

SUMMARY
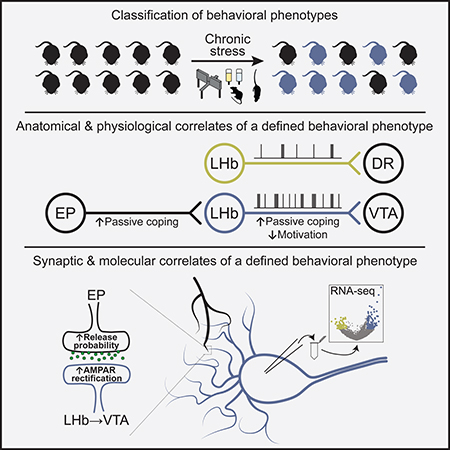
Chronic stress (CS) is a major risk factor for the development of depression. Here, we demonstrate that CS-induced hyperactivity in ventral tegmental area (VTA)-projecting lateral habenula (LHb) neurons is associated with increased passive coping (PC) but not anxiety or anhedonia. LHb→VTA neurons in mice with increased PC show increased burst and tonic firing as well as synaptic adaptations in excitatory inputs from the entopeduncular nucleus (EP). In vivo manipulations of EP→LHb or LHb→VTA neurons selectively alter PC and effort-related motivation. Conversely, dorsal raphe (DR)-projecting LHb neurons do not show CS-induced hyperactivity and are targeted indirectly by the EP. Using single-cell transcriptomics we reveal a set of genes that can collectively serve as biomarkers to identify mice with increased PC and differentiate LHb→VTA from LHb→DR neurons. Together, we provide a set of biological markers at the level of genes, synapses, cells and circuits that define a distinctive CS-induced behavioral phenotype.
eTOC
Chronic stress is a major risk factor for depression. Cerniauskas et al. identify a lateral habenula subcircuit involved in chronic stress-induced increased passive coping and reduced motivated behaviors. Single-cell transcriptomics reveal potential gene targets associated with this specific behavioral phenotype.
Graphical Abstract
INTRODUCTION
Chronic stress (CS) is a major risk factor for the development of depression in humans (Hill et al., 2012; Mazure and Maciejewski, 2003; Willner et al., 2013). This has led to the development of several animal models of depression that have been extensively used in basic research over the last few decades (Czéh et al., 2016; Nestler and Hyman, 2010; Willner, 2017). In these models, rodents are exposed to CS in order to generate a variety of behavioral changes (e.g. anhedonia, social withdrawal), which are thought to reflect some of the core symptoms seen in depressed humans (Berton et al., 2012; Monteggia et al., 2018; Nestler and Hyman, 2010; Willner et al., 1992). Although patients with depression show a highly diverse set of combination of symptoms (Carragher et al., 2009; ten Have et al., 2016; Musil et al., 2018; Willner et al., 2013), scientists often considered CS-exposed animals as a homogeneous population in their search for a pathological mechanism (Agudelo et al., 2014; Cui et al., 2018; Frisbee et al., 2015; Li et al., 2013; Moreines et al., 2017; Ramirez et al., 2015; Schweizer et al., 2009; Seo et al., 2018; Tye et al., 2012; Yang et al., 2018). However, simply dividing animals into “stressed” and “non-stressed” groups may not account for the diversity of behavioral phenotypes that arise in response to CS exposure. Since different behavioral phenotypes presumably involve discrete brain areas and circuits, it is critical to be able to differentiate these behaviors in order to study their underlying neural correlates when searching for symptom-specific therapeutic interventions. To this end, strategies that recognize the heterogeneity of CS-induced behavioral phenotypes and their biological basis remain largely underdeveloped.
In recent years, the lateral habenula (LHb) has emerged as a potential key structure in depression (Lammel et al., 2014; Lecca et al., 2014; Proulx et al., 2014; Sartorius and Henn, 2007; Yang et al., 2017). Increased neural and metabolic activity of the LHb has been observed in various animal models of depression (Li et al., 2011; Mirrione et al., 2014; Shumake et al., 2003; Tchenio et al., 2017), and elevated LHb firing and depression-related behaviors can be reversed by antidepressant treatment and by employing a deep brain stimulation (DBS)-like protocol in rodents (Li et al., 2011; Tchenio et al., 2017; Yang et al., 2018). Notably, DBS of the LHb caused a marked reduction of depression symptoms in a therapy-refractory patient, with interruption of the stimulation rapidly leading to the recurrence of depressive periods (Kiening and Sartorius, 2013; Sartorius et al., 2010). Together, these studies suggest an important role for both CS and LHb hyperactivity in depression.
Here we introduce an unbiased approach that allows us to examine the neurobiological basis of distinct CS-induced behavioral phenotypes in mice. Using this approach, we deconstructed the molecular, synaptic and circuit architecture of the LHb. The identification of biological markers that align with specific CS-induced behavioral phenotypes constitutes an important step for reducing the complexity of depression and the development of more specific treatments of this broad, heterogeneous disease.
RESULTS
Classification of chronic stress-induced behavioral phenotypes
We exposed mice to eight weeks of chronic mild stress (CMS) and subsequently analyzed them using four different behavioral assays. We first assessed anxiety-related behaviors using the elevated plus (EPM). We found that CMS mice spent significantly less time in the open arms in the elevated plus maze (EPM) compared to non-stressed control (CTRL) mice, suggesting that on average these animals develop an anxiety phenotype in response to CMS exposure (Figure 1A, left panel). Next, we used the sucrose preference test (SPT) to assesses an animal’s interest in rewarding stimuli. We found that CMS mice exhibited significantly reduced sucrose preference compared to CTRL mice, suggesting that CMS promotes anhedonia-related behavior, which is considered a core symptom of depression in humans (Figure 1B, left panel). Next, mice were tested in the tail suspension test (TST), a behavioral assay in which animals are exposed to an inescapable aversive environment. Mice initially attempt vigorous escape but then transition to a passive coping (PC) state (Koolhaas et al., 1999). PC can be modulated by genetic, behavioral, and pharmacological interventions related to depression (Andalman et al., 2019; Castagne et al., 2011; Cui et al., 2019; Warden et al., 2012; Willner, 2005; Yilmaz et al., 2002) and may be relevant to the pathological motivational impairments seen in major depression in humans. We found that CMS mice struggled significantly less in the TST when compared to CTRL mice (Figure 1C, left panel). Moreover, struggling behavior in the TST was strongly correlated with struggling behavior in the forced swim test (FST) within the same animals (Figures S1A and S1B) (Porsolt et al., 1978), suggesting that these paradigms may assess a similar behavioral phenotype. Lastly, we used a social interaction test (SIT) to assess CMS-induced deficits in sociability behavior, as social withdrawal is a common symptom of depression in humans (Nestler and Hyman, 2010). Surprisingly, we found that both CTRL and CMS mice showed a similar pattern of sociability as measured in their voluntary initiation of social interaction and their preference for social novelty, as measured by the time spent investigating a novel mouse (Figures S1C and S1D). Thus, CMS may distinguish itself from other stress paradigms (e.g. social defeat stress) in regard to the magnitude and quality of the stress response.
Figure 1. Classification of chronic stress-induced behavioral phenotypes.

(A) Left: Time spent in open arms for CTRL (green) and CMS (blue) mice in the EPM; Right: Receiver operating characteristic (ROC) curve (blue line) for EPM data. Orange line: maximum Youden’s J index (JI; *** p < 0.001, data represent means ± SEM).
(B) Left: Sucrose consumption (%) for CTRL (green) and CMS (blue) mice in the SPT; Right: ROC curve (blue line) for SPT data. Orange line indicates maximum JI (** p < 0.01, data represent means ± SEM).
(C) Left: Time spent struggling for CTRL (green) and CMS (blue) mice in the TST; Right: ROC curve (blue line) for TST data. Orange line indicates maximum JI (*** p < 0.001, data represent means ± SEM).
(D) 3D plot showing results from EPM, SPT and TST for individual CTRL (green) and CMS (blue) mice.
(E) CTRL mice positive for zero (brown), one (purple), two (jade) or three (orange) behavioral criteria. An animal was considered positive if it scored below the corresponding cutoff value (dashed line; data represent means ± SEM).
(F) CMS mice positive for zero (brown), one (purple), two (jade) or three (orange) behavioral criteria. An animal was considered positive if it scored below the corresponding cutoff value (dashed line; data represent means ± SEM).
(G) Comparison of total population of CTRL (green) and CMS (blue) mice positive for zero, one, two or three criteria (D-score of zero to three, respectively).
(H) Percentage of the total population of CTRL (top) and CMS (bottom) mice positive for zero, one, two, or three criteria (D-score of zero to three, respectively).
Our data from the behavioral screening assays showed a remarkable variability for individual test scores in both CTRL and CMS mice, making it difficult to evaluate the effects of CMS on individual animals. We therefore decided to generate receiver operating characteristic (ROC) curves, which is an objective method that has been used extensively in clinical epidemiology for the evaluation of binary classifiers (Berrar and Flach, 2012; Søreide, 2009; Zou et al., 2007). To generate ROC curves, we used the data from the three behavioral tests that showed statistically significant differences (i.e. EPM, SPT, TST). We then used the ROC curves (Figures 1A–1C, right panels) to calculate optimal cutoff values based on Youden J index (Youden, 1950), which allowed us to make an unbiased decision as to whether an individual animal is positive for a given behavioral phenotype. The cutoff values that have the combination of highest true positive and lowest false positive rates are 181.9 s for the EPM, 60.1% for the SPT and 93.5 s for the TST.
Animals may also exhibit variability being tested positive in the total number of CMS-induced behavioral phenotypes. In order to examine the number of behavioral phenotypes for individual animals, we used separate cohorts of CTRL (n = 69) and CMS (n = 107) mice that went through the three behavioral tests on three consecutive days (Figure 1D). We then separated both CTRL and CMS mice into four groups according to the number of positive criteria met and assigned them a score between zero and three (D-score). An animal was considered positive for behavioral phenotypes anxiety, anhedonia or passivity if it scored below the respective cutoff value. Strikingly, both CTRL and CMS groups contained animals that were positive for multiple criteria or showed only one or no phenotype (Figures 1E and 1F). Mice that met all three positive criteria represented 19% of the CMS group, while only 3% of CTRL mice fell into this category. Conversely, despite being exposed to CMS, a small group of animals (5%) did not test positive to any behavioral phenotype at all, whereas the percentage of CTRL mice that did not meet positive criteria was much larger (36%; Figures 1G and 1H).
Additional evidence to support our classification of mice into different subgroups is based on the robustness of behavioral phenotypes. First, the intensity of behavioral phenotypes was proportional to the number of criteria met (Figures 1E and 1F). Second, when re-testing mice on the same behavioral assays (EPM, SPT, TST) under the same experimental conditions two weeks after they performed the test for the first time, we observed a linear correlation between the first and second test in all three behavioral assays suggesting that animals exhibit persistent behavioral phenotypes that do not change over time (Figures S1E–S1G). Third, we performed bootstrapping on our experimental data shown in Figures 1A–1C to demonstrate the consistency of our cutoff values and the distribution of the D-score subgroups (Figure S2).
Together, these results suggest that rodents, like humans, show clear behavioral differences in their response to chronic stress, and that screening mice for individual behavioral phenotypes could facilitate the analysis of neural correlates underlying depression-related behaviors.
Anatomical and physiological correlates of a distinctive chronic stress-induced behavioral phenotype
Ventral tegmental area (VTA) and dorsal raphe nucleus (DR), which contain large populations of dopamine (DA) and serotoninergic neurons, respectively, represent two major downstream projection targets of LHb (Bernard and Veh, 2012). VTA-projecting LHb (LHb→VTA) neurons exhibit hyperactivity in a learned helplessness model of depression (Li et al., 2011), but whether LHb→VTA hyperactivity is associated with a specific chronic stress-induced behavioral phenotype and whether DR-projecting LHb neurons (LHb→DR) also undergo pathological adaptations is unknown. Consistent with previous anatomical studies in rats (Bernard and Veh, 2012), we found that LHb projections to the VTA and DR are organized as independent parallel projections (Figures S3A–S3C) that originate from largely separate LHb subnuclei (Figures S3D–S3G). Despite this anatomical separation, LHb→VTA and LHb→DR subpopulations may subserve similar functional roles, as optogenetic stimulation of LHb terminals in either VTA (Figures S3H–S3K; Lammel et al., 2012; Stamatakis and Stuber, 2012) or DR (Figures S3L–S3O) induced robust real-time place aversion.
Next, we performed whole-cell patch clamp recordings from retrogradely labeled LHb→VTA (Figures 2A and 2B) and LHb→DR (Figures 2H and 2I) neurons in acute slices from CMS and non-stressed CTRL mice that were behaviorally screened based on our D-score classification approach (Figure 1). Animals from each group were divided into two categories based on their individual D-scores: CTRL and CMS mice with a D-score of zero or one (CTRLD0–1 and CMSD0–1, respectively) and CTRL and CMS mice with a D-score of two or three (CTRLD2–3 and CMSD2–3, respectively). We found that under resting conditions, both LHb→VTA and LHb→DR neurons remained mostly silent and did not fire spontaneous action potentials. However, following the injection of depolarizing ramp currents, we observed remarkable differences in the firing rates of LHb→VTA neurons from animals with different D-scores. LHb→VTA neurons in CMSD2–3 mice showed very high firing rates in response to +150 pA current injection that were significantly higher when compared to CTRLD0–1 or CMSD0–1 mice. Surprisingly, the evoked firing rate of LHb→VTA neurons from CTRLD2–3 mice was only marginally lower when compared to CMSD2–3 mice, though not statistically different from CTRLD0–1 mice (Figures 2C, 2D and S3P).
Figure 2. Hyperactivity of LHb neurons is associated with projection target and behavioral phenotype.

(A) Experimental design.
(B) Injection site of beads (red) in the VTA (IPN: interpeduncular nucleus; Scale bar: 300 μm).
(C) Firing in response to +150 pA depolarizing current injection in LHb→VTA neurons from CTRL and CMS mice with different D-scores: CTRLD0–1 (top left), CMSD0–1 (top right), CTRLD2–3 (bottom left) and CMSD2–3 (bottom right) mice (Scale bars: 20 mV/0.5 s).
(D) Mean number of action potentials in response to injection of different depolarizing ramp currents recorded in LHb→VTA neurons from CTRLD0–1 (light green), CTRLD2–3 (dark green), CMSD0–1 (pink) and CMSD2–3 (blue) mice (* p < 0.05, ** p < 0.01, data represent means ± SEM).
(E-G) Mean number of action potentials in response to injection of a +150 pA depolarizing current in LHb→VTA neurons from CTRL or CMS mice that were pooled according to whether they were positive or negative for specific behavioral phenotypes (i.e. anxiety assessed in EPM (E), anhedonia assessed in SPT (F), immobility assessed in TST (G)). Animals were considered positive if they scored below the corresponding cutoff value defined in Figures 1A–1C (* p < 0.05, ** p < 0.01; data represent means ± SEM).
(H) Experimental design.
(I) Injection site of beads (red) in the DR (AQ: cerebral aqueduct; Scale bar: 400 μm).
(J) Firing in response to +150 pA depolarizing current injection in LHb→DR neurons from CTRL and CMS mice with different D-scores: CTRLD0–1 (top) and CMSD2–3 (bottom) mice (Scale bars: 20 mV/0.5 s).
(K) Mean number of action potentials in response to injection of different depolarizing ramp currents recorded in LHb→DR neurons from CTRLD0–1 (light green) and CMSD2–3 (blue) mice (data represent means ± SEM).
(L) mEPSCs recorded in LHb→DR neurons from CTRLD0–1 (top) and CMSD2–3 (bottom) mice (Scale bars: 10 pA/1 s).
(M) Mean mEPSC frequencies (left) and mEPSC amplitudes (right) recorded in LHb→DR neurons from CTRLD0–1 and CMSD2–3 mice (data represent means ± SEM).
When we separated the animals into subgroups based on whether they met individual behavioral criteria, we found that both in CTRL and CMS groups, mice with increased immobility in the TST, but not with anhedonia or anxiety, displayed increased firing (Figures 2E–2G). Strikingly, evoked firing of LHb→DR neurons was not significantly different between CMSD2–3 and CTRLD0–1 mice (Figures 2J and 2K). Furthermore, we examined excitatory transmission onto LHb→DR neurons, which is known to be altered in LHb→VTA neurons in a learned helplessness model of depression (Li et al., 2011). However, frequency and amplitude of miniature excitatory postsynaptic currents (mEPSCs) recorded from LHb→DR neurons did not significantly differ between CMSD2–3 and CTRLD0–1 mice (Figures 2L and 2M). Other electrophysiological parameters, such as membrane resistance and conductance, did not differ significantly between LHb→VTA and LHb→DR neurons (Figure S3Q) or LHb→VTA neurons in CTRLD0–1 and CMSD2–3 mice (Figures S3R).
To further investigate neural activity patterns in response to CMS, we performed in vivo tetrode recordings from LHb→VTA neurons in freely behaving mice (Figure 3A). Specifically, we tagged LHb→VTA neurons with channelrhodopsin-2 (ChR2) by injecting a retrogradely transported virus expressing Cre-recombinase (pseudotyped equine infectious anemia virus; RG-EIAV-Cre) into the VTA and a Cre-dependent adeno-associated virus (AAV) expressing ChR2-eYFP (AAV-DIO-ChR2-eYFP) into the LHb of C57BL/6 mice. In the same animals, a driveable optoelectrode (optrode) was implanted above the LHb and lowered in steps throughout the LHb during the recording sessions. CTRLD0–1 and CMSD2–3 mice were identified using our D-score classification approach. Importantly, all CMSD2–3 mice had increased immobility in the TST (i.e. time spent struggling < 93.5 s), but none of the CTRLD0–1 mice had this phenotype. RG-EIAV-Cre-mediated ChR2-eYFP expression and optrode placement in the LHb was confirmed using histological methods (Figure 3B). To identify LHb neurons that expressed ChR2 (i.e. LHb→VTA neurons), we tested whether recorded LHb cells responded with minimal latency to 473 nm light stimulation (Figures S4A–S4F). Consistent with direct excitation, all optogenetically identified LHb neurons responded with short (4.79 ± 0.25 ms, n = 12 cells) latency after the light onset. Comparison of spontaneous and light-evoked spikes revealed that stimulation did not induce detectable changes in the action potential shape (Figures S4D). After ChR2-tagged LHb→VTA neurons were identified, their spontaneous activity was recorded over a five-minute period while the mice were freely behaving in their home cages. The average action potential waveform of the LHb→VTA neurons was similar in CTRLD0–1 and CMSD2–3 mice with no significant differences in spike width (Figure 3C). Notably, LHb→VTA neurons from CMSD2–3 mice showed an overall increase in both burst activity and tonic firing compared to LHb→VTA neurons from CTRLD0–1 mice (Figures 3D–3I and S3G). As a result, the mean firing frequency was also significantly higher in CMSD2–3 than in CTRLD0–1 mice (Figure 3J). We also analyzed non-light responsive LHb cells that were concurrently recorded at the same depth and during the same session as the optogenetically-tagged LHb neurons, and found that tonic firing and burst activity of untagged LHb neurons was very similar between CMSD2–3 and CTRLD0–1 mice (Figures S4H–S4M). Taken together, LHb hyperactivity consists of both increased tonic and burst activity and is substantially defined by topographic organization, axonal projections and behavioral phenotype.
Figure 3. Chronic stress increases burst and tonic firing of LHb→VTA neurons.

(A) Experimental design.
(B) Top: Injection-site of EIAV-Cre (red) in the VTA (left) and expression of ChR2-eYFP (green) in LHb→VTA neurons (right) (IP: interpeduncular nucleus, 3V: 3rd ventricle, MHb: medial habenula; DAPI: blue; Scale bars: 300 μm (left), 250 μm (right)). Bottom: Localizations of optrodes in LHb for CTRLD0–1 (green) and CMSD2–3 (blue) mice (fr: fasciculus retroflexus, DG: dentate gyrus).
(C) Left: Action potential waveforms for LHb→VTA neurons in CTRLD0–1 (green) and CMSD2–3 (blue) mice (Scale bars: 20 μV/0.5 ms (CTRLD0–1), 30 μV/0.5 ms (CMSD2–3)). Right: Mean action potential width for LHb→VTA neurons in CTRLD0–1 (green) and CMSD2–3 (blue) mice (data represent means ± SEM).
(D) Normalized frequencies of interspike intervals for LHb→VTA neurons in CTRLD0–1 (green) and CMSD2–3 (blue) mice.
(E) Mean percentage of spikes in bursts recorded for LHb→VTA neurons in CTRLD0–1 (green) and CMSD2–3 (blue) mice (* p < 0.05, data represent means ± SEM).
(F) Mean number of spikes per burst for LHb→VTA neurons in CTRLD0–1 (green) and CMSD2–3 (blue) mice (* p < 0.05, data represent means ± SEM).
(G) Mean interburst frequencies for LHb→VTA neurons in CTRLD0–1 (green) and CMSD2–3 (blue) mice (* p < 0.05, data represent means ± SEM).
(H) Mean intraburst frequencies for LHb→VTA neurons in CTRLD0–1 (green) and CMSD2–3 (blue) mice (data represent means ± SEM).
(I) Mean tonic firing frequencies for LHb→VTA neurons in CTRLD0–1 (green) and CMSD2–3 (blue) mice (** p < 0.01, data represent means ± SEM).
(J) Mean firing frequencies for LHb→VTA neurons in CTRLD0–1 (green) and CMSD2–3 (blue) mice (** p < 0.01, data represent means ± SEM).
Distinct inputs onto LHb neurons based on their projections
We hypothesized that differences in the architecture of afferent inputs to projection-defined LHb neurons contribute to their selective CMS-induced hyperactivity phenotype. Classical anatomical tracing studies have shown that major inputs to the LHb arise from various brain regions including the entopeduncular nucleus (EP), lateral preoptic area (LPO), lateral hypothalamus (LH), DR and VTA (Herkenham and Nauta, 1977), but the connectivity of upstream brain structures with projection-defined LHb subpopulations has not yet been investigated. In order to map whole-brain monosynaptic inputs onto LHb→VTA and LHb→DR neurons, we used a rabies virus-based tracing strategy (Osakada and Callaway, 2013). We injected a retrogradely transported canine adenovirus expressing Cre-recombinase (CAV2-Cre) into either the VTA or DR of C57BL/6 mice. In addition, we injected two Cre-dependent AAVs, one expressing the avian tumor virus A receptor (AAV-DIO-TVA-mCherry) and the other expressing the rabies virus glycoprotein (AAV-DIO-RG) into the LHb. Four weeks later, we injected a modified rabies virus expressing GFP (Rabies-EnvA-ΔG-GFP) into the LHb (Figure 4A). Histological analysis revealed the specificity of the CAV2-Cre injection-sites in the VTA or DR (Figure S5A) and the localization of starter cells (i.e. cells that are both TVA-mCherry- and RV-GFP-immunopositive) in the LHb (Figures 4B, S5B and S5C). We then determined the anatomical locations as well as the number of GFP-expressing cells that synapse onto LHb→VTA and LHb→DR neurons (Figure 4C). We found that LHb→VTA and LHb→DR neurons receive qualitatively similar inputs, with dominant projections from the LH, VTA, pallidum, LPO and EP. Quantitatively, however, we found that the EP provides a significantly greater share of input to LHb→VTA neurons, while LH and VTA provide significantly more inputs to LHb→DR neurons (Figure 4D). Collectively, the EP sends substantially stronger inputs to LHb→VTA compared to LHb→DR and we speculate that chronic stress-induced synaptic adaptations in specific excitatory inputs (e.g. EP) to LHb→VTA neurons may promote their hyperactivity in depression-related states.
Figure 4. Anatomical and functional mapping of inputs to LHb subpopulations.

(A) Experimental design.
(B) Left: Anatomical distribution of starter cells in the LHb for mapping inputs to LHb→VTA neurons. Starter cells: cells that co-express RV-GFP (green) and TVA-mCherry (red; Scale bar: 150 μm). Right: Higher magnification image (DAPI: blue; Scale bar: 60 μm).
(C) Anatomical distribution of input neurons (i.e. RV-GFP-positive cells) to LHb→VTA (top) and LHb→DR (bottom) neurons in the entopeduncular nucleus (EP, left), lateral hypothalamus (LH, middle) and VTA (right; Scale bars: 200 μm (left, right), 400 μm (middle)).
(D) Quantification of inputs to LHb→VTA (purple) and LHb→DR (green) neurons (percentage of total input counted in each individual brain). See Figure S4 legend for abbreviations (*** p < 0.001, data represent means ± SEM).
(E) Experimental design.
(F) Left: EPSCs for light stimulation of EP (top; Scale bar: 50 pA/20 ms), VTA (middle; Scale bar: 50 pA/20 ms) or LH (bottom; Scale bar: 200 pA/20 ms) inputs to LHb→VTA neurons. Right: EPSCs showing dual AMPAR+NMDAR- (black), AMPAR- (jade; in 50 μM AP5) and NMDAR (purple; after digital subtraction)-mediated currents (purple) for light stimulation of EP (top; Scale bar: 20 pA/20 ms), VTA (middle; Scale bar: 40 pA/20 ms) or LH (bottom; Scale bar: 20 pA/20 ms) inputs to LHb→VTA neurons.
(G) Mean EPSC peak amplitudes and connectivity for EP, VTA and LH inputs to LHb→VTA neurons (*** p < 0.001, data represent means ± SEM).
(H) Mean decay time for dual AMPAR+NMDAR (black), AMPAR (jade) and NMDAR (purple) components for EP, VTA or LH inputs to LHb→VTA neurons (* p < 0.05, ** p < 0.01, data represent means ± SEM).
Aberrant pre- and postsynaptic plasticity in the LHb
To further examine the synaptic properties of identified excitatory inputs to LHb→VTA neurons, we injected an AAV expressing ChR2 under the control of the CaMKII promoter into the EP, VTA or LH and retrobeads into the VTA of C57BL/6 mice. We focused on EP, VTA and LH inputs because our whole brain mapping experiments revealed that these are the major inputs to LHb→VTA neurons (Figure 4D). We then performed whole-cell patch clamp recordings in acute brain slices from retrogradely labeled LHb neurons and recorded light-evoked excitatory postsynaptic currents (EPSCs; in 50 μM picrotoxin) by stimulating ChR2-expressing EP, VTA or LH terminals with blue light (Figure 4E). Our data revealed that even under control conditions the synaptic properties of individual excitatory inputs onto LHb→VTA neurons are remarkably different. Though the functional connectivity of the three inputs to LHb→VTA neurons is similar, the mean amplitude of light evoked EPSCs (at −70 mV) from the LH was significantly greater compared to the EP and VTA (Figures 4F and 4G), which is remarkably consistent with the relative input strength seen in our rabies tracing experiments (Figure 4D). Moreover, NMDA receptor (NMDAR)-mediated currents (recorded at +40 mV) evoked by stimulation of LH inputs exhibited significantly greater decay time compared to VTA and EP inputs, suggesting possible differences in the composition of NMDARs between individual inputs (Figures 4F and 4H).
Next, we compared the synaptic properties of excitatory EP, VTA and LH inputs onto LHb→VTA neurons from CTRLD0–1 and CMSD2–3 mice. While the AMPAR/NMDAR ratio was not significantly altered for any input when comparing CTRLD0–1 with CMSD2–3 mice (data not shown), we observed notable differences in the presynaptic release probability (PRP). The PRP of excitatory EP synapses was higher in CMSD2–3 compared to CTRLD0–1 mice (i.e. decreased paired pulse ratio (PPR)), whereas it was the opposite for VTA inputs (increased PPR), and no significant differences were observed for LH inputs (Figure 5A).
Figure 5. Chronic stress induces synaptic adaptations in excitatory EP inputs to LHb→VTA neurons.

(A) Left: Paired pulse EPSCs (100 ms interval; −70 mV) in response to light stimulation of EP (left), VTA (middle) or LH (right) inputs to LHb→VTA neurons in CTRLD0–1 (top) and CMSD2–3 (bottom) mice (Scale bars: 20 pA/20 ms). Right: Mean paired pulse ratios (PPR, calculated as peak2/peak1) for EP, VTA and LH inputs to LHb→VTA neurons in CTRLD0–1 (green) and CMSD2–3 (blue) mice (* p < 0.05, ** p < 0.01, data represent means ± SEM).
(B) Left: AMPAR-mediated currents at +40 mV and −70 mV (in 50 μM AP5) for EP (left), VTA (middle) or LH (right) inputs to LHb→VTA neurons from CTRLD0–1 (top) and CMSD2–3 (bottom) mice (Scale bars: 20 pA/20 ms). Right: Mean rectification index (peak amplitude. 70mV/peak amplitude+40mV) for EP, VTA and LH inputs to LHb→VTA neurons from CTRLD0. 1 (green) and CMSD2–3 (blue) mice (** p < 0.01, data represent means ± SEM).
(C) Left: EPSCs (−70 mV) for stimulation of EP inputs to LHb→VTA neurons during baseline and after wash-in of 30 μM NASPM in CTRLD0–1 (green) and CMSD2–3 (blue) mice. The amplitude of baseline EPSCs does not change over time when NASPM was not applied (black; Scale bar: 100 pA / 40 ms). Right: Normalized mean AMPAR-mediated EPSC amplitudes with and without bath application of NASPM for the three experimental groups. Arrows indicate sample traces shown on the left (* p < 0.05, data represent means ± SEM).
(D) Left: EP terminals (eYFP, green) in lateral LHb adjacent to retrogradely labeled (beads, red) LHb→VTA neurons (DAPI: blue; Scale bars: 160 μm (left), 80 μm (right)). Right: EP terminals (eYFP, green) in lateral LHb and retrogradely labeled (beads, red) LHb→DR neurons in medial LHb. Squares indicate higher magnification images (Scale bars: 160 μm (left), 40 μm (right)).
(E) Heat map representing peak response latencies of LHb→VTA (left) and LHb→DR (right) neurons in response to light stimulation of excitatory EP terminals in the LHb. Each row represents individual cells. Color code represents normalized EPSC amplitude.
(F) Left: EPSCs from LHb→DR (top) or LHb→VTA (bottom) neurons in response to light stimulation of EP terminals in the LHb (Scale bars: 40 pA/5ms (top), 60 pA/5ms (bottom)). Right: Mean peak response latencies for light stimulation of excitatory EP inputs to LHb→DR or LHb→VTA neurons (*** p < 0.001, mean ± SEM).
(G) Left: EPSCs from LHb→DR (left) or LHb→VTA (right) neurons in response to light stimulation of EP terminals at baseline (top), after bath application of TTX (middle) and TTX + 4-AP (bottom; Scale bars: 20 pA/20 ms (left), 50 pA/20 ms (right)). Right: Relative amplitudes of EPSCs recorded from LHb→DR and LHb→VTA neurons in response to light stimulation of EP terminals in the LHb at baseline and after wash-in of TTX or TTX + 4-AP (** p < 0.01, data represent means ± SEM).
Postsynaptically, AMPAR subunit composition can influence cell excitability and synaptic efficacy (Liu and Zukin, 2007). In contrast to GluR2-containing ones, GluR2-lacking AMPARs are calcium-permeable and exhibit higher single-channel conductance (Hollmann et al., 1991). We found that EP, VTA and LH inputs exhibit a characteristic inwardly rectifying AMPAR-mediated currents at +40 mV. However, only in EP inputs onto LHb→VTA neurons was the rectification index of AMPAR-mediated currents significantly increased in CMSD2–3 compared to CTRLD0–1 mice, suggesting an increase in surface expression of GluR2-lacking AMPARs (Figure 5B). To confirm an increase in GluR2-lacking AMPARs at the EP synapses, we applied 30 μM 1-naphthyl acetyl spermine (NASPM, a selective blocker of GluR2-lacking AMPARs) to brain slices prepared from CMSD2–3 and CTRLD0–1 mice. As expected, this reduced the amplitude of light-evoked AMPAR-mediated currents in both CMSD2–3 and CTRLD0–1 mice, indicating an already rectifying nature of AMPARs at the EP synapse. Importantly, the amplitude of the light-evoked AMPAR-mediated current was significantly more reduced in CMSD2–3 compared to CTRLD0–1 mice, suggesting an even stronger expression of GluR2-lacking AMPARs in response to CMS (Figure 5C). Taken together, CMS exposure predominantly affects the synaptic properties of EP synapses onto LHb→VTA neurons, which includes both increased PRP as well as increased surface expression of GluR2-lacking AMPARs.
Divergent synaptic connectivity of EP inputs with LHb subpopulations
To further examine the synaptic connectivity of EP inputs with projection-defined LHb subpopulations, we performed an additional series of experiments. First, we found that the majority of EP terminals are located in close proximity to LHb→VTA neurons, whereas there seems to be a clear anatomical separation between EP terminals and LHb→DR cells, which are mainly located in the medial LHb (Figures 5D and S3D–S3G). Second, we found that the majority of light-evoked EPSCs from excitatory EP inputs to LHb→DR neurons exhibit a delayed onset, and the peak response latency is significantly greater in LHb→DR neurons compared to LHb→VTA neurons (Figures 5E and 5F) suggesting that excitatory EP neurons may not directly target LHb→DR neurons. Third, to further test whether the EP→LHb→DR connection is indeed polysynaptic, we used a previously described technique (Cho et al., 2013; Petreanu et al., 2009) to depolarize ChR2-positive presynaptic terminals directly by blocking sodium channels using tetrodotoxin (TTX, 1 μM) and potassium channels using 4-aminopyridine (4-AP, 50 μM). As expected, bath application of TTX blocked light-evoked EPSCs in both LHb subpopulations. Subsequent application of 4-AP, however, rescued TTX-blocked EPSCs only in LHb→VTA neurons, which indicated the monosynaptic origin of these EPSCs. 4-AP did not rescue TTX-blocked polysynaptic EPSCs in LHb→DR neurons as these EPSCs would require action potential firing in local LHb neurons (Figure 5G). These results suggest that excitatory EP inputs make monosynaptic inputs onto LHb→VTA neurons, but their excitation of LHb→DR neurons primarily involves a feedforward excitation of local LHb neurons.
In vivo modulation of LHb circuitry selectively alters passive coping and effort-related motivated behavior
Because hyperactivity of the LHb→VTA pathway was specifically associated with increased immobility in the TST, but not anxiety or anhedonia (Figures 2E–2G), we hypothesized that in vivo manipulations of the LHb involving downstream VTA or upstream EP projections will selectively affect transitions from active struggling to PC behavior and that this transition can become maladaptive in response to CMS exposure. Our hypothesis is supported by a recent study which reported that optogenetic stimulation of the LHb→RMTg pathway selectively increases immobility in the FST (Proulx et al., 2018), though it is likely that FST and TST are not fully represented by the same neurocircuitry. To test this, we performed a series of behavioral experiments that involve targeting of chemogenetic or optogenetic constructs to projection-defined LHb and EP neurons. First, consistent with our hypothesis, we found that optogenetic stimulation (10 Hz, 5 ms pulses) of ChR2-expressing LHb→VTA neurons in non-stressed wildtype (C57BL/6) mice was sufficient to increase immobility in the TST; this was indicated by a significant decrease in the time mice spent struggling when compared to mice that expressed eYFP in LHb→VTA neurons. In contrast, anxiety, anhedonia and locomotor activity were not significantly altered in response to optogenetic activation of LHb→VTA neurons in ChR2 mice compared to eYFP mice (Figures S6A and S6B). Second, chemogenetic activation (4 mg/kg CNO) of LHb→VTA neurons in non-stressed wildtype mice confirmed the selective effects on passive immobility in the TST (Figures 6A and 6B). Third, we found that chemogenetic silencing of LHb→VTA neurons selectively reduced immobility in the TST in CMS mice, while other behaviors were not affected (Figures 6C and 6D). Fourth, chemogenetic activation of EP→LHb neurons selectively increased immobility in the TST in non-stressed wildtype mice (Figures 6E and 6F), whereas chemogenetic silencing of EP→LHb neurons in CMS-exposed mice selectively reduced immobility in the TST without affecting other behaviors (Figures 6G and 6H). Fifth, chemogenetic activation of LHb→VTA or EP→LHb neurons also increased immobility in the FST in non-stressed wildtype mice (Figures S6C and S6D).
Figure 6. In vivo chemogenetic modulation of LHb circuitry selectively alters passive coping and effort-related motivation.

(A) Experimental design (left) and injection-site of EIAV-Cre (green) in VTA (middle) and hM3DGq-mCherry (red) expression in LHb→VTA neurons (right) (DAPI: blue; Scale bars: 300 μm (left), 200 μm (right)).
(B) Time spent in open arms in EPM, sucrose consumption in SPT, time spent struggling in TST and total distance travelled in OFT after CNO injections for non-stressed mice expressing eYFP or hM3DGq-mCherry in LHb→VTA neurons (* p < 0.05, data represent means ± SEM).
(C) Experimental design (left) and injection-site of EIAV-Cre (green) in VTA (middle) and hM4DGi-mCherry (red) expression in LHb→VTA neurons (right) (DAPI: blue; Scale bars: left: 300 μm (left), 200 μm (right)).
(D) Time spent in open arms in EPM, sucrose consumption in SPT, time spent struggling in TST and total distance travelled in OFT after CNO injections for CMS mice expressing eYFP or hM4DGi-mCherry in LHb→VTA neurons (* p < 0.05, data represent means ± SEM).
(E) Experimental design (left) and injection-site of EIAV-Cre (green) in LHb (middle) and hM3DGq-mCherry (red) expression in EP→LHb neurons (right) (DAPI: blue; Scale bars: 300 μm (left), 200 μm (right)).
(F) Time spent in open arms in EPM, sucrose consumption in SPT, time spent struggling in TST and total distance travelled in OFT after CNO injections for non-stressed mice expressing eYFP or hM3DGq-mCherry in EP→LHb neurons (* p < 0.05, data represent means ± SEM).
(G) Experimental design (left) and injection-site of EIAV-Cre (green) in LHb (middle) and hM4DGi-mCherry (red) expression in EP→LHb neurons (right) (DAPI: blue; Scale bars: 300 μm (left), 200 μm (right)).
(H) Time spent in open arms in EPM, sucrose consumption in SPT, time spent struggling in TST and total distance travelled in OFT after CNO injections for CMS mice expressing eYFP or hM4DGi-mCherry in EP→LHb neurons (** p < 0.01, data represent means ± SEM).
The reduced ability to experience rewarding feelings and pleasure (anhedonia) and a loss of motivation are both hallmarks of depression (Nestler and Carlezon, 2006), but it is possible that these traits are represented by different neural circuits. To examine whether increased activity in LHb pathways is sufficient to decrease effort-related motivated behaviors, we tested animals in an operant task in which animals have to perform nose pokes under a fixed or progressive ratio schedule in order to receive rewards. Chemogenetic activation of LHb→VTA or EP→LHb neurons did not influence nose poking behavior of animals when the requirements to obtain a reward remained constant (i.e. under a fixed ratio; low level effort required; Figures S6E and S6G). In contrast, in the operant task where increasing the number of nose pokes was required to obtain a reward (i.e. progressive ratio), chemogenetic activation of LHb→VTA, significantly reduced nose poking behavior when compared to eYFP animals (Figure S6F). Surprisingly, activation of EP→LHb neurons did not influence effort-related motivated behaviors, which raises the possibility that other inputs or a combined activation of inputs may be necessary to elicit a behavioral response (Figure S6H). Taken together, although anxiety and anhedonia are major components of chronic stress-induced behavioral syndrome, they may not be regulated through downstream VTA and upstream EP projections of the LHb, which predominantly appear to be relevant for pathological impairments in motivated behaviors.
Molecular and physiological correlates of passive coping
We then designed a multi-level approach that describes a behavioral phenotype from circuit to physiology to gene-expression profile (Figure 7A). Specifically, we collected electrophysiological and RNA expression data from single, projection-defined LHb neurons (Figure S7A) and examined their correlation with PC. Importantly, instead of pooling animals based on the number of criteria they met and separating them into CTRL and CMS groups, we pooled all animals based solely on whether they are positive or negative for prolonged immobility in the TST using our cutoff values determined in Figure 1 (TST+ or TST−, respectively). We argue that this approach will lead to greater precision in detecting meaningful molecular biomarkers that are associated with this specific behavioral phenotype.
Figure 7. Molecular and physiological correlates of passive coping.

(A) Experimental design.
(B) Left: Firing in response to +150 pA depolarizing current injection from LHb→VTA neurons in TST-negative (TST−, top), TST-positive (TST+, middle) mice and LHb→DR neurons (TST−, bottom; Scale bars: 20 mV/0.5 s). Right: Mean number of action potentials in response to +150 pA depolarizing current injection for the three groups (* p < 0.05, ** p < 0.01, data represent means ± SEM).
(C) Current clamp recordings and pie charts showing an increased number of cells with burst firing in TST+ LHb→VTA and TST− LHb→DR compared to TST− LHb→VTA neurons. Firing was initiated with a brief, transient injection of a hyperpolarizing current (Scale bars: 20 mV/150 ms).
(D) ISI histogram with corresponding Kernel density functions for TST+ LHb→VTA, TST− LHb→VTA and TST− LHb→DR neurons.
(E) Cumulative frequency histogram displaying a shift to shorter ISIs in TST+ LHb→VTA cells compared to TST− LHb→VTA neurons. By contrast, TST− LHb→DR neurons display the shortest ISIs.
(F) Mean resting membrane potentials (RMP) for TST+ LHb→VTA, TST− LHb→VTA and TST− LHb→DR neurons (grey: cells that displayed tonic firing, black: bursting cells; * p < 0.05, ** p < 0.01, data represent means ± SEM).
(G) Number of spikes per burst is inversely correlated with RMP. Zero spikes per burst represent data from cells that displayed only tonic, but no burst firing.
(H) Volcano plots displaying differential gene expression between single LHb→VTA and LHb→DR neurons in TST− mice. Gold and brown data points denote genes that are significantly enriched in LHb→VTA versus TST− LHb→DR neurons from TST− mice, respectively. Highlighted are the ion channel-coding and synapse-related genes. Gray data points represent genes that are not significantly enriched in either category (i.e. absolute value of Log2(Fold Change) < 2 and p < 0.01).
(I) Violin plot displaying differential gene expression between single LHb→VTA neurons in TST− versus TST+ mice. Green and blue data points denote genes that are significantly enriched in cells from TST− versus TST+ mice, respectively. Gray data points represent genes that are not significantly enriched in either category (i.e. absolute value of Log2(Fold Change) < 2 and p < 0.01).
(J) Violin plots showing upregulation of Kcnc1 gene expression in single-cells from TST+ compared to TST− mice. Kcnc1 is also significantly higher expressed in TST− LHb→DR versus TST− LHb→VTA neurons, but not different between TST− LHb→DR versus TST+ LHb→VTA neurons.
(K) Regression analysis of differential gene expression between TST− LHb→VTA versus TST+ LHb→VTA neurons and between TST− LHb→DR versus TST− LHb→VTA neurons. For each gene, data points represent Log2 (Fold Change) values in both comparisons; colored data points highlight the same genes as identified in panels (H) and (I).
As expected, while the number of action potentials in response to depolarizing current injections could not be distinguished between TST− LHb→VTA and LHb→DR cells, TST+ LHb→VTA neurons consistently fired more action potentials compared to the other groups (Figures 7B and S7C). In contrast, no significant difference was observed when we compared evoked firing from animals that were positive or negative in the EPM and SPT assays (Figures S7B and S7C). As most projection-defined LHb neurons were silent in our whole-cell recordings, we transiently hyperpolarized the cells, which resulted in action potential firing under resting conditions. We then evaluated the firing modes of cells in the three groups. 62% (n = 26/42 cells) of LHb→VTA neurons in TST− mice were predominantly bursting, whereas this number increased to 77% (n = 41/53 cells) in TST+ LHb→VTA neurons, which was similar high in TST− LHb→DR neurons (85%, n = 11/13 cells; Figure 7C). We further corroborated this observation by quantifying the relative and cumulative frequency of inter spike intervals (Figures 7D and 7E). In addition, we found that TST+ LHb→VTA cells display a more hyperpolarized resting membrane potential (RMP) compared to TST− LHb→VTA cells. By contrast, the RMP in TST+ LHb→VTA and TST− LHb→DR cells was not significantly different (Figure 7F). Consistent with previous studies (Weiss and Veh, 2011; Yang et al., 2018), we found that a more hyperpolarized RMP was associated with increased burst firing (Figures 7F, 7G, S7D and S7E).
To evaluate transcriptomic differences between different LHb subpopulations, we analyzed differential gene expression between LHb→VTA and LHb→DR neurons. Among the most significant differences, we found strong enrichment of synaptic glutamate receptors Grik2 and Grid2, and the neuronal excitability-relevant potassium channel Kcnc2 (Kv3.2) in LHb→VTA (n = 16 cells, 5 mice) compared to LHb→DR neurons (n = 14 cells, 5 mice). As biomarkers, collective expression pattern of these genes sufficiently identifies the two cell populations (Figures 7H and S7F). We also analyzed differential gene expression between TST− (n = 16 cells, 5 mice) and TST+ (n = 37 cells, 10 mice) in LHb→VTA neurons (Figures 7I and S7G). We found significant upregulation of Kcnc1 in TST+ compared to TST− neurons, making its expression similar to those we found in LHb→DR cells (Figure 7J). In addition, we found downregulation of synaptic regulators Lrrtm3 and glutamate-receptor subunit Grin1 (Figures 7I and S7G). Together, these transcriptomic analyses revealed the identity of select genes that collectively can serve as biomarkers to differentiate LHb→VTA and LHb→DR (Grid2, Grik2 and Kcnc2), as well as TST− and TST+ LHb→VTA neurons (Lrrtm3, Grin1 and Kcnc1). The contrasting nature of Kcnc1 in differentiating TST− and TST+ LHb→VTA neurons prompted us to examine the full extent of genes that may follow similar patterns. For this, we performed regression analysis on the fold difference of gene expression in LHb→VTA and LHb→DR, and in TST− and TST+ LHb→VTA comparisons (Figures 7K and S7H). Surprisingly, we found that the overall gene-expression profile of TST+ LHb→VTA neurons changes towards that in TST−LHb→DR neurons. While its importance remains elusive, this finding underscores a pronounced plasticity of LHb→VTA neurons that is linked to a specific behavioral phenotype. Importantly, differential gene expression analysis made after grouping cells as CTRL (n = 26 cells, 9 mice) versus CMS (n = 27 cells, 6 mice) revealed fewer and more modest differences in LHb→VTA neurons (Figure S8), further highlighting the importance of phenotypic (TST− versus TST+) rather than experience-based (CTRL versus CMS) classification.
Together, these outcomes link a specific behavioral phenotype that involves increased PC to specific molecular, cellular and circuit characteristics. Because increased PC behavior may be related to the pathological motivation impairments seen in major depression in humans, our results may serve as a foundation for the development of symptom-specific therapeutic interventions as well as predictive biomarkers, both of which are severely lacking in current medicine.
DISCUSSION
Relevance to depression symptomatology
The clinical manifestations of depression are highly variable, and patients with different symptoms are often grouped into the same diagnostic category even if they have distinct underlying pathophysiologies (Akil et al., 2018; Drysdale et al., 2017; Waters and Mayberg, 2017). Recent years have witnessed a growing awareness of this problem, and several attempts have been made to identify valid and meaningful subtypes based on symptoms, onset, course or severity (ten Have et al., 2016; van Loo et al., 2012; Musil et al., 2018). Importantly, a recent study suggested that patients with depression can be subdivided into four symptomatic subtypes based on distinct patterns of dysfunctional connectivity in limbic and frontostriatal circuits (Drysdale et al., 2017). In basic research, this issue is particularly challenging and often neglected when working with animal models of depression (Berton et al., 2012; Monteggia et al., 2018). Over the last decades, several animal models of depression that are based on chronic stress (CS) exposure have been used (Nestler and Hyman, 2010), but these models often do not discriminate between individual behavioral phenotypes. The fact that rodents, like humans, display a high degree of individual variability in responding to CS questions whether it makes sense to consider CS-exposed animals as a homogeneous population when searching for pathophysiological mechanisms.
To address this issue, we developed an approach that involves behavioral screening and classification of both non-stressed and stressed animals before studying underlying neurobiological mechanisms. While this strategy provides insights into assigning changes in circuit function, cell firing and gene expression to discrete behavioral patterns, one should proceed with caution. First, it remains uncertain whether depression should be defined as a collection of separable behavioral symptoms or as a single disorder that manifests itself in different behavioral pathologies. Therefore, understanding how neural circuits contribute to individual depression symptoms may not solve the neural basis of depression in its entirety. However, understanding the relationship between circuit and symptom is certainly an important and much-needed step towards establishing a platform for symptom-specific treatments of depression. Second, although our behavioral screening is based on paradigms that are widely use to assess depression-related behaviors in rodents (e.g. Tye et al., 2012; Knowland et al., 2017; Yang et al., 2018; Seo et al., 2018), additional measures of depression (e.g. cortisol levels, weight gain or loss, circadian abnormalities) may be necessary to further characterize D0-D3 subgroups and their relevance to depression. It is possible that other CMS-induced pathologies that have not been assessed in this study may contribute to the cellular and circuit adaptations in the LHb. Third, it remains uncertain whether the non-stressed mice that show LHb hyperactivity and increased PC possess a bona fide depression phenotype and future studies will need to determine whether they also exhibit other depression-related phenotypic markers. However, it is conceivable that some control mice develop a depression phenotype, for example, caused by stress during transportation or fighting with littermates within the cage. Fourth, while our approach produced resilient mice that did not show a depression-related phenotype, similar to the social defeat stress model (Krishnan et al., 2007), we noticed that CMS does not induce impairments in social interaction behavior, as in social defeat stress. Conversely, social defeat stress does not affect passive immobility in the TST or FST (Krishnan et al., 2007). Thus, each animal model may recapitulate different aspects of depression. Fifth, while our approach of subdividing depression phenotypes is based on categorial differences with boundaries that are defined by statistical procedures, it neglects the fact that there is a continuum in the severity of the symptoms which lacks sharp boundaries (Figure 1D). Thus, a model with even greater precision in detecting meaningful subtypes would consider both qualitatively distinct phenotypes as well as quantitative differences in severity along an underlying continuum (ten Have et al., 2016). Despite these limitations, given that CS is a well-known trigger for depression in humans as well as for LHb hyperactivity (Figure 2; Li et al., 2011; Lecca et al., 2016; Yang et al., 2018) which can be reversed by antidepressant treatment (Yang et al., 2018; Shabel et al., 2014), our data points to the possibility that LHb→VTA hyperactivity may be linked to a specific symptom of major depression in humans that involves motivational impairments but not anxiety or anhedonia.
Circuit mechanisms of LHb hyperactivity
Although the outputs of the LHb to dopaminergic and serotoninergic neuromodulatory centers are of high biological importance to a broad range of psychiatric diseases (Hikosaka et al., 2008), relatively little is known about the effects of CS on projection-defined LHb subpopulations. While some studies suggested that depression-related LHb neuronal hyperactivity may occur throughout the LHb (Li et al., 2013; Yang et al., 2018), others pointed to specific LHb subtypes projecting to downstream structures such as the VTA (Li et al., 2011). By performing patch clamp recordings from projection-defined LHb neurons in mice, we found that CS-induced LHb hyperactivity was directly associated with the projection to downstream VTA/RMTg structures, but not the DR. Even though evoked firing and excitatory transmission was not altered in LHb→DR neurons, we cannot rule out that other CS-induced adaptations occur in these cells. Because evidence suggests a close link between the LHb and DR circuitry in depression and LHb lesions alleviate depression-related behaviors by increasing serotonin levels (Y ang et al., 2008), it may be important to further examine this LHb subtype using other types of stress (e.g. social defeat stress).
We also examined whether LHb→VTA hyperactivity can be detected in vivo in freely behaving animals (Figure 3). Importantly, our data does not support the idea put forward by Yang et al. (2018) who showed that increased burst but not tonic activity in LHb neurons is instrumental for aversion behavior and multiple depression-like symptoms such as increased PC in the FST and anhedonia in the SPT. This may explain why simply increasing the number of spikes by using low frequency optogenetic stimulation of LHb→VTA neurons was sufficient to induce both place aversion and immobility in the TST. It is possible that discrepancies are due to differences in burst detection and analysis (see methods). Alternatively, it is conceivable that pathway-specific optogenetic stimulation may generate behavioral effects that are not induced by stimulation of cell bodies independent of their projection target (Tye et al., 2011). Nonetheless, our findings are largely consistent with recordings from LHb neurons in acute brain slices demonstrating an increase in tonic firing in depression-related states (Lecca et al., 2016; Li et al., 2011; Tchenio et al., 2017). Accordingly, we propose a model in which CS-induced increased burst and tonic firing of LHb→VTA neurons directly and indirectly affects different subtypes of VTA DA neurons, which may produce symptoms that are related to reduced motivation rather than a reduced ability to experience pleasure.
Aberrant circuit-specific synaptic plasticity in the LHb
Increasing evidence suggests that imbalances in glutamate transmission play a critical role in the development of depression (Pittenger and Duman, 2008; Thompson et al., 2015). In the LHb, changes in presynaptic plasticity have been reported which involve increased release probability at glutamatergic synapses in congenitally helplessness rats (Li et al., 2011). However, the identity of the inputs that exhibit presynaptic adaptations was unknown. By combing ex vivo electrophysiology and optogenetics, we were able to selectively examine specific inputs to LHb→VTA neurons in response to CS. As a result, we could link the effects of stress-induced changes in presynaptic release probability to a specific set of excitatory inputs that originate from the EP and make direct monosynaptic connections onto LHb→VTA neurons. In addition to these presynaptic adaptations at the EP synapse, we also discovered postsynaptic changes that involve an increased surface expression of GluR2-lacking AMPARs. These results are in line with the finding that upregulation of the beta form of calcium/calmodulin-dependent protein kinase type II resulted in increased expression of GluR1 AMPAR subunits at the plasma membrane of LHb neurons (Li et al., 2013). Strikingly, by increasing or decreasing the neural activity of EP→LHb neurons in vivo, we were able to selectively modulate immobility behavior in the TST and FST, but not anhedonia or anxiety. Although our results suggest that CS induces pre- and postsynaptic adaptations of excitatory EP inputs to LHb→VTA neurons, which may contribute to hyperactivity of these cells, our in vivo manipulations targeted all EP neurons that project to the LHb. The EP also contains a separate population of cells that co-releases GABA and glutamate in the LHb and it is possible that changes in inhibitory and excitatory inputs operate in concert to promote LHb hyperactivity and depression-related behaviors (Lecca et al., 2016; Shabel et al., 2014). Because EP neurons that release only glutamate or co-release GABA and glutamate can be differentiated based on the expression of parvalbumin or somatostatin markers, respectively, (Wallace et al., 2017) future research will be needed to investigate the precise contribution of each neuronal subtype to CS-induced LHb dysfunction.
Lastly, we found that most excitatory EP inputs do not target LHb→DR neurons directly. LHb cells that interact within the LHb through local glutamatergic connections have been reported previously (Kim and Chang, 2005; Weiss and Veh, 2011), and the idea that excitation of LHb→DR neurons by EP inputs involves a local feedforward mechanism is supported by several lines of evidence. First, we found an anatomical separation of EP terminals and LHb→DR cell bodies. Second, we detected an increased peak response latency for EP inputs to LHb→DR neurons. Third, 4-AP did not rescue TTX-blocked EPSCs from EP to LHb→DR neurons. Fourth, we found a higher number of local LHb neurons that are connected with LHb→DR compared to LHb→VTA neurons. Thus, an intriguing possibility is that LHb→DR neurons lack CS-induced hyperactivity because of fundamental differences in LHb circuit architecture. An important future research direction is to examine the precise role of local glutamatergic connectivity in the LHb in light of the differential stress susceptibility of LHb subpopulations reported here.
Identification of circuit-specific biomarkers based on behavioral phenotyping
Acute and CS-induced transcriptional dysregulations have been demonstrated in several brain regions (Akil et al., 2018; Bagot et al., 2016), but the effects of CS on the regulation of gene expression in the LHb have not yet been examined. We developed a platform based on single-cell transcriptomics that allowed us to determine potential molecular biomarkers that are associated with circuit-specific neural dysfunction (hyperactivity of LHb→VTA) and specific behavioral phenotypes (Figure 7). We argue that behavioral phenotyping in response to CS is critical because simply comparing stressed versus non-stressed animals may not identify specific biomarkers due to the heterogeneity of behavioral phenotypes. Consistent with this, we find that experience dependent classification (i.e. CTRL versus CMS; Figure S8) revealed fewer and more modest differences in our gene expression analysis of LHb→VTA neurons compared to phenotypic classification. Because animals were analyzed solely based on whether they were positive or negative for TST, it remains uncertain, as mentioned earlier, whether the TST positive animals have a bona fide depression phenotype. Thus, future studies are needed to examine whether candidate genes discovered based on behavioral phenotyping can serve as true biomarkers for depression. For instance, cell type-specific editing of candidate genes using CRISPR-Cas9 may be a particularly fruitful approach towards understanding how molecular dysfunction gives rise to LHb hyperactivity and depression-related behaviors. These approaches will yield valuable insights for translational research, given that the LHb is a phylogenetically conserved structure that is present in virtually all vertebrate species and that depression-related LHb hyperactivity has been observed in both animals and humans.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Stephan Lammel (lammel@berkeley.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C57BL/6 mice (Jackson Laboratory, 25–35 g, 8–12 weeks old, male) were used for all experiments. Mice were maintained on a 12:12 hr light cycle (lights on at 07:00). All procedures complied with the animal care standards set forth by the National Institutes of Health and were approved by University of California Berkeley’s Administrative Panel on Laboratory Animal Care.
METHOD DETAILS
STEREOTAXIC SURGERIES
As previously described (Lammel et al., 2012) all stereotaxic injections were performed under general ketamine–dexmedetomidine anesthesia using a stereotaxic instrument (Kopf Instruments, Model 1900). For red/green fluorescent retrobead labeling, mice were injected unilaterally with fluorescent retrobeads (200 nL; LumaFluor Inc.) in the ventral tegmental area (VTA, bregma: −3.4 mm, lateral: 0.4 mm, ventral: 4.4 mm) or dorsal raphe nucleus (DR, bregma: −4.55 mm, lateral: 0 mm, ventral: 3.35 mm) using a 1 μL Hamilton syringe (Hamilton). The AAVs (adeno associated virus) used in this study were from the Deisseroth laboratory (AAV5-EF1α–DIO-hChR2(H134R)-eYFP; AAV5-EF1α-DIO-eYFP; AAV5-CaMKII-ChR2-eYFP; ~1012 infectious units per mL, prepared by the University of North Carolina Vector Core Facility), from the Uchida laboratory (AAV5-flex-RG; AAV5-flex-TVA-mCherry; ~1012 infectious units per mL, prepared by the University of North Carolina Vector Core Facility), or from Addgene (AAV-DIO-hM3DGq-mCherry and AAV-DIO-hM4DGi-mCherry). ΔG-Rabies-GFP and RV-EnvA-ΔG-GFP were from Salk Institute. CAV2-Cre was from Plateforme de Vectorologie de Montpellier. RG-EIAV-Cre was from the Lim laboratory (UC San Diego). For viral injection, 300–500 nL of concentrated virus solution was injected into the lateral habenula (LHb, bregma: −1.6 mm, lateral: 0.5 mm, ventral: 3.15 mm), entopeduncular nucleus (EP, bregma: −1.2 mm, lateral: 1.85 mm, ventral: 4.4 mm), lateral hypothalamus (LH, bregma: −0.8 mm, lateral: 1.0 mm, ventral: 5.1 mm), VTA or DR (same coordinates as above) using a syringe pump (Harvard Apparatus) at 150 nL/min. The injection needle was withdrawn 5 min after the end of the infusion. For in vivo optogenetic experiments, mice received unilateral (Figure S3) or bilateral (Figure S6) implantation of a chronically implanted optical fiber (NA = 0.22; Doric Lenses) dorsal to the LHb (bregma: 1.65 mm, lateral: ±0.6 mm, ventral: 2.47 mm), VTA (bregma: −3.4 mm, lateral: 0.4 mm, ventral: 3.9 mm) or DR (bregma: −4.55 mm, lateral: 0 mm, ventral: 2.85 mm). One layer of adhesive cement (C&B Metabond; Parkell) was followed by acrylic (Jet Denture Repair; Lang Dental) to secure the fiber to the skull. The incision was closed with a suture and tissue adhesive (Vetbond; 3M). The animal was kept on a heating pad until it recovered from anesthesia. Experiments were performed 6–8 weeks (for AAVs) or 3 days (for retrobeads) after stereotactic injection. Injection sites and optical fiber placements were confirmed in all animals by preparing coronal sections (100 μm) of injection and implantation sites. We routinely carried out complete serial reconstruction of the injection sites and optical fiber placements.
Although fluorescent retrobeads and retrograde viruses were targeted to the VTA, it is important to mention that the caudal VTA contains at least some parts of the rostromedial tegmental nucleus (RMTg; Jhou et al., 2009). The boundary between the VTA and RMTg is difficult to determine, particularly in the caudal VTA, which makes it difficult to determine with certainty whether retrogradely labeled LHb neurons are projecting to the VTA or RMTg. Thus, when referred to in the text, the VTA includes the RMTg, which was originally termed the ‘tail of the VTA’ (Kaufling et al., 2009).
CHRONIC MILD STRESS
The chronic mild stress (CMS) animal model of depression has been used extensively to study the pathophysiology of depression in rodents (Frisbee et al., 2015; Willner et al., 1992). This model is based on the fundamental concept that chronic exposure to stressors is an important cause for the development of depression in humans. Animals were exposed to a randomized series of mild stressors on a daily basis. Specifically, two stressors per day for 8 weeks were delivered before behavioral screening. Mice experienced one stressor during the day and a different stressor during the night. Well-validated and approved standard stressors (Frisbee et al., 2015; Willner et al., 1992; Tye et al., 2012) were randomly chosen from the following list so that they are unpredictable for the subjects: cage tilt on a 45° angle for 12 to 16 h; food deprivation for 12 to 16 h; strobe light illumination for 2 to 6 h; crowded housing for 2 to 6 h; cage shaking (100 RPM) for 2 to 6 h; individual housing for 2 to 6 h; continuous illumination for 24 to 36 h; continuous darkness for 24 to 36 h; water deprivation for 12 to 16 h; damp bedding (200 mL water poured into bedding) for 12 to 16 h; bedding removal for 12 to 16 h. When not undergoing food or water deprivation stressors, water and food were available ad libitum. Non-CMS (control, CTRL) animals were housed for 8 weeks under standard housing conditions with access to food and water ad libitum. Most CTRL and CMS mice underwent three behavioral screening tests on three consecutive days in the following order: 1. Elevated plus maze (EPM), 2. Sucrose Preference Test (SPT), 3. Tail Suspension Test (TST). However, some mice shown in Figure 1A–1C did not experience all three behavioral screening tests, and some animals also underwent a social interaction test (SIT; Figure S1). In addition, for the data shown in Figures 6 and S6, all mice also underwent an open field locomotor test (OFT), which was performed on the fourth day.
ELECTROPHYSIOLOGY
Ex vivo electrophysiology
Mice were deeply anaesthetized with pentobarbital (200 mg/kg IP; Vortech). Coronal slices (250 μm) were prepared after intracardial perfusion with ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM) 50 sucrose, 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 0.1 CaCl2, 4.9 MgCl2, and 2.5 glucose (oxygenated with 95% O2/5% CO2). After 60 min of recovery, slices were transferred to a recording chamber and perfused continuously at 2–4 mL/min with oxygenated ACSF, containing (in mM) 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 11 glucose, 1.3 MgCl2 and 2.5 CaCl2 at ~35 °C. For recording of excitatory postsynaptic currents (EPSCs), picrotoxin (50 μM, Sigma) was added to block inhibitory currents mediated by GABAa receptors. Miniature excitatory postsynaptic currents (mEPSCs) were recorded at −70 mV in the presence of 1 μM tetrodotoxin (TTX, Hello Bio) and 50 μM picrotoxin. Cells were visualized with a 40x water-immersion objective on an upright fluorescent microscope (BX51WI; Olympus) equipped with infrared-differential interference contrast video microscopy and epifluorescence (Olympus). Patch pipettes (3.2–4.4 MΩ) were pulled from borosilicate glass (G150TF-4; Warner Instruments) and filled with internal solution, which consisted of (in mM) 117 CsCH3SO3, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 TEA, 4 MgATP, 0.3 NaGTP, 5 QX314, 0.1 Spermine, pH 7.35 (270–285 mOsm). For recordings of action potential firing from retrogradely labeled LHb neurons, the internal solution contained (in mM): 135 K-gluconate, 5 KCl, 10 HEPES, 0.1 EGTA, 2 MgCl2, 2 MgATP, 0.2 NaGTP, pH 7.35 (290–300 mOsm). Electrophysiological recordings were made using a MultiClamp700B amplifier and acquired using a Digidata 1550 digitizer, sampled at 10 kHz, and filtered at 2 kHz. All data acquisition was performed using pCLAMP software (Molecular Devices).
ChR2-expressing axon terminals were stimulated by flashing 473 nm light through the light path of the microscope using an ultrahigh-powered light-emitting diode (LED) powered by an LED driver (Prizmatix) under computer control. A dual lamp house adaptor (Olympus) was used to switch between fluorescence lamp and LED light source. The light intensity of the LED was not changed during the experiments and the whole slice was illuminated (5 mW/mm2). Light-evoked EPSCs were obtained every 10 s with one pulse of 473 nm light (5 ms) with neurons voltage clamped at −70 mV, −40mV, 0 mV or +40 mV. Light-evoked EPSC amplitudes were calculated by averaging responses from 10 sweeps and then measuring the peak amplitude in a 20 ms window after the light pulse. Cells that did not show a peak in this window that exceeded the baseline noise were classified as non-responders (Figure 4G). Series resistance (5–25 MΩ) and input resistance were monitored online. Neurons were voltage-clamped at −70 mV to record AMPAR EPSCs and at +40 mV to record dual component EPSCs containing AMPAR and NMDAR EPSCs. To calculate the AMPAR/NMDAR ratio at +40 mV, an average of 10 consecutive EPSCs at +40 mV was computed before and after application of the NMDAR antagonist D-AP5 (50 μM for 5 min). NMDAR EPSCs were generated by subtracting the average EPSC in the presence of AP5 from that recorded in AP5’s absence. The peak of the AMPAR EPSC (2 ms window compared to a 2 ms window on the baseline) was divided by the peak of the NMDAR EPSC to yield an AMPAR/NMDAR ratio. The rectification index was calculated by dividing the amplitude of the AMPAR EPSCs measured at −70 mV by the amplitude at +40 mV. The decay time constant (τ) of the AMPAR and NMDAR EPSCs at +40 mV was calculated by fitting a double exponential function to each average EPSC and using the following formula τ = [(A1 × τ1) + (A2 × τ2)] / (A1 + A2); where A1 and A2 are the amplitudes and τ1 and τ2 are the decay time constants of the fast and slow components respectively. Paired-pulse ratios (PPR) were recorded at −70 mV with a 100 ms interval and calculated by dividing the mean amplitude of the second peak by the mean amplitude of the first peak (i.e. averaged responses from 10 sweeps).
For recordings of action potential firing (Figures 2 and 7), cells were held in current clamp and 2 s ramps of depolarizing currents (+50 pA, +100 pA or +150 pA) were injected. Notably, 89.4% of all recorded LHb→VTA (n = 93/104 cells) and 92.6% of all LHb→DR (n = 50/54 cells) neurons were silent and did not fire spontaneous action potentials under resting conditions. These percentages are substantially smaller than reported previously for recordings from non-projection-defined LHb neurons (Yang et al., 2018). No differences in the number of spontaneous action potentials were observed between CTRLD0–1 and CMSD2–3 animals. As most projection-defined LHb neurons were silent in our whole-cell recordings, we transiently hyperpolarized the cells, which resulted in action potential firing under resting conditions. We noticed that some cells switched between burst and tonic firing. For these cells, we defined them as bursting if > 50% of spikes in a 5 s interval were in bursts or tonic if < 50% were in bursts (Figure 7C)
For the pharmacological experiments in Figure 5C, we recorded baseline responses for 4 min and bath applied 30 μM NASPM (1-naphthyl acetyl spermine, Tocris) for 6 min, which selectively blocked GluR2-lacking AMPARs. For the experiments in Figure 5G, we first recorded baseline EPSCs and then added the voltage-gated sodium channel antagonist TTX (1 μM) and subsequently the potassium channel antagonist 4-aminopyridine (4-AP, 50 μM, Sigma) to the bath solution in order to isolate monosynaptic inputs (Petreanu et al., 2009). All electrophysiological data were analyzed offline using Clampfit (Molecular Devices) or Matlab (MathWorks).
In vivo electrophysiology and burst analysis
Animals were implanted with a custom-built driveable optoelectrode (optrode) above the LHb, which consisted of four tetrodes (12 μm polyimide-coated NiCr wire protected by silica tubing) glued to the 200 μm optical fiber using epoxy. The tetrodes protruded from the tip of the optical fiber by ~0.5 mm. Wire tips were cut flat and gold-plated to reduce electrode impedances to ~200 kΩ at 1 kHz. A small screw fixed to the skull served as a ground electrode. Data collection began one week after the optrode implantation. Neural signals were recorded using a Digital Lynx 4SX system (Neuralynx) and HS-18-MM headstage pre-amplifier (Neuralynx). Recorded signals were filtered between 0.6 and 6 kHz and sampled at 32 kHz. Spikes were sorted offline using SpikeSort 3D (Neuralynx) software. At the end of each recording session, the optrode was moved ventrally for ~80 μm. The final recording location was verified using histology after the electrolytic lesions (12 μA, 30 s).
ChR2-tagged neurons were identified by delivering 473 nm (10 mW/mm2, 5 ms pulses) light at 1 Hz frequency for 2 min (120 trials of 1s). A 2 ms bin with the highest number of spikes in the interval [0, +100 ms] around the laser pulse was identified. To test if the identified strongest response to light was higher than chance, we shuffled all the spike times in the same [0, +100 ms] interval 10,000 times and counted the highest number of spikes in a 2 ms bin for each iteration. If the number of spikes in the 2 ms bin from the real data exceed the 99.9th percentile value of the distribution of number of spikes in the most active 2 ms bin for the shuffled data, we classified the cell as light-responsive. Response latency was defined as the average response time in the most active 2 ms bin (Figure S4E; adapted from Zhang et al., 2013).
Bursts in spike trains were detected using a “Rank Surprise” burst detection algorithm (Gourévitch and Eggermont, 2007). First, all interspike intervals (ISIs) in the spontaneous spike train were assigned a rank (Rn), with the shortest ISI getting rank one and the longest ISI getting the last rank. Next, we define u as the sum of ISI ranks in a burst with q spikes, where u = Rn1 + Rn2 + … + Rq-1. We expect a burst with q number of spikes to have ISIs with short values, i.e. small Rn sums of the ISIs in that burst. We then defined Tq as the sum of q discrete uniform random variables between 1 and N, where N is the total number of ISIs in the spike train (N = q – 1). Intuitively, we would expect a burst to have smaller u compared to Tq; however, this would be a surprise if all the spike time values were independently and uniformly distributed. This degree of surprise can be defined as rank surprise (RS) statistic:
where P(Tq ≤ u) is the probability that burst in question have higher or equal ISI rank sum than the sum of random independent and uniformly distributed ISI ranks. To identify “true bursts” we intended to keep this probability low (i.e. make the RS high), so we would only identify bursts with such small ISIs sums that those sums would not be observed by summing random uniformly distributed ISI ranks. Once the RS statistic is defined, possible bursts were identified in the spike trains and compared to the RS statistic. A candidate burst that has higher RS than our defined one was identified as a burst. See (Gourévitch and Eggermont, 2007) for more details on how candidate bursts are selected from the spike train. The largest acceptable interspike interval value in a burst was set to 100 ms and the minimum significance for the surprise statistic was set to 5% (i.e. RS = −log(0.05)). The RS burst detection algorithm is preferable over a simple ISI-threshold burst detection method. In the ISI-threshold method, researchers have to define an ISI threshold for burst initiation (maximum ISI between two consecutive spikes for these spikes to be still considered as the first two spikes in the burst) and burst termination (maximum ISI between two consecutive spikes for the last of these two spikes to be still considered part of a burst). The latter parameter is particularly problematic as it heavily depends on the firing rate of a cell. A common ISI threshold for burst termination is 100 ms (Y ang et al., 2018). This means that the ISI interval between the last two spikes in the burst can be up to 100 ms. If this threshold is applied to cells with mixed tonic and burst firing and these cells fire above 10 Hz (mean firing rate of LHb→VTA cells in CMSD2–3 group: 13.8 ± 1.9 Hz), it results in many false positive additions of spikes to bursts. By chance, a cell that fires at 10 Hz may have many ISI intervals ≤ 100 ms, meaning that a lot of tonic spikes would be added to the end of identified bursts. RS burst detection solves this problem by taking the firing rate of the cell into consideration.
BEHAVIORAL ASSAYS
All behavioral tests were performed during the light phase in a temperature (68–74°F) and humidity (40–60%) controlled room that is illuminated by eight 32 W fluorescent lights each producing 2925 lumens (TST and EPM apparatus were 7 feet away from the light source; SPT and FR/PR were carried out in boxes where light source was a single 0.6 W light bulb). All behaviors were carried out between 9 am and 6 pm. SPT and FR/PR behaviors were carried out in sound proof behavioral boxes. All behaviors were carried out without the experimenter being present in the room. Behavioral equipment was cleaned with 70% EtOH between individual animals.
Tail Suspension Test (TST)
The TST (Can et al., 2012a) involves hanging the mouse by the tail using tape, where one end of the tape is secured to a horizontal bar 40 cm from the ground, thus ensuring that the animal cannot climb on other objects during the assay. Over the course of the experiment (6 min), the mouse switches from vigorous struggling behavior to increasing immobility. The experiment was recorded on video and the time spent struggling was measured by blind scoring of the video after testing was completed. Experimenters were blinded to allocation of groups and outcome assessment.
For optogenetic experiments (Figure S6), the animals’ fiberoptic implant was connected to a 473 nm DPSS laser diode (Laserglow) using a fiberoptic cable and rotary adaptor. Laser output was controlled using a Master-8 pulse stimulator (A.M.P.I.). Power output for the cable was tested using a digital power meter (Thorlabs) and was checked before and after each experimental animal; output during light stimulation was estimated at the targeted tissue 200 μm from the fiber tip (www.optogenetics.org/calc). Animals expressing ChR2 (or eYFP) received 473 nm light stimulation (10 Hz, 5–8 mW/mm2, 5 ms pulses) during the experiment.
For chemogenetic experiments (Figure 6), animals expressing DREADDs (or eYFP) were injected intraperitoneally with 4 mg/kg CNO (in 0.5% DMSO) 30 min before the start of the experiment. Injections of 4 mg/kg CNO in C57BL/6 mice did not significantly alter struggling in the TST, sucrose preference in the SPT, time in open arms in the EPM or locomotor activity in the OFT when compared to C57BL/6 mice injected with 0.9% saline of an equivalent volume (data not shown).
Sucrose Preference Test (SPT)
The SPT assesses an animal’s preference for a sweet solution (1% sucrose dissolved in water) relative to plain water, and failure to do so is indicative of anhedonia, a core symptom of depression. Volume of sucrose or water consumed was measured using a computer-controlled ‘lickometer’. Specifically, a Med Associates operant chamber was used to count every tongue contact made (‘licks’) with bottle spouts from either the 1% sucrose solution or water alone. Animals were water-restricted overnight before the experiment. Bottle side and animal group tested were counterbalanced between each trial. Testing was for 90 min and the percentage of sucrose solution over total consumption was calculated. Mice that made in total < 30 licks at any port were excluded from the experiment. Experimenters were blinded to allocation of groups and outcome assessment.
For optogenetic experiments (Figure S6), the stimulation parameters were identical to the TST (see above). For the chemogenetic experiments (Figure 6), animals expressing DREADDs (or eYFP) were injected intraperitoneally with 4 mg/kg CNO (in 0.5% DMSO) 20 min before the start of the experiment.
Elevated Plus Maze (EPM)
The EPM test is used as a measure of anxiety-related behavior in rodents (Tye et al., 2011). During the test, mice explore a plus shaped maze (length of each arm: 91 cm). Two arms were closed (wall height: 38 cm) and two arms were open. Mice typically spent time exploring all arms, but a substantial amount of time spent in the closed arms compared to open arms is indicative of anxiety-related behavior. Animals were placed in the EPM for 10 min. The movement of the mice was recorded via an automated video tracking system (Biobserve) and the time spent in open and closed arms was calculated. Experimenters were blinded to allocation of groups and outcome assessment.
For optogenetic experiments (Figure S6), the stimulation parameters were identical to the TST (see above). For the chemogenetic experiments (Figure 6), animals expressing DREADDs (or eYFP) were injected intraperitoneally with 4 mg/kg CNO (in 0.5% DMSO) 30 min before the start of the experiment. Note that two mice jumped from the EPM platform during the experiment and these animals were excluded from the data shown in Figures 6B and 6H.
Social Interaction Test (SIT)
The SIT is widely used in rodents to assess sociability and interest in social novelty. The social interaction testing apparatus was a rectangular three-chamber clear Plexiglas box (20 cm L × 40.5 cm W × 22 cm H). During the habituation phase, experimental mice were placed in the box for a 10 min session. Afterwards, an unfamiliar stranger mouse (stranger 1) was placed in a round wire cup at the corner of the box. The wire cup (7 cm L × 10 cm H) had small holes which allowed contact between mice, but prevented fighting. At the opposite corner, a second empty wire cup was located. The animals used as strangers were male C57BL/6 mice which were previously habituated to the placement in the cup. The location of strangers in the apparatus (i.e. left versus right side cups) was counterbalanced between mice and trials. In the first phase of the test (sociability test), the experimental mouse was placed in the box and allowed to explore for 10 min. The movement of the mice was recorded via an automated video tracking system (Biobserve) and the time spent exploring the wire cups was evaluated. Sociability was defined as the preference for the cup containing a novel mouse over an empty cup. At the end of the first 10 min session, each mouse was tested in a second 10 min session to evaluate the interest in social novelty. A second, unfamiliar C57BL/6 mouse (stranger 2) was placed in the empty cup. The amount of time spent exploring the wire cups was evaluated again during the second 10 min session. Social novelty indicates the preference for cup containing the unfamiliar, newly introduced second mouse compared to the familiar mouse. Experimenters were blinded to allocation of groups and outcome assessment.
Forced swim test (FST)
The FST is a behavioral challenge assay that, similar to the TST, assesses passive coping (PC; Can et al., 2012b). Mice were placed in a transparent glass beaker filled with ~25 °C tap water. The water level in the beaker was high enough so mice could not touch the bottom of the beaker while they were trying to stay afloat. The behavioral test lasted for 6 minutes and mice were recorded on video during the entire session. Typically, in the beginning of the test mice struggled vigorously, but eventually they switched to a more passive floating state. Only the last 4 minutes were analyzed due to the fact that most mice are very active in the first 2 minutes of the test. The time spent struggling was measured by blind scoring of the video after testing was completed. Experimenters were blinded to allocation of groups and outcome assessment.
For chemogenetic experiments (Figures S6C and S6D), animals expressing hM3DGq DREADDs (or mCherry) were injected intraperitoneally with 4 mg/kg CNO (in 0.5% DMSO) 30 min before the start of the experiment.
Fixed and progressive ratio operant tasks (FRT and PRT)
FRT and PRT are commonly used to evaluate effort-related motivated behavior in rodents (Hodos, 1961). In both tasks, mice were trained to make a choice between two nose pokes, one of which was rewarded with a sugar pellet. Experiments were performed in Med Associates operant chambers. Mice were food restricted to 90–95% of their body weight prior to training and testing periods. In the FRT, mice had to make one correct nose poke to receive a sucrose pellet. Subsequently, there was a 10 s timeout period where repeated correct nose pokes were not rewarded. Each FRT session lasted for 1 hr. Mice that learned to choose the correct nose poke to receive the reward were selected for the subsequent PRT. In the PRT, mice had to make a progressively higher number of correct nose pokes to receive a sucrose pellet; the number of nose pokes required to receive a sucrose pellet was calculated based on [5e(R*02)] – 5 (Richardson and Roberts, 1996). After each successful sucrose pellet delivery, there was again a 10 s timeout. Each PRT session lasted a maximum of 3 hours, or it was stopped when no correct nose pokes were made for 10 min. The total number of correct nose pokes mice made during a PRT session was used to evaluate motivated behavior. A higher number of correct nose pokes is indicative of an increased effort to obtain a reward (i.e. increased motivation). Animals expressing DREADDs (or eYFP) were injected intraperitoneally with 4 mg/kg CNO (in 0.5% DMSO) or the equivalent volume of 0.9% saline solution (in 0.5% DMSO) 20 min before the start of the experiment. Experimenters were blinded to allocation of groups and outcome assessment.
Real-time Place Preference / Aversion
Mice with fiberoptic implants were connected to a fiberoptic cable and placed in a custom-made three-compartment chamber (Lammel et al., 2012). One randomly assigned side of the chamber was assigned as the initial stimulation side (Phase 1), and after 10 min the stimulation side was switched to the previously non-stimulated side of the chamber (Phase 2). The two stimulation sides were separated by a neutral middle compartment. At the start of each session, the mouse was placed in the neutral compartment, and every time the mouse crossed to the stimulation side, 473 nm laser stimulation (10 Hz, 10 mW/mm2, 5 ms pulses) was delivered until the mouse crossed back into the neutral, non-stimulation side. There was no interruption between Phase 1 and Phase 2. The movement of the mice was recorded via a video tracking system (Biobserve) and the time spent in each area (stimulated, non-stimulated, neutral) was calculated.
Open Field Test (OFT)
The open-field test was conducted to measure the effect of optogenetic or chemogenetic manipulations on general locomotor ability. The mice were placed in a custom-made open field chamber (50 cm L × 50 cm W × 50 cm H) and their movement was recorded and analyzed for 10 min using video-tracking software (Biobserve). For optogenetic experiments (Figure S6), the stimulation parameters were identical to the TST (see above). For the chemogenetic experiments (Figure 6), animals expressing DREADDs (or eYFP) were injected intraperitoneally with 4 mg/kg CNO (in 0.5% DMSO) 30 min before the start of the experiment. Experimenters were blinded to allocation of groups and outcome assessment.
WHOLE BRAIN INPUT MAPPING
We used monosynaptic rabies virus tracing to map and characterize inputs to different LHb subpopulations (Osakada and Callaway, 2013). Specifically, mice were injected with 150 nL AAV-FLEX-TVA (i.e. a cellular receptor for subgroup A avian leukosis viruses) and 150 nL AAV-FLEX-RG (i.e. rabies virus glycoprotein) into the LHb and 300 nL CAV2-Cre into either VTA or DR. 4 weeks later, 300 nL RV-EnvA-ΔG-GFP (i.e. glycoprotein deficient, GFP expressing rabies virus with the envelope protein from avian ASLV type A) was injected into the LHb. 7 days after injection, mice were perfused with 4% PFA in PBS. Brains were stored in 10% sucrose in PBS at +4°C overnight and then processed for analysis. For input mapping, 100 μm sections of the whole brain were prepared and imaged using an Axio Imager 2 microscope (Zeiss). GFP-expressing input cells to LHb→VTA or LHb→DR neurons were counted manually. Animals were randomized and investigators were blinded to group allocation (i.e. projection target).
NEXT-GENERATION SINGLE-CELL RNA SEQUENCING
Sample collection
Procedure was described previously in Földy et al., 2016. To minimize interference with subsequent molecular experiments, only a small amount of intracellular solution (~1 μl; not autoclaved or treated with RNase inhibitor) was used in the glass pipette during electrophysiological recordings. Before and during recording, all surface areas - including manipulators, microscope knobs, computer keyboard, etc. - that the experimenter needed to contact during the experiment were cleaned with RNaseZAP solution (Sigma). After whole-cell patch-clamp recordings, the cell’s cytosol was aspirated via the glass pipette used during the recording. Although the aspirated cytosol may have contained genomic DNA, our choice of cDNA preparation, which involved poly-A based mRNA selection, virtually eliminated the possibility of genomic contamination in the RNAseq data. For sample collection, we quickly removed the pipette holder from the amplifier head stage and used positive pressure to expel samples into microtubes containing cell collection buffer while gently breaking the glass pipette tip. Cell collection microtubes were stored on ice until they were used.
cDNA library preparation
Described previously in Földy et al., 2016. Briefly, single-cell mRNA was processed using Clontech’s SMARTer Ultra Low RNA Input v4 or SMART-Seq HT kit. As a first step, cells were collected via pipette aspiration into 1 μL of 10x collection buffer, and were spun briefly and snap frozen on dry ice. Samples were stored at −80°C until further processing, which was performed according to manufacturer’s protocol. Resulting cDNA was harvested and analyzed on a Fragment Analyzer (Advanced Analytical). Library preparation was performed using Nextera XT DNA Sample Preparation Kit (Illumina) as described in the protocol. Following library preparation, cells were pooled and sequenced using NextSeq 150 high-output kit in an Illumina NextSeq 500 System with 2×75 paired-end reads.
Processing of RNA sequencing data
After sequencing, raw reads were de-multiplexed and pre-processed using Trimmomatic and Flexbar. Then, raw sequencing reads were aligned to the Ensembl GRCm38 reference transcriptome (Version-2015–06-25), using the STAR aligner with the following parameters: trimLeft=10, minTailQuality=15, minAverageQuality=20 and minReadLength=30. ‘Single-end/paired-end’ and ‘sense/antisense/both’ options. Gene counts were calculated using featureCounts. For convenience, Ensembl gene IDs were converted to gene symbols using the mouse GRCm38 gtf file (ftp://ftp.ensembl.org/pub//release86/gtf/mus_musculus/Mus_musculus.GRCm38.86.gtf.gz) as a reference. In the few cases where different Ensembl gene IDs identified the same gene symbol, average gene counts were used.
Gene categories
Altogether, expression of N = 22,800 genes were analyzed (‘All’). This list was generated using Ensemble Biomart, with the following specifications “Database=Ensembl Genes 85, Dataset=Mus Musculus genes (GRCm38.p4), Attributes=Ensemble Gene ID and Associated Gene Name”. ‘CAM’ (N = 396) and ‘ion channel’ (N = 207): these categories included genes as described previously (Földy et al., 2016).
Quality control
All data analysis was performed using Python. First, for each cell, we calculated the number of unique genes and the fraction of aligned reads. Second, we calculated the mean and standard deviation of these two values across all cells. Cells that had both values less than 1 standard deviation below the mean, or had one value above the mean and the other less than 1.5 standard deviations below the mean were considered to pass quality control. For the rest, we calculated their correlation (minimum of Pearson and Spearman) against the average of all cells within their respective cell type that passed quality control. Cells with correlation >0.4 were considered to pass quality control, otherwise they failed quality control.
Normalization of gene expression
After quality control, cells (represented by raw gene count vectors) were pooled together and normalized using scran (Lun et al., 2016), with sizes 20, 40, 60, 80, 100. Cells that had negative or zero size were removed. For further analysis, gene counts were converted into log-space by the gi’ = ln(1 + gi) transformation, where gi was the normalized gene count of the i-th gene.
HISTOLOGY AND MICROSCOPY
Immunofluorescence and confocal microscopy were performed as described previously in Lammel et al., 2012. Briefly, after intracardial perfusion with 4% paraformaldehyde in PBS, pH 7.4, the brains were post-fixed overnight and coronal brain sections (100 or 50 μm) were prepared. Sections were stained overnight in a primary antibody solution (mouse anti-Cre (1:200, Sigma). Twenty-four hours later, sections were stained for 4 hours in secondary antibody solution (goat anti-mouse Alexa Fluor 488, Thermo Fisher). Image acquisition was performed with Zeiss LSM710 laser scanning confocal microscope using 20x or 40x objectives and on a Zeiss Axio Imager 2 upright widefield fluorescence/differential interference contrast microscope with charge-coupled device camera using 5x or 10x objectives. Confocal images were analyzed using ImageJ. Sections were labeled relative to bregma using landmarks and neuroanatomical nomenclature as described in “The Mouse Brain in Stereotaxic Coordinates” (Franklin and Paxinos, 2013).
QUANTIFICATION AND STATISTICAL ANALYSIS
In order to determine whether individual animals were positive or negative for a specific behavioral phenotype as measured in the EPM, SPT and TST, we used receiver operating characteristic (ROC) curves (Zou et al., 2007). ROC curve analysis has been used extensively in clinical epidemiology for the assessment of diagnostic ability of biomarkers and imaging tests in classification of diseased from healthy subjects (Metz, 1978; Swets, 1988; Zweig and Campbell, 1993). It is considered the most objective method for evaluating and comparing classification performances and recommended for the evaluation of binary classifiers (Berrar and Flach, 2012; Søreide, 2009). ROC curves are uninfluenced by decision biases and prior probabilities and they depict a classifier’s performance over the range of thresholds for sensitivity and specificity. Youden J Index was calculated from ROC curves in order to identify the optimal cutoff value that gives the lowest false positive rate (FPR) and the highest true positive rate (TPR). Youden J index maximizes the difference between TPR (sensitivity) and FPR (1 – specificity):
Thus, by maximizing (Sensitivity + Specificity) across various cutoff points, the optimal cutoff point was calculated.
In order to determine statistical differences for anatomical, behavioral and electrophysiological data, we performed Student’s t tests (paired and unpaired), and one- and two-way ANOVAs using Prism 7 (Graphpad). Bonferroni, Tukey’s or Sidak’s post hoc analysis was applied, when applicable, to correct for multiple comparisons. Statistical significance was * p < 0.05, ** p < 0.01, *** p < 0.001. All data are presented as means ± SEM. Investigators were blinded to allocation of groups and outcome assessment for all experiments except for data shown in Figures 4E–4H, 5D–5G and S3A–S3O.
DATA AND CODE AVAILABILITY
The RNAseq datasets generated during this study are available at [NAME OF REPOSITORY] [ACCESSION CODE/WEB LINK].
Supplementary Material
Highlights.
Unbiased classification of chronic mild stress (CMS)-induced behavioral phenotypes
CMS promotes activity and synaptic changes in a lateral habenula (LHb) subcircuit
In vivo manipulations of specific LHb subcircuits affect motivated behaviors
Single-cell RNA-seq reveals genes associated with a distinct behavioral phenotype
ACKNOWLEDGEMENTS
We thank the UNC vector core for AAV viruses and the CNR Biological Imaging Facility at UC Berkeley. This work was supported by grants from the NIMH (R01MH112721), Hellman Foundation, Whitehall Foundation (2015–12-69), Shurl and Kay Curci Foundation, Rita Allen Foundation, Wayne and Gladys Valley Foundation and by a UC Berkeley Regents’ Junior Faculty Fellowship (all to S.L.), and by Schweizerischer Nationalfonds zur Forderung der Wissenschaftlichen Forschung (CRETP3_166815; to C.F.). I.C. was supported by the Boehringer Ingelheim Foundation.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agudelo LZ, Femenia T, Orhan F, Porsmyr-Palmertz M, Goiny M, Martinez-Redondo V, Correia JC, Izadi M, Bhat M, Schuppe-Koistinen I, et al. (2014). Skeletal muscle PGC-1α1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 159, 33–45. [DOI] [PubMed] [Google Scholar]
- Akil H, Gordon J, Hen R, Javitch J, Mayberg H, McEwen B, Meaney MJ, and Nestler EJ (2018). Treatment resistant depression: A multi-scale, systems biology approach. Neurosci. Biobehav. Rev 84, 272–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andalman AS, Burns VM, Lovett-Barron M, Broxton M, Poole B, Yang SJ, Grosenick L, Lerner TN, Chen R, Benster T, et al. (2019). Neuronal Dynamics Regulating Brain and Behavioral State Transitions. Cell 177, 970–985.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagot RC, Cates HM, Purushothaman I, Lorsch ZS, Walker DM, Wang J, Huang X, Schluter OM, Maze I, Pena CJ, et al. (2016). Circuit-wide Transcriptional Profiling Reveals Brain Region-Specific Gene Networks Regulating Depression Susceptibility. Neuron 90, 969–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard R, and Veh RW (2012). Individual neurons in the rat lateral habenular complex project mostly to the dopaminergic ventral tegmental area or to the serotonergic raphe nuclei. J. Comp. Neurol 520, 2545–2558. [DOI] [PubMed] [Google Scholar]
- Berrar D, and Flach P (2012). Caveats and pitfalls of ROC analysis in clinical microarray research (and how to avoid them). Brief. Bioinform 13, 83–97. [DOI] [PubMed] [Google Scholar]
- Berton O, Hahn CG, and Thase ME (2012). Are we getting closer to valid translational models for major depression? Science 338, 75–79. [DOI] [PubMed] [Google Scholar]
- Can A, Dao DT, Terrillion CE, Piantadosi SC, Bhat S, and Gould TD (2012a). The tail suspension test. J. Vis. Exp. JoVE e3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can A, Dao DT, Arad M, Terrillion CE, Piantadosi SC, and Gould TD (2012b). The mouse forced swim test. J. Vis. Exp. JoVE e3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher N, Adamson G, Bunting B, and McCann S (2009). Subtypes of depression in a nationally representative sample. J. Affect. Disord 113, 88–99. [DOI] [PubMed] [Google Scholar]
- Castagne V, Moser P, Roux S, and Porsolt RD (2011). Rodent models of depression: forced swim and tail suspension behavioral despair tests in rats and mice. Curr. Protoc. Neurosci Chapter 8, Unit 8.10A. [DOI] [PubMed] [Google Scholar]
- Cho J-H, Deisseroth K, and Bolshakov VY (2013). Synaptic Encoding of Fear Extinction in mPFC-amygdala Circuits. Neuron 80, 1491–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Yang Y, Ni Z, Dong Y, Cai G, Foncelle A, Ma S, Sang K, Tang S, Li Y, et al. (2018). Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 554, 323–327. [DOI] [PubMed] [Google Scholar]
- Cui Y, Hu S, and Hu H (2019). Lateral Habenular Burst Firing as a Target of the Rapid Antidepressant Effects of Ketamine. Trends Neurosci. 42, 179–191. [DOI] [PubMed] [Google Scholar]
- Czéh B, Fuchs E, Wiborg O, and Simon M (2016). Animal models of major depression and their clinical implications. Prog. Neuropsychopharmacol. Biol. Psychiatry 64, 293–310. [DOI] [PubMed] [Google Scholar]
- Drysdale AT, Grosenick L, Downar J, Dunlop K, Mansouri F, Meng Y, Fetcho RN, Zebley B, Oathes DJ, Etkin A, et al. (2017). Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat. Med 23, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy C, Darmanis S, Aoto J, Malenka RC, Quake SR, and Sudhof TC (2016). Single-cell RNAseq reveals cell adhesion molecule profiles in electrophysiologically defined neurons. Proc. Natl. Acad. Sci. U. S. A. 113, E5222–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, and Paxinos G (2013). Paxinos and Franklin’s The mouse brain in stereotaxic coordinates (Amsterdam: Academic Press, an imprint of Elsevier; ). [Google Scholar]
- Frisbee JC, Brooks SD, Stanley SC, and d’Audiffret AC (2015). An Unpredictable Chronic Mild Stress Protocol for Instigating Depressive Symptoms, Behavioral Changes and Negative Health Outcomes in Rodents. J. Vis. Exp. JoVE [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourévitch B, and Eggermont JJ (2007). A nonparametric approach for detection of bursts in spike trains. J. Neurosci. Methods 160, 349–358. [DOI] [PubMed] [Google Scholar]
- ten Have M, Lamers F, Wardenaar K, Beekman A, de Jonge P, van Dorsselaer S, Tuithof M, Kleinjan M, and de Graaf R (2016). The identification of symptom-based subtypes of depression: A nationally representative cohort study. J. Affect. Disord 190, 395–406. [DOI] [PubMed] [Google Scholar]
- Herkenham M, and Nauta WJ (1977). Afferent connections of the habenular nuclei in the rat. A horseradish peroxidase study, with a note on the fiber-of-passage problem. J. Comp. Neurol 173, 123–146. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Sesack SR, Lecourtier L, and Shepard PD (2008). Habenula: Crossroad between the Basal Ganglia and the Limbic System. J. Neurosci 28, 11825–11829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Hellemans KGC, Verma P, Gorzalka BB, and Weinberg J (2012). Neurobiology of chronic mild stress: parallels to major depression. Neurosci. Biobehav. Rev 36, 2085–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodos W (1961). Progressive ratio as a measure of reward strength. Science 134, 943–944. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, and Heinemann S (1991). Ca2+ permeability of KA-AMPA--gated glutamate receptor channels depends on subunit composition. Science 252, 851–853. [DOI] [PubMed] [Google Scholar]
- Jhou TC, Fields HL, Baxter MG, Saper CB, and Holland PC (2009). The Rostromedial Tegmental Nucleus (RMTg), a GABAergic Afferent to Midbrain Dopamine Neurons, Encodes Aversive Stimuli and Inhibits Motor Responses. Neuron 61, 786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufling J, Veinante P, Pawlowski SA, Freund-Mercier MJ, and Barrot M (2009). Afferents to the GABAergic tail of the ventral tegmental area in the rat. J Comp Neurol 513, 597–621. [DOI] [PubMed] [Google Scholar]
- Kiening K, and Sartorius A (2013). A new translational target for deep brain stimulation to treat depression: A new target for DBS to treat depression. EMBO Mol. Med 5, 1151–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U, and Chang S-Y (2005). Dendritic morphology, local circuitry, and intrinsic electrophysiology of neurons in the rat medial and lateral habenular nuclei of the epithalamus. J. Comp. Neurol 483, 236–250. [DOI] [PubMed] [Google Scholar]
- Knowland D, Lilascharoen V, Pacia CP, Shin S, Wang EH-J, and Lim BK (2017). Distinct Ventral Pallidal Neural Populations Mediate Separate Symptoms of Depression. Cell 170, 284–297.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolhaas JM, Korte SM, De Boer SF, Van Der Vegt BJ, Van Reenen CG, Hopster H, De Jong IC, Ruis MA, and Blokhuis HJ (1999). Coping styles in animals: current status in behavior and stress-physiology. Neurosci. Biobehav. Rev 23, 925–935. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Han M-H, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, et al. (2007). Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131, 391–404. [DOI] [PubMed] [Google Scholar]
- Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, and Malenka RC (2012). Input-specific control of reward and aversion in the ventral tegmental area. Nature 491, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Tye KM, and Warden MR (2014). Progress in understanding mood disorders: optogenetic dissection of neural circuits. Genes Brain Behav. 13, 38–51. [DOI] [PubMed] [Google Scholar]
- Lecca S, Meye FJ, and Mameli M (2014). The lateral habenula in addiction and depression: an anatomical, synaptic and behavioral overview. Eur. J. Neurosci 39, 1170–1178. [DOI] [PubMed] [Google Scholar]
- Lecca S, Pelosi A, Tchenio A, Moutkine I, Lujan R, Hervé D, and Mameli M (2016). Rescue of GABAB and GIRK function in the lateral habenula by protein phosphatase 2A inhibition ameliorates depression-like phenotypes in mice. Nat. Med 22, 254–261. [DOI] [PubMed] [Google Scholar]
- Li B, Piriz J, Mirrione M, Chung C, Proulx CD, Schulz D, Henn F, and Malinow R (2011). Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature 470, 535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Zhou T, Liao L, Yang Z, Wong C, Henn F, Malinow R, Yates JR, and Hu H (2013). PCaMKII in lateral habenula mediates core symptoms of depression. Science 341, 1016–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, and Zukin RS (2007). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134. [DOI] [PubMed] [Google Scholar]
- van Loo HM, de Jonge P, Romeijn J-W, Kessler RC, and Schoevers RA (2012). Data-driven subtypes of major depressive disorder: a systematic review. BMC Med. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, McCarthy DJ, and Marioni JC (2016). A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research 5, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazure CM, and Maciejewski PK (2003). A model of risk for major depression: effects of life stress and cognitive style vary by age. Depress. Anxiety 17, 26–33. [DOI] [PubMed] [Google Scholar]
- Metz CE (1978). Basic principles of ROC analysis. Semin. Nucl. Med 8, 283–298. [DOI] [PubMed] [Google Scholar]
- Mirrione MM, Schulz D, Lapidus KAB, Zhang S, Goodman W, and Henn FA (2014). Increased metabolic activity in the septum and habenula during stress is linked to subsequent expression of learned helplessness behavior. Front. Hum. Neurosci 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Heimer H, and Nestler EJ (2018). Meeting Report: Can We Make Animal Models of Human Mental Illness? Biol. Psychiatry 84, 542–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreines JL, Owrutsky ZL, and Grace AA (2017). Involvement of Infralimbic Prefrontal Cortex but not Lateral Habenula in Dopamine Attenuation After Chronic Mild Stress. Neuropsychopharmacology 42, 904–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil R, Seemuller F, Meyer S, Spellmann I, Adli M, Bauer M, Kronmuller K-T, Brieger P, Laux G, Bender W, et al. (2018). Subtypes of depression and their overlap in a naturalistic inpatient sample of major depressive disorder. Int. J. Methods Psychiatr. Res 27, e1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, and Carlezon WA (2006). The Mesolimbic Dopamine Reward Circuit in Depression. Biol. Psychiatry 59, 1151–1159. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, and Hyman SE (2010). Animal models of neuropsychiatric disorders. Nat. Neurosci 13, 1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakada F, and Callaway EM (2013). Design and generation of recombinant rabies virus vectors. Nat. Protoc 8, 1583–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreanu L, Mao T, Sternson SM, and Svoboda K (2009). The subcellular organization of neocortical excitatory connections. Nature 457, 1142–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger C, and Duman RS (2008). Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol 33, 88–109. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Bertin A, and Jalfre M (1978). “Behavioural despair” in rats and mice: strain differences and the effects of imipramine. Eur. J. Pharmacol 51, 291–294. [DOI] [PubMed] [Google Scholar]
- Proulx CD, Hikosaka O, and Malinow R (2014). Reward processing by the lateral habenula in normal and depressive behaviors. Nat. Neurosci 17, 1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proulx CD, Aronson S, Milivojevic D, Molina C, Loi A, Monk B, Shabel SJ, and Malinow R (2018). A neural pathway controlling motivation to exert effort. Proc. Natl. Acad. Sci 115, 5792–5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez S, Liu X, MacDonald CJ, Moffa A, Zhou J, Redondo RL, and Tonegawa S (2015). Activating positive memory engrams suppresses depression-like behaviour. Nature 522, 335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson NR, and Roberts DC (1996). Progressive ratio schedules in drug self-administration studies in rats: a method to evaluate reinforcing efficacy. J. Neurosci. Methods 66, 1–11. [DOI] [PubMed] [Google Scholar]
- Sartorius A, and Henn FA (2007). Deep brain stimulation of the lateral habenula in treatment resistant major depression. Med. Hypotheses 69, 1305–1308. [DOI] [PubMed] [Google Scholar]
- Sartorius A, Kiening KL, Kirsch P, von Gall CC, Haberkorn U, Unterberg AW, Henn FA, and Meyer-Lindenberg A (2010). Remission of major depression under deep brain stimulation of the lateral habenula in a therapy-refractory patient. Biol. Psychiatry 67, e9–e11. [DOI] [PubMed] [Google Scholar]
- Schweizer MC, Henniger MSH, and Sillaber I (2009). Chronic Mild Stress (CMS) in Mice: Of Anhedonia, ‘Anomalous Anxiolysis’ and Activity. PLoS ONE 4, e4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J-S, Zhong P, Liu A, Yan Z, and Greengard P (2018). Elevation of p11 in lateral habenula mediates depression-like behavior. Mol. Psychiatry 23, 1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabel SJ, Proulx CD, Piriz J, and Malinow R (2014). GABA/glutamate co-release controls habenula output and is modified by antidepressant treatment. Science 345, 1494–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumake J, Edwards E, and Gonzalez-Lima F (2003). Opposite metabolic changes in the habenula and ventral tegmental area of a genetic model of helpless behavior. Brain Res. 963, 274–281. [DOI] [PubMed] [Google Scholar]
- Søreide K (2009). Receiver-operating characteristic curve analysis in diagnostic, prognostic and predictive biomarker research. J. Clin. Pathol 62, 1–5. [DOI] [PubMed] [Google Scholar]
- Stamatakis AM, and Stuber GD (2012). Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidance. Nat Neurosci 15, 1105–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swets JA (1988). Measuring the Accuracy of Diagnostic Systems. Sci. New Ser 240, 1285–1293. [DOI] [PubMed] [Google Scholar]
- Tchenio A, Lecca S, Valentinova K, and Mameli M (2017). Limiting habenular hyperactivity ameliorates maternal separation-driven depressive-like symptoms. Nat. Commun 8, 1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Kallarackal AJ, Kvarta MD, Van Dyke AM, LeGates TA, and Cai X (2015). An excitatory synapse hypothesis of depression. Trends Neurosci. 38, 279–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, and Deisseroth K (2011). Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature 471, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai H-C, Finkelstein J, Kim S-Y, Adhikari A, Thompson KR, Andalman AS, et al. (2012). Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 493, 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace ML, Saunders A, Huang KW, Philson AC, Goldman M, Macosko EZ, McCarroll SA, and Sabatini BL (2017). Genetically Distinct Parallel Pathways in the Entopeduncular Nucleus for Limbic and Sensorimotor Output of the Basal Ganglia. Neuron 94, 138–152.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden MR, Selimbeyoglu A, Mirzabekov JJ, Lo M, Thompson KR, Kim S-Y, Adhikari A, Tye KM, Frank LM, and Deisseroth K (2012). A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 492, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AC, and Mayberg HS (2017). Brain-Based Biomarkers for the Treatment of Depression: Evolution of an Idea. J. Int. Neuropsychol. Soc 23, 870–880. [DOI] [PubMed] [Google Scholar]
- Weiss T, and Veh RW (2011). Morphological and electrophysiological characteristics of neurons within identified subnuclei of the lateral habenula in rat brain slices. Neuroscience 172, 74–93. [DOI] [PubMed] [Google Scholar]
- Willner P (2005). Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology 52, 90–110. [DOI] [PubMed] [Google Scholar]
- Willner P (2017). The chronic mild stress (CMS) model of depression: History, evaluation and usage. Neurobiol. Stress 6, 78–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner P, Muscat R, and Papp M (1992). Chronic mild stress-induced anhedonia: A realistic animal model of depression. Neurosci. Biobehav. Rev 16, 525–534. [DOI] [PubMed] [Google Scholar]
- Willner P, Scheel-Kruger J, and Belzung C (2013). The neurobiology of depression and antidepressant action. Neurosci. Biobehav. Rev 37, 2331–2371. [DOI] [PubMed] [Google Scholar]
- Yang L-M, Hu B, Xia Y-H, Zhang B-L, and Zhao H (2008). Lateral habenula lesions improve the behavioral response in depressed rats via increasing the serotonin level in dorsal raphe nucleus. Behav. Brain Res 188, 84–90. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang H, Hu J, and Hu H (2017). Lateral habenula in the pathophysiology of depression. Curr. Opin. Neurobiol 48, 90–96. [DOI] [PubMed] [Google Scholar]
- Yang Y, Cui Y, Sang K, Dong Y, Ni Z, Ma S, and Hu H (2018). Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 554, 317–322. [DOI] [PubMed] [Google Scholar]
- Yilmaz A, Schulz D, Aksoy A, and Canbeyli R (2002). Prolonged effect of an anesthetic dose of ketamine on behavioral despair. Pharmacol. Biochem. Behav 71, 341–344. [DOI] [PubMed] [Google Scholar]
- Youden WJ (1950). Index for rating diagnostic tests. Cancer 3, 32–35. [DOI] [PubMed] [Google Scholar]
- Zhang S-J, Ye J, Miao C, Tsao A, Cerniauskas I, Ledergerber D, Moser M-B, and Moser EI (2013). Optogenetic dissection of entorhinal-hippocampal functional connectivity. Science 340, 1232627. [DOI] [PubMed] [Google Scholar]
- Zou KH, O’Malley AJ, and Mauri L (2007). Receiver-Operating Characteristic Analysis for Evaluating Diagnostic Tests and Predictive Models. Circulation 115, 654–657. [DOI] [PubMed] [Google Scholar]
- Zweig MH, and Campbell G (1993). Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin. Chem 39, 561–577. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNAseq datasets generated during this study are available at [NAME OF REPOSITORY] [ACCESSION CODE/WEB LINK].