

Abstract
Circular RNAs (circRNAs) are potential biomarkers and therapeutic targets of coronary artery disease due to their high stability, covalently closed structure. And implied roles in gene regulation. The aim of this study was to identify and characterize circRNAs from human coronary arteries. Epicardial coronary arteries were removed during the autopsy of an 81-year-old man who died from heart attack. The natural history and histological classification of atherosclerotic lesions in coronary artery segments were analyzed by hematoxylin and eosin staining, and their circRNA expression profiles were characterized by RNA sequencing. RNA sequencing identified 1259 annotated and 381 novel circRNAs. Combined with the results of histologic examination, intersection analysis identified 54 upregulated and 12 downregulated circRNAs, representing 4.0% of the total number. Coronary artery segments with or without severe atherosclerosis showed distinctly different circRNA profiles on the basis of hierarchical clustering. Our results suggest that these 66 circRNAs contribute to the pathology underlying coronary artery atherosclerosis and may serve as diagnostic or therapeutic targets in coronary artery disease.
Keywords: Coronary artery, coronary artery disease, atherosclerosis, circular RNA, RNA sequencing
Introduction
As a single-stranded covalently closed subclass of long non-coding RNA molecules formed by the back-splicing of linear precursor RNA, circular RNAs (circRNAs) were initially thought to be splicing-associated noise [1]. Recently, a few studies demonstrated that circRNAs may be involved in microRNA sponging [2], RNA-associated protein binding [3], protein-coding gene regulation, and protein translation [4]. In addition, circRNA expression can be tissue-specific [5] and highly stable among cells, exosomes, and body fluids [6]. Therefore, with improvements in their measurement and characterization techniques, circRNAs could serve as biomarkers or therapeutic targets for various diseases. However, the association between circRNAs and coronary artery disease (CAD) have not been fully uncovered.
As the leading cause of morbidity and mortality around the world, CAD is influenced by lifestyle, genetics, and their interaction [7]. To date, however, relatively little is known about changes in gene expression in CAD [8]. This is partly because current information about the CAD transcriptome is obtained through gene expression microarray, which is a powerful methodology but has many limitations. Unlike gene expression microarray, which is limited to previously annotated transcripts, RNA sequencing (RNA-seq) is a new methodology that has revolutionized transcriptome analysis through its ability to simultaneously interrogate annotated and unannotated transcripts in an RNA sample [9]. Here, we describe the systematic detection of circRNAs in coronary artery samples from a patient who died of CAD through the pilot application of high-throughput RNA-seq technology and novel bioinformatics algorithms.
Materials and methods
Subject
Coronary artery samples were obtained from an autopsy case at the Department of Human Anatomy in Nanjing Medical University. The bereaved family gave informed consent for the research use of the samples, and the autopsy was conducted according to university guidelines. All methods were performed in accordance with approved guidelines, and all experimental protocols were approved by the ethics committees of Nanjing Medical University and First Affiliated Hospital of Nanjing Medical University. The overall research technology roadmap of the present study is shown in Figure 1.
Figure 1.

Research technology roadmap in the present study. A. Coronary artery sample preparation. B. RNA isolation, RNA-seq library preparation, and sequencing. C. Bioinformatics analysis.
Coronary artery segment preparation
An 81-year-old man died from a heart attack. Upon autopsy approximately 1 h postmortem, the epicardial coronary arteries were removed from the heart and divided into 10 segments: the proximal (p), midsegment (m), and distal (d) segment of the left anterior descending (LAD), left circumflex (LCX), and right coronary artery (RCA), respectively, and the left main trunk (LM). Each segment then divided in half, with one half used for RNA-seq and the other half used for histological analysis. Segments for RNA-seq were snap-frozen in liquid nitrogen and stored at -80°C. Segments for histological analysis were fixed overnight in 10% formalin and embedded in paraffin.
RNA isolation, RNA-seq library preparation, and sequencing
Total RNA was extracted from the coronary artery samples using Trizol (15596018, Invitrogen) following the manufacturer’s instructions, and RNA integrity number was calculated by an Agilent Bioanalyzer 2100 (Agilent Technologies, USA). RNA degradation and contamination were monitored on 1% agarose gels, and RNA purity and concentration were checked using a NanoPhotometer® spectrophotometer (IMPLEN, CA, USA).
A total of 3 µg RNA per sample was used as input material for RNA sample preparation. First, ribosomal RNA was removed using an Epicentre Ribo-zero™ rRNA Removal Kit (Epicentre, USA), and ribosomal (r) RNA-free residue was removed by ethanol precipitation. Next, sequencing libraries were generated with rRNA-depleted RNA using an NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s recommendations. Briefly, fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5×). First-strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNaseH-). Second-strand cDNA synthesis was performed using DNA polymerase I and RNase H. In the reaction buffer, dNTPs with dTTP were replaced by dUTP. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activity. After adenylation of the 3’ends of DNA fragments, NEBNext Adaptors with hairpin loop structures were ligated to prepare for hybridization. To select cDNA fragments preferentially ~150-200 bp in length, library fragments were purified using an AMPure XP system (Beckman Coulter, Beverly, USA). Next, 3 µl USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37°C for 15 min followed by 5 min at 95°C before polymerase chain reaction (PCR). PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers, and Index (X) Primer. Finally, products were purified (AMPure XP system) and library quality was assessed using a Agilent Bioanalyzer 2100 system. Sequencing libraries were sequenced on an Illumina Hiseq 4000 platform (Illumina, Inc., San Diego, CA, USA), and 150 bp paired-end reads were generated. All sequencing was performed at Genecreate Inc. (Ao-Ji Biotech, Wuhan, China). Raw data (i.e., raw reads) were first processed using a custom Perl script. Clean data (i.e., clean reads) were then obtained after removing adapter-containing, poly-N-containing, or low-quality reads from the raw data. The Q20, Q30, and GC content of the clean data were calculated. Hisat2 software (version: 2.0.4) [11,12] was used to map the sequence data to the human genome. Two bioinformatics analytic tools, Find_circ [13] and CIRI [14], were used for circRNA identification with default parameters.
Histological analysis
After decalcification with EDTA decalcification fluid (Solarbio® LIFE SCIENCES, Beijing, China) for approximately 2 weeks, coronary artery segments were fixed overnight in 10% formalin and processed for paraffin embedding. Longitudinal 5 µm consecutive sections were obtained by a rotary microtome (Leica RM2235, Leica Biosystems Nussloch GmbH, Heidelberger Str. 17-19D-69226 Nussloch, Germany) and stained with hematoxylin and eosin (H&E) for observation and morphometric evaluation. Each slide was examined by a 10× stereomicroscope (Leica DM2500 Wien, Austria) at 5-20× original magnification and digitized using an image analysis system (Leica LAS, Wetzlar, Germany).
Coronary atherosclerosis grade and stage in each coronary artery segment were analyzed by independent pathologists using American Heart Association classification guidelines [10].
Results
Natural history and histological classification of atherosclerotic lesions in coronary artery segments
The natural history and histological classification of atherosclerotic lesions in the coronary artery segments were analyzed by H&E staining (Table 1 and Figure 2). All coronary segments showed atherosclerotic changes of the intima, ranging from lesions classifiable as fatty streak tunica intima to secondary affection tunica intima. However, the LM exhibited a more serious coronary atherosclerosis grade and stage than other segments, with secondary affection tunica intima and coronary stenosis from 76 to 100%. Thus, we speculate that lesions in the LM caused the subject’s sudden cardiac arrest.
Table 1.
Natural history and histological classification of atherosclerotic lesions in human coronary artery segments
| Subject | Age | Sex | LM | LAD-p | LAD-m | LAD-d | LCX-p | LCX-m | LCX-d | RCA-p | RCA-m | RCA-d |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grade | 81 | Male | 4 | 1 | 1 | 3 | 1 | 1 | 1 | 1 | 1 | 1 |
| Stage | 4 | 1 | 1 | 2 | 2 | 2 | 1 | 1 | 1 | 2 |
LM, left main trunk; LAD-p, proximal segment of the left anterior descending; LAD-m, midsegment of the left anterior descending; LAD-d, distal segment of the left anterior descending; LCX-p, proximal segment of the left circumflex; LCX-m, midsegment of the left circumflex; LCX-d, distal segment of the left circumflex; RCA-p, proximal segment of the right coronary artery; RCA-m, midsegment of the right coronary artery; RCA-d, distal segment of the right coronary artery. Grade: 1, 0 to 25% stenosis; 2, 26 to 50% stenosis; 3, 51 to 75% stenosis; 4, 76 to 100% stenosis. Stage: 0, normal tunica intima; 1, fatty streak tunica intima; 2, fibrous plaques tunica intima; 3, atherosclerotic tunica intima; 4, secondary affection tunica intima.
Figure 2.
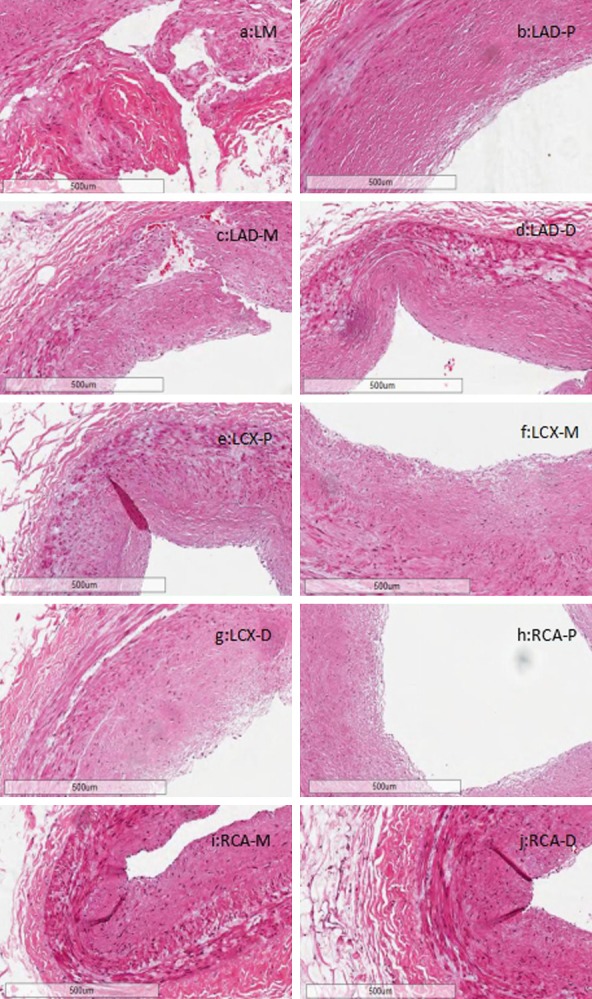
Histologic characterization of coronary artery segments via H&E staining. A. LM. B. LAD-p. C. LAD-m. D. LAD-d. E. LCX-p. F. LCX-m. G. LCX-d. H. RCA-p. I. RCA-m. J. RCA-d. All panels: 5× magnification.
CircRNA abundance in human coronary arteries
Illumina sequencing reads reached 129.52 Gb (clean reads) in total. Data analysis revealed 1640 unique circRNAs: 1259 that were previously described and 381 that were novel. Through comparison with the human genome sequence, we determined the distributions of exons, intergenic regions, and introns in the identified circRNAs across all segments (Figure S1).
Across all segments, the detected circRNAs were distributed on all human chromosomes (autosomes and sex chromosomes). However, this distribution was uneven; many more circRNAs were identified on chromosomes 1 and 2 (162 and 170 circRNAs, respectively) than on other chromosomes (Figure S2). Furthermore, the Y chromosome showed the fewest circRNAs (3 circRNAs) of all chromosomes, which was expected because the Y chromosome is the shortest and contains the smallest number of genes in the human genome. Expression levels of known and novel circRNAs in each segment were statistically analyzed and normalized by transcript per million (Figure S3).
Transcriptome profile of human coronary arteries based on RNA-seq
The transcriptomes of human coronary artery segments were characterized using RNA-seq. A total of 81.00 million, 80.93 million, 83.59 million, 79.73 million, 86.38 million, 89.71 million, 87.12 million, 91.57 million, 95.31 million, and 88.04 million reads were obtained for the LM, LAD-p, LAD-m, LAD-d, LCX-p, LCX-m, LCX-d, RCA-p, RCA-m, and RCA-d, respectively (Table 2). A total of 77.34 million (95.47%), 76.51 million (94.54%), 80.05 million (95.75%), 75.49 million (94.68%), 82.35 million (95.34%), 83.77 million (93.38%), 81.99 million (94.12%), 86.35 million (94.31%), 89.93 million (94.35%), and 82.56 million (93.78%) mapped reads were obtained for the LM, LAD-p, LAD-m, LAD-d, LCX-p, LCX-m, LCX-d, RCA-p, RCA-m, and RCA-d, respectively. Furthermore, approximately 93.35%, 92.70%, 93.69%, 92.83%, 93.62%, 90.98%, 92.34%, 92.53%, 92.54%, and 91.25% of reads for the LM, LAD-p, LAD-m, LAD-d, LCX-p, LCX-m, LCX-d, RCA-p, RCA-m, and RCA-d, respectively, could be uniquely mapped to the human reference genome. No proper-paired reads mapped to different chromosomes were found in any coronary artery segment.
Table 2.
Summary of RNA-seq data from human coronary artery segments
| Mapping Statistics | LM | LAD-p | LAD-m | LAD-d | LCX-p | LCX-m | LCX-d | RCA-p | RCA-m | RCA-d |
|---|---|---|---|---|---|---|---|---|---|---|
| Total reads | 81008982 | 80934204 | 83597902 | 79731964 | 86375208 | 89708382 | 87117902 | 91565414 | 95308074 | 88039124 |
| Total mapped | 77340347 (95.47%) | 76512757 (94.54%) | 80048831 (95.75%) | 75492316 (94.68%) | 82347313 (95.34%) | 83766351 (93.38%) | 81991150 (94.12%) | 86353960 (94.31%) | 89927162 (94.35%) | 82560897 (93.78%) |
| Multiple mapped | 1557844 (1.92%) | 1484754 (1.83%) | 1728891 (2.07%) | 1476153 (1.85%) | 1478971 (1.71%) | 2151166 (2.4%) | 1545923 (1.77%) | 1625534 (1.78%) | 1727173 (1.81%) | 2227193 (2.53%) |
| Uniquely mapped | 75782503 (93.55%) | 75028003 (92.7%) | 78319940 (93.69%) | 74016163 (92.83%) | 80868342 (93.62%) | 81615185 (90.98%) | 80445227 (92.34%) | 84728426 (92.53%) | 88199989 (92.54%) | 80333704 (91.25%) |
| Read-1 | 38333521 (47.32%) | 37891974 (46.82%) | 39555038 (47.32%) | 37421808 (46.93%) | 40857556 (47.3%) | 41342112 (46.09%) | 40612792 (46.62%) | 42792337 (46.73%) | 44536251 (46.73%) | 40639188 (46.16%) |
| Read-2 | 37448982 (46.23%) | 37136029 (45.88%) | 38764902 (46.37%) | 36594355 (45.9%) | 40010786 (46.32%) | 40273073 (44.89%) | 39832435 (45.72%) | 41936089 (45.8%) | 43663738 (45.81%) | 39694516 (45.09%) |
| Reads map to ‘+’ | 37842003 (46.71%) | 37464703 (46.29%) | 39114938 (46.79%) | 36950027 (46.34%) | 40381439 (46.75%) | 40751661 (45.43%) | 40163119 (46.1%) | 42292827 (46.19%) | 44027518 (46.19%) | 40102122 (45.55%) |
| Reads map to ‘-’ | 37940500 (46.83%) | 37563300 (46.41%) | 39205002 (46.9%) | 37066136 (46.49%) | 40486903 (46.87%) | 40863524 (45.55%) | 40282108 (46.24%) | 42435599 (46.34%) | 44172471 (46.35%) | 40231582 (45.7%) |
| Non-splice reads | 60759773 (75.00%) | 59464902 (73.47%) | 59162354 (70.77%) | 57903647 (72.62%) | 62543169 (72.41%) | 57797857 (64.43%) | 62423074 (71.65%) | 68021755 (74.29%) | 69447603 (72.87%) | 58092830 (65.99%) |
| Splice reads | 15022730 (18.54%) | 15563101 (19.23%) | 19157586 (22.92%) | 16112516 (20.21%) | 18325173 (21.22%) | 23817328 (26.55%) | 18022153 (20.69%) | 16706671 (18.25%) | 18752386 (19.68%) | 22240874 (25.26%) |
| Reads mapped in proper pairs | 73341430 (90.53%) | 72504700 (89.58%) | 75891128 (90.78%) | 71297316 (89.42%) | 78269326 (90.62%) | 78601042 (87.62%) | 77598302 (89.07%) | 81741958 (89.27%) | 84962628 (89.15%) | 77221958 (87.71%) |
| Proper-paired reads map to different chromosome | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
LM, left main trunk; LAD-p, proximal segment of the left anterior descending; LAD-m, midsegment of the left anterior descending; LAD-d, distal segment of the left anterior descending; LCX-p, proximal segment of the left circumflex; LCX-m, midsegment of the left circumflex; LCX-d, distal segment of the left circumflex; RCA-p, proximal segment of the right coronary artery; RCA-m, midsegment of the right coronary artery; RCA-d, distal segment of the right coronary artery.
Assembled transcripts of the circRNAs were further filtered based on expression levels > 0 fragments per kilobase million. Expressed circRNAs from all coronary artery segments were subject to cluster analysis. We found that the circRNAs clustered into approximately 10 different subgroups (Figure 3A), suggesting that circRNA expression profiles varied according to coronary atherosclerosis grade and stage.
Figure 3.
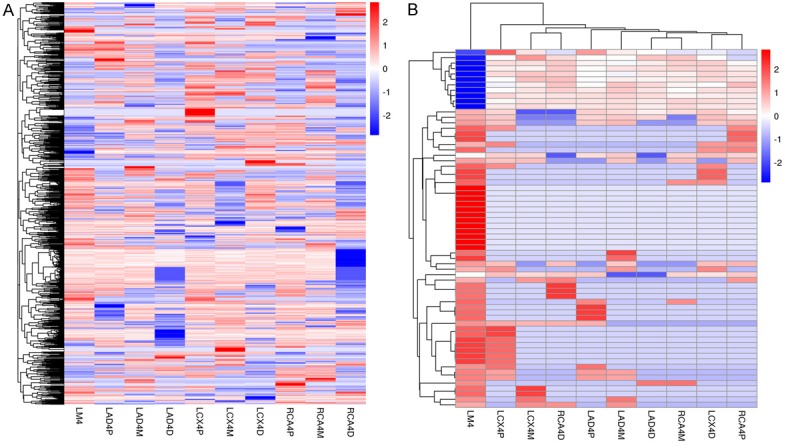
Expression profiles of circRNAs in human coronary arteries. A. Heat cluster of expressed circRNAs in human coronary arteries. B. Heat cluster of intersection expressed circRNAs in human coronary arteries. In both panels, each row represents a single circRNA, and the color of the boxes indicates its expression level in fragments per kilobase million relative to the mean center in each coronary artery segment. High expression: red, low expression: blue, mean center expression: white.
Normalized expression levels of circRNAs in the LM and other nine coronary artery segments were compared (Table 3). Upregulation was defined as expression in the LM/expression in the other segments > 3/2, and downregulation was defined as expression in the LM/expression in other segments < 2/3. We found 304 upregulated and 397 downregulated circRNAs in the LAD-p vs. LM, 359 upregulated and 358 downregulated circRNAs in the LAD-m vs. LM, 232 upregulated and 556 downregulated circRNAs in the LAD-d vs. LM, 477 upregulated and 311 downregulated circRNAs in the LCX-p vs. LM, 395 upregulated and 431 downregulated circRNAs in the LCX-m vs. LM, 362 upregulated and 396 downregulated circRNAs in the LCX-d vs. LM, 328 upregulated and 401 downregulated circRNAs in the RCA-p vs. LM, 361 upregulated and 387 downregulated circRNAs in the RCA-m vs. LM, and 201 upregulated and 628 downregulated circRNAs in the RCA-d vs. LM. Intersection analysis of these nine comparison sets revealed a total of 66 expressed circRNAs, including 54 upregulated and 12 downregulated circRNAs.
Table 3.
Summary of the comparisons of normalized circRNA expression levels in coronary artery segments
| Differential analysis | Intersection | LAD-p vs. LM | LAD-m vs. LM | LAD-d vs. LM | LCX-p vs. LM | LCX-m vs. LM | LCX-d vs. LM | RCA-p vs. LM | RCA-m vs. LM | RCA-d vs. LM |
|---|---|---|---|---|---|---|---|---|---|---|
| Upregulated and downregulated circRNAs | 66 | 701 | 717 | 788 | 788 | 826 | 758 | 729 | 748 | 829 |
| Upregulated circRNAs | 54 | 304 | 359 | 232 | 477 | 395 | 362 | 328 | 361 | 201 |
| Downregulated circRNAs | 12 | 397 | 358 | 556 | 311 | 431 | 396 | 401 | 387 | 628 |
LM, left main trunk; LAD-p, proximal segment of the left anterior descending; LAD-m, midsegment of the left anterior descending; LAD-d, distal segment of the left anterior descending; LCX-p, proximal segment of the left circumflex; LCX-m, midsegment of the left circumflex; LCX-d, distal segment of the left circumflex; RCA-p, proximal segment of the right coronary artery; RCA-m, midsegment of the right coronary artery; RCA-d, distal segment of the right coronary artery.
Cluster analysis showed that the 10 coronary artery segments clustered into two groups; nine segments including the LAD, LCX, and RCA clustered into one group, and the LM clustered into another group. Intersection expressed circRNAs across all segments clustered into two groups; 54 circRNAs clustered into an upregulated group, and 12 circRNAs clustered into a downregulated group (Figure 3B). Combined with the results of H&E staining, this cluster analysis suggests that the 66 intersection expressed circRNAs are associated with the presence and severity of coronary artery atherosclerosis.
Functional annotation of intersection expressed circRNAs
The 66 circRNAs identified in the intersection analysis were assigned one or more gene ontology (GO) terms in three major functional domains. Thirty significant GO annotations were obtained for the intersection expressed circRNAs; 19 for biological processes, 8 for molecular functions, and 3 for cellular components (Figure 4A). The top-ranking GO terms included ubiquitin-dependent protein catabolic process (GO:0006511), modification-dependent protein catabolic process (GO:0019941), modification-dependent macromolecule catabolic process (GO:0043632), early endosome membrane (GO:0031901), protein heterodimerization activity (GO:0046982), and ubiquitin protein ligase activity (GO:0061630). These results further suggest that these circRNAs are involved in coronary artery atherosclerosis.
Figure 4.

Functional annotation of intersection expressed circRNAs. A. GO annotations of circRNAs in human coronary arteries. B. KEGG pathway annotations of circRNAs in human coronary arteries.
In addition to GO term assignment, we performed Kyoto encyclopedia of genes and genomes (KEGG) pathway mapping based on the encyclopedia’s orthology terms to assess coronary artery atherosclerosis-related pathways. Twenty significantly enriched pathways with a false discovery rate < 0.05 were identified among intersection expressed circRNAs (Figure 4B). The top-ranking KEGG pathways included progesterone-mediated oocyte maturation (hsa04914), morphine addiction (hsa05032), leukocyte transendothelial migration (hsa04670), insulin signaling pathway (hsa04910), glutamatergic synapse (hsa04724), endometrial cancer (hsa05213), cGMP-PKG signaling pathway (hsa04022), cAMP signaling pathway (hsa04024), bacterial invasion of epithelial cells (hsa05100), arrhythmogenic right ventricular cardiomyopathy (hsa05412), Alzheimer’s disease (hsa05010), and adherens junction (hsa04520). These findings suggest that these circRNAs are involved in pathways associated with coronary artery atherosclerosis.
Discussion
We present the exploratory use of RNA-seq to characterize circRNA expression in human coronary arteries. RNA-seq profiling of coronary artery samples identified 1259 previously annotated circRNAs and 381 novel circRNAs. Combined with the results of H&E staining, intersection analysis identified 54 upregulated and 12 downregulated circRNAs, representing 4.0% of the total number. Moreover, coronary artery segments with or without severe atherosclerosis showed distinct differences in circRNA profiles on the basis of hierarchical clustering. Therefore, these 66 circRNAs may play pathological roles in coronary artery atherosclerosis.
Due to its resistance to exonucleases, the half-life of circRNA is longer than that of linear RNA, and circRNA can be detected in circulating plasma samples, implicating circRNAs as a promising biomarker for disease diagnosis and treatment [13]. CAD is the leading cause of cardiovascular disease-related death around the world. However, few circRNAs are known to participate in the pathological processes underlying CAD and could be employed as biomarkers. Due to the competitive edge of canonical splicing over non-canonical splicing, circRNA expression is often less than that of linear mRNA. As such, existing techniques may not be sufficiently sensitive to measure the low expression levels of circRNAs [15]. However, circRNAs are often expressed in a tissue-specific manner [16]. Based on the assumption that circRNAs are more abundant in coronary arteries than in circulating blood, we performed RNA-seq of coronary artery samples in the present study, which demonstrates the natural history and histological classification of atherosclerotic lesions and reveals that changes in circRNA expression may be associated with coronary artery atherosclerosis.
As a disease of large- and medium-sized muscular arteries, atherosclerosis is the major pathologic process resulting in cardiovascular disease in humans [17]. This autopsy study demonstrates a means of performing basic histological classification, which advances our approach to diagnosing and treating clinically significant atherosclerotic CAD. As there is no effective method of sampling coronary arteries from living people, autopsy studies may provide a unique source of human samples to further investigate atherosclerotic CAD. In this study, H&E staining of serially sectioned coronary arteries from an 81-year-old man who died suddenly of a heart attack showed the frequent presence of atherosclerotic lesions across 10 artery segments, particularly in the LM. Therefore, characterizing the expression of circRNAs across coronary artery segments combined with their histological examination increases our understanding of the molecular mechanisms of atherosclerotic CAD.
RNA-seq is a type of next-generation sequencing that can be used to reveal the presence and quantity of RNAs in certain disease states, including atherosclerotic CAD. Recently, a spatial and time-dependent transcriptome analysis of porcine myocardium suggests that ischemic postconditioning protects the coronary microvasculature from myocardial infarction [18]. A study generating high-quality sequence data shows that long non-coding RNAs may play important roles in the biological and pathological processes underlying acute myocardial infarction [19]. A whole transcriptome analysis performed using RNA-seq reports detailed changes in the macrophage transcriptome across the first week after myocardial infarction in mice [20]. Another study reveals an intrinsic interplay between CAD and type 2 diabetes using RNA-seq to identify the unique gene expression signatures of CAD, type 2 diabetes, and coexisting conditions [21]. RNA-seq analysis and quantitative real-time PCR were used to evaluate the presence of GLP-1R and GLP-2R in epicardial adipose tissue and subcutaneous fat obtained from patients with CAD and type 2 diabetes mellitus undergoing elective cardiac surgery [22]. In addition, a genome-wide RNA-seq study of human coronary artery smooth muscle cells with siRNA knockdown identified several putative TCF21 downstream pathways [23]. However, no previous studies have reported the results of RNA-seq transcriptome analysis of human coronary artery samples. In the present study, RNA-seq identified 1259 known and 381 unknown circRNAs. Considering the subject’s medical history and H&E staining results, we found that 54 upregulated and 12 downregulated circRNAs were associated with CAD. Furthermore, 30 significant GO annotations were obtained for the 66 intersection expressed circRNAs, with 19 for biological processes, 8 for molecular functions, and 3 for cellular components. In addition to GO term assignment, 20 significantly enriched KEGG pathways were identified among the intersection expressed circRNAs. These results show that gene ontology terms and pathways for these circRNAs are associated with coronary artery atherosclerosis.
Limitations
This study has several limitations. Coronary artery samples were obtained from only one subject. Future studies with large cohorts should be conducted to identify intersection expressed circRNAs in patients with coronary artery atherosclerosis using RNA-seq and verification of circRNA profiles by quantitative PCR. Additionally, as functional validation assays were not performed, we could not speculate on the specific mechanisms by which circRNAs are involved in the development of atherosclerosis.
Conclusion
We identified 1640 circRNAs in human coronary arteries, with 23% being previously unreported. Additional analysis showed that 54 upregulated and 12 downregulated circRNAs were distinctly associated with CAD. The results suggest that these circRNAs have important roles in the development of atherosclerosis in humans.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant No. 30400173, 30971257, 81170180, and 81970302) and the Priority Academic Program Development of Jiangsu Higher Education Institutions. This study received support from the National Natural Science Foundations of China (No. 81170180, 30400173, 30971257, and 81970302) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Disclosure of conflict of interest
None.
Abbreviations
- circRNAs
Circular RNAs
- H&E
Haematoxylin and Eosin
- RNA-seq
RNA-sequencing
- miRNA
microRNA
- CAD
coronary artery disease
- LAD
left anterior descending
- LCX
left circumflex
- RCA
right coronary artery
- LM
left main trunk
- AHA
American Heart Association
- TPM
transcript per million
- FPKM
fragments per kilobase million
- KEGG
Kyoto encyclopedia of genes and genomes
- ARVC
Arrhythmogenic right ventricular cardiomyopathy
- rRNA
ribosomal RNA
- NGS
next-generation sequencing
- qRT-PCR
quantitative real-time polymerase chain reaction
- HCASMC
human coronary artery SMC
Supporting Information
References
- 1.Mahmoudi E, Fitzsimmons C, Geaghan M, Shannon Weickert C, Atkins J, Wang X, Cairns MJ. Circular RNA biogenesis is decreased in postmortem cortical gray matter in schizophrenia and may alter the bioavailability of associated miRNA. Neuropsychopharmacology. 2019;44:1043–1054. doi: 10.1038/s41386-019-0348-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 3.Chen L, Zhang YH, Huang G, Pan X, Wang S, Huang T, Cai YD. Discriminating cirRNAs from other lncRNAs using a hierarchical extreme learning machine (H-ELM) algorithm with feature selection. Mol Genet Genomics. 2018;293:137–149. doi: 10.1007/s00438-017-1372-7. [DOI] [PubMed] [Google Scholar]
- 4.Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez-Hernandez D, Ramberger E, Shenzis S, Samson M, Dittmar G, Landthaler M, Chekulaeva M, Rajewsky N, Kadener S. Translation of CircRNAs. Mol Cell. 2017;66:9–21. doi: 10.1016/j.molcel.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160:1125–1134. doi: 10.1016/j.cell.2015.02.014. [DOI] [PubMed] [Google Scholar]
- 6.Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y, Wong DT, Xiao X. The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin Chem. 2015;61:221–230. doi: 10.1373/clinchem.2014.230433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lusis AJ, Mar R, Pajukanta P. Genetics of atherosclerosis. Annu Rev Genomics Hum Genet. 2004;5:189–218. doi: 10.1146/annurev.genom.5.061903.175930. [DOI] [PubMed] [Google Scholar]
- 8.Bijnens AP, Lutgens E, Ayoubi T, Kuiper J, Horrevoets AJ, Daemen MJ. Genome-wide expression studies of atherosclerosis: critical issues in methodology, analysis, interpretation of transcriptomics data. Arterioscler Thromb Vasc Biol. 2006;26:1226–1235. doi: 10.1161/01.ATV.0000219289.06529.f1. [DOI] [PubMed] [Google Scholar]
- 9.Shendure J. The beginning of the end for microarrays? Nat Methods. 2008;5:585–587. doi: 10.1038/nmeth0708-585. [DOI] [PubMed] [Google Scholar]
- 10.Stary HC. Natural history and histological classification of atherosclerotic lesions: an update. Arterioscler Thromb Vasc Biol. 2000;20:1177–1178. doi: 10.1161/01.atv.20.5.1177. [DOI] [PubMed] [Google Scholar]
- 11.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and ballgown. Nat Protoc. 2016;11:1650–67. doi: 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, Kocks C, le Noble F, Rajewsky N. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 14.Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015;16:4. doi: 10.1186/s13059-014-0571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guria A, Velayudha Vimala Kumar K, Srikakulam N, Krishnamma A, Chanda S, Sharma S, Fan X, Pandi G. Circular RNA profiling by illumina sequencing via template-dependent multiple displacement amplification. Biomed Res Int. 2019;2019:2756516. doi: 10.1155/2019/2756516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arnaiz E, Sole C, Manterola L, Iparraguirre L, Otaegui D, Lawrie CH. CircRNAs and cancer: biomarkers and master regulators. Semin Cancer Biol. 2019;58:90–99. doi: 10.1016/j.semcancer.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Willecke F, Yuan C, Oka K, Chan L, Hu Y, Barnhart S, Bornfeldt KE, Goldberg IJ, Fisher EA. Effects of high fat feeding and diabetes on regression of atherosclerosis induced by low-density lipoprotein receptor gene therapy in LDL receptor-deficient mice. PLoS One. 2015;10:e0128996. doi: 10.1371/journal.pone.0128996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukovic D, Gugerell A, Zlabinger K, Winkler J, Pavo N, Baranyai T, Giricz Z, Varga ZV, Riesenhuber M, Spannbauer A, Traxler D, Jakab A, Garamvölgyi R, Petnehazy Ö, Pils D, Tóth L, Schulz R, Ferdinandy P, Gyöngyösi M. Transcriptional alterations by ischaemic postconditioning in a pig infarction model: impact on microvascular protection. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20020344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao P, Wu H, Zhong Z, Zhang Q, Zhong W, Li B, Li C, Liu Z, Yang M. Expression profiles of long noncoding RNAs and mRNAs in peripheral blood mononuclear cells of patients with acute myocardial infarction. Medicine (Baltimore) 2018;97:e12604. doi: 10.1097/MD.0000000000012604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol. 2018;113:26. doi: 10.1007/s00395-018-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong R, Chen M, Zhang C, Chen M, Li H. A comparison of gene expression profiles in patients with coronary artery disease, type 2 diabetes, and their coexisting conditions. Diagn Pathol. 2017;12:44. doi: 10.1186/s13000-017-0630-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iacobellis G, Camarena V, Sant DW, Wang G. Human epicardial fat expresses glucagon-like peptide 1 and 2 receptors genes. Horm Metab Res. 2017;49:625–630. doi: 10.1055/s-0043-109563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nurnberg ST, Cheng K, Raiesdana A, Kundu R, Miller CL, Kim JB, Arora K, Carcamo-Oribe I, Xiong Y, Tellakula N, Nanda V, Murthy N, Boisvert WA, Hedin U, Perisic L, Aldi S, Maegdefessel L, Pjanic M, Owens GK, Tallquist MD, Quertermous T. Coronary artery disease associated transcription factor TCF21 regulates smooth muscle precursor cells that contribute to the fibrous cap. PLoS Genet. 2015;11:e1005155. doi: 10.1371/journal.pgen.1005155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.