


Abstract
DNA replication forks are intrinsically asymmetric and may arrest during the cell cycle upon encountering modifications in the DNA. We have studied real time dynamics of three DNA polymerases and an exonuclease at a single molecule level in the bacterium Bacillus subtilis. PolC and DnaE work in a symmetric manner and show similar dwell times. After addition of DNA damage, their static fractions and dwell times decreased, in agreement with increased re-establishment of replication forks. Only a minor fraction of replication forks showed a loss of active polymerases, indicating relatively robust activity during DNA repair. Conversely, PolA, homolog of polymerase I and exonuclease ExoR were rarely present at forks during unperturbed replication but were recruited to replications forks after induction of DNA damage. Protein dynamics of PolA or ExoR were altered in the absence of each other during exponential growth and during DNA repair, indicating overlapping functions. Purified ExoR displayed exonuclease activity and preferentially bound to DNA having 5′ overhangs in vitro. Our analyses support the idea that two replicative DNA polymerases work together at the lagging strand whilst only PolC acts at the leading strand, and that PolA and ExoR perform inducible functions at replication forks during DNA repair.
INTRODUCTION
DNA replication is carried out in all organisms by a multiprotein complex called the replisome (1–3). Owing to the high level of functional similarity of replication proteins in different species from bacteria and phages to eukaryotes, bacterial replication has been used as a model system (2,4,5). According to text books, DNA replication is driven by two or three major DNA polymerases in all cells (6). In eukaryotes, two different polymerases mediate replication at leading or lagging strand and polymerase I removes RNA primers at the lagging strand (5). In Escherichia coli cells, the same polymerase (PolC) acts at both strands, and polymerase I (PolA) removes RNA hybrids (2,7). The stoichiometry and architecture of the replicative DNA polymerase holoenzyme (Pol III) have been well characterized in the model organism E. coli (8). Although E. coli has served as a prototype for understanding DNA synthesis in vivo, and although some E. coli DNA replication features are conserved across species, the replisomes of many other bacterial species have a distinct organization and may operate differently (9).
The replication machineries in the Gram-positive model bacterium Bacillus subtilis, in Streptococcus pyogenes and in Staphylocuccus aureus have also provided useful model systems for understanding unique aspects of DNA replication, from both a genetic and biochemical perspective (10–12). Some features of E. coli replication are conserved (1), however, genetic studies have demonstrated that both PolC and DnaE are required for B. subtilis replication (13). In the absence of DnaE, leading strand DNA synthesis remains active whilst lagging strand synthesis ceases (14), suggesting that PolC is the leading strand replicase and DnaE the counterpart at the lagging strand (6). Indeed, only DnaE is able to extend primase-generated RNA primers at the lagging strand (1,15,16). However, recent in vitro experiments indicated that PolC also acts at the lagging strand, possibly by extending stretches synthesized by DnaE, which has relatively slow polymerase activity in vitro (1,14), and would not be able to keep up with polymerization speed set by the leading strand. Interestingly, DnaE lacks proof reading activity, and is able to bypass DNA lesions in an error-prone manner (17). Thus, an alternative scenario could be that PolC takes over a proofreading function at the lagging strand.
Structural characterization and biochemical studies of several prokaryotic DNA polymerase I (Pol I, or PolA) enzymes has established an organization into three functional domains: an N-terminal domain associated with a 5′-3′ exonuclease activity, a central domain that mediates proofreading of the 3′-5′ exonuclease activity and a C-terminal domain responsible for the polymerase activity (18). PolC also contains both DNA polymerase and proofreading 3′→5′ exonuclease activities in one polypeptide chain, whereas DnaE has no proofreading activity (19). Although short DNA fragments containing RNA accumulate in B. subtilis cells in which PolA has been inactivated (20), indicating that it removes RNA primers at the lagging strand similar to Pol I in E. coli, the polA gene is not essential, suggesting that a protein other than PolA performs the essential function of primer removal.
In spite of good knowledge on biochemical properties of the B. subtilis replicative DNA polymerases, the in vivo composition and architecture of the replisome is little investigated. Interestingly, genes encoding proteins acting on the lagging strand have evolved at a significantly higher rate than those dealing with the leading strand (13,14). All these features raise the question whether there is a division of labour between the two replicative DNA polymerases, each one being devoted to one strand (21), or if the B. subtilis replisome is more eukaryotic-like in that it relies on a two DNA polymerase system for chromosomal replication (5), using two polymerases at both strands.
DNA polymerases also play a key role in various DNA repair mechanisms, ensuring faithful chromosome replication and maintenance of their genome integrity (22). This includes the repair of oxidized, alkylated or deaminated DNA bases, as well as of DNA crosslinks and UV light-induced DNA damage (23). This variety of DNA lesions requires modular repair pathways that carry out damage recognition, damage removal, repair synthesis and ligation in sequential steps catalysed by a series of enzymes (24,25). Additionally, all repair pathway steps need to be precisely balanced to avoid accumulation of DNA intermediates that are typically more mutagenic and toxic than the original lesion (26). Rapid processing of gapped and nicked intermediates is particularly crucial (4) because they provoke lethal double-strand breaks upon encountering replication forks (27); a single such break can lead to chromosome loss and cell death. Base-excision and nucleotide-excision repair remove short sections of the damaged DNA, leaving single-stranded DNA gaps to be filled and sealed by Pol I. Indeed, single-molecule tracking experiments revealed transient binding of individual Pol I and ligase molecules in the presence of DNA methylation damage, allowing base-excision repair rates to be quantified in live E. coli cells (28).
Given that the less well-understood B. subtilis replisome appears more eukaryotic-like than the E. coli replisome but appears to be distinct from both systems, a deeper in vivo understanding of how DNA polymerase dynamics occur in B. subtilis is necessary. To quantitatively image how the replicative DNA polymerases move, bind and unbind to the replication machinery in B. subtilis and also to address the question how they deal with induced DNA damage, we applied single-molecule tracking (8,28,29) to examine the in vivo behaviour of PolC, DnaE and PolA. We also studied YpcP, which we term ExoR hereafter, which is homologous to the exonuclease domain of PolA. In contrast to E. coli Pol I, PolA is not essential in B. subtilis, which has been surprising given its evolutional conservation. Interestingly PolA becomes essential in the absence of ExoR (25,30,31), indicating that ExoR might be able to take over an essential function of PolA; whether this occurs at the replication forks is not known.
Reporting on the dynamics of PolC, DnaE, PolA and ExoR proteins within live cells and within regard to the replication machinery, we provide in vivo evidence that B. subtilis replication forks present unusual features not known from replication machineries in E. coli and eukaryotic systems.
MATERIALS AND METHODS
Bacterial strains and growth conditions
The bacterial strains and plasmids used in this study are listed in Supplementary Table S1, and the nucleotides are listed in Supplementary Table S2. Escherichia coli strain XL1-Blue (Stratagene) was used for the construction and propagation of plasmids and E. coli strain BL21 Star DE3 (Invitrogen) for the heterologous overexpression of proteins. All B. subtilis strains were derived from the wild-type strain BG214. Cells were grown in Luria-Bertani (LB) rich medium at 37°C or 30°C or in minimal medium containing S750 salts at 30°C (32). When needed, antibiotics were added at the following concentrations (in μg/ml): ampicillin, 100; chloramphenicol, 5; spectinomycin, 100; kanamycin, 30. When required, media containing mitomycin C (MMC) were prepared by adding appropriate volumes of a filter-sterilized solution 50 ng/ml.
Construction of strains
PolC, DnaE, PolA, ExoR and DnaX were visualized as a PolC-mV, DnaE-mV, PolA-mV, ExoR-mV and DnaX-mV fusion proteins expressed at the original locus. The last 500 bp coding for each gene were integrated into vector pSG1164-mVenus (33), using ApaI and EcoRI restriction sites, and BG214 cells were transformed with this construct, selecting for cm resistance (leading to strains Supplementary Table S1). For colocalization studies, DnaX-CFP was integrated at amyE locus by the use of the plasmid pSG1192 and expression was controlled by xylose addition (34). To investigate colocalization of PolC, DnaE, PolA and ExoR, the resulting strains PG3306, PG3307, PG3308 and PG3309 (see Supplementary Table S1) was transformed with chromosomal DNA of strains leading to the expression of PolC-mV, DnaE-mV, PolA-mV and ExoR-mV to DnaX-CFP. A strain with mV-tagged PolA or ExoR in an polA or exoR deletion background strains was generated by transformation of competent ΔypcP::kan trpC2 or ΔpolA::kan trpC2 cells, obtained from the Bacillus Genetic Stock Center (Columbus, Ohio) (35), with chromosomal DNA from a strain expressing either PolA-mV or ExoR-mV.
For expression of soluble 6xHis-ExoR, the coding sequence lacking the first 10 codons was amplified by polymerase chain reaction using chromosomal DNA from B. subtilis wild-type strain BG214. The fragment was further integrated in the expression vector pET28a (Novagen) by EcoRI and XhoI restriction ligation and brought into the expression host E. coli BL21 (DE3) giving rise to the strain pET28a::exoRHisTag.
Protein purification
Protein purification was performed in two consecutive steps. The purification of (His)6-ExoR initially began with affinity chromatography using an ÄKTA Prime apparatus (GE Healthcare) and Nickel-Sepharose columns (HisTrap HP 1 ml, GE Healthcare) and was continued by size-exclusion chromatography using an ÄKTA FPLC apparatus (GE Healthcare) and a gel filtration column (Superdex 75 16/60 GL, GE Healthcare). For details, see Supplementary Material.
Fluorescence microscopy
For fluorescence microscopy, B. subtilis cells were grown in S750 minimal medium at 30°C under shaking conditions until exponential growth. Conventional light microscopy was performed using a Zeiss Observer Z1 (Carl Zeiss) with an oil immersion objective (100 × magnification, 1.45 numerical aperture, alpha Plan-FLUAR, Carl Zeiss) and a CCD camera (CoolSNAP EZ, Photometrics). Data were processed using Metamorph 7.5.5.0 software (Molecular Devices, Sunnyvale, CA, USA).
Single molecule microscopy and tracking
In contrast to the wide-field illumination used in conventional epifluorescence microscopy, the excitation laser beam used in our setup is directed to underfill the back aperture of the objective lens, generating a concentrated parallel illumination profile at the level of the sample, leading to a strong excitation followed by rapid bleaching of the fluorophores. When only a few unbleached molecules are present, their movement can be tracked. In addition, freshly synthesized and folded fluorophores become visible when the sample is excited again. When an observed molecule is bleached in a single step during the imaging, it is assumed to be a single molecule (8,36). Image acquisition was done continuously during laser excitation with the electron-multiplying CCD (EMCCD) camera iXon Ultra (Andor Technology, Belfast, UK). A total of 1500 frames were taken per movie, with an exposure time of 20 ms (23 frames per second [fps]). The microscope used in the process was an Olympus IX71, with a × 100 objective (UAPON 100 × OTIRF; numerical aperture [NA], 1.49; oil immersion). A 514-nm laser diode was used as excitation source, and the band corresponding to the fluorophore was filtered out. Of note, cells continued to grow after imaging, showing that there is little to no photodamage during imaging, whilst cells stop growing when exposed to blue light (below 480 nm). Acquired streams were loaded into Fiji ImageJ (37). Automated tracking of single molecules was done using the ImageJ plugin MtrackJ, or u-track 2.1.3 (38).
Diffusion analysis of single-molecule tracks
Tracking analysis was done with u-track-2.1.3, which was specifically written for Matlab (MathWorks, Natick, MA, USA). Only trajectories consisting of a minimum of five frames were considered tracks and included for further analysis. A widely accepted method to analyse the diffusive behaviour of molecules is by using the mean squared displacement (MSD)-versus-time-lag curve (39,40). This provides an estimate of the diffusion coefficient as well as of the kind of motion, e.g. diffusive, subdiffusive or directed. However, the method requires that within a complete trajectory there be only one type of homogeneous motion and that the trajectory is preferably of infinite length. To distinguish immobile and mobile molecules from each other, we compare the frame-to-frame displacement of all molecules in x and the y directions. Using a Gaussian mixture model (GMM) to fit the probability density distribution function of all frame-to-frame displacements, determine the standard deviations σ1 and σ2, as well as the percentages F1 and F2 of the slow and the fast subfractions of molecules, respectively. Finally, the diffusion constants were calculated according 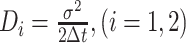 where Δt is the time interval between subsequent imaging frames.
where Δt is the time interval between subsequent imaging frames.
Generation of heat maps, analyses of molecule dwell times, and visualization of slow and fast tracks in a standardized cell are based on a custom-written Matlab script (SMTracker) that is available on request (41). SMTracker can use particle tracking tools u-track (38) and TrackMate (42) and computes the x- and y-coordinates of molecular trajectories relative to the geometry of each cell, as obtained by the cell segmentation tools MicrobeTracker (39) or Oufti (43).
Statistical data analysis
The goodness of fits of the GMMs was assessed using probability–probability plots (pp-plots) (Supplementary Figure S5). Errors on the fitted parameters are given as 95% confidence intervals, which were derived from the Jacobian matrix of the nonlinear optimization process using the MATLAB™ function nlparci. To compare fraction sizes and diffusion constants under different conditions and between different proteins, statistical hypothesis-testing was performed using a Kolmogorov–Smirnov 2-sample test to the step size distributions. Differences in dwell times distribution were tested and scored using Student's t-test in the cases where Levene test for equality of variances was not rejected. To assess the most likely number of populations for each fit, we applied the Bayesian Information Criterion (BIC), as detailed in (41). As usual, *, ** and *** stands for P-values lower than 0.1, 0.05 and 0.01, respectively, whilst n.s. stands for statistically not significant. Statistical hypothesis testing and plotting was performed using SMTracker (41) and MATLAB™ custom written scripts.
RESULTS
PolA and ExoR do not visibly assemble at the replication forks during exponential growth, but after induction of DNA damage
Cell biological studies have shown that B. subtilis DNA polymerases do not move through the cytoplasm whilst actively replicating DNA, in contrast to the E. coli replisome (44). Rather, the B. subtilis replisome has been shown to reside in a more restricted location through which template DNA is pulled in, and newly synthesized DNA is extruded towards the cell poles, which has been suggested to facilitate chromosome segregation (45). Therefore, the position of replication forks can be determined using epifluorescence microscopy and fluorescent protein fusions to components of the machinery. In order to address the question whether PolA and ExoR are also components of the replisome, we generated mVenus (mV) fusions to the C-terminus of ExoR and to the three DNA polymerases PolA, PolC and DnaE, which were integrated at the original gene locus on the chromosome. Thereby, solely fusion proteins are expressed under the control of the original promoter, ensuring the expression of wild-type levels for all proteins (Supplementary Table S1). Presence of full-length proteins (and no free mVenus or degradation products) was verified using Western blotting (Supplementary Figure S1). PolC-mV and DnaE-mV fully complemented the wild-type copies, whilst PolA-mV and ExoR-mV showed slightly reduced survival in response to treatment with 40 μg/ml mitomycin C (MMC), but not to 50 ng/ml (Supplementary Figure S2A). Because PolA-mV and ExoR-mV expressing cells were much more viable than the corresponding mutant cells (Supplementary Figure S2), both fusion proteins retain almost full functionality compared to the wild-type proteins.
Epifluorescence experiments showed that whilst PolC-mV and DnaE-mV colocalized with DnaX-CFP, a component of the clamp-loader complex, and thus visibly accumulated at the forks, neither PolA-mV nor ExoR-mV provided detectable foci during exponential growth (Figure 1). In order to find out if either of the proteins might be visible at forks in the absence of the other, we moved the FP fusions into the corresponding deletion background. Neither of the two proteins showed foci associated with DnaX-CFP but were visually localized throughout the cells (Figure 1), showing that there is no obvious visual complementation.
Figure 1.
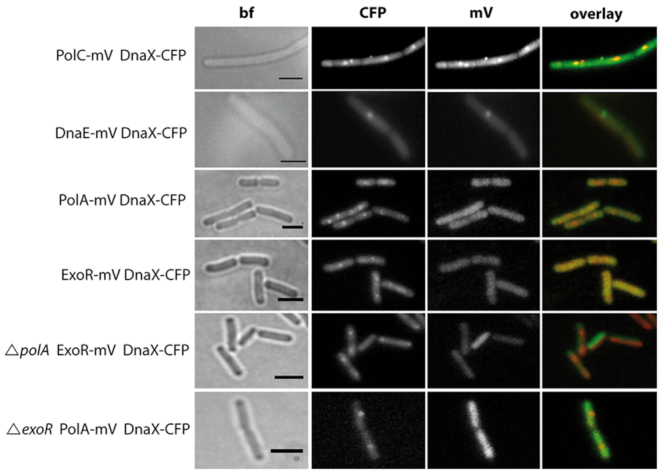
Localization of polymerases during exponential growth. Subcellular localization by epifluorescence in representative untreated live Bacillus subtilis cells. The overlay panels show DnaX-CFP in red and PolC-mV, DnaE-mV, PolA-mV and ExoR-mV in green. Scale bar: 2 μm.
We used chemical (mitomycin C, MMC) and physical (UV irradiation) damage to assess if (i) the DNA III-type polymerases PolC and DnaE remain bound to forks during DNA damage repair, and if (ii) PolA or ExoR might be recruited to the forks under these conditions. Figure 2 and Supplementary Figure S3 show that replication forks were maintained, as judged by the continued presence of single or two fluorescent DnaX-CFP foci per cells, and forks retained both, PolC and DnaE. Quantification of foci showed that 100 DnaX-CFP foci colocalized with 96 PolC-mV or 92 DnaE-mV foci after MMC treatment, showing that in spite of running into DNA interstrand crosslinks, or base dimers, replication forks rarely disassemble in vivo, or visibly lose replicative polymerases.
Figure 2.
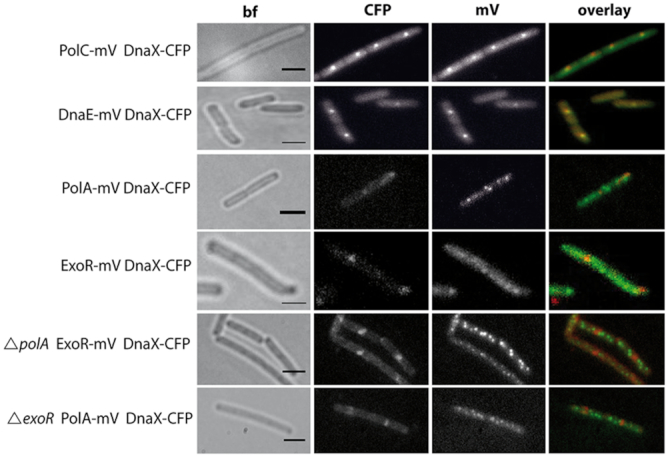
Altered localization patterns after induction of DNA damage. Localization by epifluorescence in representative live Bacillus subtilis cells after induction of DNA damage with MMC. The overlay panels show DnaX-CFP in red and PolC-mV, DnaE-mV, PolA-mV, and ExoR-mV in green. Note that DnaX-CFP foci rarely colocalize with PolA-mV or ExoR-mV foci. Scale bar: 2 μm.
Interestingly, cells contained multiple PolA-mV or ExoR-mV foci, both after treatment with MMC or after UV irradiation. In many cells, one or two foci (out of up to 10 foci in total) colocalized with DnaX-CFP, suggesting that both proteins might get recruited to forks during replication stress/DNA repair. We will come back to this point below. Clearly though, most PolA-mV or ExoR-mV foci did not colocalize with replication forks after damage induction, indicating that PolA and ExoR act at many chromosomal sites to remove DNA crosslinks and base modifications, independently from replication forks.
DnaE and PolC show similar dynamics at the single molecule level
We wished to further investigate (i) if ExoR shows a similar subcellular pattern of movement as PolA, (ii) if PolA plays its essential function at B. subtilis replication forks, (iii) if ExoR might complement a function of PolA at the forks and (iv) how PolA behaves relative to PolC and DnaE. Epifluorescence fails to visualize very short events of molecules resting at a subcellular site (dwell time) and cannot detect mobile/diffusive molecules. We therefore employed single molecule tracking (SMT), which can follow the movement of single mVenus-fused proteins. We are using YFP-type SMT, in which most mVenus fusions are bleached until few molecules remain that can be tracked. We prefer this technique over PALM tracking, because blue light (as required for PALM tracking of fluorophores PAmCherry or mEOS) arrests the B. subtilis cell cycle (unpublished results) and in our hands, B. subtilis cells show very high red auto-fluorescence under intensive excitation with 561 nm in cells lacking any FP fusion, which is not the case for 514 nm excitation required for mVenus excitation. DNA polymerase-mV fusions and ExoR-mV were tracked with 20 ms streams (50 fps), which yielded characteristic patterns of movement shown Supplementary Movies S1 and 2. Supplementary Figure S4: molecules could be static for several frames (Supplementary Figure S4A), which is defined as movement of <230 nm (Supplementary Figure S4D and E) or were mobile (Supplementary Figure S4F). The presence of at least two distinct fractions of stationary and mobile molecules can be seen in Supplementary Movies S1 and 2. No more than three signals per cell were allowed to ensure correct tracking, and single molecules were identified due to one step bleaching events (Supplementary Figure S4B and G). In order to obtain 2D patterns of movement, tracks obtained from many cells were projected into a standardized 3 × 1 μm large cell (Supplementary Figure S4D and I).
We used a GMM to fit the probability density distribution function of all displacements from frame to frame. A single, freely diffusive population of molecules would be explained by a single Gaussian distribution {see e.g. PfkA enzyme in (46)}. However, for polymerases, a superposition of two Gaussians (red curves in Figure 3 and Figure 4) was necessary to fit to the experimental displacement distributions. This was verified using pp plots (Supplementary Figure S5), where experimental data fit better to a two-population model than to a single population model. The algorithm infers two diffusion constants, D1 and D2, corresponding to one fraction of immobile and another fraction of mobile molecules (Figure 3). In case of DNA polymerases, D1 would refer to the active, tightly DNA bound molecules and D2 to molecules being in the diffusive state. From fitting the variances and respective areas under the two Gaussians, we then determined the diffusion constants and relative fractions of molecules in the mobile and immobile states. The changes in the width of the step size distributions are a convenient visual tool to see if molecules become more static (the distribution becomes narrower) or more dynamic (wider distribution). The size of the bubbles indicates the fraction sizes, and the lower bubble corresponds to the static fraction having a lower diffusion constant (shown on the y-axis).
Figure 3.

Diffusion patterns of DNA polymerases. GMM analyses of frame-to-frame displacements in x- and y-directions. (A) PolC-mV (B) DnaE-mV. Red lines represent the sum of the two Gaussian distributions. Dotted and dashed lines represent the single Gaussian distributions corresponding to the static and mobile fractions. Bubble plots show a comparison of fraction sizes (size of the bubble) and diffusion constants (y-axis), between different growth conditions: distribution in untreated cells (dark blue circles), in MMC-treated (yellow) and in UV-treated (light blue circles) cells. Step size distributions reveal two populations for each protein, a mobile (upper circles) and a static (lower circles) fraction.
Figure 4.

Diffusion patterns of PolA and of ExoR. GMM analyses of frame-to-frame displacements in x- and y-directions. (A) PolA-mV, (B) ExoR-mV, (C) PolA-mV ΔexoR (D) ExoR-mV ΔpolA. See Figure 3 for explanations.
Interestingly, PolC and DnaE showed relatively similar dynamics: for PolC 55% of the molecules were in a slow/static mode with D1,static = 0.120 ± 0.021 μm2/s compared to 51% of DnaE molecules with D1,static = 0.069 ± 0.007 μm2/s. 45% PolC molecules moved quickly, likely in a feely diffusive mode, with D2,mobile = 0.930 ± 0.160 μm2/s and 49% for DnaE with D2,mobile = 0.790 ± 0.080 μm2/s (Figure 3, Table 1). This distribution cannot be explained with PolC being exclusively engaged in continuous leading strand synthesis, because only one out of the ten or more proteins per cell would be engaged in a static mode.
Table 1.
Diffusion constants and percentages of static and mobile molecule fractions
| Strain | # cells | # tracks | D1a | F1b | D2c | F2d |
|---|---|---|---|---|---|---|
| No Damage | ||||||
| PolC-mV | 37 | 147 | 0.120 ± 0.021 | 55 ± 7.8 | 0.930 ± 0.160 | 45 ± 7.8 |
| DnaE-mV | 44 | 515 | 0.069 ± 0.007 | 51 ± 3.7 | 0.790 ± 0.080 | 49 ± 3.7 |
| PolA-mV | 40 | 3806 | 0.060 ± 0.005 | 6 ± 0.59 | 0.640 ± 0.006 | 94 ± 0.5 |
| ExoR-mV | 40 | 5217 | 0.100 ± 0.006 | 15 ± 1.1 | 0.650 ± 0.010 | 85 ± 1.1 |
| DnaX-mV | 29 | 1706 | 0.029 ± 0.002 | 46 ± 2.2 | 0.230 ± 0.008 | 54 ± 2.2 |
| ΔexoR PolA-mV | 29 | 2021 | 0.062 ± 0.003 | 22 ± 1.4 | 0.590 ± 0.017 | 78 ± 1.4 |
| ΔpolA ExoR-mV | 58 | 2770 | 0.079 ± 0.006 | 29 ± 2.3 | 0.580 ± 0.025 | 71 ± 2.3 |
| MMC-treated | ||||||
| PolC-mV | 33 | 245 | 0.120 ± 0.021 | 42 ± 7.6 | 0.930 ± 0.160 | 58 ± 7.6 |
| DnaE-mV | 44 | 278 | 0.069 ± 0.007 | 49 ± 4.0 | 0.790 ± 0.080 | 51 ± 4.0 |
| PolA-mV | 24 | 713 | 0.060 ± 0.005 | 31 ± 1.2 | 0.640 ± 0.006 | 69 ± 1.2 |
| ExoR-mV | 29 | 3160 | 0.100 ± 0.006 | 28 ± 1.4 | 0.650 ± 0.010 | 72 ± 1.4 |
| DnaX-mV | 32 | 2209 | 0.029 ± 0.002 | 33 ± 2.2 | 0.230 ± 0.008 | 67 ± 2.2 |
| ΔexoR PolA-mV | 38 | 869 | 0.062 ± 0.003 | 46 ± 1.6 | 0.590 ± 0.017 | 54 ± 1.6 |
| ΔpolA ExoR-mV | 30 | 1082 | 0.079 ± 0.006 | 45 ± 2.6 | 0.580 ± 0.025 | 55 ± 2.6 |
| UV-treated | ||||||
| PolC-mV | 31 | 170 | 0.120 ± 0.021 | 49 ± 7.6 | 0.930 ± 0.160 | 51 ± 7.6 |
| DnaE-mV | 39 | 395 | 0.069 ± 0.007 | 40 ± 3.8 | 0.790 ± 0.080 | 60 ± 3.8 |
| PolA-mV | 37 | 1517 | 0.060 ± 0.005 | 5 ± 0.66 | 0.640 ± 0.006 | 95 ± 0.6 |
| ExoR-mV | 29 | 4272 | 0.100 ± 0.006 | 18 ± 1.2 | 0.650 ± 0.010 | 82 ± 1.2 |
| DnaX-mV | 35 | 1834 | 0.029 ± 0.002 | 26 ± 2.1 | 0.230 ± 0.008 | 74 ± 2.1 |
| ΔexoR PolA-mV | 34 | 2799 | 0.062 ± 0.003 | 42 ± 1.4 | 0.590 ± 0.017 | 58 ± 1.4 |
| ΔpolA ExoR-mV | 34 | 926 | 0.079 ± 0.006 | 40 ± 2.5 | 0.580 ± 0.025 | 60 ± 2.5 |
a D 1, diffusion constant of static fraction (μm2·s−1).
b F 1, percentage of static molecules (%).
c D 2, diffusion constant of mobile fraction (μm2·s−1).
d F 2, percentage of mobile molecules (%)
PolA and ExoR dynamics change similarly after induction of DNA damage
Intriguingly, PolA and ExoR showed considerably different patterns of movement compared to PolC and DnaE: in the absence of DNA damage, PolA and ExoR consisted of a much smaller static fraction, and predominantly of mobile molecules, with PolA diffusing with a constant of D1,static = 0.060 ± 0.005 μm2/s (6%) and ExoR D1,static = 0.100 ± 0.006 μm2/s (15%) (Table 1, Figure 4A and C). These experiments reveal that PolC and DnaE have similar static fractions, whilst PolA and ExoR show only small fractions that would correspond to a DNA-bound state under exponential growth conditions. This picture changed markedly for PolA and for ExoR, when DNA damage was induced (Figure 4A and C, Table 1). Here, the static fractions of both of PolA as well as ExoR molecules increased (PolA: 31% static with D1,static = 0.060 ± 0.005 μm2/s and ExoR: 28% static with D1,static = 0.100 ± 0.006 μm2/s) after MMC-treatment. Following UV-treatment, 5% PolA enzymes remained static (D1,static = 0.060 ± 0.005 μm2/s) or 18% of ExoR (D1,static = 0.100 ± 0.006 μm2/s), which is only a moderate increase for ExoR. In agreement with the epifluorescence data, PolC and DnaE showed minor changes in their mobility upon addition of MMC or treatment with UV: 42% of PolC molecules (D1,static = 0.120 ± 0.021 μm2/s) and 49% of DnaE D1,static = 0.069 ± 0.007 μm2/s) were static (MMC-treatment) and 49% of PolC (D1,static = 0.120 ± 0.021 μm2/s) and 40% of DnaE (D1,static = 0.069 ± 0.007 μm2/s) after UV-irradiation (Figure 3A and B, Table 1), showing that there is only a moderate shift in the static fractions (i.e. replication fork bound molecules) of replicative DNA polymerase molecules during DNA damage repair under our experimental conditions (addition of 50 μg/ml of MMC leads to cell death in 25% of the cells, (47)). Thus, PolC and DnaE do not strongly change their diffusive/bound distribution after addition of MMC or after UV treatment (Figure 3), whilst the distribution of PolA and of ExoR changes in a strong and similar manner (Figure 4).
PolA and ExoR influence each other's dynamics during non-perturbed growth and after DNA damage induction
In order to investigate if PolA and ExoR change their dynamics when the other protein is missing, we performed SMT in a strain expressing PolA-mV and lacking ExoR, or vice versa. Interestingly, we observed significant differences between their mobilities: in the absence of ExoR, PolA-mV had a strongly increased static fraction (Figure 5B, blue and green circles), at the expense of the dynamic fraction (Figure 5A). This is also true for UV irradiation (Figure 5B, blue and green diamonds) and for addition of MMC (blue and green squares). For ExoR-mV, the diffusion constants of the static fractions were lower in the absence of PolA under all conditions (Figure 5B) and the static fractions were much higher (Figure 5B), with the opposite effect on the dynamic fractions (Figure 5A). These experiments show that PolA and ExoR have a considerable effect on each other, and suggest that their DNA-bound mode is increased when the other is missing. This is similar to e.g. ATPase RarA, involved in replication restart during DNA repair that shows large differences in dynamic/static (DNA-bound) fraction in mutant backgrounds lacking proteins involved in homologous recombination (48,49).
Figure 5.

Scatter plot and significance test results from dynamics and fraction sizes of polymerases. Scatter plot showing the relationship between the diffusion coefficients (Di) of PolA-mV, ExoR-mV, ΔpolA ExoR-mV and ΔexoR PolA-mV (y-axis) and comparison of fraction sizes (x-axis), of (A) fast and (B) slow-diffusing populations. (C) Results of the hypothesis testing to the differences in the fraction sizes of all proteins in terms of P-value: As usual, *, ** and *** stand for P-values lower than 0.1, 0.05 and 0.01, respectively, whilst n.s. stands for statistically not significant. The most relevant comparisons are highlighted in red.
PolC and DnaE show similar dwell times at the forks
Diffusion analyses show that PolC and DnaE have similar static and dynamic fractions, but they do not allow conclusions about exchange rates of molecules at the replication forks. To analyse this, we firstly investigated DnaX as a marker for the forks, with the rationale that DnaX dwell times at forks should be longer than those of DnaE, because the latter is expected to be exchanged with every new priming event, whilst this is likely not the case for DnaX. Figure 6 shows that DnaX has a similarly large static fraction (46% with D1,static = 0.029 ± 0.002 μm2/s) than PolC and DnaE (Table 1). We next scored the number of molecules in PolC-mV, DnaE-mV and DnaX-mV expressing cells that are immobile for a certain number of consecutive acquisition times, deduced from the probability that a molecule will remain inside a radius of 135 nm (1.35 pixels, three times our localization error) within a time frame t. Dwell times are estimated using an exponential decay mixture model, and the best result we obtained for all proteins employed a two-component model (see Supplementary Figure S6 for an example of PolA-mV), resulting in two distinct average dwell times, τ1 and τ2 (Supplementary Table S3), indicative of molecules having short dwell times (every freely diffusing molecule will stop once in a whilst) and of molecules binding somewhere in the cell: in this case to replication forks, which is the average dwell time of importance here. PolC and DnaE molecules dwelled on average PolC = 0.106 ± 0.007 s, and DnaE = 0.163 ± 0.012 s) (Supplementary Table S3 and Figure 7B), showing that they indeed come on and off the forks in a similar temporal manner. This can also be seen when their dwell times are compared with those of DnaX = 0.199 ± 0.006 s (Supplementary Table S3), because DnaX would be expected to stay longer at the forks compared to a lagging strand polymerase. Please note that determined dwell times are strong underestimates of actual dwell times in the cell, because of molecule bleaching during the acquisition. However, average dwell times are proportional between fusions and between different conditions, as all proteins were fused to the same fluorescent protein (mVenus, which has an average bleaching time of about 1.2 s (50)).
Figure 6.

Diffusion patterns of DnaX-mV compared to PolC-mV and DnaE-mV. (A) GMM analyses of frame-to-frame displacements in x- and y-directions of DnaX-mV, PolC-mV and DnaE-mV. Black lines represent the sum of the two Gaussian distributions. Dotted red and blue lines represent the single Gaussian distributions corresponding to the static and mobile fractions. (B) Bar plot with error bars shown illustrates fractions sizes in untreated cells and their error according to the 95% confidence intervals of the fit. Inside in white, each Diffusion coefficient in μm·s−2. As usual, *, ** and *** stands for p-values lower than 0.1, 0.05 and 0.01 respectively, whilst n.s. stands for statistically not significant according to a two-sample Kolmogorov–Smirnov significance test on the step size distributions. Note that the same diffusion constants for DnaE and PolC were chosen (these do not differ markedly from the actual diffusion constants), which slightly adapts the sizes of static and dynamic fractions, but allows for a direct comparison between the fraction sizes of the two proteins.
Figure 7.

Dwell times. (A) Relative differences of each strain once applied MMC and UV treatment respect to no treatment. (B) Average residence times (± standard error of the mean) of PolC-mV, DnaE-mV, PolA-mV, ExoR-mV, ExoR-mV ΔpolA and PolA-mV ΔexoR strains, before and after treatment with MMC or UV. Dwell times are estimated using an exponential decay model. Histograms show events of resting fitted by a two-component exponential function. *, ** and *** stand for P-values lower than 0.1, 0.05 and 0.01, respectively, whilst n.s. stands for statistically not significant changes on the dwell times distributions.
In agreement with some PolC and DnaE molecules leaving stalled forks in response to DNA damage, average dwell times became significantly shorter after addition of MMC for PolC from 0.106 ± 0.007 s to 0.095 ± 0.006 s (no change after UV irradiation, 0.11 ± 0.008 s), and for DnaE from 0.163 ± 0.012 s to 0.117 ± 0.009 s (MMC), or to 1.18 ± 0.005 after UV treatment) (Figure 7B). Interestingly, although DnaX continued to remain at replication forks irrespective of the induction of DNA damage (Figure 2 and Supplementary Figure S3), its dwell times became much lower (Figure 7B and Supplementary Table S3), revealing a higher subunit turnover of the clamp loader complex during the response to DNA damage.
For PolA and ExoR, average dwell times increased markedly, from 0.081 ± 0.001 s during exponential growth to 0.104 ± 0.007 s/0.096 ± 0.006 s after MMC/UV treatment for PolA (Supplementary Table S3), and in a very similar manner for ExoR (Figure 7B), revealing strongly increased DNA binding in response to DNA damage. These experiments not only verify that we can accurately detect changes in dwell times for DNA polymerases at the forks, but also underline our findings that PolA and ExoR become strongly DNA-bound, at many sites on the chromosome, during DNA repair. Average dwell times of PolA and ExoR increased in a statistically significant manner when one of the enzymes was missing (Supplementary Table S3). For ExoR and for PolA, dwell times were higher during exponential growth and during repair of UV-induced damage in the absence of each other, and for ExoR mildly during MMC induced damage (Figure 7B). These changes corroborate with the increase in the fraction of DNA-bound molecules (Figure 6B), supporting the idea that the two proteins affect each other's function in vivo and confer overlapping essential function(s).
PolA and ExoR are recruited to replication forks upon addition of DNA damage
From Figure 1, it appeared that PolA and ExoR are located throughout the cells, and are not associated with the nucleoids, whilst PolC and DnaE formed nucleoid-associated foci. In order to clarify if not even single PolA molecules are associated with the replication forks for short intervals, we tracked PolA-mV and ExoR-mV relative to DnaX-CFP. To understand how the motion varies with position within the cell, we developed a tool that allows to visualize the motion of tracked molecules relative to a defined position in the cell, in this case the replication fork (or one of the two visible DnaX-CFP foci), and mapped the step sizes of PolA-mV and ExoR-mV as a function of distance from the replisome DnaX-CFP. We first acquired the location of DnaX-CFP using 445 nm stream acquisition, which reveals the localization of static CFP molecules and then tracked mVenus fusion molecules in the same cells; representative examples of cells growing exponentially or having been treated with MMC or UV are shown in Figure 8A. Less than 5% of PolA-mV molecules showed static localization close to/at DnaX foci, corresponding to 85% of replication forks that did not show PolA-mV tracks. However, more than 90% of forks showed static PolA-mV tracks after addition of MMC or after UV treatment (Figure 8B), revealing that PolA becomes efficiently recruited to stalled replication forks. Very similarly, few to no ExoR molecules arrested at DnaX-CFP foci during unperturbed growth, whilst they clearly halted at forks after DNA damage induction (Figure 8B). The fact that most positions corresponding to a replisome show the presence of a considerable number of static PolA or ExoR molecules rules out that absence of PolA from forks during exponential growth is due to our inability to visualize all PolA molecules at a single molecule level, and supporting the findings from epifluorescence experiments that both proteins arrest at replication forks as well as at many chromosomal sites away from the replisome, during repair events. The fact that very few PolA or ExoR molecules arrest close to forks can be seen in Figure 8B, upper panels, whilst the recruitment to one, two or three visible forks can be seen in damaged cells (Figure 8B, middle and lower panels). Additionally, both PolA and ExoR become more statically positioned and less mobile after induction of DNA damage, which can be seen from the mostly flat trajectories, which is in agreement with GMM analyses. Thirdly, a histogram of the distance of trajectories to the nearest DnaX focus (Figure 8C) visualizes the strong overall shift of molecule localization towards replication forks. Therefore, PolA is not recruited to replication forks during exponential growth in a similar manner as PolC or DnaE, questioning whether it plays a significant role during unperturbed replication. If PolA was to remove RNA/DNA hybrids at the lagging strand, we would have expected to see a considerable fraction of PolA molecules at the forks. Similarly, ExoR is also transiently recruited to replication forks during active DNA repair, but rarely present during exponential growth.
Figure 8.

Localization of PolA and ExoR relative to replication forks. A) Location of DnaX-CFP and overlaid tracks from PolA-mV or from ExoR-mV in representative Bacillus subtilis cells untreated and after induction of DNA damage with MMC or UV light as indicated. (B) Distance of PolA-mV or ExoR-mV tracks relative to a DnaX-CFP focus, which is positioned at ‘0 μm’. Panels correspond to those in (A), such that the position of a second or third DnaX-CFP focus can be inferred after induction of DNA damage by the additional accumulation of static tracks far from the ‘0’ position. (C) Histogram showing the distances of PolA-mV and ExoR-mV trajectories to the location of DnaX-CFP in 50 cells, treated or untreated.
Heat maps can help to detect significant changes in binding patterns between different cellular states. Therefore, all localization events from hundreds of cells were plotted into a standardized 3 × 1 μm large cell. Supplementary Figure S7 shows that whilst PolC and DnaE mostly reside on the central part of the cells that is occupied by the nucleoid, PolA and ExoR are observed more diffusely throughout the cell without clear accumulations, except when DNA damage is induced; here, many more foci reside at places within the central part (i.e. the nucleoids) of the cells.
ExoR is a magnesium-dependent exonuclease and shows highest binding affinity to 5′ overhangs in dsDNA
ExoR is a small conserved protein of previously unknown function, and its sequence similarity is restricted to the N-terminal domain of PolA, which contains a 3′→5′ exonuclease motif shown to be functional in Streptococcus pneumoniae and E. coli (25,51,52). We performed structure modelling (53) based on the homology of ExoR to known exopolymerases, and found that ExoR can be superimposed with the exonuclease domain of Pol I (Supplementary Figure S8), revealing a close structural relation. For biochemical characterization, a 6His-ExoR construct was purified to homogeneity by Nickel-Sepharose affinity chromatography followed by size-exclusion chromatography (see ‘Materials and Methods’ section and Supplementary Figure S9). ExoR showed magnesium-dependent nuclease activity on double stranded DNA (Supplementary Figure S10), in agreement with its proposed structure. We wished to gain insight into preferred substrates of ExoR, and therefore performed DNA binding assays, in the absence of magnesium. Linear DNA fragments carrying regions of 68 bp or less (dsDNA, ssDNA and RNA) were mixed, and increasing amounts of ExoR of 0 to 750 nM molar excess with respect to DNA were added to the different substrates. As shown in the EMSA assays in Figure 9, well-defined complexes were detected even at the lowest amount of ExoR used. Defined and single shifted bands were observed on all dsDNA, ssDNA and RNA molecules, indicating a distinct number of ExoR molecules interacting with the substrate, similarly to all polymerases containing the C-terminal exonuclease domain (54). ExoR had a clear preference for dsDNA with a 5′ overhang (Figure 9A) over 3′ overhangs (Figure 9E), over blunt end dsDNA (Figure 9D). Interestingly, it bound more readily to an ssDNA template (Figure 9B) than to the corresponding dsDNA, and also to ssRNA (Figure 9C), whilst affinity to a DNA/RNA hybrid with a 5′ overhang was higher than to ssRNA or dsDNA (Figure 9F). Figure 9G shows a plot of the measured relative intensity of the bands in each gel, showing the highest affinity of ExoR for the 5′ overhang substrates. These results clearly show binding of ExoR to dsDNA is influenced by the DNA structure and support the idea that ExoR could extend single stranded gaps in DNA. Previous functional genomics studies have suggested that DNA polymerase I (PolA) is essential in S. pneumoniae (52), E. coli (51) and Salmonella enterica (55) but not in B. subtilis (56), S. aureus (57) or Haemophilus influenzae (58), but that a double exoR polA deletion is lethal. Our data suggest that exonuclease activity of either PolA or of ExoR must be present in B. subtilis to overcome damage-induced replication stress.
Figure 9.

DNA binding of ExoR. EMSAs showing ExoR binding to different nucleotide substrates. ExoR binding specifically to dsDNA panels A, D and E or ssDNA (panel B), or to RNA (panel C and F). EMSAs were performed with increasing amounts (0–750 nM) of purified ExoR and fragments of 68 bp, containing either DNA or RNA, generated by annealing of custom-made oligonucleotides. Samples were mixed with loading buffer and analysed through 6% (v/v) native polyacrylamide gels. Lanes labelled ‘0’ show the control substrate in the absence of protein. ExoR shows highest binding affinity to 5′ overhangs in dsDNA (panel A). Lines below each panel represent DNA in blue or RNA in red. (G) Plot of the measurement of the relative intensities of the two bands in the EMSA gels for each DNA/RNA substrate.
DISCUSSION
Many critical cell-cycle processes are achieved by multienzyme complexes, in which the timely interplay of components is crucial for the correct functioning of the machinery. This is especially true for replication forks, where the synthesis of two DNA strands occurs in parallel, however in a continuous and discontinuous manner for leading and lagging strand. The molecular mechanism of DNA synthesis has been intensively investigated in vitro (59,60), revealing distinct mechanisms of leading and lagging strand synthesis. The advent of SMT has made it possible to visualize the dynamics of single enzymes in real time, i.e. follow diffusion and binding/unbinding events in millisecond intervals, and with high spatial precision (down to 20 nm resolution). We have applied SMT to study B. subtilis replication forks to study the dynamics of two replicative polymerases, a subunit of the clamp loader complex (DnaX), polymerase I (PolA) and a putative exonuclease, ExoR (YpcP).
Our experiments document that PolC and DnaE are recruited to and released from the replisome in a time scale of few seconds, with both polymerases exchanging with similar kinetics, but slower than DnaX, a part of the clamp loader complex (Supplementary Figure S11). If PolC was exclusively involved in leading strand synthesis, we would have expected much longer dwell times than those of DnaE and a much lower fraction of proteins being involved in replication. Thus, our findings are in agreement with genetic and biochemical experiments showing that PolC and DnaE work together at the lagging strand. Because only DnaE can extend RNA primers but is a relatively slow and error-prone enzyme in vitro (1,14,61), PolC must take over DNA extensions made by DnaE. It had been speculated that DnaE only briefly carries out primer extension, and leaves elongation to PolC, which has proofreading activity. Our findings that PolC and DnaE exchange in a similar manner support the idea that DnaE is involved in both, RNA primer extension as well as in elongation, which becomes less error-prone in the presence of PolC, which directly interacts with DnaE (62). Our data show that PolC and DnaE operate with balanced dynamics at replication forks, supporting the findings of coordinated activity at the lagging strand (bearing in mind that leading strand activity and exchange of PolC will hardly affect the measured in vivo exchange rates).
The measured slow polymerase activity of DnaE at the lagging strand is increased in the presence of DnaN and of PolC (62), so PolC has been proposed to speed up lagging strand synthesis to keep up with the fast speed of leading strand synthesis. The current model of a hand-over of short DnaE-generated extension of RNA primers (generated by primase) to PolC for rapid further extension (Supplementary Figure S11) is supported by our in vivo experiments. Interestingly, in vitro single molecule experiments with E. coli replication forks have shown that leading strand polymerase activity comprises stochastic stopping and slowing down events, so lagging strand synthesis can catch up, and furthermore that DNA helicase can adapt its activity to slowed-down synthesis at the lagging strand (63).
A second important finding is an apparent stability of replication forks in response to the induction of chemical damage to DNA, which is thought to lead to stalling of replication forks. We found that only about 20% of PolC and DnaE are displaced from the forks during repair, under a condition where 25% of the cells die (47). Although we have no direct measure for how many replication forks are blocked under our experimental conditions, our results suggest that the replication machinery is relatively robust against chemical insults, and that in response to conditions leading to severe loss of viability, 20% of forks will exchange PolC and/or DnaE for translesion DNA polymerases, which can polymerize over non-canonical base pairs that are induced by e.g. UV irradiation (64). Interestingly, DnaE can also operate in an error-prone mode (17), and may even switch template, to a stalled leading strand to bypass lesions (65). We would like to point out that in case the fluorescent protein fusions generated for this study were not fully functional, we would have expected high loss of PolC and DnaE from the forks, but the opposite was true, strongly suggesting that our observations closely reflect actual processes in vivo.
A third major conceptual insight from our study is the scarce presence of polymerase I (PolA) at replication forks during normal replication (Supplementary Figure S11). If PolA were to remove RNA hybrids left in Okazaki fragments like Pol I in E. coli cells, we would have expected a behaviour of PolA at least partially similar to that of DnaE. However, we found that few PolA molecules dwell at the replication machinery during exponential growth, and a vast majority is either statically positioned at various sites on the chromosome of diffusing through the cell. Conversely, PolA become significantly enriched at forks after induction of DNA damage, be it caused by chemical or physical (UV) insult. These data strongly suggest that an enzyme other than PolA removes RNA primers at the lagging strand, and that PolA performs an inducible repair function at stalled forks.
A fourth important contribution of our study is the finding that similar to PolA, ExoR showed a considerably different pattern of movement and binding events in response to DNA damage. Whilst during exponential growth, ExoR was mostly diffusive or occasionally arrested at random places on the nucleoids, engaged in repair processes that occur due to spontaneous lesions, the protein was recruited to replication forks after damage induction, concomitant with a strong increase in average dwell time and in the fraction of immobile molecules. Heat maps of PolA and of ExoR showed a higher concentration on the nucleoids during unperturbed growth, suggesting that many molecules are engaged in a DNA-scanning mode for DNA lesions. After induction of interstrand crosslinks, or to a minor degree after UV irradiation, both proteins showed an increase in statically positioned molecules on the nucleoids, including replication forks (Supplementary Figure S11). Our data suggest that ExoR and PolA perform a redundant essential function at the replication forks, which becomes more heavily required when DNA damage has occurred. To better characterize the previously poorly characterized protein ExoR (YpcP), which likely has a similar structure as the exonuclease domain of Pol I/PolA, we purified ExoR to shows that it indeed has DNA nuclease activity, dependent on the presence of magnesium and has highest affinity to dsDNA with 5′ overhangs but can also binds to DNA/RNA hybrids. These experiments indicate that ExoR could work at DNA nicks or remove excess ssDNA during homologous recombination. Because ExoR and PolA differ significantly in their bound/mobile fractions or in their average dwell times in the absence of each other, the proteins appear to act in a partially redundant but interconnected manner. Based on the conservation of ExoR between Gram-positive and Gram-negative bacteria, ExoR could play an important role in DNA repair in a large variety of bacteria.
DnaE has been suggested to be involved in DNA repair (61) in addition to its vital contribution to the elongation phase of the lagging strand (14). However, if DnaE had a substantial role in DNA repair following damage with MMC or UV, we would have expected to observe increased dwells for DnaE. Because DnaE becomes less static in response to DNA damage, our findings suggest that the contribution of DnaE to DNA repair synthesis is only minor, compared to that of PolA.
In toto, we provide in vivo evidence that B. subtilis replication forks present interesting features of dynamics of replication proteins, which suggest that forks operate differently from the E. coli or eukaryotic systems, with a possible scenario being depicted in Supplementary Figure S11. It will be interesting to study the molecular basis of the division of labour at the lagging strand between the two replicative polymerases, and the exact function of ExoR at replication forks.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: R. H.-T. performed all experiments, evaluated data and co-wrote the manuscript, L.M.O.B. devised of SMT methods and helped analyse the data, G.F. conceived of the GMM methods, helped in data analyses and co-wrote the manuscript, P.L.G. conceived of the study and co-wrote the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The National Council of Science and Technology of Mexico [229388]; LOEWE Program of the state of Hessen for the Center for Synthetic Microbiology, SYNMIKRO, at the Philipps-Universität Marburg; Deutsche Forschungsgemeinschaft (DFG)-funded Research Consortium [TRR 174]. Funding for open access charge: DFG.
Conflict of interest statement. None declared.
REFERENCES
- 1. Sanders G.M., Dallmann H.G., McHenry C.S.. Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol. Cell. 2010; 37:273–281. [DOI] [PubMed] [Google Scholar]
- 2. Henrikus S.S., van Oijen A.M., Robinson A.. Specialised DNA polymerases in Escherichia coli: roles within multiple pathways. Curr. Genet. 2018; 64:1189–1196. [DOI] [PubMed] [Google Scholar]
- 3. Reyes-Lamothe R., Possoz C., Danilova O., Sherratt D.J.. Independent positioning and action of Escherichia coli replisomes in live cells. Cell. 2008; 133:90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Senejani A.G., Dalal S., Liu Y., Nottoli T.P., McGrath J.M., Clairmont C.S., Sweasy J.B.. Y265C DNA polymerase beta knockin mice survive past birth and accumulate base excision repair intermediate substrates. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:6632–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kunkel T.A. Balancing eukaryotic replication asymmetry with replication fidelity. Curr. Opin. Chem. Biol. 2011; 15:620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dervyn E., Suski C., Daniel R., Bruand C., Chapuis J., Errington J., Janniere L., Ehrlich S.D.. Two essential DNA polymerases at the bacterial replication fork. Science. 2001; 294:1716–1719. [DOI] [PubMed] [Google Scholar]
- 7. Yang Y., LiCata V.J.. Pol I DNA polymerases stimulate DNA end-joining by Escherichia coli DNA ligase. Biochem. Biophys. Res. Commun. 2018; 497:13–18. [DOI] [PubMed] [Google Scholar]
- 8. Reyes-Lamothe R., Sherratt D.J., Leake M.C.. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science. 2010; 328:498–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Georgescu R.E., Kurth I., O’Donnell M.E.. Single-molecule studies reveal the function of a third polymerase in the replisome. Nat. Struct. Mol. Biol. 2011; 19:113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barnes M.H., Miller S.D., Brown N.C.. DNA polymerases of low-GC gram-positive eubacteria: identification of the replication-specific enzyme encoded by dnaE. J. Bacteriol. 2002; 184:3834–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bruck I., O’Donnell M.. The DNA replication machine of a gram-positive organism. J. Biol. Chem. 2000; 275:28971–28983. [DOI] [PubMed] [Google Scholar]
- 12. Bruck I., Georgescu R.E., O’Donnell M.. Conserved interactions in the Staphylococcus aureus DNA PolC chromosome replication machine. J. Biol. Chem. 2005; 280:18152–18162. [DOI] [PubMed] [Google Scholar]
- 13. Titok M., Suski C., Dalmais B., Ehrlich S.D., Janniere L.. The replicative polymerases PolC and DnaE are required for theta replication of the Bacillus subtilis plasmid pBS72. Microbiology. 2006; 152:1471–1478. [DOI] [PubMed] [Google Scholar]
- 14. Seco E.M., Ayora S.. Bacillus subtilis DNA polymerases, PolC and DnaE, are required for both leading and lagging strand synthesis in SPP1 origin-dependent DNA replication. Nucleic Acids Res. 2017; 45:8302–8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bruck I., Goodman M.F., O’Donnell M.. The essential C family DnaE polymerase is error-prone and efficient at lesion bypass. J. Biol. Chem. 2003; 278:44361–44368. [DOI] [PubMed] [Google Scholar]
- 16. Paschalis V., Le Chatelier E., Green M., Nouri H., Kepes F., Soultanas P., Janniere L.. Interactions of the Bacillus subtilis DnaE polymerase with replisomal proteins modulate its activity and fidelity. Open Biol. 2017; 7:170146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bruck I., Goodman M.F., O’Donnell M.. The essential C family DnaE polymerase is error-prone and efficient at lesion bypass. J. Biol. Chem. 2003; 278:44361–44368. [DOI] [PubMed] [Google Scholar]
- 18. Patel P.H., Suzuki M., Adman E., Shinkai A., Loeb L.A.. Prokaryotic DNA polymerase I: evolution, structure, and “base flipping” mechanism for nucleotide selection. J. Mol. Biol. 2001; 308:823–837. [DOI] [PubMed] [Google Scholar]
- 19. Rocha E.P. The replication-related organization of bacterial genomes. Microbiology. 2004; 150:1609–1627. [DOI] [PubMed] [Google Scholar]
- 20. Tamanoi F., Okazaki T., Okazaki R.. Persistence of RNA attached to nascent short DNA pieces in Bacillus subtilis cells defective in DNA polymerase I. Biochem. Biophys. Res. Commun. 1977; 77:290–297. [DOI] [PubMed] [Google Scholar]
- 21. Goswami A., Roy Chowdhury A., Sarkar M., Saha S.K., Paul S., Dutta C.. Strand-biased gene distribution, purine assymetry and environmental factors influence protein evolution in Bacillus. FEBS Lett. 2015; 589:629–638. [DOI] [PubMed] [Google Scholar]
- 22. Friedberg E.C. A history of the DNA repair and mutagenesis field: The discovery of base excision repair. DNA Repair (Amst). 2016; 37:A35–A39. [DOI] [PubMed] [Google Scholar]
- 23. Duigou S., Ehrlich S.D., Noirot P., Noirot-Gros M.F.. DNA polymerase I acts in translesion synthesis mediated by the Y-polymerases in Bacillus subtilis. Mol. Microbiol. 2005; 57:678–690. [DOI] [PubMed] [Google Scholar]
- 24. Million-Weaver S., Samadpour A.N., Merrikh H.. Replication restart after replication-transcription conflicts requires reca in bacillus subtilis. J. Bacteriol. 2015; 197:2374–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thomaides H.B., Davison E.J., Burston L., Johnson H., Brown D.R., Hunt A.C., Errington J., Czaplewski L.. Essential bacterial functions encoded by gene pairs. J. Bacteriol. 2007; 189:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fu D., Calvo J.A., Samson L.D.. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer. 2012; 12:104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kuzminov A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8241–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Uphoff S., Reyes-Lamothe R., Garza de Leon F., Sherratt D.J., Kapanidis A.N.. Single-molecule DNA repair in live bacteria. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:8063–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stracy M., Kapanidis A.N.. Single-molecule and super-resolution imaging of transcription in living bacteria. Methods. 2017; 120:103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sarkar B., Cao G.J., Sarkar N.. Identification of two poly(A) polymerases in Bacillus subtilis. Biochem. Mol. Biol. Int. 1997; 41:1045–1050. [DOI] [PubMed] [Google Scholar]
- 31. Duigou S., Ehrlich S.D., Noirot P., Noirot-Gros M.F.. Distinctive genetic features exhibited by the Y-family DNA polymerases in Bacillus subtilis. Mol. Microbiol. 2004; 54:439–451. [DOI] [PubMed] [Google Scholar]
- 32. Jaacks K.J., Healy J., Losick R., Grossman A.D.. Identification and characterization of genes controlled by the sporulation-regulatory gene spo0H in Bacillus subtilis. J. Bacteriol. 1989; 171:4121–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lucena D., Mauri M., Schmidt F., Eckhardt B., Graumann P.L.. Microdomain formation is a general property of bacterial membrane proteins and induces heterogeneity of diffusion patterns. BMC Biol. 2018; 16:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feucht A., Lewis P.J.. Improved plasmid vectors for the production of multiple fluorescent protein fusions in Bacillus subtilis. Gene. 2001; 264:289–297. [DOI] [PubMed] [Google Scholar]
- 35. Koo B.M., Kritikos G., Farelli J.D., Todor H., Tong K., Kimsey H., Wapinski I., Galardini M., Cabal A., Peters J.M. et al.. Construction and analysis of two genome-scale deletion libraries for bacillus subtilis. Cell Syst. 2017; 4:291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Plank M., Wadhams G.H., Leake M.C.. Millisecond timescale slimfield imaging and automated quantification of single fluorescent protein molecules for use in probing complex biological processes. Integr. Biol. (Camb). 2009; 1:602–612. [DOI] [PubMed] [Google Scholar]
- 37. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al.. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012; 9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jaqaman K., Loerke D., Mettlen M., Kuwata H., Grinstein S., Schmid S.L., Danuser G.. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods. 2008; 5:695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Michalet X. Mean square displacement analysis of single-particle trajectories with localization error: brownian motion in an isotropic medium. Phys. Rev. E. 2011; 82:041914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vestergaard C.L., Blainey P.C., Flyvbjerg H.. Optimal estimation of diffusion coefficients from single-particle trajectories. Phys. Rev. E. 2014; 89:022726. [DOI] [PubMed] [Google Scholar]
- 41. Rosch T.C., Oviedo-Bocanegra L.M., Fritz G., Graumann P.L.. SMTracker: a tool for quantitative analysis, exploration and visualization of single-molecule tracking data reveals highly dynamic binding of B. subtilis global repressor AbrB throughout the genome. Sci. Rep. 2018; 8:15747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tinevez J.Y., Perry N., Schindelin J., Hoopes G.M., Reynolds G.D., Laplantine E., Bednarek S.Y., Shorte S.L., Eliceiri K.W.. TrackMate: an open and extensible platform for single-particle tracking. Methods. 2017; 115:80–90. [DOI] [PubMed] [Google Scholar]
- 43. Paintdakhi A., Parry B., Campos M., Irnov I., Elf J., Surovtsev I., Jacobs-Wagner C.. Oufti: an integrated software package for high-accuracy, high-throughput quantitative microscopy analysis. Mol. Microbiol. 2016; 99:767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Berkmen M.B., Grossman A.D.. Spatial and temporal organization of the Bacillus subtilis replication cycle. Mol. Microbiol. 2006; 62:57–71. [DOI] [PubMed] [Google Scholar]
- 45. Graumann P.L. Chromosome architecture and segregation in prokaryotic cells. J. Mol. Microbiol. Biotechnol. 2014; 24:291–300. [DOI] [PubMed] [Google Scholar]
- 46. El Najjar N., El Andari J., Kaimer C., Fritz G., Rosch T.C., Graumann P.L.. Study of DNA translocases in Bacillus subtilis by single molecule tracking reveals strikingly different dynamics of SftA, SpoIIIE and FtsA. Appl. Environ. Microbiol. 2018; 84:e02610-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kidane D., Graumann P.L.. Dynamic formation of RecA filaments at DNA double strand break repair centers in live cells. J. Cell Biol. 2005; 170:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Romero H., Rosch T.C., Hernandez-Tamayo R., Lucena D., Ayora S., Alonso J.C., Graumann P.L.. Single molecule tracking reveals functions for RarA at replication forks but also independently from replication during DNA repair in Bacillus subtilis. Sci. Rep. 2019; 9:1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hernandez-Tamayo R., Graumann P.L.. Bacillus subtilis RarA forms damage-inducible foci that scan the entire cell. BMC Res. Notes. 2019; 12:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cranfill P.J., Sell B.R., Baird M.A., Allen J.R., Lavagnino Z., de Gruiter H.M., Kremers G.J., Davidson M.W., Ustione A., Piston D.W.. Quantitative assessment of fluorescent proteins. Nat. Methods. 2016; 13:557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Joyce C.M., Grindley N.D.F.. Method for determining whether a gene of escherichia-coli is essential—application to the pola gene. J. Bacteriol. 1984; 158:636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Diaz A., Lacks S.A., Lopez P.. The 5′ to 3′ exonuclease activity of DNA-polymerase-I is essential for streptococcus-pneumoniae. Mol. Microbiol. 1992; 6:3009–3019. [DOI] [PubMed] [Google Scholar]
- 53. Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.E.. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015; 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Theobald D.L., Mitton-Fry R.M., Wuttke D.S.. Nucleic acid recognition by OB-fold proteins. Annu. Rev. Biophys. Biomol. Struct. 2003; 32:115–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Knuth K., Niesalla H., Hueck C.J., Fuchs T.M.. Large-scale identification of essential Salmonella genes by trapping lethal insertions. Mol. Microbiol. 2004; 51:1729–1744. [DOI] [PubMed] [Google Scholar]
- 56. Kobayashi K., Ehrlich S.D., Albertini A., Amati G., Andersen K.K., Arnaud M., Asai K., Ashikaga S., Aymerich S., Bessieres P. et al.. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:4678–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Forsyth R.A., Haselbeck R.J., Ohlsen K.L., Yamamoto R.T., Xu H., Trawick J.D., Wall D., Wang L.S., Brown-Driver V., Froelich J.M. et al.. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol. Microbiol. 2002; 43:1387–1400. [DOI] [PubMed] [Google Scholar]
- 58. Akerley B.J., Rubin E.J., Novick V.L., Amaya K., Judson N., Mekalanos J.J.. A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Burgers P.M.J., Kunkel T.A.. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017; 86:417–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Skarstad K., Katayama T.. Regulating DNA replication in bacteria. Cold Spring Harb. Perspect. Biol. 2013; 5:a012922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Le Chatelier E., Becherel O.J., d’Alencon E., Canceill D., Ehrlich S.D., Fuchs R.P.P., Janniere L.. Involvement of DnaE, the second replicative DNA polymerase from Bacillus subtilis, in DNA mutagenesis. J. Biol. Chem. 2004; 279:1757–1767. [DOI] [PubMed] [Google Scholar]
- 62. Paschalis V., Le Chatelier E., Green M., Nouri H., Kepes F., Soultanas P., Janniere L.. Interactions of the Bacillus subtilis DnaE polymerase with replisomal proteins modulate its activity and fidelity. Open Biol. 2017; 7:170146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Graham J.E., Marians K.J., Kowalczykowski S.C.. Independent and stochastic action of DNA polymerases in the replisome. Cell. 2017; 169:1201–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hubscher U., Maga G.. DNA replication and repair bypass machines. Curr. Opin. Chem. Biol. 2011; 15:627–635. [DOI] [PubMed] [Google Scholar]
- 65. Rannou O., Le Chatelier E., Larson M.A., Nouri H., Dalmais B., Laughton C., Janniere L., Soultanas P.. Functional interplay of DnaE polymerase, DnaG primase and DnaC helicase within a ternary complex, and primase to polymerase hand-off during lagging strand DNA replication in Bacillus subtilis. Nucleic Acids Res. 2013; 41:5303–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.