

Abstract
As a potential antitumor drug and chemotherapeutic sensitizer, disulfiram combined with Copper (DSF/Cu2+) does not exert considerable antitumor effects on an immunocompetent hepatocellular carcinoma (HCC) model. In this article, we will explore the mechanism underlying the resistance to DSF in HCC. We analyzed the antitumor effect of DSF/Cu2+ in vivo studies. Tumor and immune cells collected from mice were analyzed by flow cytometry. Then, we analyzed the transcriptional changes in liver cancer cells after DSF/Cu2+ treatment by transcriptional expression profiling. The expression of PD-L1 was detected by real-time PCR, Western blotting and flow cytometry. The expression of PARP1 and GSK3β was knocked down by small interfering RNAs (siRNAs). A subcutaneous Hepa1-6 tumor model was used for single-drug or combined-drug studies. Tissue chips (268 samples of liver cancer tissue) were used to analyze the relationship among PARP1, p-GSK3β and PD-L1. We found that DSF/Cu2+ failed to inhibit HCC tumor growth in C57BL/6 mice. DSF/Cu2+ upregulated PD-L1 expression by inhibiting PARP1 activity and enhancing GSK3β phosphorylation at Ser9 and ultimately inhibited T cell infiltration. The combination of DSF/Cu2+ and an anti-PD-1 antibody produced an additive effect that slowed HCC growth in mice. In addition, we observed negative associations between PARP1 and p-GSK3β (Ser9) or PD-L1 expression in tumor tissue samples from HCC patients. Through in vitro and in vivo studies, we found that DSF/Cu2+ could restrain GSK3β activity by inhibiting PARP1, leading to the upregulation of PD-L1 expression. Combination therapy with DSF/Cu2+ and an anti-PD-1 antibody showed much better antitumor efficacy than monotherapy.
Keywords: PD-L1, hepatocellular carcinoma, PARP1, GSK3, disulfiram
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer in the world and the second leading cause of cancer-related death [1]. Current treatments for early-stage HCC include surgical resection, liver transplantation, and local radiofrequency (RF) ablation [2], but their effects still need to be improved. Molecular targeted therapies such as the small-molecule multikinase inhibitors sorafenib (first-line use) [3], regorafenib (second-line use) [4] and lenvatinib (first-line use) [5] have been approved by the US Food and Drug Administration (FDA) for the treatment of advanced HCC. However, these drugs extend median overall survival for less than 4 months in patients with advanced HCC, and the overall response rate is extremely low [6]. Immune checkpoint blockade (ICB) therapy has shown considerable clinical benefit in patients with various cancers by enhancing the T cell response and maintaining prolonged antitumor activity [7-10]. Anti-PD1 therapy, which is approved for treatment of HCC, achieves about 20% response rate [11]. However, pembrolizumab and nivolumab failed to meet the primary endpoints in the KEYNOTE-240 and CheckMate-459 HCC clinical trials. Therefore, improving the therapeutic effect of ICB treatment and developing more effective combination therapies for HCC are urgently needed.
The expression levels of PD-L1 within the tumor microenvironment can predict treatment response to ICB therapy that blocking the PD-L1/PD-1 axis in different tumor types [12,13], which are reported being regulated in a highly complex manner and being influenced by transcriptional and posttranslational regulation [14,15]. A number of transcription factors, including MYC, STAT3, NF-kB and IRF1, have been shown to be involved, pointing to their important roles in the evasion of the immune system by cancer cells. Multiple studies have indicated that active STAT3 can act as a activating factor, which directly acts on the promoter of PD-L1 to enhance PD-L1 expression in human lymphoma and head and neck squamous cell carcinoma cells [16,17]. Similarly, NF-kB, a family of transcription factors, is demonstrated being activated in cancers by oncogenic mutations or inflammatory cytokines produced in the tumor microenvironment. Inhibition of the NF-kB pathway can lead to a decrease of PD-L1 expression in immune cells such as natural killer (NK)/T cell lymphomas, primary monocytes and in tumor cells such as melanoma cells [18-20]. Moreover, increasing evidences have shown that PD-L1 also undergoes different posttranslational protein modifications that affect its stability, such as ubiquitination [21], deubiquitination [22], phosphorylation [23], glycosylation [24] and Palmitoylation [25,26]. For example, glycogen synthase kinase-3β (GSK-3β), a serine/threonine protein kinase, has been shown to induce phosphorylation-dependent proteasomal degradation of PD-L1 to regulate anticancer immunity [27,28]. GSK-3β directly binds with the C-terminal domain of PD-L1 and then enhances the phosphorylation of PD-L1 at T180 and S184, ultimately promoting PD-L1 poly-ubiquitination and degradation [23]. Besides, COP9 signalosome 5 (CSN5) was shown as a deubiquitinating enzyme in PD-L1 deubiquitination, which regulates the stabilization of PD-L1 to affect T cell suppression [22]. Indeed, researches in investigating and elucidating the molecular mechanisms that regulate the PD-1/PD-L1 pathway will be important steps in developing novel therapeutic strategies to overcome anti-PD1 or anti-PD-L1 resistance and improve the therapeutic efficacy for cancer therapy. However, recent studies have reported that the upregulation of PD-L1 expression in tumor cells mediates immune tolerance and reduces the killing ability of tumor-infiltrating T cells, which may be responsible for drug resistance to molecularly targeted drugs [21,27,28]. In our previous study, we found that a MET inhibitor could reduce the phosphorylation of GSK3β at Y56, inactivating the kinase and leading to enhanced expression of PD-L1, which induced immune tolerance and led to treatment failure in HCC. The combination of the MET inhibitor with anti-PD-1 and anti-PD-L1 antibodies produced an additive effect that inhibited the growth of HCC in mice [28]. Because the effects of current chemotherapeutic drugs on liver cancer are still poor and developing new drugs takes a long time and requires a substantial expenditure, it is very important to repurpose well-known and well-characterized noncancer drugs for new uses in oncology [29].
Disulfiram (DSF), which was approved for use as an alcohol aversion therapy over 6 decades ago, showed anticancer activity in a broad spectrum of malignancies as early as 40 years ago [30]. DSF/Cu2+ treatment leads to the downregulation of PTEN protein expression and activation of AKT along with the induction of cell death, facilitating phosphoinositide 3-Kinase inhibition in human breast cancer cells in vitro and in vivo [31]. DSF/Cu2+ can also bind to NPL4 and induce its aggregation, consequently disrupting the vital p97-NPL4-UFD1 pathway and resulting in a complex cellular phenotype that leads to cells death [32]. Furthermore, DSF/Cu2+ could overcome cytarabine (Ara-C) and bortezomib (BTZ) resistance in cell lines from Down syndrome-associate acute megakaryoblastic leukemia (DS-AMKL) patients and induce apoptosis by inhibiting the proteasome system and Poly (ADP-ribose) polymerase 1 (PARP1) activity in tumor cells [33,34]. However, the effect of DSF/Cu2+, a promising anticancer drug, on antitumor immunity has barely been studied. Therefore, understanding the underlying effect of DSF/Cu2+ on the immune system may identify a new effective treatment for HCC.
Poly (ADP-ribose) polymerases (PARPs) compose a large family of enzymes with diverse functions, and some of these enzymes are important for the repair of single-strand breaks (SSBs) in DNA via the base-excision repair (BER) pathway [35]. High PARP expression can lead to chemotherapeutic drug resistance [36], and the downregulation of PARP1 expression has been proved to enhance the expression of PD-L1, leading to tumor-associated immunosuppression [27].
In the present study, we found that DSF/Cu2+ treatment could inhibit proliferation in immunodeficient mice but failed in immunocompetent mice, and we unexpectedly discovered that DSF/Cu2+ could upregulate PD-L1 expression by inhibiting the activity of PARP1 and inactivating GSK3β, which allowed HCC to escape T cell-mediated killing. These findings may explain the ineffectiveness of DSF/Cu2+ in HCC, suggesting that adding anti-PD-1/PD-L1 therapy has the potential to significantly improve the therapeutic efficacy of DSF/Cu2+ against this disease.
Methods and materials
Antibody and reagents
The antibodies listed below were used in Western blotting, immunohistochemical and flow cytometry analyses: anti-PARP1 (#9532; Cell Signaling Technology, Danvers, MA, USA; ab227244, Abcam), anti-PAR (#83732; Cell Signaling Technology, Danvers, MA, USA), anti-phospho-GSK3β (Ser9, AF2016; Affinity Biosciences, Cincinnati, OH, USA), anti-GSK3β (AF5016; Affinity Biosciences, Cincinnati, OH, USA), anti-K48 linkage-specific polyubiquitin (#8081; Cell Signaling Technology, Danvers, MA, USA); anti-STAT3 (#9139; Cell Signaling Technology, Danvers, MA, USA), anti-p-STAT3 (Tyr705) (#9145; Cell Signaling Technology, Danvers, MA, USA); anti-PD-L1 (#13684T, Cell Signaling Technology, Danvers, MA, USA; 329702, BioLegend, San Diego, CA, USA; ab205921, Abcam, Cambridge, UK; 564715, BD Biosciences), anti-granzyme B (ab4059; Abcam), anti-CD8 (ab22378; Abcam; 560776; BD Biosciences), anti-PD-1 (551892; BD Biosciences), anti-CD11b (557395; BD Biosciences), anti-NK1.1 (557391; BD Biosciences), anti-CD19 (553785; BD Biosciences), and anti-CD4 (550954; BD Biosciences). DSF (#S1680) and olaparib (#S1060) were purchased from Selleck Chemicals (Houston, TX, USA), and copper gluconate (#344419) was purchased from Sigma-Aldrich.
Cell culture and transfection
The HCC cell lines Hep3B and Hepa1-6 were obtained from the Liver CancerInstitute, Fudan University, Shanghai, China. The cells were cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetalbovine serum (FBS; Gibco) and penicillin-streptomycin at 37°C and in a humidified atmosphere with 5% CO2. Small interfering RNAs (siRNAs) targeting human PARP1 and GSK3β were synthesized by Genomeditech (Shanghai, China). The synthesized siRNA sequences were as follows: 5’-UCACAACCUCCUAGAAAGAGUAGA-3’ (siPARP1-NC); 5’-CGACCUGAUCUGGAACAUCAATT-3’ (siPARP1#1); 5’-GCAGCUUCAUAACCGAAGAUUTT-3’ (siPARP1#2); 5’-CACUGGUCACGUUUGGAAATT-3’ (siGSK3β-NC); and 5’-CACUGGUCACGUUUGGAAATT-3’ (siGSK3β).
Real-time PCR assay
Total RNA was isolated by using Trizol reagent (Invitrogen, Carlsbad, CA, USA). Equal amounts of RNA were reverse transcribed into cDNA and amplified by PCR according to the manufacturer’s protocol (Takara). qRT-PCR was performed using SYBR-Green PCR Master mix (Yeasen Biotechnology Co., Ltd.) according to the manufacturer’s protocol. The primers used were as follows: human PD-L1 forward, 5’-GCTGCACTAATTGTCTATTGGGA-3’ and reverse, 5’-AATTCGCTTGTAGTCGGCACC-3’; human GAPDH forward, 5’-TGACTTCAACAGCGACACCCA-3’ and reverse, 5’-CACCCTGTTGCTGTAGCCAAA-3’; human PARP1 forward, 5’-AAGGCGAATGCCAGCGTTAC-3’ and reverse, 5’-GGCACTCTTGGAGACCATGTCA-3’; mouse PD-L1 forward, 5’-GCTCCAAAGGACTTGTACGTG-3’ and reverse, 5’-TCCTTTTCCCAGTACACCACTA-3’; and mouse PARP1 forward, 5’-GTGACTTTTTAGCGGAGTACGC-3’ and reverse, 5’-CCAGCGGTCAATCATACCCAG-3’.
Immunoprecipitation (IP) and Western blot analysis
IP and Western blot analysis were performed as described previously [28]. In brief, for IP, liver cancer cells were lysed in a buffer (50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 5 mM ethylenediaminetetraacetic acid; and 0.5% Nonidet P-40). After removing cell debris, the indicated antibodies were added to clear the lysates with 25 μl of protein A/G agarose beads (#3159558; EMD Millipore Corp., USA). The samples were incubated on a rotating wheel overnight at 4°C. The washed beads were boiled in a 5 × SDS-polyacrylamide gel electrophoresis sample buffer. For Western blot analysis, band intensity quantitation for Western blotting was performed using ImageJ (National Institutes of Health, Bethesda, MD, USA). PVDF membranes were blocked with 5% milk, incubated with primary antibodies for 12-16 h at 4°C and then incubated with HRP-conjugated anti-mouse/rabbit secondary antibodies for 2 h after 3 washes with TBST. Low-abundance proteins were visualized with an enhanced chemiluminescence detection reagent (Thermo Fisher Scientific, Waltham, MA, USA).
Immunohistochemistry
Tissue microarrays containing tumor and matched nontumor liver tissue samples were constructed as described previously [28]. Briefly, tumor specimens were collected from HCC patients who underwent surgical resection from August 2001 to November 2007 in Liver Surgery Department of Zhongshan Hospital, Fudan University, Shanghai, China. All patients signed the informed consents and the protocols were approved by the Research Ethics Committee of Zhongshan Hospital. Paraffin-embedded implanted tumors were cut into 5-μm sections. Immunohistochemical (IHC) staining of the HCC samples was performed as described previously. In brief, each sample was stained with indicated antibodies and then incubated with an avidin-biotin-peroxidase complex. Visualization of the target protein was performed using the chromogen 3-amino-9-ethylcarbazole. The H-score method was used to score the samples by combining the values for immunoreaction intensity and percentage of tumor cell staining. The hybrid score formula was as follows: (% cells of 1 + intensity score × 1) + (% cells of 2 + intensity score × 2) + (% cells of 3 + intensity score × 3). The following four groups were created according to the histological scores: high (+++), medium (++), low (+), and negative (-).
In vivo tumor experiments
Mouse Hepa1-6 liver cancer cells were injected (107 cells transplanted subcutaneously (s.c.)) to grow tumors in C57BL/6 mice and NOD-SCID mice (male, 5-6 weeks old, weighing 20-22 g). This study was approved by the Shanghai Medical Experimental Animal Care Committee and performed according to the National Institutes of Health “Guide for the Care and Use of Laboratory Animals”. The mice were randomly divided into groups, each containing 5 mice, after the tumors grew to 108-171.5 mm3 on average and were treated as follows: for antibody-based drug intervention, 250 μg of anti-PD-1 antibody (RMP1-14; Bio X Cell, West Lebanon, NH, USA) or rat IgG (control; Bio X Cell) was injected intraperitoneally every 3 days. For drug-based intervention, mice were given daily oral DSF plus copper gluconate (50 mg/kg DSF; 0.15 mg/kg Cu2+). Subcutaneous tumors were measured using a caliper twice a week. Tumor volumes were calculated using the formula: tumor volume = length × width2/2. At the end of the experiment, the mice were euthanized by cervical dislocation, and the tumors were obtained for subsequent histological and flow cytometric analyses.
Results
DSF/Cu2+ upregulated PD-L1 expression in HCC cells and induced T cell suppression
To test the therapeutic efficacy of DSF/Cu2+ in HCC, we subcutaneously inoculated Hepa1-6 cells into immunocompetent (C57BL/6) and immunodeficient (NOD-SCID/CrlSlac) mice. We found that drug intervention with DSF/Cu2+ reduced tumor progression in the NOD-SCID/CrlSlac mice (Figure 1A) but failed to inhibit tumor growth in the C57BL/6 mice (Figure 1B). These results suggest that an intact immune system compromises the therapeutic efficacy of DSF/Cu2+. Previous studies have shown that the immune system may compromise the antitumor effects of targeted molecular drugs [21,27,28]. To investigate how antitumor immunity affects the therapeutic efficacy of DSF/Cu2+, we analyzed the immunophenotypes of tumor-infiltrating T cells and tumor cells in Hepa1-6 tumors treated with DSF/Cu2+ or left untreated by flow cytometry. We observed that PD-L1 expression increased substantially in the tumor region with a concomitant decrease in CD8+ T cell activity when the mice received DSF/Cu2+ treatment (Figure 1C).
Figure 1.

DSF/Cu2+ inhibited proliferation and upregulated PD-L1 expression in HCC tumor cells. A. The growth of tumors generated by Hepa1-6 cells in NOD-SCID/CrlSlac immunodeficient mice following drug intervention with DSF/Cu2+. Tumors were measured at the indicated time points. B. Hepa1-6 tumor growth in C57BL/6 mice following drug intervention with DSF/Cu2+. C. DSF/Cu2+-treated mice. Tumor-infiltrating immune cells and inflammatory cells, including T cells (CD8+), natural killer (NK) cells (NK1.1+), and B lymphocytes (CD19+), as well as PD-L1+ tumor cells were detected by flow cytometry. D. PD-L1 protein expression after DSF/Cu2+ treatment. Hep3B and Hep1-6 cells were treated with DSF/Cu2+ (0, 5, or 10 μM DSF in Hep3B cells and 0, 2.5, or 5 μM DSF in Hep1-6 cells; 1 µM Cu2+), and PD-L1 protein levels were analyzed by Western blotting. E. Quantitative analysis of PD-L1 expression after DSF/Cu2+ treatment through ImageJ intensity measurements. F. Cell-surface PD-L1 expression with DSF/Cu2+ treatment in both Hep3B cells and Hep1-6 cells. Cell-surface PD-L1 levels were measured by flow cytometry. Error bars: mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
To verify the upregulation of PD-L1 expression induced by DSF/Cu2+ in HCC cells, we treated Hep3B and Hepa1-6 cells with DSF/Cu2+, and immunoblot analysis showed that PD-L1 expression was upregulated in the Hep3B and Hep1-6 cells after DSF/Cu2+ treatment (Figure 1D, 1E). We also measured PD-L1 levels by flow cytometry, and the results showed that cell-surface PD-L1 expression was significantly increased after DSF/Cu2+ treatment (Figure 1F). Together, these results indicate that DSF/Cu2+ may affect antitumor immunity by upregulating PD-L1 expression in HCC cells.
DSF/Cu2+ induced PD-L1 expression via the PARP1/GSK3β pathway
Next, we investigated the mechanisms by which DSF/Cu2+ regulates PD-L1 in HCC cells. PD-L1 mRNA expression was quantified by real-time PCR after DSF/Cu2+ treatment. The results showed that PD-L1 mRNA expression was not affected by DSF/Cu2+ treatment in Hep3B and Hepa1-6 cells (Supplementary Figure 1A), implying that the regulation does not occur at the transcriptional level but may occur at the posttranslational level.
We then detected the pattern of changes in the transcriptional level of Hep3B cells after DSF/Cu2+ treatment by gene chip analysis, and we found that PARP1 was one of the DNA damage repair-related genes with the most downregulated expression (Figure 2A). Chemotherapeutic drugs have been reported to induce DNA damage and then regulate PD-L1 expression in tumor cells, thereby affecting the local immune status of the tumor microenvironment [37,38]. As a potential chemotherapeutic drug, DSF/Cu2+ may also affect the expression of PD-L1 by inducing DNA damage in liver cancer cells. We then validated the activity of PARP1 after DSF/Cu2+ treatment. We found that the activity of PARP1 was decreased in a dose-dependent manner, with reduced levels of PARP1 but the expression level of PD-L1 was increased (Figure 2B, 2C and Supplementary Figure 1B-G). PARP1, a DNA BER enzyme, has been reported to regulate the stability of PD-L1 [27]. To determine whether PARP1 inhibition could upregulate PD-L1 expression, we tested PD-L1 expression in Hep3B cells exposed to PARP1-specific siRNA. As predicted, PD-L1 expression was substantially higher in the treated cells than in control cells (Figure 2D, 2E). SiRNA-mediated PARP1 inhibition and PARP inhibitor (PARPi) treatment have been reported to inactivate GSK3β by phosphorylation at Ser9, decreasing PD-L1 ubiquitination and ultimately upregulating PD-L1 expression [23,27]. Therefore, we asked whether DSF/Cu2+ stabilizes PD-L1 through GSK3β-mediated PD-L1 ubiquitination. The results showed that PD-L1 ubiquitination in the presence of MG132 was abolished by DSF/Cu2+ treatment in Hep3B cells (Figure 2F). Furthermore, we examined the status of GSK3β phosphorylation at Ser9 in response to DSF/Cu2+ in Hep3B cells by immunoblotting. The results showed that DSF/Cu2+ treatment induced high GSK3β phosphorylation at Ser9 (Figure 2G, 2H). We also observed that PD-L1 expression could not be upregulated by DSF/Cu2+ treatment in the presence of siRNA-mediated GSK3β expression knockdown (Figure 2I, 2J), suggesting that the upregulation of PD-L1 expression induced by DSF/Cu2+ was GSK3β dependent. A previous study demonstrated that silencing PARP1 could enhance the transcription of PD-L1 by activating STAT3 phosphorylation [39]. To determine whether this mechanism is also involved after DSF/Cu2+ treatment, we evaluated STAT3 activity in our system. Our data showed that DSF/Cu2+ inhibited the phosphorylation of STAT3 at Tyr705 (Supplementary Figure 1H, 1I), and this finding was opposite to the previous results, suggesting that STAT3 did not participate in this process. Taken together, our results suggest that PARP-mediated GSK3β inactivation is required for DSF/Cu2+-induced upregulation of PD-L1 expression.
Figure 2.

DSF/Cu2+ promoted PD-L1 expression via the PARP1/GSK3β pathway. A. Heatmap of a selected list of DNA damage repair-related genes showing fold changes in expression between DSF/Cu2+-treated and control Hep3B cells. B. Activity of PARP1 after DSF/Cu2+ treatment. PAR expression in Hep3B cells was detected by Western blot analysis after DSF/Cu2+ treatment for 16 h (2.5, 5, 10, or 15 μM DSF and 1 µM Cu2+). C. Quantitative analysis of PAR expression after DSF/Cu2+ treatment through ImageJ intensity measurements. D. PARP1 and PD-L1 expression after treatment with PARP1-specific siRNA. PARP1 and PD-L1 protein expression in Hep3B cells was evaluated by Western blotting after treatment with PARP1-specific siRNA. E. Quantitative analysis of PARP1 and PD-L1 expression after treatment with PARP1-specific siRNA through ImageJ intensity measurements. F. Ubiquitination assay evaluating PD-L1 in Hep3B cells. Cell lysates were immunoprecipitated with an anti-PD-L1 antibody and subjected to Western blot analysis with an antibody against K48-linked ubiquitin. The cells were treated with DSF/Cu2+ or MG132 prior to the ubiquitination analysis. G, I. PD-L1 and p-GSK3β (Ser9) expression after treatment with DSF/Cu2+, olaparib or GSK3β-specific siRNA. PD-L1 and p-GSK3β (Ser9) protein expression in Hep3B cells was evaluated by Western blotting after treatment with DSF/Cu2+ (5 or 10 μM DSF and 1 µM Cu2+), olaparib (10 μM for 24 h) or GSK3β-specific siRNA. H, J. Quantitative analysis of PD-L1 and p-GSK3β (Ser9) expression after treatment with DSF/Cu2+, olaparib or GSK3β-specific siRNA through ImageJ intensity measurements. Error bars: mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Anti-PD-1 therapy in combination with DSF/Cu2+ improved antitumor activity
Our data have already demonstrated that DSF/Cu2+ stabilizes PD-L1 to support immune evasion, which may be the underlying factor contributing to the ineffectiveness of DSF/Cu2+ in our immunocompetent HCC model. To determine whether PD-L1 pathway blockade can further enhance DSF/Cu2+ antitumor efficacy in vivo, we treated mice bearing subcutaneous Hepa1-6 tumors with DSF/Cu2+ or an anti-PD-1 antibody alone or in combination with and detected tumor growth (Figure 3A). The results showed that DSF/Cu2+ failed to significantly reduce the tumor burden, the anti-PD-1 antibody slightly restricted tumor growth, and the combination treatment showed much better antitumor efficacy than control treatment or each treatment alone (Figure 3B, 3C). In addition, compared with each treatment alone, the combination treatment with DSF/Cu2+ and the anti-PD-1 antibody substantially prolonged the overall survival of the mice bearing subcutaneous Hepa1-6 tumors (Figure 3D). IHC analysis showed that PD-L1 expression was upregulated, while PARP1 expression was downregulated in tumor tissue samples from the mice treated with DSF/Cu2+ alone or in combination with the anti-PD-1 antibody (Figure 3E, 3F). In addition, the size of the activated tumor-infiltrating CD8+ T cell population and the expression of granzyme B were increased in the mice treated with the combination of DSF/Cu2+ and the anti-PD-1 antibody (Figure 3E, 3F), indicating that the combination treatment improved antitumor immunity in mice. Taken together, these results illustrated that DSF/Cu2+ induced immunosuppression through the upregulation of PD-L1 expression and that the combination of DSF/Cu2+ and the anti-PD-1 antibody showed potential therapeutic benefits.
Figure 3.

Anti-PD-1 therapy in combination with DSF/Cu2+ improved antitumor activity. A. Schematic diagram of the drug intervention protocol for disulfiram plus copper gluconate and/or anti-PD-1 antibody treatment of C57BL/6 mice. B. The growth of subcutaneous Hepa1-6 tumors in disulfiram plus copper gluconate and/or anti-PD-1 antibody-treated C57BL/6 mice. Tumors were measured at the indicated time points. C. Tumor weights after the drug intervention endpoints. D. Survival of mice bearing Hepa1-6 tumors following treatment with disulfiram plus copper gluconate and/or anti-PD-1 antibody. Significance was evaluated using the log-rank test. E. Immunohistochemical staining for PD-L1, PARP1, CD8, and granzyme B protein expression patterns in Hepa1-6 tumors. Scale bar, 50 μm. F. The positive cells of PD-L1, PARP1, CD8, and granzyme B in Hepa1-6 tumors following treatment with disulfiram plus copper gluconate and/or anti-PD-1 antibody. Error bars: mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Correlations among the expression of PARP1, p-GSK3β (Ser9) and PD-L1 in human tumor tissue samples
To further validate our findings in human cancer patient samples, we analyzed the correlations among the expression of PARP1, p-GSK3β (Ser9) and PD-L1 by performing IHC staining of 268 HCC samples (Figure 4A). As expected, the expression level of PARP1 was inversely correlated with that of PD-L1 (P < 0.001) and p-GSK3β (Ser9) (P < 0.0001) (Figure 4B). Specifically, approximately 75% of the tumor samples with low expression of PARP1 showed strong PD-L1 staining, and 68.9% of those with high expression of PARP1 showed weak PD-L1 staining or no PD-L1 staining. Taken together, these data indicate that low expression of PARP1 is associated with high p-GSK3β (Ser9) and PD-L1 expression in clinical HCC samples.
Figure 4.

Correlation among the expression of PARP1, p-GSK3β (Ser9) and PD-L1 in human tumor tissue samples. A. Representative pictures of IHC staining of HCC tumors for PARP1, PD-L1 and p-GSK3β (Ser9) in HCC tumors. Patient tissue samples were stained for PARP1, PD-L1 and p-GSK3β (Ser9). B. The correlations between PARP1 and PD-L1 or p-GSK3β (Ser9) expression levels in liver cancer patients. P, Pearson chi-square test; -/+, negative or low expression; ++/+++, medium or high expression. Scale bar, 100 μm.
On the basis of these findings, we proposed the model shown in Figure 5. DSF/Cu2+ inhibited the activity of PARP1 and induced the phosphorylation of GSK3β at Ser9, thus inactivating GSK3β to reduce the degradation of PD-L1, which led to the suppression of antitumor immunity. The combination of DSF/Cu2+ and an anti-PD-1 monoclonal antibody may boost antitumor immunity efficacy.
Figure 5.
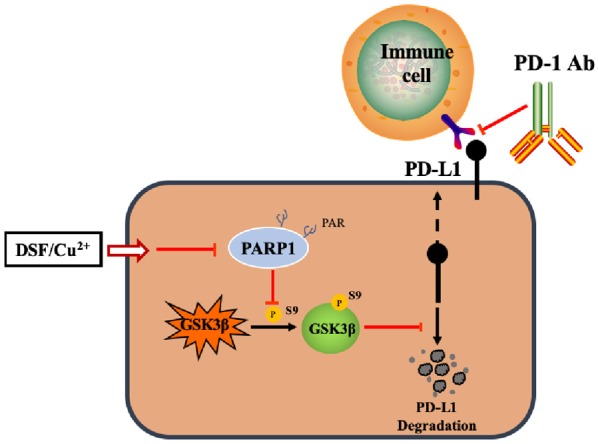
An illustration of the proposed working model. The phosphorylation of GSK3β at Ser9 induced by PARP inhibition is a key step in stabilizing PD-L1.
Discussion
DSF/Cu2+ has shown anticancer activity against a broad spectrum of malignancies, but the effect of DSF/Cu2+ on antitumor immunity is still largely unknown. In our study, we determined that DSF/Cu2+ induced antitumor immunosuppression in HCC cells in a mouse tumor model by upregulating PD-L1 expression, which was dependent on PARP-mediated GSK3β inactivation. Our results provide more evidence of crosstalk between cytotoxic anticancer agents and cancer-associated immunity, which may lead to more efficient combinatorial regimens for cancer therapy.
PARP1 is an enzyme that plays a critical role in mediating DNA BER and catalyzes the covalent attachment of polymers of ADP-ribose (PAR) moieties to itself and its target proteins. PARP1 inhibition limits tumor xenograft growth and prevents tumor vasculogenesis, while high PARP1 expression has been reported to lead to chemotherapeutic drug resistance [36,40]. PARP1 has also been reported to regulate PD-L1 expression by either transcriptional or posttranscriptional mechanisms [27,37,39]. In this study, PARP1 was one of the DNA damage repair-related genes with the most downregulated expression after DSF/Cu2+ treatment (Figure 2A). In addition, low PARP1 expression was associated with high p-GSK3β (S9) and PD-L1 expression in clinical HCC samples (Figure 4A, 4B). Mechanistically, inhibiting PARP1 activity increased GSK3β phosphorylation at Ser9, leading to a reduction in GSK3β activity and then decreased PD-L1 degradation, as shown in the current report (Figure 5). Our study revealed that GSK3β inhibition may be related to the inhibition of PARP1 activity, which was consistent with previous studies showing that PARPi treatment induced high levels of GSK3β phosphorylated at Ser9, the inactivated form of GSK3β. In contrast, PARP1 has been shown to be a negative regulator of the STAT3-mediated transcription of PD-L1 in various cancer cells. PARP1 poly(ADP-ribosyl)ates STAT3, inhibiting STAT3 phosphorylation and transcriptional activity and thus attenuating the expression of PD-L1 [39]. To investigate whether STAT3 and its phosphorylation levels are affected by DSF/Cu2+, we examined STAT3 Tyr705 phosphorylation levels and found that STAT3 phosphorylation at Tyr705 was inhibited after DSF treatment (Supplementary Figure 1H, 1I). Taken together, our data indicate that the upregulation of PD-L1 expression induced by DSF/Cu2+ is dependent on the PARP1/GSK3β pathway, suggesting that we can simultaneously target PARP1 and the PD-1/PD-L1 axis to inhibit both the activity of PARP1 and immunosuppression induced by the upregulation of PD-L1 expression to achieve improved antitumor efficacy.
GSK-3β, a serine/threonine protein kinase, is a complex regulator of numerous cellular functions, including many metabolic and signaling pathways as well as the modification of structural proteins [41], which has also been reported to play significant roles in tumorigenesis and regulating cancer cell mesenchymal-epithelial transition, apoptosis, chromosome stability and cancer immunosuppression by inducing phosphorylation-dependent proteasomal degradation of Snail [42], Mcl-1 [43], EZH2 [44] and PD-L1 [23,27,28]. In this study, we revealed that DSF/Cu2+ enhanced GSK3β phosphorylation at Ser9, leading to a reduction in GSK3β activity and a decrease in PD-L1 degradation, which induced immunosuppression. This evidence of crosstalk between cytotoxic anticancer agents and cancer-associated immunity may lead us to develop more efficient combinatorial regimens for cancer therapy.
Chemotherapy, that was originally thought to be solely immunosuppressive, can shape the tumor microenvironment to promote antitumor immunity which may be beneficial in combination with immunotherapy [37,38,45]. For example, in patients with DNA damage-repair gene mutations or deletions, PARPi and anti-PD-1 monoclonal antibodies can exert strong antitumor effects. PARPi can exogenously increase the infiltration of immune cells [46], and upregulate the expression of PD-L1 in tumor cells, thereby leading to an increase in the response rate to immune checkpoint blockade [27,37,38]. At present, many clinical trials have explored whether combining PARP inhibitors and anti-PD-1 monoclonal antibodies can increase clinical benefits [47-49]. In this paper, we found that PARP1 inhibition and upregulated PD-L1 expression as well as a decreased CD8+ T cell ratio were observed with DSF/Cu2+ treatment alone (Figure 3E, 3F). The combination of DSF/Cu2+ and an anti-PD-1 monoclonal antibody inhibited PARP1 and the PD-1/PD-L1 axis, which not only reduced the viability of tumor cells but also increased the number of infiltrating T cells in the tumor, achieving a two-pronged effect. Our data may at least partially indicate that the combination of PARP inhibitors and an anti-PD-1 monoclonal antibody could produce powerful antitumor effects.
In conclusion, our current finding demonstrates that DSF/Cu2+ upregulates PD-L1 expression via PARP1/GSK3β inhibition and produces improved antitumor activity in combination with an anti-PD-1 antibody. DSF combined with copper may be repurposed for cancer therapy.
Acknowledgements
We want to thank Shizhe Zhang, Xiaolong Li, Kangshuai Li, Longhai Feng and Qianni Ma for their great work in improving the experiment design. This work was supported by Shanghai Sailing Program (19YF1407600) and the National Natural Science Foundation of China (81572301, 81502487, 81802893, 81871924).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 3.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 4.Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, Gerolami R, Masi G, Ross PJ, Song T, Bronowicki JP, Ollivier-Hourmand I, Kudo M, Cheng AL, Llovet JM, Finn RS, LeBerre MA, Baumhauer A, Meinhardt G, Han G. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66. doi: 10.1016/S0140-6736(16)32453-9. [DOI] [PubMed] [Google Scholar]
- 5.Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, Baron A, Park JW, Han G, Jassem J, Blanc JF, Vogel A, Komov D, Evans TRJ, Lopez C, Dutcus C, Guo M, Saito K, Kraljevic S, Tamai T, Ren M, Cheng AL. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163–1173. doi: 10.1016/S0140-6736(18)30207-1. [DOI] [PubMed] [Google Scholar]
- 6.Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, Cicin I, Merle P, Chen Y, Park JW, Blanc JF, Bolondi L, Klumpen HJ, Chan SL, Zagonel V, Pressiani T, Ryu MH, Venook AP, Hessel C, Borgman-Hagey AE, Schwab G, Kelley RK. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med. 2018;379:54–63. doi: 10.1056/NEJMoa1717002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018;18:153–167. doi: 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- 9.Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, Aren Frontera O, Havel L, Steins M, Garassino MC, Aerts JG, Domine M, Paz-Ares L, Reck M, Baudelet C, Harbison CT, Lestini B, Spigel DR. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, Donskov F, Bono P, Wagstaff J, Gauler TC, Ueda T, Tomita Y, Schutz FA, Kollmannsberger C, Larkin J, Ravaud A, Simon JS, Xu LA, Waxman IM, Sharma P. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling TH Rd, Meyer T, Kang YK, Yeo W, Chopra A, Anderson J, Dela Cruz C, Lang L, Neely J, Tang H, Dastani HB, Melero I. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbe C, Charles J, Mihalcioiu C, Chiarion-Sileni V, Mauch C, Cognetti F, Arance A, Schmidt H, Schadendorf D, Gogas H, Lundgren-Eriksson L, Horak C, Sharkey B, Waxman IM, Atkinson V, Ascierto PA. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, Srinivas S, Retz MM, Grivas P, Joseph RW, Galsky MD, Fleming MT, Petrylak DP, Perez-Gracia JL, Burris HA, Castellano D, Canil C, Bellmunt J, Bajorin D, Nickles D, Bourgon R, Frampton GM, Cui N, Mariathasan S, Abidoye O, Fine GD, Dreicer R. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Dang F, Ren J, Wei W. Biochemical aspects of PD-L1 regulation in cancer immunotherapy. Trends Biochem Sci. 2018;43:1014–1032. doi: 10.1016/j.tibs.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Atsaves V, Tsesmetzis N, Chioureas D, Kis L, Leventaki V, Drakos E, Panaretakis T, Grander D, Medeiros LJ, Young KH, Rassidakis GZ. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia. 2017;31:1633–1637. doi: 10.1038/leu.2017.103. [DOI] [PubMed] [Google Scholar]
- 17.Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, Wang HY, Wysocka M, Cheng M, Ruggeri BA, Wasik MA. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1) Proc Natl Acad Sci U S A. 2008;105:20852–20857. doi: 10.1073/pnas.0810958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bi XW, Wang H, Zhang WW, Wang JH, Liu WJ, Xia ZJ, Huang HQ, Jiang WQ, Zhang YJ, Wang L. PD-L1 is upregulated by EBV-driven LMP1 through NF-kappaB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J Hematol Oncol. 2016;9:109. doi: 10.1186/s13045-016-0341-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS One. 2015;10:e0123410. doi: 10.1371/journal.pone.0123410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang G, Wen Q, Zhao Y, Gao Q, Bai Y. NF-kappaB plays a key role in inducing CD274 expression in human monocytes after lipopolysaccharide treatment. PLoS One. 2013;8:e61602. doi: 10.1371/journal.pone.0061602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, Tan Y, Ci Y, Wu F, Dai X, Guo J, Huang YH, Fan C, Ren S, Sun Y, Freeman GJ, Sicinski P, Wei W. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–95. doi: 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, Chang SS, Lin WC, Hsu JM, Hsu YH, Kim T, Chang WC, Hsu JL, Yamaguchi H, Ding Q, Wang Y, Yang Y, Chen CH, Sahin AA, Yu D, Hortobagyi GN, Hung MC. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–939. doi: 10.1016/j.ccell.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, Khoo KH, Chang SS, Cha JH, Kim T, Hsu JL, Wu Y, Hsu JM, Yamaguchi H, Ding Q, Wang Y, Yao J, Lee CC, Wu HJ, Sahin AA, Allison JP, Yu D, Hortobagyi GN, Hung MC. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, Cha JH, Xia W, Chan LC, Kim T, Chang SS, Lee HH, Chou CK, Liu YL, Yeh HC, Perillo EP, Dunn AK, Kuo CW, Khoo KH, Hsu JL, Wu Y, Hsu JM, Yamaguchi H, Huang TH, Sahin AA, Hortobagyi GN, Yoo SS, Hung MC. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33:187–201. e110. doi: 10.1016/j.ccell.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, Wei Y, Xia W, Hou J, Qiu Y, Hung MC. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2019;29:83–86. doi: 10.1038/s41422-018-0124-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, Lu H, Fang C, Zhang Y, Liang L, Zhou X, Wang C, Xue Y, Cui Y, Xu J. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng. 2019;3:306–317. doi: 10.1038/s41551-019-0375-6. [DOI] [PubMed] [Google Scholar]
- 27.Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, Hsu JL, Yu WH, Du Y, Lee HH, Li CW, Chou CK, Lim SO, Chang SS, Litton J, Arun B, Hortobagyi GN, Hung MC. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23:3711–3720. doi: 10.1158/1078-0432.CCR-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Li CW, Li X, Ding Q, Guo L, Liu S, Liu C, Lai CC, Hsu JM, Dong Q, Xia W, Hsu JL, Yamaguchi H, Du Y, Lai YJ, Sun X, Koller PB, Ye Q, Hung MC. MET inhibitors promote liver tumor evasion of the immune response by stabilizing PDL1. Gastroenterology. 2019;156:1849–1861. doi: 10.1053/j.gastro.2019.01.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pantziarka P, Verbaanderd C, Sukhatme V, Rica Capistrano I, Crispino S, Gyawali B, Rooman I, Van Nuffel AM, Meheus L, Sukhatme VP, Bouche G. ReDO_DB: the repurposing drugs in oncology database. Ecancermedicalscience. 2018;12:886. doi: 10.3332/ecancer.2018.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iljin K, Ketola K, Vainio P, Halonen P, Kohonen P, Fey V, Grafstrom RC, Perala M, Kallioniemi O. High-throughput cell-based screening of 4910 known drugs and drug-like small molecules identifies disulfiram as an inhibitor of prostate cancer cell growth. Clin Cancer Res. 2009;15:6070–6078. doi: 10.1158/1078-0432.CCR-09-1035. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Chen D, Ringler J, Chen W, Cui QC, Ethier SP, Dou QP, Wu G. Disulfiram treatment facilitates phosphoinositide 3-kinase inhibition in human breast cancer cells in vitro and in vivo. Cancer Res. 2010;70:3996–4004. doi: 10.1158/0008-5472.CAN-09-3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skrott Z, Mistrik M, Andersen KK, Friis S, Majera D, Gursky J, Ozdian T, Bartkova J, Turi Z, Moudry P, Kraus M, Michalova M, Vaclavkova J, Dzubak P, Vrobel I, Pouckova P, Sedlacek J, Miklovicova A, Kutt A, Li J, Mattova J, Driessen C, Dou QP, Olsen J, Hajduch M, Cvek B, Deshaies RJ, Bartek J. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature. 2017;552:194–199. doi: 10.1038/nature25016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bista R, Lee DW, Pepper OB, Azorsa DO, Arceci RJ, Aleem E. Disulfiram overcomes bortezomib and cytarabine resistance in Down-syndrome-associated acute myeloid leukemia cells. J Exp Clin Cancer Res. 2017;36:22. doi: 10.1186/s13046-017-0493-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006;66:10425–10433. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- 35.Sonnenblick A, de Azambuja E, Azim HA Jr, Piccart M. An update on PARP inhibitors--moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12:27–41. doi: 10.1038/nrclinonc.2014.163. [DOI] [PubMed] [Google Scholar]
- 36.Yuan K, Sun Y, Zhou T, McDonald J, Chen Y. PARP-1 regulates resistance of pancreatic cancer to TRAIL therapy. Clin Cancer Res. 2013;19:4750–4759. doi: 10.1158/1078-0432.CCR-13-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, Lamb A, Henon C, Dorvault N, Rouanne M, Marlow R, Bajrami I, Cardenosa ML, Konde A, Besse B, Ashworth A, Pettitt SJ, Haider S, Marabelle A, Tutt AN, Soria JC, Lord CJ, Postel-Vinay S. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest. 2019;129:1211–1228. doi: 10.1172/JCI123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L, Fan Y, Yang Y, Wang J, Glisson BS, Wistuba II, Sage J, Heymach JV, Gibbons DL, Byers LA. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9:646–661. doi: 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding L, Chen X, Xu X, Qian Y, Liang G, Yao F, Yao Z, Wu H, Zhang J, He Q, Yang B. PARP1 suppresses the transcription of PD-L1 by Poly(ADP-Ribosyl)ating STAT3. Cancer Immunol Res. 2019;7:136–149. doi: 10.1158/2326-6066.CIR-18-0071. [DOI] [PubMed] [Google Scholar]
- 40.Quiles-Perez R, Munoz-Gamez JA, Ruiz-Extremera A, O’Valle F, Sanjuan-Nunez L, Martin-Alvarez AB, Martin-Oliva D, Caballero T, Munoz de Rueda P, Leon J, Gonzalez R, Muntane J, Oliver FJ, Salmeron J. Inhibition of poly adenosine diphosphate-ribose polymerase decreases hepatocellular carcinoma growth by modulation of tumor-related gene expression. Hepatology. 2010;51:255–266. doi: 10.1002/hep.23249. [DOI] [PubMed] [Google Scholar]
- 41.Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- 42.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 43.Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, Hung MC. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27:4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ko HW, Lee HH, Huo L, Xia W, Yang CC, Hsu JL, Li LY, Lai CC, Chan LC, Cheng CC, Labaff AM, Liao HW, Lim SO, Li CW, Wei Y, Nie L, Yamaguchi H, Hung MC. GSK3β inactivation promotes the oncogenic functions of EZH2 and enhances methylation of H3K27 in human breast cancers. Oncotarget. 2016;7:57131–57144. doi: 10.18632/oncotarget.11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dosset M, Vargas TR, Lagrange A, Boidot R, Vegran F, Roussey A, Chalmin F, Dondaine L, Paul C, Lauret Marie-Joseph E, Martin F, Ryffel B, Borg C, Adotevi O, Ghiringhelli F, Apetoh L. PD-1/PD-L1 pathway: an adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology. 2018;7:e1433981. doi: 10.1080/2162402X.2018.1433981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang J, Wang L, Cong Z, Amoozgar Z, Kiner E, Xing D, Orsulic S, Matulonis U, Goldberg MS. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(-/-) murine model of ovarian cancer. Biochem Biophys Res Commun. 2015;463:551–556. doi: 10.1016/j.bbrc.2015.05.083. [DOI] [PubMed] [Google Scholar]
- 47.Heinhuis KM, Ros W, Kok M, Steeghs N, Beijnen JH, Schellens JHM. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol. 2019;30:219–235. doi: 10.1093/annonc/mdy551. [DOI] [PubMed] [Google Scholar]
- 48.Karzai F, VanderWeele D, Madan RA, Owens H, Cordes LM, Hankin A, Couvillon A, Nichols E, Bilusic M, Beshiri ML, Kelly K, Krishnasamy V, Lee S, Lee MJ, Yuno A, Trepel JB, Merino MJ, Dittamore R, Marte J, Donahue RN, Schlom J, Killian KJ, Meltzer PS, Steinberg SM, Gulley JL, Lee JM, Dahut WL. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer. 2018;6:141. doi: 10.1186/s40425-018-0463-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, Van Le L, Sachdev JC, Chapman-Davis E, Colon-Otero G, Penson RT, Matulonis UA, Kim YB, Moore KN, Swisher EM, Farkkila A, D’Andrea A, Stringer-Reasor E, Wang J, Buerstatte N, Arora S, Graham JR, Bobilev D, Dezube BJ, Munster P. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019 doi: 10.1001/jamaoncol.2019.1048. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.