. Nat. Protoc. 2017; 12:1951–1961. [DOI] [PubMed] [Google Scholar]
- 25. Afgan E., Baker D., van den Beek M., Blankenberg D., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Eberhard C. et al.. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016; 44:W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S. . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boeva V., Lermine A., Barette C., Guillouf C., Barillot E.. Nebula–a web-server for advanced ChIP-seq data analysis. Bioinformatics. 2012; 28:2517–2519. [DOI] [PubMed] [Google Scholar]
- 30. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010; 28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ye T., Krebs A.R., Choukrallah M.A., Keime C., Plewniak F., Davidson I., Tora L.. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 2011; 39:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cong R., Das S., Ugrinova I., Kumar S., Mongelard F., Wong J., Bouvet P.. Interaction of nucleolin with ribosomal RNA genes and its role in RNA polymerase I transcription. Nucleic Acids Res. 2012; 40:9441–9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ranes M., Boeing S., Wang Y., Wienholz F., Menoni H., Walker J., Encheva V., Chakravarty P., Mari P.O., Stewart A. et al.. A ubiquitylation site in Cockayne syndrome B required for repair of oxidative DNA damage, but not for transcription-coupled nucleotide excision repair. Nucleic Acids Res. 2016; 44:5246–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cleaver J.E., Brennan-Minnella A.M., Swanson R.A., Fong K.W., Chen J., Chou K.M., Chen Y.W., Revet I., Bezrookove V.. Mitochondrial reactive oxygen species are scavenged by Cockayne syndrome B protein in human fibroblasts without nuclear DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:13487–13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pascucci B., Lemma T., Iorio E., Giovannini S., Vaz B., Iavarone I., Calcagnile A., Narciso L., Degan P., Podo F. et al.. An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell. 2012; 11:520–529. [DOI] [PubMed] [Google Scholar]
- 37. Andrade L.N., Nathanson J.L., Yeo G.W., Menck C.F., Muotri A.R.. Evidence for premature aging due to oxidative stress in iPSCs from Cockayne syndrome. Hum. Mol. Genet. 2012; 21:3825–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scheibye-Knudsen M., Tseng A., Borch Jensen M., Scheibye-Alsing K., Fang E.F., Iyama T., Bharti S.K., Marosi K., Froetscher L., Kassahun H. et al.. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:12502–12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lake R.J., Boetefuer E.L., Tsai P.F., Jeong J., Choi I., Won K.J., Fan H.Y.. The sequence-specific transcription factor c-Jun targets Cockayne syndrome protein B to regulate transcription and chromatin structure. PLos Genet. 2014; 10:e1004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smerdon M.J. DNA repair and the role of chromatin structure. Curr. Opin. Cell Biol. 1991; 3:422–428. [DOI] [PubMed] [Google Scholar]
- 41. Epanchintsev A., Costanzo F., Rauschendorf M.A., Caputo M., Ye T., Donnio L.M., Proietti-de-Santis L., Coin F., Laugel V., Egly J.M.. Cockayne's syndrome A and B proteins regulate transcription arrest after genotoxic stress by promoting ATF3 degradation. Mol. Cell. 2017; 68:1054–1066. [DOI] [PubMed] [Google Scholar]
- 42. Consortium, E.P. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004; 306:636–640. [DOI] [PubMed] [Google Scholar]
- 43. Bai P., Canto C., Oudart H., Brunyanszki A., Cen Y., Thomas C., Yamamoto H., Huber A., Kiss B., Houtkooper R.H. et al.. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011; 13:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ljungman M., Hanawalt P.C.. Efficient protection against oxidative DNA damage in chromatin. Mol. Carcinog. 1992; 5:264–269. [DOI] [PubMed] [Google Scholar]
- 45. Takata H., Hanafusa T., Mori T., Shimura M., Iida Y., Ishikawa K., Yoshikawa K., Yoshikawa Y., Maeshima K.. Chromatin compaction protects genomic DNA from radiation damage. PLoS One. 2013; 8:e75622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roginskaya M., Bernhard W.A., Razskazovskiy Y.. Protection of DNA against direct radiation damage by complex formation with positively charged polypeptides. Radiat. Res. 2006; 166:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Storch K., Eke I., Borgmann K., Krause M., Richter C., Becker K., Schrock E., Cordes N.. Three-dimensional cell growth confers radioresistance by chromatin density modification. Cancer Res. 2010; 70:3925–3934. [DOI] [PubMed] [Google Scholar]
- 48. Ocampo A., Reddy P., Martinez-Redondo P., Platero-Luengo A., Hatanaka F., Hishida T., Li M., Lam D., Kurita M., Beyret E. et al.. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell. 2016; 167:1719–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laugel V. Cockayne syndrome: the expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013; 134:161–170. [DOI] [PubMed] [Google Scholar]
- 50. Natale V. A comprehensive description of the severity groups in Cockayne syndrome. Am. J. Med. Genet. A. 2011; 155A:1081–1095. [DOI] [PubMed] [Google Scholar]
- 51. Fousteri M., Vermeulen W., van Zeeland A.A., Mullenders L.H.. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol. Cell. 2006; 23:471–482. [DOI] [PubMed] [Google Scholar]
- 52. Zarkovic G., Belousova E.A., Talhaoui I., Saint-Pierre C., Kutuzov M.M., Matkarimov B.T., Biard D., Gasparutto D., Lavrik O.I., Ishchenko A.A.. Characterization of DNA ADP-ribosyltransferase activities of PARP2 and PARP3: new insights into DNA ADP-ribosylation. Nucleic Acids Res. 2018; 46:2417–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martin-Hernandez K., Rodriguez-Vargas J.M., Schreiber V., Dantzer F.. Expanding functions of ADP-ribosylation in the maintenance of genome integrity. Semin. Cell Dev. Biol. 2017; 63:92–101. [DOI] [PubMed] [Google Scholar]
- 54. Wei H., Yu X.. Functions of PARylation in DNA damage repair pathways. Genomics Proteomics Bioinformatics. 2016; 14:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stevnsner T., Muftuoglu M., Aamann M.D., Bohr V.A.. The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mech. Ageing Dev. 2008; 129:441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hinz J.M., Czaja W.. Facilitation of base excision repair by chromatin remodeling. DNA Repair (Amst.). 2015; 36:91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hansel-Hertsch R., Beraldi D., Lensing S.V., Marsico G., Zyner K., Parry A., Di Antonio M., Pike J., Kimura H., Narita M. et al.. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016; 48:1267–1272. [DOI] [PubMed] [Google Scholar]
- 58. Mozzetta C., Boyarchuk E., Pontis J., Ait-Si-Ali S.. Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat. Rev. Mol. Cell Biol. 2015; 16:499–513. [DOI] [PubMed] [Google Scholar]
- 59. Luijsterburg M.S., de Krijger I., Wiegant W.W., Shah R.G., Smeenk G., de Groot A.J.L., Pines A., Vertegaal A.C.O., Jacobs J.J.L., Shah G.M. et al.. PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol. Cell. 2016; 61:547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Adam S., Polo S.E., Almouzni G.. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell. 2013; 155:94–106. [DOI] [PubMed] [Google Scholar]
- 61. Sun Q.Y., Ding L.W., Xiao J.F., Chien W., Lim S.L., Hattori N., Goodglick L., Chia D., Mah V., Alavi M. et al.. SETDB1 accelerates tumourigenesis by regulating the WNT signalling pathway. J. Pathol. 2015; 235:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rodriguez-Paredes M., Martinez de Paz A., Simo-Riudalbas L., Sayols S., Moutinho C., Moran S., Villanueva A., Vazquez-Cedeira M., Lazo P.A., Carneiro F. et al.. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene. 2014; 33:2807–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang H., Cai K., Wang J., Wang X., Cheng K., Shi F., Jiang L., Zhang Y., Dou J.. MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells. 2014; 32:2858–2868. [DOI] [PubMed] [Google Scholar]
- 64. Regina C., Compagnone M., Peschiaroli A., Lena A.M., Melino G., Candi E.. DeltaNp63alpha modulates histone methyl transferase SETDB1 to transcriptionally repress target genes in cancers. Cell Death Discov. 2016; 2:16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen K., Zhang F., Ding J., Liang Y., Zhan Z., Zhan Y., Chen L.H., Ding Y.. Histone methyltransferase SETDB1 promotes the progression of colorectal cancer by inhibiting the expression of TP53. J. Cancer. 2017; 8:3318–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fei Q., Shang K., Zhang J., Chuai S., Kong D., Zhou T., Fu S., Liang Y., Li C., Chen Z. et al.. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat. Commun. 2015; 6:8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wong C.M., Wei L., Law C.T., Ho D.W., Tsang F.H., Au S.L., Sze K.M., Lee J.M., Wong C.C., Ng I.O.. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology. 2016; 63:474–487. [DOI] [PubMed] [Google Scholar]
- 68. Reid-Bayliss K.S., Arron S.T., Loeb L.A., Bezrookove V., Cleaver J.E.. Why Cockayne syndrome patients do not get cancer despite their DNA repair deficiency. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:10151–10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kamenisch Y., Fousteri M., Knoch J., von Thaler A.K., Fehrenbacher B., Kato H., Becker T., Dolle M.E., Kuiper R., Majora M. et al.. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J. Exp. Med. 2010; 207:379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Scheibye-Knudsen M., Croteau D.L., Bohr V.A.. Mitochondrial deficiency in Cockayne syndrome. Mech. Ageing Dev. 2013; 134:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sun N., Youle R.J., Finkel T.. The mitochondrial basis of aging. Mol. Cell. 2016; 61:654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fang E.F., Kassahun H., Croteau D.L., Scheibye-Knudsen M., Marosi K., Lu H., Shamanna R.A., Kalyanasundaram S., Bollineni R.C., Wilson M.A. et al.. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016; 24:566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Koide S., Oshima M., Takubo K., Yamazaki S., Nitta E., Saraya A., Aoyama K., Kato Y., Miyagi S., Nakajima-Takagi Y. et al.. Setdb1 maintains hematopoietic stem and progenitor cells by restricting the ectopic activation of nonhematopoietic genes. Blood. 2016; 128:638–649. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the raw and processed ChIP sequencing and ADPr-ChAP sequencing data reported in this paper is GEO: GSE133176.