


Abstract
The development of synthetic biological systems requires modular biomolecular components to flexibly alter response pathways. In previous studies, we have established a module-swapping design principle to engineer allosteric response and DNA recognition properties among regulators in the LacI family, in which the engineered regulators served as effective components for implementing new cellular behavior. Here we introduced this protein engineering strategy to two regulators in the TetR family: TetR (UniProt Accession ID: P04483) and MphR (Q9EVJ6). The TetR DNA-binding module and the MphR ligand-binding module were used to create the TetR-MphR. This resulting hybrid regulator possesses DNA-binding properties of TetR and ligand response properties of MphR, which is able to control gene expression in response to a molecular signal in cells. Furthermore, we studied molecular interactions between the TetR DNA-binding module and MphR ligand-binding module by using mutant analysis. Together, we demonstrated that TetR family regulators contain discrete and functional modules that can be used to build biological components with novel properties. This work highlights the utility of rational design as a means of creating modular parts for cell engineering and introduces new possibilities in rewiring cellular response pathways.
INTRODUCTION
In synthetic biology, one of the primary goals is to program genetic response behavior to biological and chemical cues in a wide range of biological systems (1–4). To flexibly alter those pathways between signal detection and cellular response, synthetic biologists have engineered various biological components that enable the use of multiple molecular signals to regulate the activity of a particular DNA-based promoter for driving gene expression, including the Tango system (5), chimeric antigen receptors (6), scaffold-based two-component systems (7), synNotch receptors (8), MESA receptors (9) and the dCas9-SynR system (10). The underlying reason for developing these biological components is that rewiring of signal detection-cellular response connections is essential for many practical applications (6,11,12).
Most of the abovementioned modular parts are designed for a targeted purpose and thus, they can only be used to develop genetic circuits in highly specific biological systems. Many of the modular receptors mentioned above are membrane-bound proteins, which constrains their use in cells with different membrane architectures. Additionally, these sensors detect signals that are relevant to highly specific physiological conditions, such as the presence of unique antigens or cell–cell interactions. These modular parts are ideal for their intended applications but might not be applicable to new hosts under different environments. Building a genetic circuit in a new type of cell usually requires extensive searching and engineering of circuit parts, which hampers the engineering of many organisms that have great potential for practical applications. This problem can potentially be solved by developing modular sensors that function universally in a wide range of organisms for wiring molecular inputs and genetic outputs.
Allosterically regulated transcriptional regulators from prokaryotes can be used to develop universal parts. Many of these regulators comprise an N-terminal domain that interacts with promoters (DNA-binding) and a C-terminal domain that senses molecular signals (ligand-binding). They regulate transcription by binding to a promoter in response to ligands that are permeable to most types of cells. With such a simple mechanism of action, the resulting inducible expression systems do not involve additional biological components from the host, which allows them to be functional in many organisms and types of cell. Indeed, some regulators, such as LacI and TetR, have been used to control gene expression in yeast (13), human cells (14), plants (15), animals (16) and many microbial organisms (17).
In previous studies, we established a module-swapping strategy to develop hybrid regulators with LacI family proteins, in which the resulting regulators possess desirable and predictable combinations of DNA-binding and ligand-binding properties originated from different native regulators (18,19). Based on this discovery, we constructed a set of hybrid regulators that enable flexible connections between small molecule sensing and promoter control. These engineered parts were harnessed to establish two novel circuit designs with potential biotechnological applications, including a Passcode kill switch (18), and a system of multiple toggle switches with a master OFF signal (19). These studies show that transcriptional regulators are promising for creating robust, modular parts that facilitate cell engineering. However, this module-swapping strategy has not been applied to any other regulator families and LacI family proteins mostly detect sugars and primary metabolites that are degraded easily by most organisms, which inhibits their use as reliable signaling molecules for controlling genetic circuits. To expand the types of signals for genetic control, it is essential to extend the module-swapping strategy to other families of regulators.
In this study, we expand the development of modular sensors to transcriptional regulators in the TetR family. TetR is a well-characterized regulator and has been used extensively to construct genetic circuits (20–22). Previous studies demonstrate that TetR family regulators can be engineered to change DNA recognition and allosteric response properties (23–25). The large amount of knowledge concerning the TetR family provides a strong foundation for investigating the feasibility of the module-swapping strategy for this family of regulators. Additionally, these regulators are able to detect a wide range of drug molecules and therefore, sensors developed from the TetR family are expected to be useful in biomedical research. Here we use a DNA-binding module from TetR (UniProt Accession ID: P04483) and a ligand-binding module from MphR (Q9EVJ6) to build a TetR-MphR hybrid regulator with desirable properties, in which the engineered regulator binds to the DNA sequence recognized by TetR and responds to the inducer of MphR. By demonstrating that TetR family regulators are competent for generating modular sensors, this study aims to facilitate the creation of biological circuit components that are key within synthetic genetic network.
MATERIALS AND METHODS
Chemicals and reagents
All DNA oligonucleotides were synthesized by Eurofins Genomics (Louisville, KY, USA). All enzymes and reagents for cloning were obtained from New England Biolabs (Ipswich, MA, USA). LB medium broth, inducers, and antibiotics were obtained from VWR (Radnor, PA, USA).
Analysis of protein sequences and crystal structures
Protein sequence analyses were carried out with CLC sequence viewer and Clustal Omega (26,27). Proteins analyzed include TetR (Uniprot ID: P04483), MphR (Q9EVJ6), QacR (P0A0N3), EthR (P9WMC1), TtgR (Q9AIU0), AcnR (Q8NQ97), KstR (P96856) and IcaR (Q5HKQ1). Protein crystal structures were analyzed with the software PyMol 1.5.x. Structures studied include PDB IDs: 3ZQI, 4AC0, 2TRT, 3FRQ, 3G56, 3BTJ, 1T56, 2UXU, 4AF5, 3MNL and 2ZCM.
Structural alignment and monomeric interaction comparison of TetR and MphR
The structural similarity analysis on TetR and MphR was computed by rigid-body structural alignment. Here we used a template modeling (TM)-score based algorithm, the TM-align method, developed by Zhang's lab (28) to align the 3D-structure of TetR and MphR (https://zhanglab.ccmb.med.umich.edu/TM-align/). TM-align combines TM-score rotation matrix and dynamic programming and exhibits high accuracy in identifying structural alignment. It supports the well-established similarity measurements including TM-score and root-mean square deviation (RMSD), and also provides the corresponding structure-based sequence alignment (Supplementary Figure S3). We also computed the monomeric residue contacts from the PDB files of these two proteins. The distances between the alpha-carbon atoms of any two residues were computed from the coordinates in the PDB files and a cutoff of 10Å was used to extract those interacting residues.
Construction of plasmids to express TetR-MphR hybrid regulator genes
Construction of hybrid genes required overlap extension polymerase chain reaction (PCR) to fuse the DNA-binding region of tetR with the ligand-binding region of mphR. First, the two regions were cloned separately with PCR. For cloning the DNA-binding module, the reverse primer complementing the 3′ end contained a 5′ overhang of about 25 nucleotides, complementing the 5′ end of the ligand-binding module of the hybrid. Similarly, the forward primer complementing the 5′ end of the ligand-binding module comprised a 5′ overhang that is identical to the last ∼25 nucleotides at the 3′ end of the DNA recognition region. Thus, in the two PCR products, the last ∼50 bp at the 3′ end of the DNA-binding module fragment matched the 5′ end of the ligand-binding module fragment. Then, these two DNA fragments were mixed to perform PCR again to generate a full-length hybrid gene. The primers used to generate each hybrid gene (TM44 to TM58) are listed in Supplementary Table S1, and the nucleotide sequences of these genes are provided in Supplementary Table S2.
To construct tetR-mphR mutants described in Table 1 and Supplementary Table S4, we also used overlap extension PCR, in which primers containing a mutated codon were used to generate gene fragments that are on the N-terminal and C-terminal sides of the mutation. The two PCR products contained an ∼50-bp overlap and they were mixed to perform PCR again to generate a full-length mutant gene.
Table 1.
Characterization of mutants derived from the TM52 hybrid regulator
| GFP fluorescence levels | |||
|---|---|---|---|
| Mutant | 0 mM Erythromycin | 1 mM Erythromycin | Fold-change |
| TM52 L14T | 24931 ± 199 | 28179 ± 539 | 1.13 ± 0.02 |
| TM52 R54A | 191 ± 14 | 209 ± 18 | 1.09 ± 0.09 |
| TM52 M55L | 1071 ± 32 | 8423 ± 190 | 7.87 ± 0.29 |
| TM52 R54A/M55L | 196 ± 64 | 217 ± 36 | 1.19 ± 0.37 |
| TM52 L14T/R54A | 25497 ± 507 | 27712 ± 467 | 1.10 ± 0.04 |
| TM52 L14T/M55L | 25077 ± 577 | 28210 ± 416 | 1.12 ± 0.03 |
| TM52 (no mutations) | 390 ± 5 | 3299 ± 92 | 8.90 ± 0.19 |
Each data point represents mean ± S.D. of six biological replicates.
Rogers et al. have constructed an Escherichia coli-compatible plasmid, pJKR-H-mphR (Addgene number 62559), which contains a constitutively expressing mphR gene, a gfp gene driven by a MphR-regulator promoter and an ampicillin-resistant gene (29). We removed the gfp gene on this plasmid and replaced the mphR gene with a hybrid regulator gene (TM44 to TM58) by using the Gibson assembly methods (30). These plasmids are able to constitutively express a hybrid TetR-MphR regulator derivative in E. coli cells. The sequence of the plasmid containing TM52 (pCC-TM52) is available in the GenBank database.
Relative quantification of hybrid regulator expression with a western blot method
For the gene of each hybrid regulator derivative (TM44 to TM58) in the expression system described above, a HIS-tag was inserted to the C-terminus. Each plasmid containing a resulting HIS-tagged gene was transformed into E. coli MG1655 and the cells were grown in LB medium with 100 μg/ml ampicillin. At OD600 of 0.8–1.0, 1 ml of each culture was harvested by centrifugation and the cell pellet was stored at −80°C. Each pellet was resuspended in 400 μl of a 1 × BugBuster buffer (MilliporeSigma; Burlington, MA, USA) with a protease inhibitor cocktail (Sigma-Aldrich; St Louis, MO, USA) and cells were lysed with ultrasonication. After using centrifugation to remove cell debris, each supernatant containing the same amount of total proteins (determined with Bradford assay analysis) was mixed with a Laemmli sample buffer (Bio-rad, Hercules, CA, USA). Each sample was incubated at a 100°C water bath for 10 min before loading to a Mini-PROTEAN®TGX Stain-Free™ gel for sodium dodecyl sulphate-polyacrylamide gelelectrophoresis (SDS-PAGE). Proteins were then transferred to a PVDF membrane by using a Trans-Blot®Turbo™ transfer system (Bio-rad). The blot was imaged on an UV imager (BioRad Chemi-Doc™ MP Imaging System) to determine the efficiency of blot transfer (Supplementary Figure S6) and it was further stained with Amido Black 10B (Alfa Aesar; Haverhill, MA, USA). The blot was then incubated in 5% Carnation® milk (Nestle; Vevey, Switzerland) in 1 × PBST buffer (Teknova; Hollister, CA, USA) for 2 h to block non-specific protein interactions, before incubated with the primary antibody for the target proteins for 1 h. For the quantification of GAPDH (housekeeping protein) and HIS-tagged hybrid regulators, GAPDH Loading Control Antibody (Catalog #MA5-15738-HRP) and Anti-His C-term-HRP Antibody (Catalog #46-0707) were used, respectively (Invitrogen; Carlsbad, CA, USA). The Clarity Max™ Western ECL Substrate (Bio-rad) was then used to react with the antibodies for 3 min and the same UV imager was used to detect the target protein at the chemiluminescent mode.
Construction of transcriptional reporter plasmids
We previously developed a plasmid, pZA22-GFP, which contains a gfp gene driven by a pLlacO-1 promoter and a kanamycin-resistant gene (18,31). To generate each new promoter, we purchased two single-stranded DNA oligomers that hybridize to form a double-stranded DNA fragment with the 5′- and 3′-ends identical to sticky ends produced by restriction site digestions with XhoI and EcoRI, respectively. The promoter, pLlacO-1, was then removed from pZA22-GFP by these two restriction enzymes, and was replaced by the synthetic DNA fragment by ligation. With this method, we created those transcriptional reporters illustrated in Figures 2D, 3A, 4A, and 5A. Sequences of these promoter fragments are provided in Supplementary Table S1. The sequences of plasmids containing a 0-bp, 1-bp, 2-bp and 3-bp operator (pCC-0, pCC-1, pCC-2 and pCC-3, respectively), as well as the one containing three 2-bp operators (pCC-3op) are available in the GenBank database.
Figure 2.

The design principle of operators for hybrid regulators was established based on structural analyses of multiple TetR family members. (A) A conserved mechanism of action among regulators in the TetR family is illustrated in this panel. The ligand-binding module governs distance between the two DNA-binding domains that interact with the two protein-binding sequences to facilitate regulator-operating association. Thereby, a spacer with a specific length is required to align the two protein-binding DNA sequences to the two DNA-binding domains, such that regulator-operator binding is favorable. An effector molecule triggers conformational changes in the regulator, which alters the distance between the two DNA-binding domains and hampers interactions between the regulator and the operator. (B) If the DNA-binding domain of regulator B is replaced by homologous domains of regulator A, the hybrid regulator is expected to interact with protein-binding DNA sequences from operator A. However, since the DNA-binding domains’ interdistance is controlled by the ligand-binding module B, regulator-operator association requires the length of spacer to be the same as that in operator B. (C) Based on this rationale, we engineered an operator for the TetR-MphR hybrid regulator that is composed of a DNA-binding module from TetR and a ligand-binding module from MphR. This engineered operator contains two TetR-binding DNA sequences separated by a spacer from the MphR operator. (D) The engineered operator was inserted downstream of the −10 region of a pL promoter; binding of the regulator to the operator is expected to repress gene expression driven by the pL promoter.
Figure 3.

A series of TetR-MphR hybrid regulators were characterized to determine the boundaries of a DNA-binding module from TetR and a ligand-binding module from MphR. (A) In Escherichia coli cells, the expression of a gfp gene is driven by the engineered promoter containing the hybrid operator. A TetR-MphR derivative was also expressed in each strain of cells to test its functionality in regulating GFP expression (left panel). To develop these TetR-MphR derivatives, N-terminal regions of TetR were used to replace homologous regions in MphR; the borders between regions from TetR and MphR are illustrated on the top of the right panel. When these strains of cells were exposed to 1 mM erythromycin, the inducer of MphR, for 3 h, cells containing TM49 and TM52 exhibited statistically significant increases in the levels of GFP. Each data point represents mean ± S.D. of six biological replicates. Experimental procedures are described in the ‘Materials and Methods’ section. (B) The TM52 protein with a C-terminal HIS-tag was purified with nickel affinity chromatography. The purified protein (right lane) was analyzed with SDS-PAGE. (C) A gel shift assay was used for invitro characterization of DNA-binding and allosteric response properties of TM52. An amount of 0.5 nmol of an operator DNA fragment was mixed with different concentrations of purified TM52 and erythromycin (Ery) as indicated above the gel images. DNA bands were visualized by staining with ethidium bromide.
Figure 4.
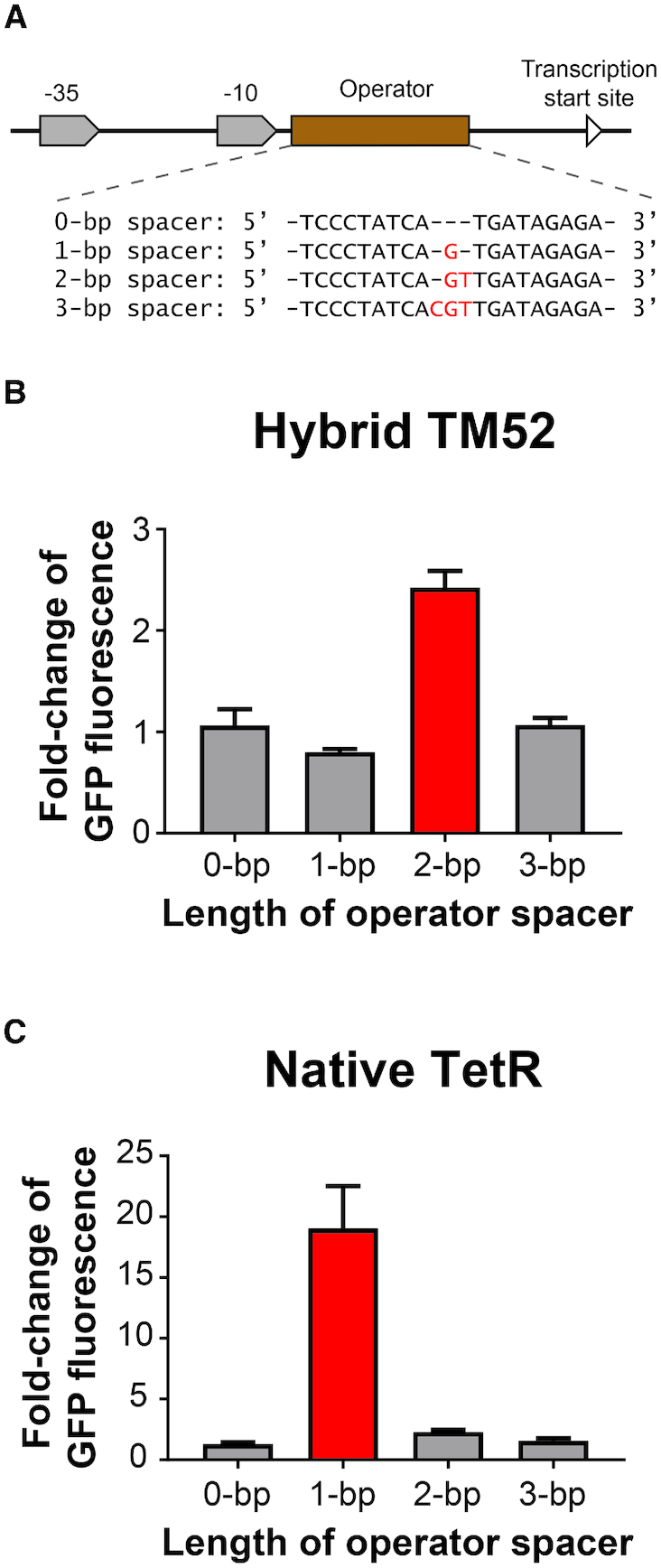
The specificity of the length of spacers was further investigated. (A) We modified the operator by separating the two TetR-binding DNA sequences with spacers with 0, 1, 2 or 3 bp. These modified promoters were used to control GFP expression in Escherichia coli cells containing (B) the TM52 hybrid regulator or (C) the native TetR. We then compared GFP expression in these cells in their uninduced and induced states as described in the ‘Materials and Methods’ section. For TM52-containing strains, cells were exposed to 0 and 1 mM erythromycin and for TetR-containing strains, cells were exposed to 0 and 100 nM of anhydrotetracycline. Data are represented as ratios of GFP fluorescence in induced cells to that in uninduced cells. Each data point represents mean ± S.D. of six biological replicates.
Figure 5.
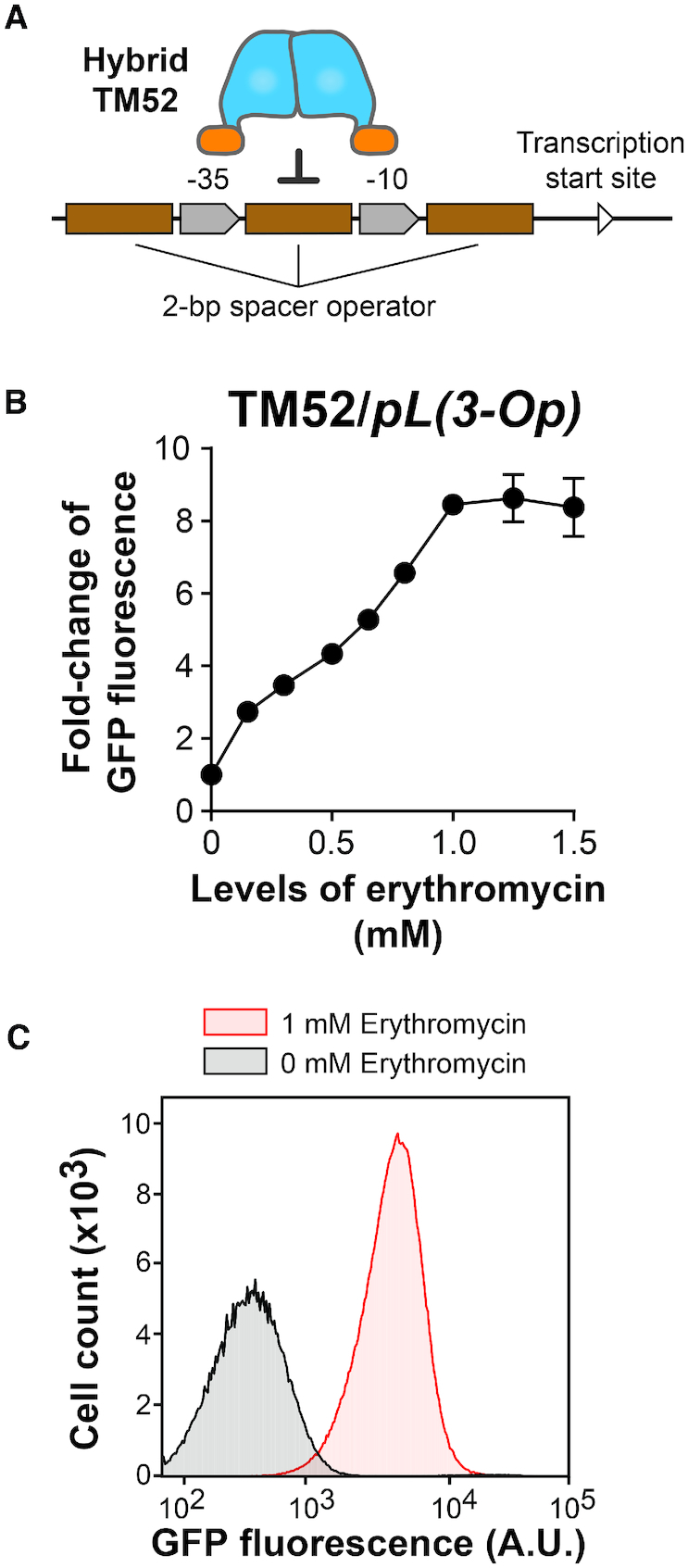
The TetR-MphR system was modified to improve the dynamic range of gene expression upon induction. (A) Three copies of the TetR-MphR operator were incorporated into the promoter for controlling GFP expression. (B) Cells containing this modified expression system responded to erythromycin dose-dependently to reach above 8-fold of induction at 1 mM. Each data point represents mean ± S.D. of six biological replicates. Representative raw data of these experiments, acquired with a flow cytometer, are illustrated in panel (C).
Characterization of inducible expression systems in E. coli cells
Each expression system was tested with the E. coli MG1655 strain. In each experiment, a regulator gene-containing plasmid and a transcriptional reporter plasmid were co-transformed into the cells. By acquiring these two plasmids, the cells gained resistance toward both ampicillin and kanamycin. We picked single colonies formed on LB agar plate with 100 μg/ml ampicillin and 50 μg/ml kanamycin and inoculated them in LB medium with the same antibiotics at 37° C and 200 rpm for cell growth.
At OD600 of 0.2–0.3, a volume of 200 μl of the culture was transferred to the wells of a 96-well plate. An inducer (erythromycin or anhydrotetracycline) was added to reach the concentrations as described in Figures 3–5. After 3 h of exposures to the inducer, flow cytometry data were obtained using an ACEA NovoCyte 2030YB flow cytometer (ACEA Biosciences, Inc.) The geometric means of GFP fluorescence were calculated using the NovoExpress® software.
Protein purification of TM52
The C-terminal HIS-tagged TM52 gene was cloned into a plasmid pZA12 via the restriction sites, KpnI and HindIII (31). The plasmid pZA12 contains an ampicillin-resistance gene and a pLlacO-1 promoter and thus, the resulting plasmid (pZA12-TM52HIS) allows inducible expression of the HIS-tagged TM52 protein in E. coli cells in response to Isopropyl β-D-1-thiogalactopyranoside (IPTG) as an inducer. This plasmid was transformed into E. coli MG1655Pro (32), which constitutively express LacI to control TM52 expression. The cells were grown in LB medium with 100 μg/ml ampicillin at 37°C and 200 rpm overnight. The saturated culture was diluted 100-fold in two 4-l flasks, each containing 1 l of the same type of cultural medium, and these cells were grown at the same conditions. At OD600 reached 0.5–0.6, a final concentration of 1 mM of IPTG was then added to induce TM52 expression. After 3 h of incubation at the same conditions, cells were collected with centrifugation at 4°C. After the removal of supernatant, cell pellets were stored at −80°C.
The overexpressed protein, TM52-HIS, was then purified with nickel affinity chromatography. As we observed that several native proteins undesirably co-purified with our target protein (data not shown), we performed the purification at a denaturing condition to eliminate the binding of these proteins to the affinity column. All subsequent steps were performed at 4°C. Cell pellets from the 2-l culture were resuspended in 30 ml of lysis buffer (20 mM Tris–HCL, 100 mM NaCl, 10 mM β-mercaptoethanol (BME), pH 8.0) with 6 M guanidine HCl and 0.1 M phenylmethylsulfonyl fluoride and they were then lysed with ultrasonification. Proteins from these lysed cells were denatured by incubating overnight with stirring. Cell debris was then removed by centrifugation at 20 000 × g for 45 min. Supernatant was loaded to a 5-ml nickel affinity column that was equilibrated with 20 ml of 6 M guanidine HCl in wash buffer (50 mM Tris–HCl, 300 mM NaCl, 10 mM BME, 10% glycerol, pH 8.0). The column was then washed serially with three buffers, which were 20 ml of wash buffer with 8 M urea and 20 mM imidazole, 20 ml of wash buffer with 4 M urea and 40 mM imidazole, and 20 ml of wash buffer with 0 M urea and 40 mM imidazole. At last, HIS-tagged TM52 was eluted with wash buffer with 250 mM imidazole and the protein elution was monitored with absorbance at 280 nm. Collected fractions were analyzed with SDS-PAGE to evaluate protein purity. Imidazole was then removed from the sample with dialysis (25 mM Tris–HCl, 5 mM NaCl, 2 mM dithiothreitol, 30% glycerol, pH 7.4). The purified TM52 protein was stored at −80°C.
Gel shift assay for in vitro characterization of TM52
Serial quantities of TM52 and erythromycin were mixed with 0.5 nmol of DNA (containing the operator site; see Supplementary Table 1 for the DNA sequence) to reach a final volume of 15 μl in the complex buffer (20 mM Tris–HCl, 5 mM MgCl2, pH 8.0) and incubated at room temperature for 15 min. The samples were then analyzed with electrophoresis, using a Bio-rad Criterion™ set. Samples were loaded on a native acrylamide gel that contained a 4% stacking gel and an 8% resolving gel, which were prepared with the TBM buffer (90 mM Tris–HCl, 90 mM boric acid, 5 mM MgCl2, pH 8.0). The gel was first ran at 50 V for 15 min and then at 80 V for 1 h and 45 min. It was then placed in 50 ml of TBM buffer containing ethidium bromide for 45 min to stain the DNA. The stained gel was observed with an UV imager to characterize DNA-TM52 association.
Characterization of candidates from the TetR-MphR mutant library
To construct a TetR-MphR mutant library with a random mutation at amino acid residue position 14, we performed overlap extension PCR again by using primers that contain three randomized nucleotides that code for residue 14 of the gene. These gene fragments were used to replace the native mphR gene in pJKR-H-mphR. We then replaced the promoter regulated by MphR with the one regulated by TetR-MphR as shown in Figure 5A. Primers used are listed in Supplementary Table S1.
Plasmids containing the tetR-mphR gene with a randomly mutated residue 14 were transformed into cells. A total of 180 colonies from this transformation were picked and grown in 200 μl of LB media with 50 μg/ml of kanamycin in 96-well plates. At OD600 = 0.2, 50 μl of culture from each candidate was mixed with 150 μl of fresh LB media with kanamycin and erythromycin at a final concentration of 0 or 1 mM. These plates were incubated at 37°C and 1000 rpm for 3 h. OD600 and GFP florescence of these samples were measured by using a BioTek Synergy H1M microplate reader. Fold-change of GFP florescence per OD600 for each candidate in response to the inducer was calculated to assess mutant performance (Supplementary Table S4).
RESULTS AND DISCUSSION
Selection of feasible candidates for applying the module-swapping strategy
To gain insight into whether regulators in the TetR family are competent for module-swapping, we analyzed the sequences and crystal structures of eight members of this family generated from previous studies, including TetR, MphR, QacR, EthR, TtgR, AcnR, KstR and IcaR (33–40). These TetR family members are functioning as homodimers, in which two DNA-binding domains are located at the N-terminal end of the monomers (Supplementary Figure S1). Each DNA-binding domain contains three α-helices (α1, α2 and α3 on each monomer), which includes a helix-turn-helix motif. Helix α4 connects the DNA-binding domain to the ligand-binding domain that mediates ligand-binding and dimerization.
Among these members, we selected TetR and MphR as our targets because they are structurally homologous and their biological activities have been well-characterized (29). We used the Template Modeling (TM)-align method (28) to compare the 3D-structures of TetR and MphR. Proteins with a TM-score >0.5 are considered as possessing the same protein folding architecture and those with a TM-score <0.3 are expected to be structurally unrelated. For TetR and MphR, TM-score is 0.58 when it is normalized based on the MphR sequence and RMSD is 4.27 Å, which suggests that they have high structural similarity (Figure 1A). Specifically, our TM-align analysis shows that the DNA-binding domains of TetR and MphR are structurally homologous (TM-score = 0.70; RMSD = 1.34; Supplementary Figure S2). The ligand-binding domains of TetR and MphR are also structurally similar, which contain α-helices 4–10 of TetR and α-helices 4–9 of MphR. The alignment of the two ligand-binding domains generated a TM-score of 0.55 and a RMSD of 4.26 Å, in which this relatively low TM-score is mainly due to an additional α-helix in TetR (α-helix 9) that does not exist in MphR. For the region around the C-terminus of these proteins, α-helices 7, 8 and 10 of TetR align well with α-helices 7, 8 and 9 of MphR (Supplementary Figure S2).
Figure 1.

Structural analyses of TetR and MphR suggest that they are feasible for module-swapping. (A) The results of TM-align analysis (top) support that TetR (PDB ID: 3ZQI) and MphR (3G56) have a high structural similarity; the TM-scores are above 0.5 when they are normalized by both the sequences of TetR and MphR, and the RMSD is relatively small (4.27 Å). The graphical ribbon representation of the alignment between monomers of TetR and MphR are shown at the bottom. (B) Domain–domain interactions are also structurally conserved between TetR and MphR. Monomeric residue contact maps were generated for TetR and MphR based on the proximity between residues, with a distance cutoff at 10 Å between alpha-carbon atoms. Interactions between the DNA-binding domain and the ligand-binding domain are indicated with the boxes. The residues involved are located at the same aligned position in both TetR and MphR according to the structural-based sequence alignment (Supplementary Figure S3), suggesting that the domain interfaces of TetR and MphR have similar structural features.
In addition to structural similarity between TetR and MphR, our analysis on residue interactions between the DNA-binding domain and ligand-binding domain also supports that module-swapping is feasible to these two proteins. Based upon the structural features (Figure 1A), TM-align also provided a sequence alignment of TetR and MphR to assign corresponding residues between the two proteins (Supplementary Figure S3). A monomer contact map was then generated for each protein based on residue proximity, which illustrates potential residue-residue interactions within the protein monomer (Figure 1B). We then searched the maps for residues involved in inter-domain interactions and the results imply that these interactions are also structurally conserved. In both TetR and MphR, the DNA-binding domain (aligned positions 5–25) interacts with residues at aligned positions 50–60 and 90–100 from the ligand-binding domain. These results suggest that the interfaces between the DNA-binding domain and the ligand-binding domain are highly similar between TetR and MphR.
Our analyses demonstrate that TetR and MphR share many structural features, including spatial conformation and interface between domains driven by inter-domain residual interactions. Intriguingly, previous studies suggested that the sequence diversity between ligand-binding domains of these two proteins is significant and they were not considered to be in the same domain family in the Pfam database (41). However, based on structural analyses, we proposed that functional regulators can be generated by swapping the DNA-binding module and ligand-binding module between these two proteins.
Rational design of an operator for the TetR-MphR hybrid regulator
After selecting TetR and MphR as the target for this study, we developed a DNA-based operator for a hybrid regulator that contains the TetR DNA-binding module and the MphR ligand-binding module. The engineered operator was designed based on the molecular mechanism of action of these regulators, which is illustrated in Figure 2A. TetR family members are homodimers with each monomer possessing a DNA-binding domain. Each regulator binds to an operator that has an internal palindromic symmetry with a specific number of base pairs at the center (spacer; Figure 2A). Each of the two DNA-binding domains interacts with one of the symmetric fragment, which is the protein-binding sequence recognized by the regulator. Binding of an inducer to the regulatory domain triggers a series of conformational changes, which lead α4 in each monomer to move away from each other like a pincer. As a result of this movement, protein-DNA interactions are perturbed due to the increase in distance between the two DNA-binding domains.
Based upon this model, DNA–protein association not only depends on DNA recognition by the two DNA-binding domains but also the length of spacer that separates the two protein-binding DNA sequences. Previous studies show that each TetR family regulator binds to a different operator with highly specific length of spacer (42,43). Ligand-binding domains dominantly affect distance between the two DNA-binding domains and thus, they determine the required length of spacer in an operator. As a result, a hybrid transcriptional regulator in the TetR family may only interact with a hybrid operator that contains protein-binding sequences and a spacer originated from two different operators. As illustrated in Figure 2B, regulators A and B are members of the TetR family. If the DNA-binding module of regulator A is fused with the ligand-binding module of regulator B, the resulting hybrid regulator is expected to recognize a new operator that is composed of two protein-binding sequences A separated by a spacer B.
To test this hypothesis, we developed a hybrid operator that is expected to be recognized by a TetR-MphR regulator—a hybrid regulator containing a DNA-binding module from TetR and a ligand-binding module from MphR. We predicted that the spacer length in operators for TetR and MphR are 1 bp and 2 base-pairs, respectively (Figure 2C), based on palindromic symmetry of these DNA fragments. We then replaced the spacer in the tet operator with that in the mph operator. In the resulting operator, the two protein-binding sequences are separated by 2 bp (Figure 2C). This engineered operator was incorporated downstream of the −10 region of a pL promoter (31); binding of the TetR-MphR to this location is expected to repress gene expression driven by this promoter (Figure 2D). We also demonstrated that this promoter is biologically active in expressing the gfp gene in E. coli cells (Supplementary Figure S4).
Development of a functional TetR-MphR hybrid regulator
In parallel to designing the hybrid operator, we constructed the hybrid regulator, TetR-MphR, which contains the DNA-binding module from TetR and the ligand-binding module from MphR. TetR amino acids 1 to 45 form α-helices 1 to 3 that bind to DNA; additionally, Tyr48 at the N-terminal end of α-helix 4 interacts with a phosphate group at the backbone of the operator (44). Indeed, a previous study confirmed that helices 1–3 and the N-terminal part of α4 are essential for DNA binding (45) and thus, we hypothesized that in TetR, the boundary between a DNA-binding module and a ligand-binding module is at the N-terminal region of α-helix 4.
To identify the TetR DNA-binding module, we chose regions stretching from the N-terminus of TetR to a group of residues near the α3-α4 interface (amino acids 44–58), and then used these fragments to replace the homologous regions from MphR (Figure 3 and Supplementary Figure S1). The resulting hybrids (designated TM44–TM58; Figure 3A) were expressed in a strain of E. coli cells that harbors a GFP transcriptional reporter driven by the promoter described in Figure 2D. Cells containing each hybrid derivative were exposed to erythromycin, which is an inducer of the native MphR; and GFP expression in each strain of cells was measured afterward. As illustrated in Figure 3A, TM49 and TM52 generated significant increases in GFP expression in response to erythromycin, which implies that the two hybrid regulators possess allosteric response and DNA recognition properties of MphR and TetR, respectively. We also used a western blot method to show that the difference in transcriptional control activities was not caused by an alteration in protein expression among these TetR-MphR derivatives. A HIS-tag was inserted to the C-terminal end of each TM derivative gene, which did not affect the transcriptional control function of the wild-type TetR and the TM52 hybrid regulator (Supplementary Figure S5). The cellular levels of these HIS-tagged regulators were relatively quantified with a western blot method, which demonstrates that cellular levels of these TM derivatives were similar (Supplementary Figure S6). These results imply that TM49 and TM52 possess expected DNA-binding and allosteric response properties.
In addition to the transcriptional reporter assay, we also tested the hybrid regulator TM52 in vitro. Results from gel shift assays further confirm that TM52 possesses the expected DNA-binding and allosteric response properties. We purified the HIS-tagged TM52 protein with nickel affinity chromatography (Figure 3B). A range of quantities of the purified protein was mixed with a DNA fragment containing the operator sequence, and the samples were analyzed on native polyacrylamide gels. As shown on Figure 3C, a protein quantity-dependent shift of the DNA band was observed, which implies that TM52 was able to bind to the operator site. We then characterized how erythromycin may affect DNA operator-TM52 protein association and we observed that the DNA band was shifted back to the free DNA location when erythromycin concentration was above 0.5 mM (Figure 3C), which supports that TM52 responds to erythromycin as an inducer.
Together, these experiments support that our module-swapping strategy is feasible for engineering regulators from the TetR family. All other hybrids, including TM44 to TM48 and TM53 to TM58, did not respond to erythromycin. Intriguingly, basal GFP levels under different hybrids varied drastically (Supplementary Table S3), suggesting that each TetR-MphR derivative interacted with the hybrid operator to a different extent.
Investigation on the role of spacer in the hybrid operator
We constructed a TetR-MphR hybrid regulator by using a DNA-binding module from TetR and a ligand-binding module from MphR (Figure 3), and this engineered regulator is capable of recognizing a hybrid operator that contains protein-binding sequences from TetR and a spacer from MphR (Figure 2D). These results led us to ask whether the length of spacer is critical for the hybrid regulator-operator association and also, whether the ligand-binding module determines this length. To investigate these two questions, we built a series of promoters that contain an operator with 0 to 3 bp to serve as the spacer between palindromic protein-binding sequences (Figure 4A). All four versions of engineered promoters were capable of activating gfp expression in E. coli (Supplementary Figure S4). We then use a TetR-MphR hybrid (TM52) and a native TetR to control these four promoters for driving GFP expression in E. coli cells (Figure 4B and C).
For the TetR-MphR hybrid, the regulator only repressed GFP expression driven by the promoter with a 2-bp spacer (Supplementary Table S3). Upon inducer exposure, GFP expression only increased when the gene was under this 2-bp spacer promoter (Figure 4B). These results from the TetR-MphR hybrid regulator suggest that the hybrid regulator only bound to the operator with a 2-bp spacer, which implies that length of spacer in the operator plays a key role in facilitating interactions between the hybrid regulator and the operator.
For the native TetR, it only regulated GFP expression driven by the 1-bp spacer promoter and did not interact with the other three promoters (Figure 4C and Supplementary Table S3). While the TetR-MphR and the native TetR contain the same DNA-binding module, the two regulators associated specifically to operators with different spacer length. These results imply that ligand-binding modules define the essential spacer length for regulator-operator association.
Improvement on the dynamic range of inducible expression by TetR-MphR
In vivo and in vitro characterization shows that the TM52 hybrid regulator is biologically active. However, the TM52 inducible system only generated a dynamic range of 2.5-fold in GFP expression, which is less efficient compared to using the native TetR to control a promoter with similar architecture (19.0-fold; Supplementary Figure S7). These results led us to seek improvements on the TetR-MphR system, aiming to expand the dynamic range of gene expression that the system is capable to modulate. This may be achieved by increasing the number of operator sites in the promoter. In our expression system, the binding of the regulator to the promoter represses gene expression. However, at equilibrium, a portion of the promoters are not associated to the regulator, which leads to basal GFP expression. Potentially, multiple operators can be incorporated into a promoter, in which the binding of a regulator to any operator sites represses gene expression. As a consequence, an increase in the number of operators reduces basal expression by elevating the portion of promoters that are associated to a regulator. Recently, Lee et al. showed that this strategy is efficient in eliminating leaky expression from an inducible system (46).
Based upon this rationale, we incorporated a total of three operator sites to the promoter—one located upstream of the −35 region, another one between the −35 and −10 regions, and the last one downstream of the −10 region (Figure 5A). By using this modified TetR-MphR inducible system to control GFP expression, the basal GFP levels indeed reduced ∼10-fold (Supplementary Table S3). When cells containing this new system were exposed to 0–1.5 mM erythromycin, GFP expression increased dose-dependently to reach 8.9 ± 0.6-fold of induction and started to plateau when erythromycin concentrations were above 1 mM (Figure 5B and Supplementary Table S3). With this increased dynamic range of inducible expression, the TetR-MphR system is more proficient to serve as components for cell engineering.
Characterization of the functional role of residues 14, 54 and 55 in TetR-MphR
As discussed above, sequences of ligand-binding modules in TetR and MphR are not conserved even they are structurally homologous, which led us to ask whether our module-swapping perturbs any residue-specific interactions between the DNA-binding module and ligand-binding module. Based on crystal structures of TetR and MphR, we speculated that hybrid protein residues at positions 14, 54 and 55 play a key role in facilitating interactions between the DNA-binding module and the ligand-binding module. In the native TetR, leucine 14 is located at α-helix 1 and it is at close proximity to residues at α-helix 4, such as alanine 54 and leucine 55, which belong to the ligand-binding module (Supplementary Figure S8). These residues are expected to form hydrophobic interactions that stabilize the protein structure and fixate the orientation between the DNA-binding module and α-helix 4. In the native MphR, the type of interactions between α-helices 1 and 4 appears to be different because the positions that are homologous to TetR leucine 14, alanine 54 and leucine 55 are occupied by threonine, arginine and methionine, respectively, which are expected to generate polar interactions (Supplementary Figure S8). These observations suggest that the type of interactions between DNA-binding module and ligand-binding module is not conserved between TetR and MphR. In the TetR-MphR hybrid regulator, since α-helices 1 and 4 are originated from TetR and MphR, respectively, interactions between the two helices are expected to be hampered. This may reduce structural stability and thus, diminish the performance of the resulting hybrid regulator in transcriptional control.
To test whether amino acid residues 14, 54 and 55 play a key role in module–module interactions within the TetR-MphR hybrid regulator, we mutated L14 on TM52 to a threonine, aiming to imitate the α1–α4 interactions in the native MphR, and generated three other mutants, including R54A, M55L and R54A/M55L, to imitate the native interactions in TetR. These mutants were used to control GFP expression in cells, which are under a similar system illustrated in Figure 5A. As shown in Table 1, cells containing the L14T mutant possessed high-basal GFP expression and did not respond to erythromycin, suggesting that the mutant lost the DNA-binding function. This result implies that L14 is essential for DNA binding. In contrast, the R54A mutant led to decreased basal GFP levels but the expression was not inducible by erythromycin, which implies that the mutant remained active in DNA-binding but became inactive in allosteric response. The residue R54 is originated from MphR, and for native TetR, a mutation at the adjacent homologous position (TetR L55S) is also reported as induction deficient (47); the similarity in mutant phenotype suggests that TetR and MphR have similar molecular mechanism for allostery. There are at least two plausible explanations for this R54A mutant phenotype: (i) residue R54 is involved in maintaining a rigid confirmation between the DNA-binding module and α-helix 4. The R54A mutation increases the degrees of freedom of the DNA-binding module, which allows it to bind to a DNA operator even at the induced state. (ii) R54 plays a role in facilitating the conformational change during allosteric response and the mutation at this position hinders the resulting protein to respond to the inducer. Results from our phenotypic analysis of two additional mutants, M55L and R54A/M55L, favor the first hypothesis, in which R54 contributes to module–module interactions. For cells with the M55L mutant, the basal GFP expression increased ∼3-fold but their fold-change in GFP fluorescence was similar to cells containing non-mutated TetR-MphR upon induction (Table 1), which suggests that residue 55 contributes to maintaining a desirable protein conformation for DNA-binding and it does not affect allosteric response. If R54 only involves in ligand binding, a R54A/M55L mutant should generate an increased basal expression, similar to the singly mutated M55L regulator. However, the double mutation, R54A/M55L, generated a mutant that tightly associated to the operator but lost the erythromycin response activity, which was similar to the singly mutated R54A regulator. The mutation M55L did not affect DNA-binding in the presence of R54A, which implies that R54 plays a critical role in interacting with the DNA-binding module. It is likely that R54A has a dominating role in facilitating module-module interactions and thus, the R54A mutation eliminated the effect of M55L on the DNA-binding module. In addition, both mutants, L14T/R54A and L14T/M55L, provided similar effect as the single mutation L14T, in which these mutants all did not repress gene expression. The dominating effect of L14T strongly supports that residue 14 directly involves in the DNA binding function. Overall, we have illustrated that key residues at the module–module interface also play critical roles in maintaining a range of protein activities, in which residues 14 and 55 support DNA-protein association and residue 54 enables allosteric response.
The L14T mutation harms the DNA-binding activity of TetR-MphR and this led us to ask whether leucine at this position is essential for the protein function. We created a mutant library by randomly mutating the codon for residue 14 of the tetR-mphR gene, and assessed the regulation activity of GFP expression by 180 mutant candidates. As shown in Supplementary Table S4, most mutants had reduced or no activity—their basal GFP fluorescence levels were high and the fold-changes in GFP expression were decreased upon induction. We sequenced those candidates within the top 5% in fold-change of GFP expression upon induction (eight candidates) and intriguingly, all of them contained a leucine residue at position 14 (Supplementary Table S4). These results suggest that L14 is an essential residue for the functional activity of the TetR-MphR hybrid regulator.
CONCLUSION
Building modular transcriptional regulators from TetR family proteins will establish many new connections between signal detection and promoter control. Given that the TetR family contains over 73 000 members that detect a wide range of molecules, our approach has the potential of creating a vast array of biomolecular sensing elements for building cellular response pathways. In this study, we have demonstrated that a module-swapping strategy is feasible for engineering transcriptional regulators in the TetR family. Specifically, we have determine a part of the TetR protein that can be used to replace the homologous region in the MphR for changing the DNA-binding specificity of the resulting protein. This shows that the DNA-binding function and allosteric response function can be modular within the TetR family. Additionally, our study shows that the spacer in an operator plays a key role in facilitating protein–DNA association and the ligand-binding module dominantly control the optimal length of the spacer.
The performance of the TetR-MphR hybrid is relatively low in reference to the native TetR, in which based upon our recent study, this can be the result of incompatibility among residues that facilitate interactions between the DNA-binding module and the ligand-binding module (19). To further our understanding on interactions between the TetR DNA-binding module and the MphR ligand-binding module, we studied three amino acid residues at the module–module interface by using mutant analysis and we showed that these residues are also critical for DNA-binding and allosteric response activities of the hybrid regulator. Due to the high complexity of their functional roles, it is difficult to modify these residues rationally to enhance the performance of the hybrid regulator. Instead of using rational design, we may achieve the same goal with a directed evolution approach; recently, Meyer et al. used this approach to improve the efficiency of multiple transcriptional regulators, where they used rounds of positive and negative selections to screen repressor mutant libraries, identifying candidates that provide expression systems with low background, high dynamic range, high ligand sensitivity and low crosstalk with other expression systems (48). A similar strategy may be capable of evolving the TetR-MphR hybrid, which improves the efficiency of the inducible system.
As another future direction to promote the development of modular regulators from the TetR family, we may use computational methods to predict highly compatible modules from its family members. A feature of the TetR family is the conservation of its DNA-binding module (Pfam PF00440) while the ligand-binding module exhibits diversity in sequence and structure but shares common architectural and generic features, allowing us to study these systems from an evolutionary point of view. Additionally, coevolutionary constraints encode interactions between DNA-binding module and ligand-binding module, which are responsible for the induction of conformational changes upon ligand binding through inter-domain contacts (49). We have recently built a coevolutionary model to predict the compatibility of a hybrid repressor with DNA-binding and ligand-binding modules from different LacI family members (19). The model predicts the compatibility and functionality of a new hybrid repressor from the variation of the coupling strength of co-evolving amino acid residue pairs in two modules after module-swapping, at an accuracy of 87.5%. This model demonstrates the critical role of co-evolution in defining compatibility of engineered regulators. With the current study revealing that the module-swapping strategy is feasible for regulators in the TetR family, it is promising for us to use a similar coevolutionary modeling approach to identify TetR family members that are compatible for constructing hybrid regulators.
Furthermore, the mode of DNA-binding can be mechanistically different for different TetR family regulators, which provides an additional challenge in designing operators for hybrid regulators. For some TetR family regulators, only one protein molecule binds to an operator for repressing gene expression. However, some other members require two regulator molecules to cooperatively bind to an operator, one on the distal side of the operator and the other one on the proximal side (50). The mode of regulator-operator association has not been fully characterized for many TetR family members. To engineer operators that are optimal to hybrid regulators, it would be important to fully understand the architecture of regulator–DNA complexes.
Overall, this work and our previous results show that the module-swapping strategy can be applied to regulators in both the TetR and LacI families, which suggests that it might be a general strategy for many regulator families, such as the families of GntR, LysR and AraC. In fact, this notion is supported by a recent study by Schmidl et al., in which they demonstrated that swapping the DNA-binding domains of regulators in the OmpR and NarL families led to rational alteration of DNA-binding specificity of the engineered proteins (51). In our future studies, we will extend our studies to other TetR family members, as well as regulators in other protein families, to further test the feasibility of our engineering approach and to create an extensive toolset for cell engineering.
DATA AVAILABILITY
DNA sequences are available in GenBank database under accession numbers MH026045, MH026046, MH026047, MH026048, MH026049 and MH026050.
Supplementary Material
ACKNOWLEDGEMENTS
Authors’ contributions: R.P.D., B.R.J. and C.T.Y.C. performed construction of genes and inducible expression systems, and all in vivo experiments. X.J., F.M. and C.T.Y.C. performed structural and computational analyses. R.P.D. and D.P.P. performed protein purification and gel shift assay experiments. B.R.J. and J.S.G. performed western blot characterization. R.P.D., X.J., C.M., F.M. and C.T.Y.C. composed the manuscript. C.T.Y.C. designed the overall research.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [MCB-1914538]; National Institutes of Health [NIGMS-7R15GM119118-02]; UT Tyler Internal Research Grants program (to C.T.Y.C.); The University of Texas System Rising STARs Program [# 802-1053-T000674F to C.T.Y.C. and # 802-1053-T171053F to J.S.G]; Welch Foundation [# BP-0037]; The School of Natural Sciences and Mathematics at The University of Texas at Dallas (to F.M.). Funding for open access charge: The University of Texas at Tyler and National Science Foundation [MCB-1914538].
Conflict of interest statement. None declared.
REFERENCES
- 1. Nielsen A.A., Segall-Shapiro T.H., Voigt C.A.. Advances in genetic circuit design: novel biochemistries, deep part mining, and precision gene expression. Curr. Opin. Chem. Biol. 2013; 17:878–892. [DOI] [PubMed] [Google Scholar]
- 2. Arkin A.P. A wise consistency: engineering biology for conformity, reliability, predictability. Curr. Opin. Chem. Biol. 2013; 17:893–901. [DOI] [PubMed] [Google Scholar]
- 3. Purnick P.E., Weiss R.. The second wave of synthetic biology: from modules to systems. Nat. Rev. Mol. Cell Biol. 2009; 10:410–422. [DOI] [PubMed] [Google Scholar]
- 4. Lu T.K., Khalil A.S., Collins J.J.. Next-generation synthetic gene networks. Nat. Biotechnol. 2009; 27:1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barnea G., Strapps W., Herrada G., Berman Y., Ong J., Kloss B., Axel R., Lee K.J.. The genetic design of signaling cascades to record receptor activation. PNAS. 2008; 105:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H.. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011; 365:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Whitaker W.R., Davis S.A., Arkin A.P., Dueber J.E.. Engineering robust control of two-component system phosphotransfer using modular scaffolds. PNAS. 2012; 109:18090–18095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morsut L., Roybal K.T., Xiong X., Gordley R.M., Coyle S.M., Thomson M., Lim W.A.. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. 2016; 164:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schwarz K.A., Daringer N.M., Dolberg T.B., Leonard J.N.. Rewiring human cellular input-output using modular extracellular sensors. Nat. Chem. Biol. 2017; 13:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baeumler T.A., Ahmed A.A., Fulga T.A.. Engineering synthetic signaling pathways with programmable dCas9-Based chimeric receptors. Cell Rep. 2017; 20:2639–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roybal K.T., Williams J.Z., Morsut L., Rupp L.J., Kolinko I., Choe J.H., Walker W.J., McNally K.A., Lim W.A.. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell. 2016; 167:419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Porter D.L., Kalos M., Zheng Z., Levine B., June C.. Chimeric antigen receptor therapy for B-cell malignancies. J. Cancer. 2011; 2:331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mazumder M., McMillen D.R.. Design and characterization of a dual-mode promoter with activation and repression capability for tuning gene expression in yeast. Nucleic Acids Res. 2014; 42:9514–9522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gomez-Martinez M., Schmitz D., Hergovich A.. Generation of stable human cell lines with Tetracycline-inducible (Tet-on) shRNA or cDNA expression. J.Vis. Exp. 2013; 73:e50171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ulmasov B., Capone J., Folk W.. Regulated expression of plant tRNA genes by the prokaryotic tet and lac repressors. Plant Mol. Biol. 1997; 35:417–424. [DOI] [PubMed] [Google Scholar]
- 16. Cronin C.A., Gluba W., Scrable H.. The lac operator-repressor system is functional in the mouse. Genes Dev. 2001; 15:1506–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Licht A., Preis S., Brantl S.. Implication of CcpN in the regulation of a novel untranslated RNA (SR1) in Bacillus subtilis. Mol. Microbiol. 2005; 58:189–206. [DOI] [PubMed] [Google Scholar]
- 18. Chan C.T., Lee J.W., Cameron D.E., Bashor C.J., Collins J.J.. ‘Deadman’ and ‘Passcode’ microbial kill switches for bacterial containment. Nat. Chem. Biol. 2016; 12:82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dimas R.P., Jiang X., Alberto de la Paz J., Morcos F., Chan C.T.Y.. Engineering repressors with coevolutionary cues facilitates toggle switches with a master reset. Nucleic Acids Res. 2019; 47:5449–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gardner T.S., Cantor C.R., Collins J.J.. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000; 403:339–342. [DOI] [PubMed] [Google Scholar]
- 21. Elowitz M.B., Leibler S.. A synthetic oscillatory network of transcriptional regulators. Nature. 2000; 403:335–338. [DOI] [PubMed] [Google Scholar]
- 22. Brophy J.A., Voigt C.A.. Principles of genetic circuit design. Nat. Methods. 2014; 11:508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Backes H., Berens C., Helbl V., Walter S., Schmid F.X., Hillen W.. Combinations of the alpha-helix-turn-alpha-helix motif of TetR with respective residues from LacI or 434Cro: DNA recognition, inducer binding, and urea-dependent denaturation. Biochemistry. 1997; 36:5311–5322. [DOI] [PubMed] [Google Scholar]
- 24. Henssler E.M., Bertram R., Wisshak S., Hillen W.. Tet repressor mutants with altered effector binding and allostery. FEBS J. 2005; 272:4487–4496. [DOI] [PubMed] [Google Scholar]
- 25. Werten S., Schneider J., Palm G.J., Hinrichs W.. Modular organisation of inducer recognition and allostery in the tetracycline repressor. FEBS J. 2016; 283:2102–2114. [DOI] [PubMed] [Google Scholar]
- 26. Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. et al.. Clustal W and Clustal X version 2.0. Bioinformatics. 2007; 23:2947–2948. [DOI] [PubMed] [Google Scholar]
- 27. Sievers F., Higgins D.G.. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014; 1079:105–116. [DOI] [PubMed] [Google Scholar]
- 28. Zhang Y., Skolnick J.. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005; 33:2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rogers J.K., Guzman C.D., Taylor N.D., Raman S., Anderson K., Church G.M.. Synthetic biosensors for precise gene control and real-time monitoring of metabolites. Nucleic Acids Res. 2015; 43:7648–7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A. 3rd, Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 31. Lutz R., Bujard H.. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997; 25:1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Callura J.M., Dwyer D.J., Isaacs F.J., Cantor C.R., Collins J.J.. Tracking, tuning, and terminating microbial physiology using synthetic riboregulators. PNAS. 2010; 107:15898–15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hinrichs W., Kisker C., Duvel M., Muller A., Tovar K., Hillen W., Saenger W.. Structure of the Tet repressor-tetracycline complex and regulation of antibiotic resistance. Science. 1994; 264:418–420. [DOI] [PubMed] [Google Scholar]
- 34. Schumacher M.A., Miller M.C., Grkovic S., Brown M.H., Skurray R.A., Brennan R.G.. Structural mechanisms of QacR induction and multidrug recognition. Science. 2001; 294:2158–2163. [DOI] [PubMed] [Google Scholar]
- 35. Zheng J., Sagar V., Smolinsky A., Bourke C., LaRonde-LeBlanc N., Cropp T.A.. Structure and function of the macrolide biosensor protein, MphR(A), with and without erythromycin. J. Mol. Biol. 2009; 387:1250–1260. [DOI] [PubMed] [Google Scholar]
- 36. Dover L.G., Corsino P.E., Daniels I.R., Cocklin S.L., Tatituri V., Besra G.S., Futterer K.. Crystal structure of the TetR/CamR family repressor Mycobacterium tuberculosis EthR implicated in ethionamide resistance. J. Mol. Biol. 2004; 340:1095–1105. [DOI] [PubMed] [Google Scholar]
- 37. Peters K.M., Schuman J.T., Skurray R.A., Brown M.H., Brennan R.G., Schumacher M.A.. QacR-cation recognition is mediated by a redundancy of residues capable of charge neutralization. Biochemistry. 2008; 47:8122–8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alguel Y., Meng C., Teran W., Krell T., Ramos J.L., Gallegos M.T., Zhang X.. Crystal structures of multidrug binding protein TtgR in complex with antibiotics and plant antimicrobials. J. Mol. Biol. 2007; 369:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ho N.A., Dawes S.S., Crowe A.M., Casabon I., Gao C., Kendall S.L., Baker E.N., Eltis L.D., Lott J.S.. The structure of the transcriptional repressor KstR in complex with CoA thioester cholesterol metabolites sheds light on the regulation of cholesterol catabolism in Mycobacterium tuberculosis. J. Biol. Chem. 2016; 291:7256–7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jeng W.Y., Ko T.P., Liu C.I., Guo R.T., Liu C.L., Shr H.L., Wang A.H.. Crystal structure of IcaR, a repressor of the TetR family implicated in biofilm formation in Staphylococcus epidermidis. Nucleic Acids Res. 2008; 36:1567–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A. et al.. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016; 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grkovic S., Brown M.H., Schumacher M.A., Brennan R.G., Skurray R.A.. The staphylococcal QacR multidrug regulator binds a correctly spaced operator as a pair of dimers. J. Bacteriol. 2001; 183:7102–7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wissmann A., Meier I., Hillen W.. Saturation mutagenesis of the Tn10-encoded tet operator O1. Identification of base-pairs involved in Tet repressor recognition. J. Mol. Biol. 1988; 202:397–406. [DOI] [PubMed] [Google Scholar]
- 44. Orth P., Schnappinger D., Hillen W., Saenger W., Hinrichs W.. Structural basis of gene regulation by the tetracycline inducible Tet repressor-operator system. Nat. Struct. Biol. 2000; 7:215–219. [DOI] [PubMed] [Google Scholar]
- 45. Aleksandrov A., Schuldt L., Hinrichs W., Simonson T.. Tet repressor induction by tetracycline: a molecular dynamics, continuum electrostatics, and crystallographic study. J. Mol. Biol. 2008; 378:898–912. [DOI] [PubMed] [Google Scholar]
- 46. Lee J.W., Gyorgy A., Cameron D.E., Pyenson N., Choi K.R., Way J.C., Silver P.A., Del Vecchio D., Collins J.J.. Creating single-copy genetic circuits. Mol. Cell. 2016; 63:329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Muller G., Hecht B., Helbl V., Hinrichs W., Saenger W., Hillen W.. Characterization of non-inducible Tet repressor mutants suggests conformational changes necessary for induction. Nat. Struct. Biol. 1995; 2:693–703. [DOI] [PubMed] [Google Scholar]
- 48. Meyer A.J., Segall-Shapiro T.H., Glassey E., Zhang J., Voigt C.A.. Escherichia coli “Marionette” strains with 12 highly optimized small-molecule sensors. Nat. Chem. Biol. 2019; 15:196–204. [DOI] [PubMed] [Google Scholar]
- 49. Cuthbertson L., Nodwell J.R.. The TetR family of regulators. Microbiol. Mol. Biol. Rev. 2013; 77:440–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schumacher M.A., Miller M.C., Grkovic S., Brown M.H., Skurray R.A., Brennan R.G.. Structural basis for cooperative DNA binding by two dimers of the multidrug-binding protein QacR. EMBO J. 2002; 21:1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schmidl S.R., Ekness F., Sofjan K., Daeffler K.N., Brink K.R., Landry B.P., Gerhardt K.P., Dyulgyarov N., Sheth R.U., Tabor J.J.. Rewiring bacterial two-component systems by modular DNA-binding domain swapping. Nat. Chem. Biol. 2019; 15:690–698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequences are available in GenBank database under accession numbers MH026045, MH026046, MH026047, MH026048, MH026049 and MH026050.