


Abstract
Histone variants, present in various cell types and tissues, are known to exhibit different functions. For example, histone H3.3 and H2A.Z are both involved in gene expression regulation, whereas H2A.X is a specific variant that responds to DNA double-strand breaks. In this study, we characterized H4G, a novel hominidae-specific histone H4 variant. We found that H4G is expressed in a variety of human cell lines and exhibit tumor-stage dependent overexpression in tissues from breast cancer patients. We found that H4G localized primarily to the nucleoli of the cell nucleus. This localization was controlled by the interaction of the alpha-helix 3 of the histone fold motif with a histone chaperone, nucleophosmin 1. In addition, we found that modulating H4G expression affects rRNA expression levels, protein synthesis rates and cell-cycle progression. Our data suggest that H4G expression alters nucleolar chromatin in a way that enhances rDNA transcription in breast cancer tissues.
INTRODUCTION
Eukaryote genomic DNA is tightly packaged into chromatin structure composed of discrete nucleosome subunits. The nucleosome core particle is a histone core octamer wrapped by a 147 base pairs (bp) DNA. Each histone octamer comprises two copies of the core histones H2A, H2B, H3 and H4. In addition to the core histones, histone H1 is a linker histone that binds to the linker DNA, which connects adjacent nucleosomes. Histone H1 is in close proximity to the canonical histone octamer region, helping to further compact nucleosomal DNA in the chromatin fiber (1). Epigenetic regulators, including histone variants and histone post-translational modifications (PTMs), change chromatin structure and regulate important biological functions such as DNA replication and repair, transcription (2).
Epigenetic alterations, such as changes in histone PTMs and DNA methylation, are integral to regulating gene expression patterns and maintaining genome stability (3–5). Changes in the expression patterns of histone variants have also been reported to affect the genome instability observed in cancer cells (6). For example, H2A.X phosphorylation is essential for DNA double-strand break repair, and H2A.X-knockout mice show immunodeficiency, radio-sensitivity and a high susceptibility to cancer (7–9). The expression of MacroH2A, a transcription repressor involved in X-chromosome inactivation (10), is downregulated in both breast and colon cancers (11). Decrease of MacroH2A has been reported to promote tumor progression through senescence bypass (11,12). Increase in the H2A.Z expression levels has also been observed in many cancer types, including colorectal, breast and prostate cancers (11,13). Although increasing evidence suggests potential links between histone variant expression and tumor progression, the mechanisms by which histone variants control cellular proliferation and growth remain poorly understood.
Many types of histone variants are present in humans, including H2A.Z, H2A.X, H2A.Bbd, macroH2A, TH2B, H2BFWT, H3.3, H3.4, H3.5, H1t, H1.X and H1foo. Histone variants have been shown to have cell type and tissue-specific localizations, indicating that they are functionally unique (14,15). Holmes et al. studied the expression of histone H4 genes (16) and found that, compared with histones H1, H2A, H2B and H3, histone H4 is the only histone that does not have functionally distinct variants. However, in this study, we describe the function of a previously uncharacterized H4 variant: H4G. The H4G protein lacks five amino acids in the C-terminal tail region of the canonical human histone H4, which is a region shown to be required for cell viability in yeast (17,18) and shares only 85% identity with H4 in the remaining 98 amino acids (19). Several residues are changed throughout the protein at the N-terminal tail region, α1, α2 and α3 regions. The human gene HIST1H4g (h4g) is located in the histone cluster 1 on chromosome 6, with many other histone genes encoded nearby and is present only in hominids (16,19). Several transcriptomic data have shown that the expression of h4g is increased in T-cell prolymphocytic leukemia, HuR silenced thyroid carcinoma BCPAP cell line and an endometrioid carcinoma TOV112 cell line (20–22). However, the functional role of H4G remains uncharacterized.
We found that h4g expression is elevated in the human breast cancer cell line MCF7 compared to the normal breast epithelial cell line MCF10A or HEK293T cells. Moreover, we detected an elevated h4g level in breast tissues from human breast cancer patients and h4g expression level is correlated to breast cancer stage progression. We also found that H4G is primarily localized to the nucleoli, and its expression positively regulates the transcription of rDNA. H4G depletion in MCF7 breast cancer cells decreased cellular proliferation rates as a result of reduced rRNA and protein synthesis. Furthermore, using a mouse xenograft model, we found that H4G expression promotes the growth of the breast cancer cells. At last, consistent with a nucleolar function for this histone variant, we identify NPM1 (nucleophosmin1), a nucleolar histone chaperone involved in ribosomal biogenesis and in tumor progression (23–26), as an H4G interacting protein that preferentially recognizes the α-helix 3 of the H4G histone fold domain.
MATERIALS AND METHODS
Plasmids
Human H4, H4G, H4sN, H4GsN, H4sC, H4GsN+C, H4sα1, H4sα2, H4sα3, H4Gsα3, H4D85A, H4A89V, H4GA85D and H4GV89A sequences were inserted into the p3xFLAG-CMV-14 vector (Sigma-Aldrich). For stable cell line construction, the human H4G gene with the C-terminal 3xFLAG sequence was inserted into the pLVX-Tet-On-Puro vector (Clontech). The N-terminal FLAG H4G was cloned into pFLAG-CMV-2 vector (Sigma-Aldrich) and the C-terminal EGFP H4G was cloned into pEGFP-N3 vector (Clontech). For in vitro nucleosome reconstitution, the h4 and h4g genes were inserted into the pGEX4T3 vector, and the canonical histones H2B, H2A, and H3 were cloned into the pET11a vector.
In vitro nucleosome reconstitution, MNase digestion and pulldown assay
The human histones H2A, H2B, H3, GST-H4 and GST-H4G were expressed in Escherichia coli BL21-codonplus (DE3), and they were purified from the inclusion bodies (27). The H3-GST-H4, H3-GST-H4G and H2A-H2B complexes were reconstituted as follows: Purified GST–H4 or GST–H4G was mixed with H3 at a 1:1 molar ratio in unfolding buffer (20 mM Tris–HCl [pH 7.5], 7 M guanidine hydrochloride and 20 mM 2-mercaptoethanol), and the mixture was dialyzed against refolding buffer (10 mM Tris–HCl [pH 7.5], 2 M NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA) and 5 mM 2-mercaptoethanol). The H3-GST-H4/H3-GST-H4G and H2A-H2B complexes were mixed and further mixed with the 206 bp DNA containing the 601 nucleosome positioning sequence in a solution containing 2 M NaCl. The nucleosomes were then reconstituted by the salt-dialysis method (28,29), and the reconstituted nucleosomes were purified by non-denaturing 4% polyacrylamide gel using a Prep Cell apparatus (Biorad). A total of 20 μl of the purified reconstituted nucleosome was digested by the indicated amount of MNase (Worthington) at room temperature for 5 min. Samples were loaded onto 6% non-denaturing acrylamide gel without DNA extraction. For the MNase digestion with deproteinization assay, proteinase K (NEB) was added for the digestion of proteins. For pulldown assays of His-SUMO-H4 and -H4G with H3, His-SUMO-H4 and -H4G were purified using Ni-NTA resin following manufacturer's instruction (Qiagen) under urea denaturing condition. His-SUMO-H4 or -H4G was mixed with H3 at a 1:1 molar ratio in unfolding buffer (20 mM Tris–HCl [pH 7.5], 7 M guanidine hydrochloride and 5 mM dithiothreitol) and the mixture was dialyzed against refolding buffer (10 mM Tris–HCl [pH 7.5], 2 M NaCl, 0.1 mM EDTA and 5 mM 2-mercaptoethanol) before being subjected to pulldown assay.
Cell culture
The MCF-7, LCC1, LCC2, MCF-10A and HEK293T cell lines were purchased from ATCC and cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C with 5% CO2. Plasmid transfection was performed using Lipofectamine 2000 (Thermo Fisher Scientific) or Polyethylenimine (Polysciences).
Immunofluorescence
Cells were grown on coverslips and transfected with indicated plasmids using PEI (Polyscience). Twenty-four hours after transfection, cells were collected for immunofluorescence. Cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) for 10 min and permeabilized with PBS-T (0.2% Triton X-100 in phosphate-buffered saline (PBS)) for 10 min. The cells were then blocked for 30 min with 5% bovine serum albumin resolved in PBST (0.1%TWEEN-20 in PBS) and incubated with Flag M2 antibody (Sigma-Aldrich) for 1 h in PBST. Cells were washed in PBS for 15 min (3 × 5 min) followed by incubation with secondary antibody (Invitrogen) and Phalloidin (Invitrogen) in PBST for 1 h. Cells were then stained with Hoechst 33258 (Thermo Fisher Scientific) for 10min and washed in PBS for 15 min (3 × 5 min). Cells were either imaged using a Nikon TE2000E-PFS fluorescence microscope or in Leica sp8 confocal microscope.
Immunoprecipitation
HEK293T and MCF7 cells transfected with p3xFLAG-CMV-14 H4 or H4G were collected 48 h after transfection. For immunoprecipitation, cell extracts were prepared at 4°C in a lysis buffer containing 10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 420 mM NaCl, 0.5% NP-40, 5 mM 2-mercaptoethanol and protease inhibitor cocktail (Roche) with sonication. Anti-FLAG M2-coupled beads (Sigma-Aldrich) were added to the extracts and incubated at 4°C for 2 h. Subsequently, the beads were extensively washed in lysis buffer, and the bound proteins were analyzed by immunoblotting against FLAG M2 antibody (Sigma-Aldrich) and NPM1 (ab15440 from Abcam).
Mass spectrometry analysis and data processing
Anti-FLAG immunoprecipitation was performed as described above. After immunoprecipitation, bound proteins were eluted by incubating the beads with the FLAG peptide (1 μg/μl). Eluted proteins were resolved using sodium dodecyl sulphate-polyacrylamide gelelectrophoresis (SDS-PAGE) and stained with SYPRO Ruby (Thermo Fisher Scientific). For protein identification, gels that contained the interacting proteins were cut and analyzed using an LTQ Velos liner ion-trap LC-MS system in HKUST BioCRF. The acquired tandem mass spectra were then subjected to gene database searches using the MASCOT search engine (Matrix Science).
Knockout and rescue cell line establishment
To construct a knockout H4G line in MCF7 cells, sgRNA (5′-CACCGTTCGGGGCAAGGCCGGAAA-3′) that targets HIST1H4G was inserted into the PX459 plasmid (pSpCas9(BB)-2A-Puro (PX459) V2.0 was a gift from Prof. Feng Zhang (Addgene plasmid #62988) (30). Transfection plasmids in MCF7 cells were carried out with the FuGENE® HD Transfection Reagent (Promega) according to the manufacturer's instructions. Genomic DNA was extracted and subjected to polymerase chain reaction (PCR) using a forward primer (5′-GGACGAATTCTCCCGCCTTTCCTGGTCTTTCAG-3′) and reverse primer (5′-GTTAGGATCCCAGGGTTCTTCCCTGGCGTT-3′) to generate a product spanning the targeted region. The pLVX-TetOn-Puro H4G 3xFLAG plasmid was then transfected into the KO cell lines and selected with puromycin for rescue cell line establishment.
RNA extraction and quantitative real-time PCR
Except where specifically indicated in the text, RNA isolation was performed using the RNeasy Mini Kit (Qiagen). cDNA was generated using Superscript III First-Strand Synthesis System (Thermo Fisher Scientifics) using a random hexamer primer. A negative control was generated by replacing reverse transcriptase with water in the cDNA synthesis process to exclude genomic DNA contamination. Messenger RNA (mRNA) expression was measured using quantitative real-time PCR (q-RT-PCR) assays using gene-specific primers (SYBR Green assay) by a LightCycler® detector (Roche). The relative fold change for each gene was calculated using the ΔΔCT method as previously described (31) and the Student's t-test was used to determine statistical significance. Primers used for q-RT-PCR were H4G forward (5′-TGTGATCTGGTACGCCGTG-3′) and reverse (5′-CTGGCGTTTGAGCACGTAGA-3′); H4D forward (5′-GGAAAATGTAATCCHCHATGC-3′) and reverse (5′-CCATAAAGAGTGCGTCCCTG-3′); H4E forward (5′-GTCTACGCGCTGAAGAGACA-3′) and reverse (5′-AGTCGAGATGCTGAGTCGTT-3′); GAPDH forward (5′- CTCCTGCACCACCAACTGCT-3′) and reverse (5′-GGGCCATCCACAGTCTTCTG-3′); and ETS forward (5′-GAACGGTGGTGTGTCGTT-3′) and reverse (5′-GCGTCTCGTCTCGTCTCACT-3′). For quantification of rRNA, total RNA was normalized by GAPDH mRNA and loaded on agarose gels. The 18S and 28S rRNA were quantified using ImageJ software.
Protein synthesis assay
MCF7 cells were stained with the Click-iT® Plus OPP Alexa Fluor 488 Protein Synthesis Assay Kit (Life Technologies). OPP was added at a 10 μM final concentration, and cells were incubated for 30 min. Cells were then fixed with 70% ethanol and processed as instructed in the OPP assay kit. Cells were analyzed using flow cytometry.
Cell-cycle analysis
MCF7 cells were synchronized by serum starvation for 24 h and induced to re-enter the cell cycle by the addition of serum. Cells were then harvested for propidium iodide staining and analyzed by fluorescence-activated cell sorting (FACS) using a Becton Dickinson FACSAria™ III flow cytometer (BD Biosciences) to determine the cell-cycle fraction. Data were analyzed using FCS Express software (De Novo Software).
Subcutaneous Xenograft tumor models
The Xenograft experiment was performed by Guangzhou Jennio Biotech Co., Ltd. Female NOG recipient mice 4 weeks of age were embedded with 0.25 mg β-estradiol pellet 1 week before implantation and maintained under specific pathogen free (SPF) conditions. Cells (1 × 107) were implanted subcutaneously in a Matrigel matrix under the left shoulder of each mouse and allowed to grow to reach at a minimum size of ∼30–40 mm3. All mice were handled according to the Guide for the Care and Use of Laboratory Animals.
RESULTS
The human genome contains three histone clusters where multiple copies of the major core and linker histones as well as many histone variants are located (15). The canonical histone genes encode proteins of highly conserved amino acid sequences, and they are expressed primarily during the S phase of the cell cycle. h4g is encoded in histone cluster 1 located at the chromosome 6p22.1–22.2 region in the human genome (16). In this study, we searched the genomes of other species, and we were only able to identify the h4g gene in hominid genomes (Figure 1A). H4 is known as one of the most evolutionarily conserved histones (32,33). Surprisingly H4G shares only 85% amino acid identity with human canonical H4 (Figure 1A) (19), lacking the last five amino acids of the C-terminal tail and harboring several different amino acids within the N-terminal tail and the α-helix 1, 2 and 3 domains of the histone fold (Figure 1A) (19). In addition, in comparison with its canonical histone counterpart, H4G has a higher hydrophobicity due to amino acid changes in the N-terminal tail and α-helix 3 of the core domain (Figure 1A and Supplementary Figure S1).
Figure 1.

H4G expression in breast cancer patient tissues. (A) Amino acids sequence alignment between primate histone H4G and canonical H4 in primates. Unique amino acids to H4G in comparison with canonical human H4 are highlighted in yellow. (B) Relative H4G expression (mean ± SEM) obtained by quantitative real-time PCR in breast, colon, kidney, liver, lung, ovary, prostate and thyroid normal versus corresponding tumor tissues using the Cancer Survey qPCR panel (#CSRT101, Origene Technologies). *P < 0.05 and **P < 0.01 (Student's t-test). (C) The relative H4G expression obtained from quantitative real-time PCR in breast cancer patient breast tissues and normal breast tissues (#BCRT302, Origene Technologies). The black lines represent the mean expression level of h4g. (D) The relative H4G expression in breast cancer stages (#BCRT302, Origene Technologies). The black lines represent the mean expression level of h4g.
To study the function of H4G, we first checked h4g expression levels in several cultured cell lines. We found that overall h4g expression is low compared to canonical histone H4 genes: HIST1H4d (h4d) and HIST1H4e (h4e) (Supplementary Figure S2). In general, the h4g gene is expressed in breast cancer cell lines at low levels, consistent with the expression data from database (The Human Protein Atlas). Notably, the expression of h4g in breast cancer cell lines, including MCF7, LCC1 and LCC2 cells was higher than the expression of h4g in the noncancerous breast epithelial cell line, MCF10A; or the embryonic kidney cell line, HEK293T (Supplementary Figure S2). To confirm the presence at protein level, we have done the LC-MS in MCF7 cell. The H4G is present in MCF7 cell, however the amounts were low (Supplementary Table S1).
We then checked the expression levels of h4g using the human cancer survey cDNA panel (ORIGENE) containing eight different tumors (breast, colon, kidney, liver, lung, ovary, prostate and thyroid). The results showed that the expression levels of h4g vary among tissues (Figure 1B, blue bars) and h4g expression is significantly increased in breast cancer and colon cancer tissues compared to normal tissue control (Figure 1B). We further evaluated the expression of h4g using the human breast cancer cDNA panel (ORIGENE, Figure 1C and D). We again observed that the expression of h4g gene was significantly increased in breast cancer tissues compared to normal breast tissues (Figure 1C). Importantly, the h4g expression level was progressively increased as tumors progressed to more advanced stages (Figure 1D). Note that the expression of canonical h4d and h4e increased at a much less amplitude than that of h4g in breast cancer patients (Supplementary Figure S3).
H4G is unable to form a nucleosome in vitro
Histone variants are known to affect nucleosomal stability; such effects underlie their functions in regulating processes such as transcription and DNA repair (34). Three regions of H4G are divergent from H4. Frist, H4G is truncated in comparison with canonical H4, lacking five C-terminal residues that form a β-sheet structure between H4 and H2A (35). Second, the N-terminal H4G sequence is different from that of H4; this region also directly interacts with H2A (36–38). At last, amino acid sequence differences are also found in the alpha helix 2 and alpha helix 3 domains of the histone fold leading to a significant increase in hydrophobicity for H4G (Figure 1A and Supplementary Figure S1). In the nucleosome it has been shown that the H2A-H2B dimers interact with the H3-H4 tetramer through a 4-helix bundle that involves these H2B and H4 alpha helical domains (39). These changes in H4G might impair these critical interactions that underpin nucleosome assembly.
To test if sequence differences in H4G affect nucleosome formation, we monitored the assembly of nucleosome particles using the salt gradient approach and the 601 nucleosome positioning sequence (28,40) (Figure 2). We expressed H4 or H4G as GST-fusions, which significantly increased the protein expression and solubility of H4G. Nucleoprotein complexes generated with H4G displayed increased sensitivity to MNase digestion when compared to those generated from H4 (Figure 2B arrowhead). While the DNA extracted from MNase digestion of H4-reconstituted complexes showed the characteristic 147 bp species expected from canonical nucleosome core particles, the H4G-reconstituted complexes did not (Figure 2C arrowhead). At last, SDS-PAGE of these nucleoprotein complexes revealed that, while nucleosomes assembled with H4 contain the expected stoichiometric amounts of core histones, complexes reconstituted with H4G do not (Figure 2D). These experiments demonstrate that H4G cannot form a stable nucleosome using this salt-dialysis method. We further compared the ability of H4 and H4G to interact with H3 through pulldown assays using His-SUMO-H4G and His-SUMO-H4 proteins and found that both proteins bind to H3 (Supplementary Figure S4), suggesting that H4G might affect the assembly of H2A/H2B and H3/H4G complexes. Taken together, we conclude that under the salt reconstitution approach used here, H4G is excluded from the reconstituted nucleoprotein complexes and that it lacks an intrinsic ability to form nucleosomes, at least under these conditions. These results support the hypothesis that H4G likely has distinct modes of action compared to H4.
Figure 2.

H4G interferes with the formation of nucleosome. (A) SDS-PAGE of the canonical histone mixture and the H4G mixture for nucleosome loading. Asterisk (*) represents the GST-H4 and GST-H4G proteins. (B) MNase treatment assay for the GST-H4 and the GST-H4G nucleosomes without DNA extraction. (C) MNase treatment assay for the GST-H4 and the GST-H4G nucleosomes with DNA extraction. 147 represents 147 bp PCR amplified DNA. (D) SDS-PAGE of reconstituted nucleoprotein complexes.
The α−helix 3 domain of H4G is responsible for its nucleolar localization
The different nuclear localization pattern of histone variants can be an indicator of their distinct regulatory roles in different cellular processes. For instance, CENP-A localizes to the centromeric region, whereas H3.3 localizes to the promoters of active genes (34). We found that when FLAG-tagged H4G was expressed in MCF7 cells, it localized to the nucleolus, whereas the overexpressed FLAG-tagged canonical H4 was localized to the nucleus, as expected (Figure 3). This localization pattern was also observed in LCC1 and LCC2 cells (Figure 3). This nucleolar localization pattern of H4G was not affected by the N-terminal FLAG or the C-terminal EGFP (Supplementary Figure S5A and B), as was observed in other cell lines such as MCF10A, HeLa and HEK293T (Supplementary Figure S5C–E).
Figure 3.
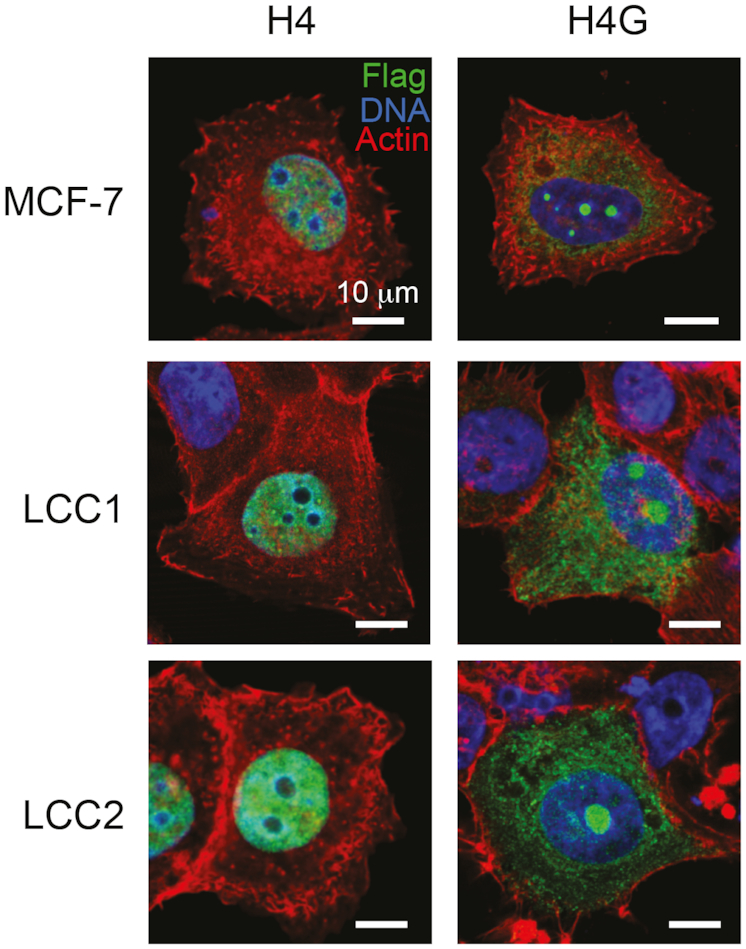
H4G localizes to the nucleolar in breast cancer cells. The cellular localization of H4 and H4G in MCF7, LCC1 and LCC2 cells. Each image is representative of MCF7 H4: N = 192 (100%) and MCF7 H4G: N = 110 (75%). The scale bars represent 10 μm.
To further dissect the structural determinants of the H4G nucleolar localization, we made a series of H4G mutants, in which we swapped different H4 and H4G regions and analyzed their localization patterns (Figure 4A–H). To study the effect of the N-terminal tail, we made constructs containing the H4 histone fold with the H4G N-terminal tail (H4sN; s stands for swapping) and a construct containing the H4G histone fold with the H4 N-terminal tail (H4GsN) (Figure 4). The H4sN was found to localize to the nucleus like H4, whereas the H4GsN was found to localize to the peripheral region of the nucleoli in comparison with the uniform nucleolar localization of H4G (Figures 4B and Supplementary Figure S6). For the analysis of the C-terminal tail, we made constructs of H4sC with a deletion of the last five amino acids of the original H4 construct and H4G with both N- and C-terminal tails of H4 (H4GsN+C). The H4sC localization was similar to that of H4, whereas that of H4GsN+C localization was similar to that of H4GsN (Figure 4A, B and D). These results indicate that the C-terminal tail of H4G does not mediate H4G nucleolar localization. We further found that the H4 histone containing the H4G α-helix 3 domain protein (residues 85 and 89) exhibited a nucleolar localization similar to that of H4G (Figure 4G). These findings were confirmed using reverse-swapping in which the α-helix 3 of H4G was replaced with the α-helix 3 of the canonical H4, leading to the complete absence of nucleolar localization (Figure 4H). Together, these data suggest that H4G α-helix 3 domain is responsible to the nucleolar localization of H4G.
Figure 4.

The α-helix 3 of H4G is important for the nucleolar localization. (A–H) The cellular localizations of the H4G/H4 hybrid constructs in MCF7 cells. Each image is representative of (A) H4sN: N = 131 (99%), (B) H4GsN: N = 160 (88%), (C) H4sC: N = 105 (100%), (D) H4GsN+C: N = 123 (100%), (E) H4sα1: N = 207 (97%), (F) H4sα2: N = 208 (96%), (G) H4sα3: N = 126 (31%) and (H) H4Gsα3: N = 132 (100%). The scale bars represent 10 μm.
The localization of H4G in the nucleolus is likely mediated by other proteins such as histone chaperones. We conducted an anti-FLAG immunoprecipitation assay using HEK293T cells which overexpress FLAG-tagged H4 or H4G, and analyzed interacting proteins by LC-MS. By comparing the interacting proteins of H4 and H4G, we found that the nucleolar histone chaperone protein, NPM1 (nucleophosmin/B23), specifically interacts with H4G but not with H4 (Table 1) (26). These interactions were confirmed by immunoprecipitation in both HEK293T and MCF7 cells (Supplementary Figure S7). NPM1 was previously reported to interact with H3 (26), which we also observed as a control group in our co-IP assay (Supplementary Figure S7). Interestingly, NPM1 appears to interact more strongly with H4G, and this interaction is likely mediated by the α-helix 3 in H4G, as suggested by the disappearance of the interaction in H4Gsα3 and the presence of the interaction in H4sα3 (Figure 5). Because amino acids 85 and 89 are different in the α-helix 3 between H4 and H4G, we repeated the immunoprecipitation study to determine the role of these amino acids. We found that both positions are likely important for the interaction of H4G with NPM1, and mutation of the position 85 (D in H4 and A in H4G) appears to have a greater impact on disrupting the interaction than mutation of position 89 (A in H4 and V in H4G) (Figure 5). Another H3/H4 binding chaperone, Asf1, has also been reported to bind to both H3 and H4 (41,42). The C-terminal region of histone H4 has been shown to be necessary to function with Asf1 from H3/H4 tetramers (35). The deletion of H4 C-terminal tail from 97 to 102 causes lethality in yeast (17,18) and the H4Y98H mutation fails to form histone octamers (43). Because H4G lacks the last five amino acids at its C-terminal end, we examined whether H4G would be able to interact with Asf1. Mass spectrometry analyses showed that Asf1 interacts only with H4 but not with H4G (Table 1 and Supplementary Figure S8). Rbap46 (a part of the histone acetyltransferase HAT1 complex) and Rbap48 (a component of replication coupled histone chaperone CAF1 complex) have also been shown to interact with the α-helix 1 of H4 (44). In this study, we found that Rbap46 and Rbap48 were able to interact with both H4 and H4G; however, the mass spectrometry signal of H4G was much lower than that of H4 (Table 1). Collectively, these results indicate that the α-helix 3 of H4G mediates its interaction with NPM1, which is critical for its nucleolar localization.
Table 1.
Proteins interacting with H4G and H4 identified by LC-MS
| Protein name | MW (kDa) | Uniprot ID | Score in H4G-IP | Score in H4-IP |
|---|---|---|---|---|
| Chromatin binding proteins | ||||
| SP16H | 119.9 | Q9Y5B9 | 82 | 71 |
| SSRP1 | 81.1 | Q08945 | 52 | 69 |
| RBAP46 | 47.8 | Q16576 | 56 | 297 |
| RBAP48 | 47.7 | Q09028 | 56 | 451 |
| NASP | 85.2 | P49321 | 43 | 621 |
| ASF1A | 23 | Q9Y294 | N/A | 162 |
| CAF1B | 61.5 | Q13112 | N/A | 108 |
| CAF1A | 106.9 | Q13111 | N/A | 50 |
| Nucleolar proteins | ||||
| NOP58 | 59.6 | Q9Y2X3 | 67 | N/A |
| NPM1 | 32.6 | P06748 | 70 | N/A |
Figure 5.
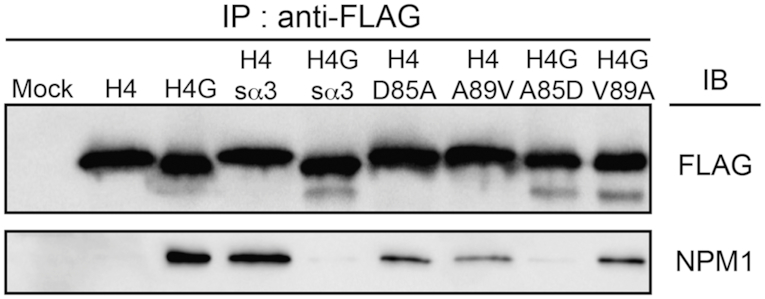
H4G strongly interacts with NPM1 and this interaction is mediated by α-helix 3 domain. Immunoprecipitation using FLAG-proteins to identify the interaction of H4G with the nucleolar histone chaperone NPM1.
H4G regulates the synthesis of rRNA and cell growth
In eukaryotic cells, rDNA transcription occurs in the nucleolus (45). We next assessed whether the nucleolar localization of H4G is linked with rDNA transcription or ribosome biogenesis. We treated H4G-transfected MCF7 cells with a low concentration of actinomycin D, which primarily inhibits transcription by RNA polymerase I (46,47) and observed that the nucleolar localization of H4G was lost with H4G dispersed throughout the nucleus (Supplementary Figure S9). In contrast, in non-transfected control cells, the addition of actinomycin D did not change the localization of H4. Similar results were obtained in other cell lines, including HeLa, LCC1 and LCC2 cells (data not shown). In addition, H4G localization did not change when H4G-transfected MCF7 cells were treated with rapamycin, an mTOR signaling inhibitor. mTOR signaling has been shown to regulate the synthesis of ribosome components, pre-rRNA and 5S rRNA without affecting rDNA transcription (48) (Supplementary Figure S9). These results suggest that the nucleolar localization of H4G may be dependent on ongoing rDNA transcription.
We next examine whether the nucleolar localization of H4G has a functional role in the regulation of rDNA transcription. We produced H4G knockout (KO) MCF7 cell lines through CRISPR-Cas9 technique. We found that, in the H4GKO cells, the amount of total rRNA production was reduced (Figure 6A). To examine whether transcription or RNA processing were affected by H4G levels, we compared rRNA levels by Q-PCR using primers for the external transcribed spacers (ETS) at the 5′ UTR of the rRNA. Our results obtained with the ETS primer were similar with that of the total rRNA (Figure 6A). To confirm the phenotype of H4GKO cells is not caused by off-target effects, we produced the H4G rescue lines by transfecting the pLVX-TetOn-Puro H4G 3xFLAG plasmid into H4GKO cells. The amount of ETS rRNA in the H4G rescue lines was similar to the amount observed in the WT MCF7 line (Supplementary Figure S10). These data indicate that H4G promotes the bulk transcription of rDNA. Because H4G positively contributes to rRNA synthesis, we further examined whether H4G may affect protein synthesis in general. We quantified the amounts of protein synthesis, as measured by OPP (O-propargyl-puromycin) incorporation, and found that protein synthesis rates were reduced by ∼10% in H4GKO cells in comparison with WT cells (N = 3; Figure 6B). Moreover, we quantified cell-cycle progression at 18 h after releasing cell synchronized from G0 (Figure 6C and D) and found that the overall cell cycle was significantly delayed in the H4GKO cells, a likely consequence from dampened protein synthesis (Figure 6D). The cell-cycle delay may be linked to the amounts of rRNA and the decrease of the protein synthesis speed. This may also be due to the decrease of the restarting efficiency from G0/G1. Furthermore, to investigate the growth effects in vivo, the xenograft NOG mice of MCF7, MCF7-KO3 and MCF7-KO9 were created. This xenograft experiment showed that both H4GKO lines generated tumors with sizes half of WT MCF7 cell tumors (Figure 6E and F; Supplementary Figure S11). These results suggest that H4G has a positive role in promoting rDNA transcription, cell growth and proliferation.
Figure 6.

H4G is involved in rRNA transcription. (A) Comparison of rRNA amounts among WT and H4G knockout (H4GKO) MCF7 cell lines. Total RNA was normalized by GAPDH mRNA and loaded onto agarose gels. Error bars represent SD in triplicate experiments. *P < 0.05 and **P < 0.01 (Student's t-test). (B) The relative protein synthesis rate among WT and H4GKO MCF7 cell lines. Error bars represent SD. **P < 0.01. (C) Cell-cycle histograms acquired by flow cytometry and quantification of the subpopulation fraction of the histogram among WT and H4GKOs MCF7 cells. FACS plots and data are representative of at least three separate experiments. (D) The percentage of S phase cells among WT and H4GKOs MCF7 cells. Error bars represent SD. *P < 0.05, ***P < 0.001 (Student's t-test). (E) Representative photographs of the tumors from mouse xenograft model obtained with WT and H4GKO MCF7 cells. The scale bars represent 1 mm. (F) Tumor weight of the MCF7 cell and H4GKO MCF7 cells mouse xenograft models. The black lines represent the mean of tumor weight.
DISCUSSION
In this study, we have characterized H4G, a novel hominidae-specific histone H4 variant. Although this variant has initially been reported to be a replication-dependent histone (16), we were able to obtain cDNA for H4G from polyA-mRNA. Indeed, the presence of poly A+ in histone mRNA is characteristic of replication-independent histone variants (15). A cell cycle-independent, intron-containing, histone H4 gene h4r has been previously identified in Drosophila. However, it encodes a protein identical to that of the cell cycle-dependent H4 gene counterpart (49). The reason(s) why Drosophila has cell cycle-dependent and cycle-independent H4 genes remains unclear. However, the situation is different in primate H4G, in which the amino acid sequence shares only 85% sequence identity with the canonical H4 counterpart. The ensuing 15% sequence difference severely impairs the ability of H4G to form a canonical nucleosome upon in vitro reconstitution (Figure 2). At present the structural molecular details involved in such disruption remain to be elucidated. A simple explanation is that in the presence of adequate chaperoning assistance in vivo, an assembly of a defined H4G-H3/H2A-H2B-containing nucleoprotein complex could take place. Certainly, given that NPM1 interacts with H4G (Figure 5 and Supplementary Figure S7), this chaperone is reasonable candidate for the H4G chaperone. Alternately, it is tempting to speculate that the chromatin disruptive activity described here might serve to further enhance the well-known nucleosome depleted organization of transcriptionally poised rRNA genes in the nucleolus, where H4G variant is targeted (50). Changes in the rate of rRNA synthesis in the nucleolus play a critical role in the regulation of cell proliferation during cancer progression as it will be described below (51).
The amino acid sequences of the N-terminal tail of H4 and H4G differ by four amino acids (Figure 1A). The N-terminal tail of H4 is important for the compaction for chromatin fiber by interacting with the acidic patch on an adjacent nucleosome and with chromatin remodeler ISWI-family and histone H4K5 K8 acetyltransferase HAT1 (37,39,52–54). Our mass spectrometry data showed that both H4 and H4G interact with Hat1 (a catalytic subunit of HAT1), however, neither sample contains the ISWI-family proteins. This is probably due to variation of the immunoprecipitation conditions. More interestingly, we noticed that the core domain of H4G is mainly responsible for its altered cellular localization and interaction proteins. For example, it is known that Rbap46 and Rbap48 interact with H4 alpha 1 domain (44). Rbap46 and Rbap48 can interact with H4G as well, however the interaction efficiency was lower (Table 1). Even though Rbap48 is a subunit of histone chaperone CAF1, H4G does neither interact with CAF1A and CAF1B nor with Asf1. This is most likely the reason why H4G does not localize to the nucleoplasm. Further studies including the mass spectrometry assay using the amino acid swapping between H4 and H4G will provide a more detailed understanding of the H4G function.
In addition to nucleolar histone chaperone NPM1, several other nucleolar histone chaperones have been identified, including factor Spt2 and nucleolin. Nucleolin directly binds to H2A-H2B dimers in order to facilitate nucleosome assembly (55), whereas the transcription factor Spt2 functions as a nucleolar histone chaperone by interacting with the H3/H4 tetramer (56,57). The Spt2 interaction with H4 involves the alpha 3 domain of H4 (56), which is a diverging sequence from that of H4G. Our H4G affinity interaction assay was not able to detect nucleolin nor Spt2, likely due to the stringent washing conditions used.
Of special interest is the expression of the h4g gene in cancer: we found that h4g expression is increased in breast cancer cell lines and in breast cancer patient tissues. Furthermore, the expression was correlated to the progression of the breast cancer stages. This stage dependent expression pattern might be related to the presence of decreasing proportion of normal tissue during cancer progression. Further work is required to understand this breast cancer stage correlation. Our functional analyses further revealed that H4G is likely involved in rDNA transcription. The knockout cell lines showed a decrease of rRNA and protein synthesis, and dysregulated cell-cycle progressions, consistent with previous findings of rRNA and protein synthesis rate might affect cell-cycle progression (58). Furthermore, the xenograft mouse experiment showed that the tumors from H4G knockout lines are much smaller than the tumors from WT MCF7 cell tumors. All of these data suggest that H4G enhances rDNA transcription, which is an important molecular alternation in cancer cells (58).
One well-studied histone variant involved in cancer progression is H3.3 and its K27M mutation, which occurs in pediatric high-grade glioma (59,60). This mutation blocks the polycomb repressor complex 2 methylation activity of H3K27, which is a marker for gene silencing (61–64). Indeed, the reduction of H3K27 trimethylation is associated with cancer-associated genes such as p16INK4A and CDK6 and contributes to tumorigenesis (65).
Other histone variants also show expression changes in human cancer progression, including macroH2A, H2A.Z, H2A.X and H3.3 [for more details see the reviews by Vardabasso et al. (11) and Buschbeck and Hake (66)]. Indeed, the expression of H2A.Z is increased in colorectal, breast, lung and bladder cancers. The elevated expression of H2A.Z is also significantly associated with metastasis to lymph nodes and shorter survival (11,66). However, for many of the histone variants, it is not clear whether altered expressions are a consequence of the cancer state or if they act as oncogenic factors. Even though H4G is a histone variant expressed in cancer patients, several differences are noteworthy. First, while the expressions of other histone variants are altered in cancer patients and cells, they are also present in normal cells whereas H4G appears to be preferentially expressed in breast cancer cell lines and breast tissues from breast cancer patients. Second, H4G is uniquely localized to the nucleoli, where it enhances rRNA expression. How h4g gene expression is triggered by tumorigenicity is intriguing and remains to be elucidated.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Joyce Pui Shuen Wong (HKUST BioCRF) for help with Mass spectrometory analysis and Dorothy Pui San Ieong for her technical assistance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Research Grants Council of the Hong Kong SAR [16104917, 26100214, C-7029-15G, C6009-15G, C7007-17G to T.I.]; Canadian Institutes of Health Research (CIHR) Grant [MOP-130417 to J.A.]. Funding for open access charge: Research Grants Council of the Hong Kong SAR.
Conflict of interest statement. None declared.
REFERENCE
- 1. Workman J.L., Kingston R.E.. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 1998; 67:545–579. [DOI] [PubMed] [Google Scholar]
- 2. Goldberg A.D., Allis C.D., Bernstein E.. Epigenetics: a landscape takes shape. Cell. 2007; 128:635–638. [DOI] [PubMed] [Google Scholar]
- 3. Di Cerbo V., Schneider R.. Cancers with wrong HATs: the impact of acetylation. Brief. Funct. Genomics. 2013; 12:231–243. [DOI] [PubMed] [Google Scholar]
- 4. Baylin S.B. Resistance, epigenetics and the cancer ecosystem. Nat. Med. 2011; 17:288–289. [DOI] [PubMed] [Google Scholar]
- 5. Lo P.K., Sukumar S.. Epigenomics and breast cancer. Pharmacogenomics. 2008; 9:1879–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Skene P.J., Henikoff S.. Histone variants in pluripotency and disease. Development. 2013; 140:2513–2524. [DOI] [PubMed] [Google Scholar]
- 7. Bassing C.H., Suh H., Ferguson D.O., Chua K.F., Manis J., Eckersdorff M., Gleason M., Bronson R., Lee C., Alt F.W.. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003; 114:359–370. [DOI] [PubMed] [Google Scholar]
- 8. Celeste A., Petersen S., Romanienko P.J., Fernandez-Capetillo O., Chen H.T., Sedelnikova O.A., Reina-San-Martin B., Coppola V., Meffre E., Difilippantonio M.J. et al.. Genomic instability in mice lacking histone H2AX. Science. 2002; 296:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Celeste A., Difilippantonio S., Difilippantonio M.J., Fernandez-Capetillo O., Pilch D.R., Sedelnikova O.A., Eckhaus M., Ried T., Bonner W.M., Nussenzweig A.. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003; 114:371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chadwick B.P., Willard H.F.. Multiple spatially distinct types of facultative heterochromatin on the human inactive X chromosome. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:17450–17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vardabasso C., Hasson D., Ratnakumar K., Chung C.Y., Duarte L.F., Bernstein E.. Histone variants: emerging players in cancer biology. Cell Mol. Life Sci. 2014; 71:379–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang R., Poustovoitov M.V., Ye X., Santos H.A., Chen W., Daganzo S.M., Erzberger J.P., Serebriiskii I.G., Canutescu A.A., Dunbrack R.L. et al.. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell. 2005; 8:19–30. [DOI] [PubMed] [Google Scholar]
- 13. Dryhurst D., McMullen B., Fazli L., Rennie P.S., Ausio J.. Histone H2A.Z prepares the prostate specific antigen (PSA) gene for androgen receptor-mediated transcription and is upregulated in a model of prostate cancer progression. Cancer Lett. 2012; 315:38–47. [DOI] [PubMed] [Google Scholar]
- 14. Ausio J. Histone variants–the structure behind the function. Brief. Funct. Genomic. Proteomic. 2006; 5:228–243. [DOI] [PubMed] [Google Scholar]
- 15. Cheema M.S., Ausio J.. The structural determinants behind the epigenetic role of histone variants. Genes (Basel). 2015; 6:685–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Holmes W.F., Braastad C.D., Mitra P., Hampe C., Doenecke D., Albig W., Stein J.L., van Wijnen A.J., Stein G.S.. Coordinate control and selective expression of the full complement of replication-dependent histone H4 genes in normal and cancer cells. J. Biol. Chem. 2005; 280:37400–37407. [DOI] [PubMed] [Google Scholar]
- 17. Kayne P.S., Kim U.J., Han M., Mullen J.R., Yoshizaki F., Grunstein M.. Extremely conserved histone H4 N terminus is dispensable for growth but essential for repressing the silent mating loci in yeast. Cell. 1988; 55:27–39. [DOI] [PubMed] [Google Scholar]
- 18. Santisteban M.S., Arents G., Moudrianakis E.N., Smith M.M.. Histone octamer function in vivo: mutations in the dimer-tetramer interfaces disrupt both gene activation and repression. EMBO J. 1997; 16:2493–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Singh R., Bassett E., Chakravarti A., Parthun M.R.. Replication-dependent histone isoforms: a new source of complexity in chromatin structure and function. Nucleic Acids Res. 2018; 46:8665–8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Durig J., Bug S., Klein-Hitpass L., Boes T., Jons T., Martin-Subero J.I., Harder L., Baudis M., Duhrsen U., Siebert R.. Combined single nucleotide polymorphism-based genomic mapping and global gene expression profiling identifies novel chromosomal imbalances, mechanisms and candidate genes important in the pathogenesis of T-cell prolymphocytic leukemia with inv(14)(q11q32). Leukemia. 2007; 21:2153–2163. [DOI] [PubMed] [Google Scholar]
- 21. Jutras S., Bachvarova M., Keita M., Bascands J.L., Mes-Masson A.M., Stewart J.M., Gera L., Bachvarov D.. Strong cytotoxic effect of the bradykinin antagonist BKM-570 in ovarian cancer cells–analysis of the molecular mechanisms of its antiproliferative action. FEBS J. 2010; 277:5146–5160. [DOI] [PubMed] [Google Scholar]
- 22. Baldan F., Mio C., Allegri L., Conzatti K., Toffoletto B., Puppin C., Radovic S., Vascotto C., Russo D., Di Loreto C. et al.. Identification of tumorigenesis-related mRNAs associated with RNA-binding protein HuR in thyroid cancer cells. Oncotarget. 2016; 7:63388–63407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Herrera J.E., Savkur R., Olson M.O.. The ribonuclease activity of nucleolar protein B23. Nucleic Acids Res. 1995; 23:3974–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murano K., Okuwaki M., Hisaoka M., Nagata K.. Transcription regulation of the rRNA gene by a multifunctional nucleolar protein, B23/nucleophosmin, through its histone chaperone activity. Mol. Cell Biol. 2008; 28:3114–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Di Matteo A., Franceschini M., Chiarella S., Rocchio S., Travaglini-Allocatelli C., Federici L.. Molecules that target nucleophosmin for cancer treatment: an update. Oncotarget. 2016; 7:44821–44840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Finn R.M., Ellard K., Eirin-Lopez J.M., Ausio J.. Vertebrate nucleoplasmin and NASP: egg histone storage proteins with multiple chaperone activities. FASEB J. 2012; 26:4788–4804. [DOI] [PubMed] [Google Scholar]
- 27. Wittmeyer J., Saha A., Cairns B.. DNA translocation and nucleosome remodeling assays by the RSC chromatin remodeling complex. Methods Enzymol. 2004; 377:322–343. [DOI] [PubMed] [Google Scholar]
- 28. Lowary P.T., Widom J.. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998; 276:19–42. [DOI] [PubMed] [Google Scholar]
- 29. Thåström A., Lowary P.T., Widom J.. Measurement of histone-DNA interaction free energy in nucleosomes. Methods. 2004; 33:33–44. [DOI] [PubMed] [Google Scholar]
- 30. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 32. Kamakaka R.T., Biggins S.. Histone variants: deviants. Genes Dev. 2005; 19:295–310. [DOI] [PubMed] [Google Scholar]
- 33. Van Holde K.E. Chromatin. 1989; NY: Springer. [Google Scholar]
- 34. Henikoff S., Smith M.M.. Histone Variants and Epigenetics. 2006; NY: Cold Spring Harbor Laboratory Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chavez M.S., Scorgie J.K., Dennehey B.K., Noone S., Tyler J.K., Churchill M.E.. The conformational flexibility of the C-terminus of histone H4 promotes histone octamer and nucleosome stability and yeast viability. Epigenetics Chromatin. 2012; 5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kan P.Y., Caterino T.L., Hayes J.J.. The H4 tail domain participates in intra- and internucleosome interactions with protein and DNA during folding and oligomerization of nucleosome arrays. Mol. Cell Biol. 2009; 29:538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dorigo B., Schalch T., Bystricky K., Richmond T.J.. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 2003; 327:85–96. [DOI] [PubMed] [Google Scholar]
- 38. Zhou B.R., Feng H., Ghirlando R., Kato H., Gruschus J., Bai Y.. Histone H4 K16Q mutation, an acetylation mimic, causes structural disorder of its N-terminal basic patch in the nucleosome. J. Mol. Biol. 2012; 421:30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luger K., Mader A.W., Richmond R.K., Sargent D.F., Richmond T.J.. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997; 389:251–260. [DOI] [PubMed] [Google Scholar]
- 40. Tatchell K., Van Holde K.E.. Reconstitution of chromatin core particles. Biochemistry. 1977; 16:5295–5303. [DOI] [PubMed] [Google Scholar]
- 41. English C.M., Adkins M.W., Carson J.J., Churchill M.E., Tyler J.K.. Structural basis for the histone chaperone activity of Asf1. Cell. 2006; 127:495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Natsume R., Eitoku M., Akai Y., Sano N., Horikoshi M., Senda T.. Structure and function of the histone chaperone CIA/ASF1 complexed with histones H3 and H4. Nature. 2007; 446:338–341. [DOI] [PubMed] [Google Scholar]
- 43. Flaus A., Rencurel C., Ferreira H., Wiechens N., Owen-Hughes T.. Sin mutations alter inherent nucleosome mobility. EMBO J. 2004; 23:343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murzina N.V., Pei X.Y., Zhang W., Sparkes M., Vicente-Garcia J., Pratap J.V., McLaughlin S.H., Ben-Shahar T.R., Verreault A., Luisi B.F. et al.. Structural basis for the recognition of histone H4 by the histone-chaperone RbAp46. Structure. 2008; 16:1077–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cooper G.M., Housman R.E.. The Cell: A Molecular Approach. 2015; 7th ednSunderland, MA: Sinauer Associates. [Google Scholar]
- 46. Ochs R.L. Methods used to study structure and function of the nucleolus. Methods Cell Biol. 1998; 53:303–321. [DOI] [PubMed] [Google Scholar]
- 47. Bensaude O. Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity. Transcription. 2011; 2:103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mayer C., Grummt I.. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006; 25:6384–6391. [DOI] [PubMed] [Google Scholar]
- 49. Akhmanova A., Miedema K., Hennig W.. Identification and characterization of the Drosophila histone H4 replacement gene. FEBS Lett. 1996; 388:219–222. [DOI] [PubMed] [Google Scholar]
- 50. Derenzini M., Pasquinelli G., O’Donohue M.F., Ploton D., Thiry M.. Structural and functional organization of ribosomal genes within the mammalian cell nucleolus. J. Histochem. Cytochem. 2006; 54:131–145. [DOI] [PubMed] [Google Scholar]
- 51. Nguyen l.X., Raval A., Garcia J.S., Mitchell B.S.. Regulation of ribosomal gene expression in cancer. J. Cell Physiol. 2015; 230:1181–1188. [DOI] [PubMed] [Google Scholar]
- 52. Racki L.R., Naber N., Pate E., Leonard J.D., Cooke R., Narlikar G.J.. The histone H4 tail regulates the conformation of the ATP-binding pocket in the SNF2h chromatin remodeling enzyme. J. Mol. Biol. 2014; 426:2034–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mueller-Planitz F., Klinker H., Ludwigsen J., Becker P.B.. The ATPase domain of ISWI is an autonomous nucleosome remodeling machine. Nat. Struct. Mol. Biol. 2013; 20:82–89. [DOI] [PubMed] [Google Scholar]
- 54. Ludwigsen J., Pfennig S., Singh A.K., Schindler C., Harrer N., Forne I., Zacharias M., Mueller-Planitz F.. Concerted regulation of ISWI by an autoinhibitory domain and the H4 N-terminal tail. Elife. 2017; 6:e21477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Angelov D., Bondarenko V.A., Almagro S., Menoni H., Mongelard F., Hans F., Mietton F., Studitsky V.M., Hamiche A., Dimitrov S. et al.. Nucleolin is a histone chaperone with FACT-like activity and assists remodeling of nucleosomes. EMBO J. 2006; 25:1669–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen S., Rufiange A., Huang H., Rajashankar K.R., Nourani A., Patel D.J.. Structure-function studies of histone H3/H4 tetramer maintenance during transcription by chaperone Spt2. Genes Dev. 2015; 29:1326–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Osakabe A., Tachiwana H., Takaku M., Hori T., Obuse C., Kimura H., Fukagawa T., Kurumizaka H.. Vertebrate Spt2 is a novel nucleolar histone chaperone that assists in ribosomal DNA transcription. J. Cell Sci. 2013; 126:1323–1332. [DOI] [PubMed] [Google Scholar]
- 58. Drygin D., Rice W.G., Grummt I.. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 2010; 50:131–156. [DOI] [PubMed] [Google Scholar]
- 59. Schwartzentruber J., Korshunov A., Liu X.Y., Jones D.T., Pfaff E., Jacob K., Sturm D., Fontebasso A.M., Quang D.A., Tonjes M. et al.. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012; 482:226–231. [DOI] [PubMed] [Google Scholar]
- 60. Wu G., Broniscer A., McEachron T.A., Lu C., Paugh B.S., Becksfort J., Qu C., Ding L., Huether R., Parker M. et al.. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012; 44:251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bender S., Tang Y., Lindroth A.M., Hovestadt V., Jones D.T., Kool M., Zapatka M., Northcott P.A., Sturm D., Wang W. et al.. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013; 24:660–672. [DOI] [PubMed] [Google Scholar]
- 62. Lewis P.W., Müller M.M., Koletsky M.S., Cordero F., Lin S., Banaszynski L.A., Garcia B.A., Muir T.W., Becher O.J., Allis C.D.. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013; 340:857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Venneti S., Garimella M.T., Sullivan L.M., Martinez D., Huse J.T., Heguy A., Santi M., Thompson C.B., Judkins A.R.. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 2013; 23:558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chan K.M., Fang D., Gan H., Hashizume R., Yu C., Schroeder M., Gupta N., Mueller S., James C.D., Jenkins R. et al.. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013; 27:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kallappagoudar S., Yadav R.K., Lowe B.R., Partridge J.F.. Histone H3 mutations–a special role for H3.3 in tumorigenesis. Chromosoma. 2015; 124:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Buschbeck M., Hake S.B.. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017; 18:299–314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.