

Abstract
Background
The clinical application of methotrexate (MTX), an efficacious cytotoxic drug, is restricted due to its associated liver toxicity. Ellagic acid (EA), a natural polyphenol, possesses hepatoprotective, antioxidant and anti-inflammatory properties.
Objectives
The present study seeks to address the hepatoprotective effects of Ellagic acid (EA) against MTX-mediated oxidative stress (OS) and widen our current knowledge of the underlying molecular mechanisms of MTX toxicity.
Methods
Wistar rats were orally given EA (5 mg/kg and 10 mg/kg) for 10 successive days and at the end of the third day they were administered a single dose of MTX (20 mg/kg i.p).
Results
After performing biochemical analysis, liver enzymes and malondialdehyde were significantly higher in the MTX group, indicating hepatic oxidative damage. MTX-induced OS was further confirmed with observation of events such as reactive oxygen species (ROS) overproduction, mitochondrial outer membrane potential decrease, mitochondrial swelling, cytochrome c release and caspase-3/9 increase, resulting in apoptosis. Furthermore, overexpression of pro-inflammatory factors such as nuclear factor kappa B (NF-ĸB) and interleukin 6 (IL-6) indicated the MTX-induced inflammation in MTX-treated group. Interestingly, EA was able to significantly prevent OS, mitochondrial dysfunction, apoptosis and inflammation induced by MTX. Also, EA-treated rats demonstrated significant upregulation of both nuclear factor erythroid 2-related factor 2 (Nrf2) and hemoxygenase-1 (HO-1), which were considerably downregulated in MTX-treated rats.
Conclusions
EA protects rats against MTX-induced apoptosis and mitochondrial dysfunction via up-Regulating Nrf2 and HO-1 expression and inhibiting the NF-κB signaling pathway. Therefore, EA may protect patients against MTX-induced hepatotoxicity and encourage its clinical application.
Graphical abstract.

Beneficial effect of Ellagic acid (EA) on Methotrexate (MTX)-induced liver injury: molecular mechanism.
Keywords: Ellagic acid, Mitochondrial dysfunction, Oxidative stress, Inflammation, Apoptosis
Introduction
Methotrexate (MTX) is a highly effective drug for a range of malignant and autoimmune diseases such as osteosarcoma, lymphoma, rheumatoid arthritis and psoriasis [1]. The main mechanism of action of MTX is irreversible inhibition of dihydrofolate reductase (DHFR) that causes a decrease in tetrahydrofolate production as well as thymidylate and purine nucleotides, which result in inhibition of DNA repair, synthesis and consequently, cellular arrest [2]. Some new studies have shown that MTX is also able to affect folate metabolism-independent cellular mechanisms. For example, MTX exerts anti-proliferative and pro-apoptotic properties by altering the intracellular reactive oxygen species (ROS) levels [3, 4]. Despite the importance of MTX as an advantageous treatment option, the potential MTX-associated adverse effects have attracted considerable attention [5].
The exact mechanism of MTX-induced toxicity remains to be elucidated. It is suggested that MTX takes down the cellular antioxidant machinery and paves the way for the oxidative stress-induced damage to the hepatic cells [6]. MTX-induced OS easily damages the mitochondrion, because this organelle is the main source of cellular ROS [7]. MTX also causes the further production of ROS through inhibition of the mitochondrial electron transport chain’s complex, which leads to the formation of mitochondrial permeability transition (MPT) pores. Altogether, these events disrupt the mitochondrial membrane potential, induce the mitochondrial matrix swelling and cytochrome c release and plays a key role in early apoptosis [8].
The MTX-induced mitochondrial dysfunction and the subsequent overgeneration of ROS activate nuclear factor kappa B (NF-ĸB). This protein complex promotes the production of pro-inflammatory cytokines such as tumor necrosis factor (TNF-α) and interleukins (ILs), primarily, IL-1 and IL-6. The mentioned events, altogether, cause the production of other pro-inflammatory cytokines and increase inflammation in different organs [9].
Ellagic acid (EA) is a natural phenolic compound found in grapes, green tea, nuts, blackcurrants, strawberries, raspberries, pomegranates, and the stem and bark of Eucalyptus. EA possesses strong anti-inflammatory, anti-mutagenic, antioxidant, anti-apoptotic, hepatoprotective, antiviral and antibacterial properties [10–12]. Having four hydroxyl (H bond acceptors) and two lactone (H bond donor) functional groups, gives EA the ability to scavenge free radicals [13]. Direct scavenging of reactive species, such as hydroxyl radicals, peroxyl radicals and NO2 radicals is the primary antioxidant mechanism of EA. In addition, EA has multiple indirect mechanisms against OS. EA can noticeably increase antioxidant enzymes such as Heme Oxygenase-1 (HO-1) and Superoxide dismutase (SOD) through up-regulation of nuclear factor erythroid 2-related factor 2 (Nrf2) and can also dramatically decrease the cytoplasmic stability of Keap1 [14].
This experimental study investigates the possible protective role of EA against MTX-induced hepatotoxicity. We also aimed to elucidate the mechanism of action behind acute MTX hepatotoxicity and address EA’s plausible protective effect against this incident. Therefore, we studied the alterations in ROS generation, mitochondrial membrane potential (MMP), mitochondrial swelling and cytochrome c release upon treatment of rats with MTX, alone or in combination with EA. These pathways are linked to mitochondrial injury, the MTX-mediated OS and inflammation-dependent NF-ĸB activation, and also the primary antioxidant response triggered by Nrf2 activation. This multidimensional approach was designed to reach an integrated pattern of MTX-induced toxicity and EA’s ameliorative ability in the rat liver that might explain the possible hepatoprotective effects of EA in MTX-treated rats, focusing on its regulatory function in OS and Nrf2/HO-1 pathway.
Materials and methods
Reagents
Methotrexate (MTX), Ellagic acid (EA), bovine serum albumin (BSA), trichloroacetic acid (TCA), 2-nitrobenzoic acid (DTNB), Tris, ethylenediaminetetraacetic acid (EDTA), Glycine, KH2PO4, KCl and thiobarbituric acid (TBA) were purchased from Sigma chemicals (St. Louis, MO, USA). All reagents were supplied as the analytical grade from different commercial suppliers.
Animals and experimental design
Thirty-two 7-weeks-old male Wistar rats (210 ± 20 g) were selected for this study (Animal House, Pharmacy Faculty, TUMS, Iran). The rats were housed under standard laboratory conditions with 12-h light/dark cycles at 25 ± 3 °C (water and standard food ad libitum). All the experimental procedures followed the guidelines of the Tehran University of Medical Sciences’ Ethics committee for the Use and Care of Laboratory Animals (code number: IR.TUMS.PSRC.REC.1396.4444).
The animals were kept for one week before the experiments to get acclimated to the new environment and then were divided randomly into 4 major groups (n = 8) (Fig. 1):
Group A (Control): 10 days of oral corn oil (0.5%) + a single dose of saline (ip).
Group B (MTX): 10 days of oral corn oil (0.5%) + a single 20 mg/kg dose of MTX at the end of the third day (ip).
Group C (5 mg EA & MTX): 5 mg/kg oral EA for 10 consecutive days + a single 20 mg/kg dose of MTX (ip) at the end of the third day.
Group D (10 mg EA & MTX): 10 mg/kg oral EA for 10 consecutive days + a single 20 mg/kg dose of MTX (ip) at the end of the third day.
Fig. 1.

Scheme of the experimental design. MTX, methotrexate; EA, Ellagic acid
Samples preparation
The doses were chosen based on previous works and guidelines [15]. The anesthetized animals were euthanized by cervical dislocation before separating their serum from blood samples. Then liver tissue was moved to a Teflon homogenizer filled with ice-cold aqueous PBS to get dissected, washed and homogenized. After centrifugation of homogenates, clear supernatant was obtained and used for following biochemical analyses. Small pieces of liver tissue were prepared for histopathological examination by fixation in 10% buffered formalin solution. We kept extra samples at −80 °C for the purpose of western blotting.
Measurement of liver function indicators
The levels of serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were examined using automatic analyzer (Olympus AU600 analyzer; Japan).
Malondialdehyde (MDA) measurement
The level of Malondialdehyde (MDA) was evaluated using thiobarbituric acid reactive substances (TBARS) test to measure the extent of lipid peroxidation (LPO) [16]. In this assay, MDA attaches to thiobarbituric acid (TBA) forming a colorimetrically and fluorometrically-quantifiable complex (MDA-TBA). Briefly, we mixed 100 μL of the supernatant solution with the reaction mixture composed of TBA 0.67%, 0.1 M Hydrochloric acid, Sodium dodecyl sulfate 7% and Phosphotungstic acid 10%. Then, the mixture was boiled for one hour in a water bath of 95–98 °C and then cooled under tap water to the temperature of room, before adding 150 μL n-butanol. Finally, the supernatant’s fluorescence intensity (excitation and emission at 530 nm and 575 nm) was measured after 10 min centrifugation of the mixture at 3000 g.
Measurement of glutathione
We carried out the procedure described by Tietze in order to measure the level of reduced glutathione (GSH) [17]. In this regard, we centrifuged the liver tissue homogenates for 10 min at 15000 g and 4 °C to obtain a clear supernatant fraction. Then, 100 μL of this clear fraction was transferred to a 96-well microplate before adding the Ellman’s reagent (200 μL, 2-nitrobenzoic acid in sodium citrate 0.25%) to each well. Finally, the absorbance was read at 412 nm by BioTek microplate reader (Winooski, VT, USA).
Measurement of superoxide dismutase (SOD) activity
We used a commercial kit (Cayman Chemicals Inc. Ann Arbor, MI, USA) to measure the SOD activity. The assay is based on the ability of superoxide anion to reduce the tetrazolium salt and form the water-soluble formazan dye in an inversely proportional relation with SOD activity. As per the kit protocol, after homogenization of the liver tissue, the supernatant was removed. Next, a volume of 10 μL from the sample or the standard and 200 μL from the assay buffer (50 mM Tris-HCl, pH = 8, 0.1 mM Pentetic acid and 0.1 mM hypoxanthine) were transferred to the 96-well microplate. Then, 20 μL xanthine oxidase was added to each well and after half an hour the BioTek absorbance reader was used to read the optical density at 440–460 nm.
Mitochondrial preparation
We used differential centrifugation to isolate mitochondria from rats’ livers [18]. Briefly, the liver tissue homogenates were centrifuged for 10 min at 1500 g and 4 °C. Then, the supernatant was collected and re-centrifuged for 10 min at 7000 g and 4 °C. At this point, the upper fluid fraction was thrown away and the pellet containing mitochondria was added in the isolation medium and centrifuged for 10 min at 10000 g and 4 °C. The supernatant from the last centrifugation was discarded and using the bicinchoninic acid (BCA) assay the protein concentration was evaluated [19].
Evaluation of mitochondrial ROS production
The production of ROS in the mitochondria was determined using the fluorescent probe Dichloro-dihydro-fluorescein diacetate (DCFH-DA). This dye goes under de-acetylation by cellular esterases and forms DCFH which is non-fluorescent. The DCF, a highly potent fluorescent agent is formed through oxidization of DCFH by ROS, hence, the level of ROS generation could be determined by measuring DCF’s fluorescence intensity. In this regard, a respiration buffer (50 μM egtazic acid, 5 mM sodium succinate, 0.5 mM MgCl2, 0.32 mM sucrose, 10 mM Tris, 20 mM MOPS and 0.1 mM KH2PO4 (pH = 7.4)) was used to re-suspend mitochondrial extracts [20]. Next, 100 μL of DCFH-DA (10 μM) was mixed with mitochondria and after 10 min of incubation, the mixture’s fluorescence intensity was read with excitation and emission wavelengths at 488 and 540 nm, respectively.
Evaluation of the mitochondrial membrane potential (MMP)
The mitochondrial membrane potential was assessed using Rhodamine 123 (RH-123), a cationic fluorescent dye. This fluorescent probe crosses the mitochondria with no trouble and accumulates in their membrane. Briefly, 10 μM RH-123 was added to mitochondrial fractions, which were suspended in an MMP assay buffer (220 mM sucrose, 68 mM D-mannitol, 10 mM KCl, 5 mM KH2PO4, 2 mM MgCl2, 50 μM EGTA, 5 mM sodium succinate, 10 mM HEPES, 2 μM Rotenone). Finally, the mixture’s fluorescence intensity was measured by a fluorescence spectrophotometer at the excitation and emission wavelength of 490 nm and 520 nm, respectively [20].
Evaluation of mitochondrial swelling
Changes in absorbance at 540 nm (30 °C) were analyzed to evaluate the mitochondrial swelling rate [18]. There is an inverse relationship between the mitochondrial swelling rate and the absorbance intensity. A swelling buffer (70 mM sucrose, 230 mM mannitol, 3 mM HEPES, 2 mM tris-phosphate, 1 μM rotenone and 5 mM of succinate) was used to suspend mitochondrial fractions before measuring the absorbance at 10 min intervals with the BioTek microplate reader.
Evaluation of cytochrome c release
Cytochrome c release was assayed using an immunoassay kit (R and D Systems Inc. Minneapolis, MN, USA). The kit was delivered with a cytochrome c specific monoclonal antibody (MAB)-precoated 96-well microplate. Briefly, 75 μL of cytochrome c specific Horseradish peroxidase (HRP) conjugated MAB and 50 μL of the test or control group were mixed gently in each microplate well. The microplate was incubated for 2 h and then to eliminate any remaining liquid, each well was aspirated one time, followed by four times of washing. Then, the substrate solution (100 μL) was added to the wells and incubated for half an hour. Finally, the reaction was stopped by adding stop solution (100 μL) before reading the absorbance every half an hour at 450 nm by the aforementioned BioTek reader.
Western blotting
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was implemented in 0.1 (w/v) polyacrylamide gel using same amounts of protein in each lane. The electrophoresed proteins were transferred to polyvinylidene difluoride (PVDF) membrane. The membrane is blocked with 3% BSA diluted in PBS for an hour and a half. Next, the membrane was kept overnight at 4 °C with primary antibodies against Bax (mouse monoclonal 1:500; Santa cruz), Bcl-2 (rabbit monoclonal 1:300; santa cruz) and NF-κB (rabbit monoclonal 1:300; santa cruz), Nrf2 (rabbit polyclonal 1:100; santa cruz), HO-1(rabbit polyclonal 1:100; santa cruz). Proteins were detected using HRP-conjugated secondary antibodies (1:5000) and the enhanced chemiluminescence (ECL) system. Proteins were normalized to β-actin as an internal control for loading of equal quantities. For western blotting study, 3 samples (from different animals) ran in 3 different gels. The bands density was measured by ImageJ software (version 1.44p, NIH).
Evaluation of caspase-3 and caspase-9 activity
The activity of caspase-3 was assessed by an ELISA kit purchased from Cell Signaling (Danvers, MA, USA). The kit was delivered with a cleaved caspase-3 rabbit MAB-precoated 96-well microplate. Briefly, the microplate was kept for 2 h at 37 °C after adding cell lysates to each well. Washing buffer 1X (200 μL) was used to wash wells. Next, 100 μL from biotin-conjugated rabbit monoclonal anti-caspase-3 antibody was added to the washed wells and the microplate was kept at 37 °C for an hour. Further, 100 μL from HRP-conjugated second antibody was put into wells and the microplate was kept at 37 °C for half an hour. Then, Tetramethylbenzidine ELISA substrate (100 μL) was added and the microplate was kept at 37 °C for 10 min. Lastly, the absorbance was read at 450 nm after the reaction was terminated by adding 100 μL stop solution.
Ac-LEHD-pNA (Caspase-9 Chromogenic Substrate) was used to assess caspase-9 activity. The assay is based on evaluation of color generated from cleaved pNA (pnitroanilide) which is in a direct relationship with the caspase-9 activity. First, a lysis buffer was used for suspension of liver extracts. Then, the suspension was centrifuged for 20 min at 20000 g and the supernatant was removed and transferred into caspase buffer. To this, Ac-LEHD-pNA was added and the solution was kept at 37 °C for 4 h. Finally, the absorbance was read at 405 nm by BioTek microplate reader.
Histopathological analysis
The liver tissues were soaked in 10% buffered formaldehyde phosphate prior to dehydration by graded ethyl alcohol and embedment in paraffin wax. Next, the fixed samples were cut into 5 μm thick sections and treated with hematoxylin and eosin (H&E) before observation under light microscopy. The Ishak Modified HAI system was used for histopathological examination and scoring the pathological changes [21].
Evaluation of protein content
The protein concentration was evaluated by standard BCA protein assay (Parstous Inc., Mashhad, Iran). Under alkaline conditions, Cu2+ turns into Cu+, which reacts with BCA and forms a purple-colored product. This process is the basis of BCA method for estimation of protein content. Briefly, 1:50 ratio of Copper reagent (4% copper (II) sulfate) and BCA reagent (Na2CO3, NaHCO3, BCA and sodium tartrate in 0.1 M NaOH) were mixed together. Then, 75 μl of the mentioned solution and 25 μl of test or control were dispensed into each well of a 96-well microplate. The microplate was put into the at 60 °C incubator for an hour prior to reading the solution’s absorbance at 562 nm.
Statistical analysis
All analyses were performed using Graph Pad Prism 5.04 (GraphPad Software, Inc. Cal, USA). One-way analysis of variance (ANOVA) and the Newman Keuls’ post hoc test were carried out for data analysis. Data are presented as means and standard deviation (SD) values that were calculated for each group and a value of P < 0.05 was chosen to indicate statistical significance.
Results
Effects of MTX and EA treatment on the activity of serum marker enzymes
The hepatic marker enzymes activities are represented in Table 1. ALT and AST were considerably promoted in MTX group compared with control group (P < 0.001); However, EA pre-treatment prevented this increase.
Table 1.
Effect of EA on serum ALT and AST in MTX-induced rats
| Parameter | Groups | |||
|---|---|---|---|---|
| Control | MTX | MTX + EA5mg/kg | MTX + EA10mg/kg | |
| ALT (IU/L) | 34.91 ± 3.09 | 50.95 ± 3.37*** | 41.15 ± 3.45## | 36.9 ± 4.92### |
| AST (IU/L) | 51.16 ± 3.77 | 67.01 ± 2.67*** | 57.45 ± 4.31## | 55.91 ± 3.41### |
Data are presented as means ± SD; n = 5 for each treatment group***P < 0.001, significantly different from the control group. ##P < 0.01, ###P < 0.001 significantly different from the MTX group. EA, Ellagic acid; MTX, methotrexate; ALT, alanine transaminase; AST, aspartate transaminase
EA prevents MTX-mediated oxidative stress (OS)
The variations in oxidative stress (OS) indicators such as MDA, GSH content and SOD activity between experiment’s different groups are depicted in Fig. 2. Hepatic MDA level is equivalent to the scale of LPO in the liver, which is the main indicator of OS (Fig. 2a). MTX meaningfully raised the liver MDA level (P < 0.001) in comparison to the control. Although, EA and MTX co-treatment meaningfully and dose-dependently held down the MDA level at the approximate level of control.
Fig. 2.
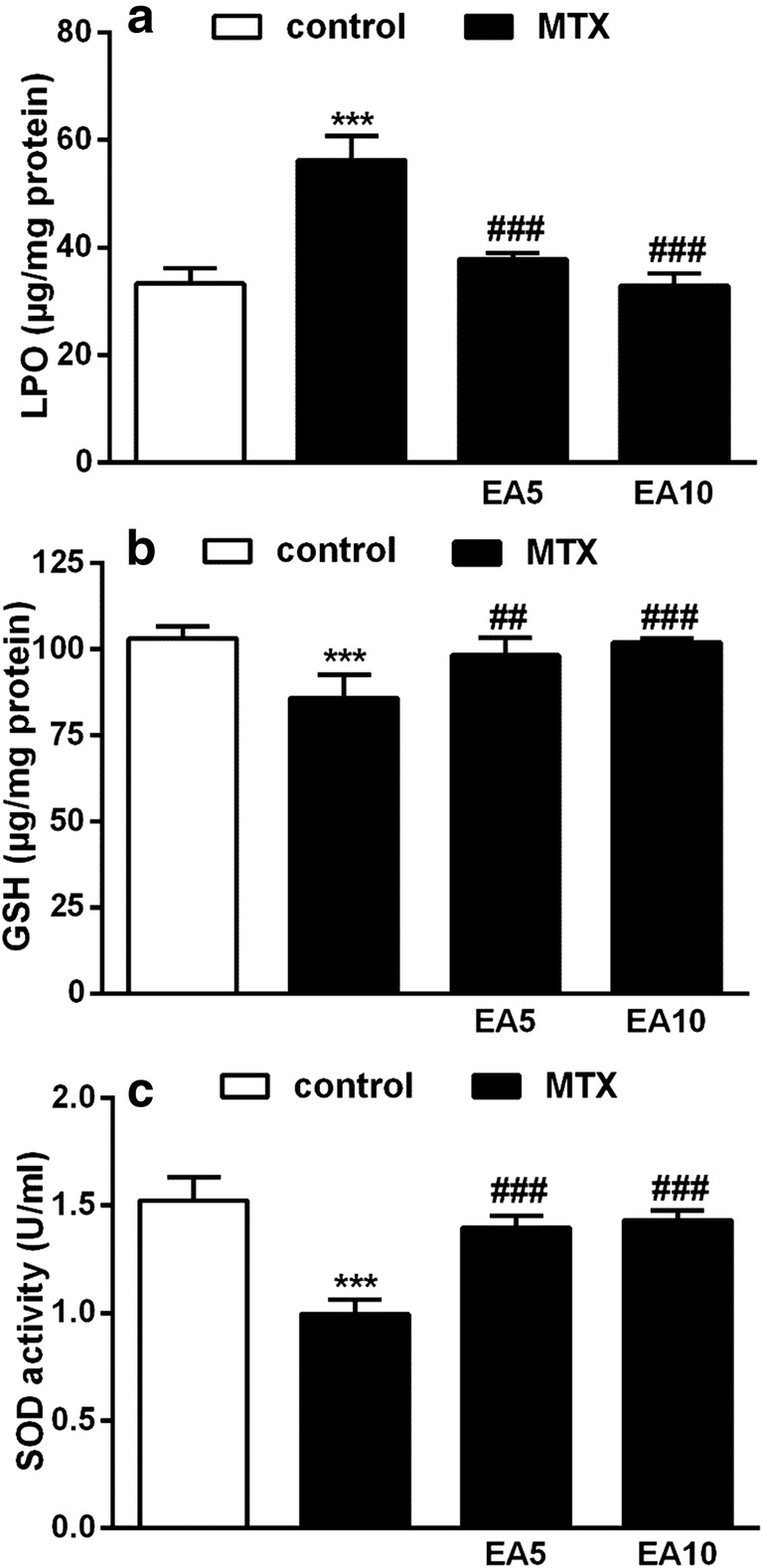
Effects of EA on hepatic OS. MDA level (a), Total thiols (b), SOD activity (c) in control, MTX and EA co-administrated with MTX. Data are expressed as means and standard deviation (SD) values (n = 5). ***P < 0.001 compared with control. ##P < 0.01, ###P < 0.001 compared with MTX-treated group. EA, Ellagic acid; MTX, Methotrexate; MDA, Malondialdehyde; OS, oxidative stress; SOD, Superoxide dismutase
Figure 2b exhibits the liver GSH content among all groups. Treatment with MTX significantly depleted GSH (P < 0.001) compared with control. In comparison with MTX group, both EA treatment groups showed significantly enhanced GSH content (5 mg/kg (P < 0.01), 10 mg/kg (P < 0.001)).
SOD is responsible for the dismutation of O2− into hydrogen peroxide and plays a key role in protecting cells against MTX-mediated OS. The activity of SOD was meaningfully suppressed in the liver of rats treated with MTX (P < 0.001) in comparison to the control group. Although, the activity of SOD was significantly maintained in both groups treated with EA plus MTX (Fig. 2c). To sum up, MTX caused elevation of MDA level and suppressed both enzymatic and non-enzymatic liver antioxidants activities leading to hepatic OS. The induction of OS was significantly prevented in EA plus MTX groups.
EA attenuates MTX-induced mitochondrial dysfunction
Generation of hepatic mitochondrial ROS was significantly higher in MTX group (P < 0.001) compared with the control group (Fig. 3a). Conversely, the observed increase in ROS formation was significantly blocked in groups treated with EA along MTX (P < 0.001).
Fig. 3.

Effects of EA on ROS formation (a), MMP collapse (b), Mitochondrial swelling (c), cytochrome c release from mitochondria (d) in in experimental groups. Data are expressed as means and standard deviation (SD) values (n = 5). **P < 0.01, ***P < 0.001 compared with control.##P < 0.01, ###P < 0.001 compared with MTX-treated group. EA, Ellagic acid; MTX, Methotrexate; ROS, Reactive oxygen species; MMP, Mitochondrial membrane potential
In the rats treated with MTX, the mitochondrial membrane potential (MMP) was significantly reduced (P < 0.001) in comparison to the control group. Although, the MTX-mediated disturbance of MMP was notably prevented in EA plus MTX-administered groups (P < 0.001) (Fig. 3b).
Figure 3c displays the rate of mitochondrial swelling, which is reversely proportional to the absorbance intensity. In comparison to the control group, MTX group showed a greater rate of mitochondrial swelling (P < 0.001). This increase was significantly inhibited in the both EA groups (P < 0.001).
The cytochrome c release into the cytoplasm is an indicator of severe mitochondrial damage. As displayed in Fig. 3D, cytochrome c release was clearly increased in MTX group (P < 0.01) compared with the control group. However, EA treatment (5 mg/kg and 10 mg/kg) effectively inhibited the MTX-induced cytochrome c release (P < 0.01).
EA prevents MTX-induced apoptosis in liver rats
Intrinsic or mitochondrial pathway of apoptosis is initiated upon activation of a proteolytic cascade including both caspase-9 and caspase-3, respectively. As shown in Fig. 4a, initiator caspase-9 activity was significantly escalated by 26.66% (P < 0.001) in hepatic cells of MTX group in comparison to hepatic cells of the control group. Additionally, initiator caspase-3 activity was remarkably increased by 45.88% (P < 0.001) compared to the control group. On the other hand, EA (5 mg/kg and 10 mg/kg) along MTX considerably prevented the increase in the activity of these apoptotic markers in comparison with the MTX-treated rats.
Fig. 4.

Effects of EA on hepatic apoptotic markers. Alterations in activities of caspase 3 (a) and caspase 9 (b) in control, MTX and EA co-administered with MTX. Data are expressed as means and standard deviation (SD) values (n = 5). ***P < 0.001 compared with control. ##P < 0.01, ###P < 0.001 compared to MTX-treated group. EA, Ellagic acid; MTX, Methotrexate
Effects of MTX and EA on the protein expression of Bcl-2 and Bax
Figure 5 indicates that EA kept liver cells safe from MTX-mediated apoptotic death. We studied the expression of apoptosis-related genes. We showed that the Bax and Bcl-2 genes expression was altered upon MTX administration (Fig. 5a). The intracellular level of Bax protein was meaningfully raised in the MTX group in comparison to the control group, while the Bcl-2 level was reduced in the MTX group. Remarkably, EA treatment blocked both MTX-induced increase in Bax and decrease in Bcl-2. The MTX-treatment led to a 2.5-fold increase of the Bax/Bcl-2 ratio compared to the control, whereas this ratio remained unaffected in the EA groups (Fig. 5b). These results indicate that MTX disrupted the balance between pro and anti-apoptotic genes expression and EA brought this vital balance back.
Fig. 5.

Effects of treatments on Bax and Bcl-2 proteins expression. Western blot analysis of Bax and Bcl-2 protein expressions in the liver of rats treated with MTX and EA. Expression of β-Actin was used as internal control (a). As shown above, MTX treatment resulted in higher Bax/Bcl-2 (b). Representative bar diagram showing quantitative results for relative levels of Bax/Bcl-2 ratio. Data are expressed as means and standard deviation (SD) values (n = 5). ***P < 0.001 compared with control. ###P < 0.001 compared to MTX-treated group. EA, Ellagic acid; MTX, Methotrexate; Bax, Bcl-2-like protein 4; Bcl-2, B cell lymphoma 2
EA up-regulates Nrf2/HO-1 pathway in liver of MTX-induced rats
The effects of EA on hepatic Nrf2 protein expression in MTX-treated rats are displayed in Fig. 6a, b. We observed a major down-regulation of Nrf2 expression in MTX group in comparison to the control. The 5 mg/kg and 10 mg/kg dose of EA with MTX caused a significant up-regulation in Nrf2 expression (P < 0.05).
Fig. 6.

Effect of EA on Nrf2 protein (a & b) and HO-1 protein expression (c & d) in liver of MTX-treated group. Data are expressed as means and standard deviation (SD) values (N = 3). *P < 0.05 versus Control. #P < 0.05 versus MTX. EA, Ellagic acid; MTX, Methotrexate; Nrf2, Nuclear factor erythroid 2- related factor 2; HO-1, Heme oxygenase-1
We also evaluated the expression of HO-1 in our experimental groups. The HO-1 expression level was significantly decreased in MTX group compared to control (P < 0.05). As displayed in Fig. 6C&D, both doses of EA when administered along MTX led to a meaningful increase in HO-1 expression (P < 0.05). The HO-1 results were in line with our above results for Nrf2 protein expression.
EA suppresses liver inflammation and NF-κB activation induced by MTX
Pro-inflammatory cytokines and factors were analyzed in rat serum and liver to determine the protective effects of EA against MTX-induced inflammation. As NF-κB signaling pathway is important in MTX-induced inflammatory response, we assayed the expression levels of NF-κB in rat liver via Western blotting. Total NF-κB p65 was significantly elevated in the liver of MTX group in comparison with the Control (Fig. 7a, b). However, EA administration prevented MTX-mediated NF-κB activation (Fig. 7a, b). As depicted in Fig. 7c, d, IĸB was phosphorylated and degraded in the MTX group, contrary to the rats treated with EA.
Fig. 7.

Effects of EA on liver inflammatory markers. Alterations in NF-ĸB p65 protein expression (a and b), p-IĸBα protein expression (c and d) and levels of IL-6 (E) in the liver of control, MTX and EA co-administered with MTX. Data are expressed as means and standard deviation (SD) values (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001 compared to control group. #P < 0.05, ##P < 0.01, ###P < 0.001 compared with MTX group. EA, Ellagic acid; MTX, Methotrexate; NF-ĸB, Nuclear factor-kappaB; IĸBα, inhibitor of NFĸB; p-IĸBα, Phosphorylated-inhibitor of NFĸB; IL-6, Interleukin-6
In order to evaluate the anti-inflammatory properties of EA, level of IL-6 which is a serum pro-inflammatory cytokine, was assessed. As shown in Fig. 7c, we observed a marked elevation in the serum IL-6 levels when comparing MTX-treated rats with the control (P < 0.001). Both EA doses significantly prevented the MTX-induced upsurge in circulating IL-6 levels (P < 0.001). Our findings indicated that EA ameliorates MTX-induced liver injury through suppression of NF-κB activation and inhibits MTX-induced pro-inflammatory factor expressions in both serum and liver.
Effects of MTX and EA treatment on histopathological changes
Table 2 displays histopathological liver tissue alterations. We observed major histopathological irregularities (score 5) in MTX-treated rats including necrosis, degeneration and inflammation (Fig. 8b). In contrast, injurious effects of MTX were considerably prevented with either doses of EA (Fig. 8c, d). We observed only a minor injury (score 2) in the group receiving 10 mg/kg EA, therefore it seems that the mechanism in which EA aids the liver tissue recovery is dose-dependent (Fig. 8d).
Table 2.
Effect of EA on MTX-induced liver histological changes. EA, Ellagic acid; MTX, methotrexate
| Groups | Histological score | Degenerative changes |
|---|---|---|
| Control | 1 | No abnormality |
| MTX | 5 | Extensive pathologic changes such as prominent granular degeneration, inflammation, eosinophilic cells, vacuolation etc. |
| MTX + EA 5 mg/kg | 3 | Moderate pathologic changes such as Inflammation |
| MTX + EA 10 mg/kg | 2 | Mild pathological changes |
1: No abnormality 2: Mild degenerative changes (10%). 3: Moderate degenerative changes (25%). 4: Severe degenerative changes (50%). 5: Extensive and marked changes (>75%)
Fig. 8.

Photographs of liver tissue. We observed the Hepatoprotective effect of EA against MTX-induced liver injury (n = 3). (a) Control group showed normal architecture. (b) Extensive necrosis, degeneration and inflammation of hepatocytes in MTX group. (c) Co-treatment of MTX and EA 5 showed moderate inflammation. (d) EA 10 treatment caused just minor pathological alterations in MTX-administered rats. EA, Ellagic acid; MTX, Methotrexate. Arrow indicates Inflammation, Arrow head indicates eosinophilic cells, Asterisk indicates vacuolization of cells. Original magnification ×20
Discussion
Despite the important role of MTX in prophylactic and therapeutic regimens of various diseases such as inflammatory disorders, malignancies and auto-immune diseases, its ability to induce liver toxicity broadly restricts its clinical use [22]. Several investigations have highlighted the mechanistic role of OS in MTX-induced liver damage. Therefore, using antioxidants seems a plausible approach to manage its toxicity [23]. In this study, we tried to study the effects of EA, as a polyphenol antioxidant, on MTX-induced liver toxicity. The main cause of EA’s protective ability is believed to relate to its antioxidant feature, but several studies have highlighted the ability of polyphenols to stimulate or inhibit signaling pathways through regulation of both cellular gene expression and signaling molecules phosphorylation [24].
Aminotransferases are enzymes found mainly in the hepatocytes. An elevation in serum ALT and AST is either related to damage or destruction of tissues rich in the aminotransferases, especially liver cells, or to changes in cell membrane permeability that permit leakage into the circulation [25]. Here, MTX treatment considerably promoted the serum activities of ALT and AST. This is in good agreement with previous findings in the literature. Safaei et al. showed that 24 h after MTX administration, their experimental rats developed hepatotoxicity, which was reflected by a marked elevation in the levels of AST and ALT [26]. Interestingly, level of ALT and AST in the MTX plus EA-treated groups were significantly lower than the MTX-treated animals. In the same context, the hepatoprotective effect of EA have been supported in a study by Girish et al. which displayed the prevention of serum transaminases increase in mice with paracetamol-mediated hepatotoxicity [27]. Following intravenous administration of liver toxicant at high dose, levels of serum ALT can rise to 10 to 20 times the upper limit of normal (ULN) within 12 to 48 h, but then rapidly fell to normal levels with rare instances of jaundice or symptoms of liver injury [28].
The peroxidative breakdown of polyunsaturated fatty acids by free radicals has been implicated in many types of liver diseases and most importantly in the liver damage induced by any toxic substances such as MTX [29]. In our investigation, it seems that, because of the overproduction of free radicals, hepatic MDA levels in MTX group were significantly elevated in comparison to the control. Furthermore, hepatic LPO was effectively ameliorated in EA groups. This could be associated with the prevention of MTX-mediated cellular membrane damage due to the ROS-scavenging function of EA [20]. GSH is a non-enzymatic antioxidant and the master of cellular ROS scavengers that protects cellular membranes and lipoproteins from peroxidation [30]. SOD is also an important antioxidant enzyme in nearly all living organisms, which are exposed to oxygen. This enzyme catalyzes the reaction of superoxide (O2−) radicals into less toxic hydrogen peroxide (H2O2) or molecular oxygen (O2) [31]. In the present study, the content of GSH and activity of SOD were decreased in MTX-treated animals. The observed protection provided by EA was signified by maintaining GSH levels and SOD activity in MTX-treated animals. This protective effect may be related to EA’s free radical scavenging ability that prevented ROS-induced both depletion of GSH and reduction of SOD activity [32].
Mitochondria, essential cytoplasmic organelles, have important physiological functions in the liver cells. They are the primary targets of ROS-induced OS due to being their main source [33]. The MTX through ROS overproduction results in mitochondrial damage and the cell may enter apoptotic phase if ROS is continuously produced. In our study, MTX-treatment significantly increased mitochondrial ROS formation in liver of rats. Subsequently, MTX through raising ROS generation triggered a cascade of events leading to the induction of apoptosis including disruption of the mitochondrial outer membrane, disturbance of MMP and translocation of cytochrome c. Mitochondrial overproduction of ROS, also results in the oxidization of sulfhydryl groups surrounding MPT pore and causes the MPT pore to open. Consequently, cytochrome c is released into the cytosol and mitochondrial swelling and caspase cascade is initiated. However, EA significantly suppressed the ROS formation level in the mitochondria of MTX-treated rats. In our study, MTX-induced mitochondrial swelling, MMP loss and cytochrome c release were prevented by EA administration. Our result complies with our earlier work that showed the ability of EA to mitigate the mitochondrial dysfunction and oxidative damage in the kidney of rats exposed to Gentamicin [20].
Bax/Bcl-2 expression ratio is believed to play a crucial role in the regulation of cell apoptosis [34, 35]. We found that the MTX-induced increase in Bax protein and reduction in Bcl-2 protein were effectively blocked by EA treatment. Bax and Bcl-2 proteins are integral parts of the apoptotic cascade, hence, it is not unexpected that regulating these proteins ratio by EA treatment can prevent the occurrence of apoptotic cascade and thereby apoptosis. Seemingly, EA through modification of Bax and Bcl-2 protein expression eventually stopped the initiation of mitochondria-mediated downstream events such as cytochrome c release and caspase-3 activation. In good agreement with present findings, Mahmoud et al. showed that MTX administration is associated with increased Bax expression and decreased Bcl-2 expression in the liver of rats [9]. Furthermore, El-Sheikh et al. reported the increase in apoptotic protein caspase-3 expression in the hepatic cells of MTX-treated rats [36].
Another important player in the pathophysiology of liver inflammation, fibrosis, and drug-mediated toxicity is the transcription factor Nrf2, which contributes to normal function of the liver. ROS overproduction is recognized as being able to activate Nrf2 by degrading its associated protein, Keap1. Consequently, Nrf2 migrates to the nucleus and promotes the expression of proteins operating in cell defense and protection such as Heme Oxygenase-1 (HO-1), glutathione-S-transferase, SOD, CAT, and Glutathione peroxidase [37]. In light of this, we assessed the expression level of Nrf2 in order to elucidate the mechanism behind EA’s protective ability against MTX-mediated OS and inflammation. In our study, protein expression levels were down-regulated in hepatic cells of rats exposed to MTX. In line with this, previous investigations signified that the intensive ROS formation leads to down-regulation of Nrf2 in endothelial cells [38], liver [39] and kidney [40]. Co-administration of EA and MTX significantly promoted the Nrf2 and HO-1 upregulation, which were associated with less OS and improved antioxidant defenses in the liver of rats. Nrf2/HO-1 cascade has been reported to inhibit apoptosis via its effect on apoptosis-related proteins such as Bcl-2 and Bax. Our finding substantiates previous findings in the literature that showed the positive impact of EA on atherosclerosis and endothelial dysfunction through upregulation of Nrf2 [41].
These results can be due to the ability of MTX to induce production of ROS and pro-inflammatory cytokines. Increase in OS level also activates the transcription factor NF-κB, which in cytoplasm is bound to its inhibitor, Iκ-B. Excessive intracellular ROS level can degrade the Iκ-B, allowing NF-κB to translocate to the nucleus [42]. These events may lead to hepatic injury and recruitment of apoptotic machinery [36]. Our results highlighted that expression levels of the NF-κB p65 subunit were increased following exposure to MTX, indicating that MTX induced the accumulation of ROS in the liver and then caused the upregulation of NF-κB. Unlike the MTX-treated group, administration of EA plus MTX inhibited the NF-κB nuclear translocation in hepatic cells. Soares et al. showed that HO-1 inhibited the TNF-α dependent activation of NF-κB in endothelial cells [43]. Our results are in line with a previous study, which indicated administration of EA for 16 weeks significantly inhibited NF-ĸb mediated ROS production and synthesis of proinflammatory cytokines [44]. Investigations have signified that HO-1 reduces the activity of NF-ĸB through prevention of IĸB degradation [45]; specially, by preventing its phosphorylation, hence, leads to reductions in NF-ĸb translocation to the nucleus and the transcription of its downstream inflammatory genes. Furthermore, NF-ĸb inactivation through HO-1-mediated glutathionylation of its p65 subunit [46] highlights the anti-inflammatory capacity of EA, which is likely mediated through Nrf2/HO-1 signaling pathway. In addition, Lavrovsky et al. stated that NF-κB binds to the HO-1 promoter region and thus contributes to defense against cellular injury [47]. EA through boosting the antioxidant enzymes, suppressing NF-κB, upregulating Nrf2 expression and modulating the crosstalk between these two transcription factors employ beneficial effects against oxidative liver injury in rats.
Conclusion
Our study shows that MTX-induced mitochondrial dysfunction is related to the progress of toxic effects upon triggering early inflammatory processes and Nrf2 inhibition. Our work has led us to conclude that disturbance of the sophisticated association between NF-κB and Nrf2, for instance in a condition like MTX exposure, results in disruption of the balance between ROS level and antioxidant enzymes like HO-1 [48]. Interestingly, in our study EA was able to bring back the balance between Nrf2 and NF-κB, resulting in the prevention of MTX-induced apoptosis in the hepatocytes and inhibition of deleterious actions of NF-κB, IL-6 and caspases. It seems that EA exerts its hepatoprotective and antioxidant effects mainly through regulation of Nrf2 and NF-κB activities. Future studies is required to determine the possible differences between EA and pharmaceutical products of EA such as nanoformulation. In this regard, dose, formulation and molecular mechanism that cause possible beneficial effects or enhance therapeutic efficacy should be considered.
Acknowledgements
This work was supported by Vice Chancellor for Research, Tehran University of Medical Sciences, Tehran, Iran under Grant (96-03-169-36364).
Compliance with ethical standards
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this article.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Emery P, Breedveld FC, Hall S, Durez P, Chang DJ, Robertson D, Singh A, Pedersen RD, Koenig AS, Freundlich B. Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet. 2008;372(9636):375–382. doi: 10.1016/S0140-6736(08)61000-4. [DOI] [PubMed] [Google Scholar]
- 2.Gangjee A, Kurup S, Namjoshi O. Dihydrofolate reductase as a target for chemotherapy in parasites. Curr Pharm Des. 2007;13(6):609–639. doi: 10.2174/138161207780162827. [DOI] [PubMed] [Google Scholar]
- 3.Neradil J, Pavlasova G, Veselska R. New mechanisms for an old drug; DHFR- and non-DHFR-mediated effects of methotrexate in cancer cells. Klin Onkol. 2012;25(Suppl 2):2S87–2S92. [PubMed] [Google Scholar]
- 4.Sramek M, Neradil J, Veselska R. Much more than you expected: the non-DHFR-mediated effects of methotrexate. Biochim Biophys Acta Gen Subj. 2017;1861(3):499–503. doi: 10.1016/j.bbagen.2016.12.014. [DOI] [PubMed] [Google Scholar]
- 5.Cetiner M, Sener G, Sehirli AO, Eksioglu-Demiralp E, Ercan F, Sirvanci S, Gedik N, Akpulat S, Tecimer T, Yegen BC. Taurine protects against methotrexate-induced toxicity and inhibits leukocyte death. Toxicol Appl Pharmacol. 2005;209(1):39–50. doi: 10.1016/j.taap.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Elango T, Dayalan H, Gnanaraj P, Malligarjunan H, Subramanian S. Impact of methotrexate on oxidative stress and apoptosis markers in psoriatic patients. Clin Exp Med. 2014;14(4):431–437. doi: 10.1007/s10238-013-0252-7. [DOI] [PubMed] [Google Scholar]
- 7.Paul M, Hemshekhar M, Thushara RM, Sundaram MS, NaveenKumar SK, Naveen S, Devaraja S, Somyajit K, West R, Basappa NSC, Zakai UI, Nagaraju G, Rangappa KS, Kemparaju K, Girish KS. Methotrexate promotes platelet apoptosis via JNK-mediated mitochondrial damage: alleviation by N-Acetylcysteine and N-Acetylcysteine amide. PLoS One. 2015;10(6):e0127558. doi: 10.1371/journal.pone.0127558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al Maruf A, O'Brien PJ, Naserzadeh P, Fathian R, Salimi A, Pourahmad J. Methotrexate induced mitochondrial injury and cytochrome c release in rat liver hepatocytes. Drug Chem Toxicol. 2018;41(1):51–61. doi: 10.1080/01480545.2017.1289221. [DOI] [PubMed] [Google Scholar]
- 9.Mahmoud AM, Hussein OE, Hozayen WG, Abd El-Twab SM. Methotrexate hepatotoxicity is associated with oxidative stress, and down-regulation of PPARgamma and Nrf2: protective effect of 18beta-Glycyrrhetinic acid. Chem Biol Interact. 2017;270:59–72. doi: 10.1016/j.cbi.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Usta C, Ozdemir S, Schiariti M, Puddu PE. The pharmacological use of ellagic acid-rich pomegranate fruit. Int J Food Sci Nutr. 2013;64(7):907–913. doi: 10.3109/09637486.2013.798268. [DOI] [PubMed] [Google Scholar]
- 11.Oveissi V, Ram M, Bahramsoltani R, Ebrahimi F, Rahimi R, Naseri R, Belwal T, Devkota HP, Abbasabadi Z, Farzaei MH. Medicinal plants and their isolated phytochemicals for the management of chemotherapy-induced neuropathy: therapeutic targets and clinical perspective. Daru. 2019;27:389–406. doi: 10.1007/s40199-019-00255-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bahramsoltani R, Ebrahimi F, Farzaei MH, Baratpourmoghaddam A, Ahmadi P, Rostamiasrabadi P, Rasouli Amirabadi AH, Rahimi R. Dietary polyphenols for atherosclerosis: a comprehensive review and future perspectives. Crit Rev Food Sci Nutr. 2019;59(1):114–132. doi: 10.1080/10408398.2017.1360244. [DOI] [PubMed] [Google Scholar]
- 13.Chen P, Chen F, Zhou B. Antioxidative, anti-inflammatory and anti-apoptotic effects of ellagic acid in liver and brain of rats treated by D-galactose. Sci Rep. 2018;8(1):1465. doi: 10.1038/s41598-018-19732-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hseu YC, Chou CW, Senthil Kumar KJ, Fu KT, Wang HM, Hsu LS, Kuo YH, Wu CR, Chen SC, Yang HL. Ellagic acid protects human keratinocyte (HaCaT) cells against UVA-induced oxidative stress and apoptosis through the upregulation of the HO-1 and Nrf-2 antioxidant genes. Food Chem Toxicol. 2012;50(5):1245–1255. doi: 10.1016/j.fct.2012.02.020. [DOI] [PubMed] [Google Scholar]
- 15.Mukherjee S, Ghosh S, Choudhury S, Adhikary A, Manna K, Dey S, Sa G, Das T, Chattopadhyay S. Pomegranate reverses methotrexate-induced oxidative stress and apoptosis in hepatocytes by modulating Nrf2-NF-kappaB pathways. J Nutr Biochem. 2013;24(12):2040–2050. doi: 10.1016/j.jnutbio.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Razavi-Azarkhiavi K, Ali-Omrani M, Solgi R, Bagheri P, Haji-Noormohammadi M, Amani N, Sepand MR. Silymarin alleviates bleomycin-induced pulmonary toxicity and lipid peroxidation in mice. Pharm Biol. 2014;52(10):1267–1271. doi: 10.3109/13880209.2014.889176. [DOI] [PubMed] [Google Scholar]
- 17.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27(3):502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 18.Bodaghi-Namileh V, Sepand MR, Omidi A, Aghsami M, Seyednejad SA, Kasirzadeh S, Sabzevari O. Acetyl-l-carnitine attenuates arsenic-induced liver injury by abrogation of mitochondrial dysfunction, inflammation, and apoptosis in rats. Environ Toxicol Pharmacol. 2018;58:11–20. doi: 10.1016/j.etap.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150(1):76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 20.Sepand MR, Ghahremani MH, Razavi-Azarkhiavi K, Aghsami M, Rajabi J, Keshavarz-Bahaghighat H, Soodi M. Ellagic acid confers protection against gentamicin-induced oxidative damage, mitochondrial dysfunction and apoptosis-related nephrotoxicity. J Pharm Pharmacol. 2016;68(9):1222–1232. doi: 10.1111/jphp.12589. [DOI] [PubMed] [Google Scholar]
- 21.Brunt EM. Grading and staging the histopathological lesions of chronic hepatitis: the Knodell histology activity index and beyond. Hepatology. 2000;31(1):241–246. doi: 10.1002/hep.510310136. [DOI] [PubMed] [Google Scholar]
- 22.Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 2003;349(5):474–485. doi: 10.1056/NEJMra021844. [DOI] [PubMed] [Google Scholar]
- 23.Cetinkaya A, Bulbuloglu E, Kurutas EB, Kantarceken B. N-acetylcysteine ameliorates methotrexate-induced oxidative liver damage in rats. Med Sci Monit. 2006;12(8):BR274–BR278. [PubMed] [Google Scholar]
- 24.Labrecque L, Lamy S, Chapus A, Mihoubi S, Durocher Y, Cass B, Bojanowski MW, Gingras D, Beliveau R. Combined inhibition of PDGF and VEGF receptors by ellagic acid, a dietary-derived phenolic compound. Carcinogenesis. 2005;26(4):821–826. doi: 10.1093/carcin/bgi024. [DOI] [PubMed] [Google Scholar]
- 25.Nyblom H, Berggren U, Balldin J, Olsson R. High AST/ALT ratio may indicate advanced alcoholic liver disease rather than heavy drinking. Alcohol Alcohol. 2004;39(4):336–339. doi: 10.1093/alcalc/agh074. [DOI] [PubMed] [Google Scholar]
- 26.Safaei F, Mehrzadi S, Khadem Haghighian H, Hosseinzadeh A, Nesari A, Dolatshahi M, Esmaeilizadeh M, Goudarzi M. Protective effects of gallic acid against methotrexate-induced toxicity in rats. Acta Chir Belg. 2018;118(3):152–160. doi: 10.1080/00015458.2017.1394672. [DOI] [PubMed] [Google Scholar]
- 27.Girish C, Koner BC, Jayanthi S, Ramachandra Rao K, Rajesh B, Pradhan SC. Hepatoprotective activity of picroliv, curcumin and ellagic acid compared to silymarin on paracetamol induced liver toxicity in mice. Fundam Clin Pharmacol. 2009;23(6):735–745. doi: 10.1111/j.1472-8206.2009.00722.x. [DOI] [PubMed] [Google Scholar]
- 28.Saukkonen JJ, Cohn DL, Jasmer RM, Schenker S, Jereb JA, Nolan CM, Peloquin CA, Gordin FM, Nunes D, Strader DB, Bernardo J, Venkataramanan R, Sterling TR, Subcommittee ATSHoAT An official ATS statement: hepatotoxicity of antituberculosis therapy. Am J Respir Crit Care Med. 2006;174(8):935–952. doi: 10.1164/rccm.200510-1666ST. [DOI] [PubMed] [Google Scholar]
- 29.Aghsami M, Sharifzadeh M, Sepand MR, Yazdankhah M, Seyednejad SA, Pourahmad J. A cAMP analog attenuates beta-amyloid (1-42)-induced mitochondrial dysfunction and spatial learning and memory deficits. Brain Res Bull. 2018;140:34–42. doi: 10.1016/j.brainresbull.2018.03.016. [DOI] [PubMed] [Google Scholar]
- 30.Jahovic N, Cevik H, Sehirli AO, Yegen BC, Sener G. Melatonin prevents methotrexate-induced hepatorenal oxidative injury in rats. J Pineal Res. 2003;34(4):282–287. doi: 10.1034/j.1600-079X.2003.00043.x. [DOI] [PubMed] [Google Scholar]
- 31.Kopff M, Zakrzewska I, Czernicki J, Klem J, Strzelczyk M, Chmielewski H. Red blood cell superoxide dismutase and catalase activities in patients suffering from multiple sclerosis treated with adrenocorticotropic hormone. Pol J Pharmacol. 1996;48(4):441–445. doi: 10.1111/j.2042-7158.1996.tb05949.x. [DOI] [PubMed] [Google Scholar]
- 32.Rios JL, Giner RM, Marin M, Recio MC. A pharmacological update of Ellagic acid. Planta Med. 2018;84(15):1068–1093. doi: 10.1055/a-0633-9492. [DOI] [PubMed] [Google Scholar]
- 33.Haghi-Aminjan H, Asghari MH, Goharbari MH, Abdollahi M. A systematic review on potential mechanisms of minocycline in kidney diseases. Pharmacol Rep. 2017;69(4):602–609. doi: 10.1016/j.pharep.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 34.Keshavarz-Bahaghighat H, Sepand MR, Ghahremani MH, Aghsami M, Sanadgol N, Omidi A, Bodaghi-Namileh V, Sabzevari O. Acetyl-L-Carnitine attenuates arsenic-induced oxidative stress and hippocampal mitochondrial dysfunction. Biol Trace Elem Res. 2018;184(2):422–435. doi: 10.1007/s12011-017-1210-0. [DOI] [PubMed] [Google Scholar]
- 35.Haghi-Aminjan H, Asghari MH, Farhood B, Rahimifard M, Hashemi Goradel N, Abdollahi M. The role of melatonin on chemotherapy-induced reproductive toxicity. J Pharm Pharmacol. 2018;70(3):291–306. doi: 10.1111/jphp.12855. [DOI] [PubMed] [Google Scholar]
- 36.El-Sheikh AA, Morsy MA, Hamouda AH. Protective mechanisms of Thymoquinone on methotrexate-induced intestinal toxicity in rats. Pharmacogn Mag. 2016;12(Suppl 1):S76–S81. doi: 10.4103/0973-1296.176106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Copple IM, Goldring CE, Kitteringham NR, Park BK. The Nrf2-Keap1 defence pathway: role in protection against drug-induced toxicity. Toxicology. 2008;246(1):24–33. doi: 10.1016/j.tox.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 38.Kovac S, Angelova PR, Holmstrom KM, Zhang Y, Dinkova-Kostova AT, Abramov AY. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta. 2015;1850(4):794–801. doi: 10.1016/j.bbagen.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aleksunes LM, Manautou JE. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol Pathol. 2007;35(4):459–473. doi: 10.1080/01926230701311344. [DOI] [PubMed] [Google Scholar]
- 40.Abdo S, Zhang SL, Chan JS. Reactive oxygen species and nuclear factor Erythroid 2-related factor 2 activation in diabetic nephropathy: a hidden target. J Diabetes Metab. 2015;6(6). 10.4172/2155-6156.1000547. [DOI] [PMC free article] [PubMed]
- 41.Ding Y, Zhang B, Zhou K, Chen M, Wang M, Jia Y, Song Y, Li Y, Wen A. Dietary ellagic acid improves oxidant-induced endothelial dysfunction and atherosclerosis: role of Nrf2 activation. Int J Cardiol. 2014;175(3):508–514. doi: 10.1016/j.ijcard.2014.06.045. [DOI] [PubMed] [Google Scholar]
- 42.Sheibani M, Faghir-Ghanesefat H, Dehpour S, Keshavarz-Bahaghighat H, Sepand MR, Ghahremani MH, Azizi Y, Rahimi N, Dehpour AR. Sumatriptan protects against myocardial ischaemia-reperfusion injury by inhibition of inflammation in rat model. Inflammopharmacology. 2019;27:1071–1080. doi: 10.1007/s10787-019-00586-5. [DOI] [PubMed] [Google Scholar]
- 43.Soares MP, Seldon MP, Gregoire IP, Vassilevskaia T, Berberat PO, Yu J, Tsui TY, Bach FH. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol. 2004;172(6):3553–3563. doi: 10.4049/jimmunol.172.6.3553. [DOI] [PubMed] [Google Scholar]
- 44.Ahad A, Ganai AA, Mujeeb M, Siddiqui WA. Ellagic acid, an NF-kappaB inhibitor, ameliorates renal function in experimental diabetic nephropathy. Chem Biol Interact. 2014;219:64–75. doi: 10.1016/j.cbi.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 45.Jun CD, Kim Y, Choi EY, Kim M, Park B, Youn B, Yu K, Choi KS, Yoon KH, Choi SC, Lee MS, Park KI, Choi M, Chung Y, Oh J. Gliotoxin reduces the severity of trinitrobenzene sulfonic acid-induced colitis in mice: evidence of the connection between heme oxygenase-1 and the nuclear factor-kappaB pathway in vitro and in vivo. Inflamm Bowel Dis. 2006;12(7):619–629. doi: 10.1097/01.ibd.0000225340.99108.8a. [DOI] [PubMed] [Google Scholar]
- 46.Yeh PY, Li CY, Hsieh CW, Yang YC, Yang PM, Wung BS. CO-releasing molecules and increased heme oxygenase-1 induce protein S-glutathionylation to modulate NF-kappaB activity in endothelial cells. Free Radic Biol Med. 2014;70:1–13. doi: 10.1016/j.freeradbiomed.2014.01.042. [DOI] [PubMed] [Google Scholar]
- 47.Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci U S A. 1994;91(13):5987–5991. doi: 10.1073/pnas.91.13.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andreadi CK, Howells LM, Atherfold PA, Manson MM. Involvement of Nrf2, p38, B-Raf, and nuclear factor-kappaB, but not phosphatidylinositol 3-kinase, in induction of hemeoxygenase-1 by dietary polyphenols. Mol Pharmacol. 2006;69(3):1033–1040. doi: 10.1124/mol.105.018374. [DOI] [PubMed] [Google Scholar]