

Abstract
The current pharmacological therapies for osteoarthritis (OA) are mainly focused on symptomatic relief of pain and inflammation through the use of nonsteroidal anti-inflammatory drugs (NSAIDs). Etoricoxib is a cyclooxygenase-2 (COX-2) selective NSAID with a higher cyclooxygenase-1 (COX-1) to COX-2 selectivity ratio than the other COX-2 selective NSAIDs and a lower risk of gastrointestinal toxicity compared to traditional NSAIDs. In this study, we first evaluated the anti-inflammatory and chondro-protective effects of etoricoxib on interlecukin-1β-stimulated human osteoarthritic chondrocytes. We found that etoricoxib not only inhibited the expression of inflammation mediators COX-2, prostaglandin E2 (PGE2), and nitric oxide, but also had a similar chondro-protective effect to celecoxib through down-regulating matrix degrading enzymes matrix metalloproteinase-13 (MMP-13) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS-5). We then used PLGA-PEG-PLGA triblock copolymeric nanoparticles (NPs) as a drug delivery system to locally deliver etoricoxib into the articular cavity to reduce the risk of cardiovascular toxicity of etoricoxib when administered systemically or orally. The etoricoxib-loaded NPs showed a sustained drug release over 28 days in vitro; in rat OA model, the intra-articular injection of etoricoxib-loaded NPs alleviated the symptoms of subchondral bone, synovium, and cartilage. In conclusion, our study confirmed the chondro-protective role of etoricoxib in OA, and proved the curative effects of etoricoxib-loaded PLGA-PEG-PLGA NPs in vivo.
Keywords: Osteoarthritis, etoricoxib, chondrocytes, PLGA-PEG-PLGA triblock copolymers, polymeric nanoparticles
Introduction
Osteoarthritis (OA) is a multifactorial degenerative joint disorder associated with structural changes of joint tissues, including loss of articular cartilage, remodeling of subchondral bone, and inflammation of the synovium. OA causes progressive disabilities in the elderly due to its irreversible outcomes [1]. The treatments for patients suffering from OA are currently very limited because the pathological mechanism of OA remains poorly understood [2]. To date, OA pharmacological therapies are mainly focused on symptomatic relief of pain and inflammation using nonsteroidal anti-inflammatory drugs (NSAIDs). The American College of Rheumatology (ACR) recommends administering either a traditional NSAID or a cyclooxygenase-2 (COX-2) selective inhibitor for pain relief in patients with OA [3,4]. However, traditional NSAIDs are linked to a higher risk of gastrointestinal (GI) toxicity due to their inhibition of cyclooxygenase-1 (COX-1) enzyme, while COX-2 selective inhibitors are associated with an elevated risk of cardiovascular (CV) events [5,6].
Etoricoxib is a COX-2 selective NSAID with a higher COX-1 to COX-2 selectivity ratio than the other COX-2 selective NSAIDs and a lower risk of GI toxicity compared to traditional NSAIDs [7]. In OA treatment, etoricoxib has shown a comparable efficacy to traditional NSAIDs and a greater efficacy than placebo [8,9]. However, it has been reported recently that etoricoxib induces CV toxicity when administered systemically or orally [10,11]. Direct intra-articular (IA) injection can minimize systemic side effects by reducing the blood concentration of etoricoxib, but repeated (e.g. weekly) injection increases the risk of joint infection. Meanwhile, unformulated molecules within the injected drugs are cleared rapidly from the IA space [12]. In order to reduce the distribution of etoricoxib in non-target organs and prolong the drug retention in the joint cavity, IA drug delivery system (DDS) that delivers etoricoxib in a controlled manner in the joint cavity for a longer duration of time would be a better option.
An ideal IA DDS should offer controlled release of the therapeutic agent with extended bioavailability and joint retention, have no or minimal safety concerns, promise a disease-modifying effect and/or cartilage regeneration, and be readily translatable. So far, the only FDA-approved IA DDS is poly(lactic-co-glycolic acid) (PLGA) [13]. Block co-polymers containing PLGA and polyethyleneglycol (PEG) blocks are gaining attention in controlled release due to their thermogelling and biocompatible properties [14,15]. The triblock copolymer PLGA-PEG-PLGA is one the most widely studied polymer, and is very suitable for delivering hydrophobic drugs such as etoricoxib in a controlled manner.
In this study, we first investigated the effects of etoricoxib treatment on human osteoarthritic chondrocytes; we then used PLGA-PEG-PLGA triblock copolymer to prepare the etoricoxib-loaded nanoparticles (NPs); we finally delivered the etoricoxib-loaded NPs in rat OA model, and tested their efficacy in vivo.
Materials and methods
Isolation and culture of human osteoarthritic chondrocytes
Cartilage specimens were obtained intraoperatively from patients (4 male and 14 female, average age 65±10 years) undergoing total knee arthroplasties, with approval (No: 201802060029) from Ethics Committee of Southwest Hospital (Chongqing, China). Chondrocytes were isolated according to our previous protocol [16]. Briefly, cartilage pieces were digested with 0.2% (w/v) collagenase type II (Sigma) for 10 h at 37°C. The resulting cell suspension was filtered through a 40 μm cell strainer (BD Biosciences), and centrifuged at 400 g for 5 min. The collected cells were resuspended in high glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (TBD). Finally, cells were incubated in a humidified atmosphere of 5% CO2 at 37°C. The medium was changed every 2-3 days. Chondrocytes at passage 1 were used in the following experiments.
Live/dead cell staining and cell viability assay
The effects of etoricoxib on the viability of chondrocytes were evaluated using a Live/Dead Staining kit (Yeasen). Briefly, Chondrocytes were treated with etoricoxib at different concentrations (0, 10, 50, 100, 500, or 1000 μM) for 48 h, then the cells were incubated in Assay Buffer containing 2 µM Calcein-AM and 1.5 µM propidum iodine (PI) for 15 min at 37°C. Labeled cells were visualized using a confocal microscope (Olympus IX71). Live cells were stained green, whereas dead cells were stained red.
To further evaluate the cytotoxicity of etoricoxib, Cell Counting Kit-8 (CCK-8; Dojindo Laboratories) was used. Chondrocytes were cultured in 96-well plates at a density of 7×103 cells per well for 24 h. Then, cells were treated with etoricoxib at different concentrations (0, 10, 50, 100, 500, or 1000 μM) for 48 h. After that, 10 μl of CCK-8 solution was added to each well, and the mixture was incubated at 37°C for 2 h. The optical density (OD) was read at a wavelength of 450 nm with a microplate reader (Thermo Fisher Scientific).
Treatment of human OA chondrocytes with etoricoxib and IL-1β
Chondrocytes at passage 1 were seeded in high glucose DMEM supplemented with 2% FBS overnight and were then stimulated with IL-1β (10 ng/ml) for 2 h followed treatment with etoricoxib (10 μM). Chondrocytes treated with high glucose DMEM supplemented with 2% FBS were served as control.
Flow cytometry of cell apoptosis detection
Annexin V-FITC-PI kit (4A biotech) was used to detect cell apoptosis. Briefly, cell suspension (1×106 cells in a 1.5 ml-centrifuge tube) was centrifuged at 300 g for 5 min, and the supernatant was discarded. Afterwards, 1 ml of PBS was added to the cell pellet, and the samples were centrifuged at 300 g for 5 min. This washing procedure was repeated twice. Subsequently, 100 μl of binding buffer, 5 μl of Annexin V-FITC, and 10 μl of PI were added, and the samples were incubated at room temperature (RT) for 15 min in the dark. Finally, 400 μl of binding buffer was added to the samples, and cell apoptosis was detected by flow cytometry.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted using RNA pure Total RNA Kit (Bioteke), and the RNA concentration was determined using a Nanodrop-2000 spectrophotometer (Thermo Scientific). cDNA was synthesized using Transcriptor-First-Strand-cDNA-Synthesis-Kit (Roche) according to the manufacturer’s instructions. Quantitative real-time PCR based on FastStart Essential DNA Green Master (Roche) was performed. Relative gene expression was calculated by the ΔΔCt method. The ΔCt was calculated using the reference gene β-actin. Target gene primers were presented in Table 1.
Table 1.
Target gene primers
| Name | Forward | Reverse |
|---|---|---|
| COL2A1 | TGCTGCCCAGATGGCTGGAGGA | TGCCTTGAAATCCTTGAGGCCC |
| ACAN | TCGAGGACAGCGAGGCC | TCGAGGGTGTAGCGTGTAGAGA |
| SOX9 | GACTTCCGCGACGTGGAC | GTTGGGCGGCAGGTACTG |
| MMP-13 | TCCTGGCTGCCTTCCTCTTCTTG | AGTCATGGAGCTTGCTGCATTCTC |
| ADAMTS-5 | GACCGATGGCACTGAATGTAGGC | TCTCCTCCACATACTCCGCACTTG |
| COX-2 | TGGTCTGGTGCCTGGTCTGATG | CCTGCTTGTCTGGAACAACTGCTC |
| iNOS | GACTTTCCAAGACACACTTCAC | TTCGATAGCTTGAGGTAGAAGC |
| β-Actin | CCTGGCACCCAGCACAAT | GGGCCGGACTCATAC |
Enzyme-linked immunosorbent assay (ELISA)
The supernatant of the culture medium was collected after 72 h treatment of etoricoxib. The expression of COX-2 and Prostaglandin E2 (PGE2) was detected using ELISA kits (CUSABIO). According to the kit instructions, 100 μl standard or sample was added to each well and incubated for 2 h at 37°C. Then, 100 μl of Biotin antibody (1×) was added to each well and incubated for 1 h at 37°C. After three washes, 100 μl of HRP-avidin (1×) was added to each well and incubated for 1 h at 37°C. After five washes, 90 μl of TMB Substrate was added to each well and incubated for 20 min at 37°C in the dark. Finally, 50 μl of Stop Solution was added to each well. The OD value of each well was measured at a wavelength of 450 nm. This was positively correlated to its respective concentration of COX-2 and PGE2. The experiment was repeated three times.
Measurement of nitric oxide
The nitric oxide (NO) levels in the culture medium were determined by the Griess reaction. Briefly, 50 μl of the cell culture supernatant was reacted with an equal volume of Griess reagent (Beyotime) in 96-well plates for 10 min at RT in the dark. Nitrite levels were determined by measuring the absorbance at 540 nm using a spectrophotometer. The levels of nitrite were normalized to standard values.
Western blot
The cells were lysed in RIPA with 1% PMSF (Beyotime). Proteins were separated in 8% to 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (according to the molecular weights) and transferred to a PVDF membrane (Beyotime) at 200 mA for 1.5 h at 4°C. The blot was blocked with QuickBlock™ Blocking Buffer (Beyotime) for 2 h at RT and incubated separately with the following primary antibodies: type II collagen (1:5000; Abcam), aggrecan (1:100; Abcam), SOX9 (1:1000; Abcam), MMP-13 (1:3000; Abcam), a disintegrin and metalloproteinase with thrombospondin motifs-5 (ADAMTS-5; 1:250; Abcam), β-actin (1:200; San-ta Cruz) for overnight at 4°C, followed by incubation with horseradish peroxidase-conjugated secondary antibody (Goat Anti-Mouse or Goat Anti-Rabbit, 1:2000, Proteintech) for 1 h at RT.
Immunofluorescence staining
Cells were washed with PBS and fixed with 4% formaldehyde for 10 min at RT. Then, cells were washed three times with cold PBS, and treated with Triton X-100 (Beyotime) for 10 min at RT. Cells were washed again three times with PBS and blocked 1 h with Blocking Buffer (Beyotime) at RT followed by incubation with primary antibodies: Collagen II (1:200; Abcam), Aggrecan (1:100; Abcam), SOX9 (1:200; Abcam) for overnight at 4°C. Next, the cells were washed three times with PBST, incubated with secondary antibody for 1 h at RT. Cell nuclei were counterstained with 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI; Beyotime) for 5 min at 37°C. The images were obtained by confocal fluorescent microscope (LSM710, Carl Zeiss).
Preparation of etoricoxib-loaded PLGA-PEG-PLGA triblock copolymer NPs
Etoricoxib (A3405, APExBIO)-loaded NPs were prepared by Oil in Water (O/W) emulsion solvent evaporation technique with a modification of literature procedure [17]. 15 mg of the PLGA3000-PEG2000-PLGA3000 triblock copolymers (Ruixi Biological, PL:GA=1:1, PDI<1.2) and 7 mg of etoricoxib were dissolved in 1.5 mL of dichloromethane (DCM), and the solutions were added drop by drop to 10 mL of 0.5% poly vinyl alcohol (PVA) aqueous solution for emulsification at 2500 rpm using magnetic stirrers. The resulting emulsion was further stirred for 3 h at 2500 rpm to remove excess DCM and harden the formed NPs. The etoricoxib-loaded NPs dispersion was filtered through a 0.45 μm strainer to remove the larger particles and again centrifuged at 15000 g for 30 min. The supernatant was removed, and the NPs were washed with deionized water three times, and freeze-dried.
Characterization of NPs
The spectra of the PLGA-PEG-PLGA, etoricoxib, NPs, and etoricoxib-loaded NPs were determined using Fourier transform infrared spectroscopy (FTIR) spectrometer (PerkinElmer, US). The spectra between the wavelengths of 400 and 4000 cm-1 were recorded. The morphology of the NPs was studied using a field-emission scanning electron microscopy (SEM; S-3400N II, Hitachi) and a transmission electron microscope (TEM; TECNAI 10, Philips). Samples were mixed with Milli-Q water and a drop of the resulting NP suspension was placed on a glass slide, dried in air, and gold sputter-coated before SEM observation. For TEM analysis, a drop of aqueous solution containing the NPs was placed on carbon-coated copper grids. The samples were dried and kept under a desiccator before being loaded onto a specimen holder, operated at 200 kV. Samples were dispersed at 0.1 mg/ml in Milli-Q water in a fresh polystyrene cuvette, and were subjected to a minimum of 12 runs after equilibrating at the desired temperature for 5 min. The mean diameter, PDI, and Zeta potential of NPs were determined by dynamic light scattering (DLS).
In vitro release of etoricoxib from NPs
The drug release from the NPs was done using dialysis membrane method [18] in PBS. 10 mg of the drug-loaded NPs were dispersed in 2 ml of PBS, and then transferred to a dialysis bag (MWCO 3500), suspended in 500 ml of PBS at 37°C with continuous agitation. 1 ml buffer was removed and replaced with the same amount of fresh buffer at 1, 2, 3, 7, 14, 21, and 28 days. The amount of free etoricoxib in the supernatant was measured by UV-Vis spectrophotometer (NanoDrop 2000, Thermo Scientific).
Cytotoxicity test of NPs
The cytotoxicity of etoricoxib NPs was evaluated by the CCK-8 assay. Briefly, passage 1 chondrocytes were plated in 96-well plates at a density of 7×103 cells per well, and then treated with NPs that can respectively maximally release etoricoxib to 10 μM according to the release curve. After incubation for 2 days with the NPs, the absorbance was measured using a spectrophotometric reader at 450 nm.
OA induction in rats
Animal experiments were approved by Animal Ethics Committee of Third Military Medical University (No. 201805300061). A total of 20 healthy female Sprague-Dawley rats (8-week-old) were supplied by the animal experiment center of Third Military Medical University. OA was induced by anterior cruciate ligament transection (ACLT) as previously described [19]. Rats were anesthetized with 1% pentobarbital sodium, and the right knee joint was exposed through a medial parapatellar approach. The patella was dislocated laterally, and the knee was placed in full flexion followed by ACLT with micro-scissors.
IA injections
The rats were randomly divided into the following four groups (n=5/group): (1) ACLT+ saline (Control, IA injections of 100 μl saline); (2) ACLT+ etoricoxib (ETX, IA injections of 10 μM etoricoxib in 100 μl saline); (3) ACLT+NPs (NPs, IA injections of 6.93 μg NPs in 100 μl saline); (4) ACLT+ etoricoxib NPs (ETX-NPs, IA injections of 6.93 μg ETX-NPs in 100 μl saline). IA injections were performed at 3, 6, and 9 weeks. Rats were sacrificed for analysis at 12 weeks after OA induction.
Microtomography and histology
High-resolution microtomography (μCT) scanning (viva CT40; Scanco, Switzerland) was performed to measure subchondral bone of knee joints at 12 weeks after OA induction. The knees were fixed in 4% paraformaldehyde for 48 h. The epiphysis of the tibia was manually chosen as the region of interest in 3D analysis of the subchondral bone. After reconstruction, the following parameters were measured: relative bone volume to total volume (BV/TV), trabecula number (Tb. N), trabecula separation (Tb. Sp), trabecula thickness (Tb. Th).
The joints were fixed in 4% (v/v) paraformaldehyde for 48 h, and tissues were decalcified in 10% wt EDTA disodium salt dehydrate (GRM1195, neofroxx, Germany) solution for 3 weeks at RT. After serial dehydration, the samples were embedded in paraffin and sectioned at 4 μm thickness. The sections were stained with hematoxylin and eosin and safranin-O/fast green. Osteoarthritis Research Society International (OARSI) grading system was used to evaluate the degenerative status [20]. Scoring was done by two blinded observers (TL and XW). Immunohistochemistry (IHC) was performed using Biotin-Streptavidin HRP-based SPlink Detection Kits (ZSGB-BIO). After deparaffinization and hydration with distilled water, the antigen repair was conducted at 100°C for 10 min. Then the tissue slices were treated with H2O2 for about 20 min and then blocked for 30 min avoiding the homologous serum. The diluted (1:100-400) primary antibodies were used to incubate the slices for overnight at 4°C. After three times wash with PBS, the tissue slices were incubated with biotinylated Goat Anti-Mouse IgG for 15 min at RT. Subsequently, the slices were treated with streptavidin avidin-biotin enzyme complex for 15 min. Proteins were visualized by the DAB solution kit (ZSGB-BIO).
Statistical analysis
Results were represented as mean ± standard deviation (mean ± SD). The cytotoxicity of the NPs to human chondrocytes was analyzed using t-tests, the other results were analyzed using one-way analysis of variance (ANOVA). *P<0.05; **P<0.01; ***P<0.001.
Results
Effects of etoricoxib on human OA chondrocytes viability and apoptosis
The human OA chondrocytes were treated with various concentrations of etoricoxib (0, 10, 50, 100, 500, and 1000 μM) for 48 h. Live-dead cell staining results (Figure 1A) showed that, treatment with 500 μM and 1000 μM etoricoxib resulted in a lower density of live cells (green) and a greater distribution of dead cells (red). Consistently, the CCK-8 assay (Figure 1B) showed that treatment with 500 μM or 1000 μM etoricoxib reduced the cell viability to 81.56±4.43% and 67.93±3.16%, respectively, both significantly lower than control. The concentration of 10 μM was used for subsequent experiments because it is the maximum pharmacological plasma concentration of etoricoxib [21].
Figure 1.
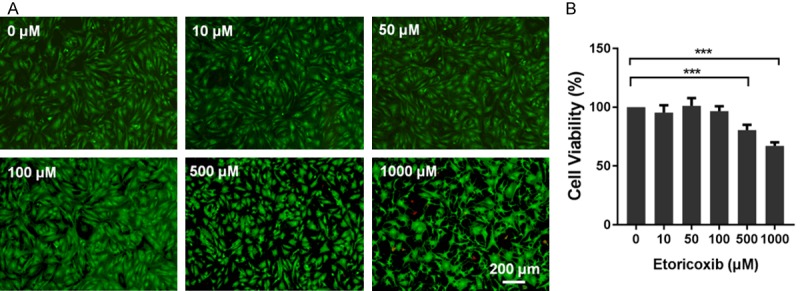
Effect of etoricoxib on viability of human osteoarthritic chondrocytes. Cells were cultured with increasing concentrations of etoricoxib (0, 10, 50, 100, 500, 1000 μM) for 48 h. The cell viability was determined by the Live-Dead Cell Staining (A) and CCK-8 assay (B). (n=5, mean ± SD, ***P<0.001 in comparison with the control group).
Flow cytometry was used to detect the effect of etoricoxib on chondrocyte apoptosis. Annexin V/PI double staining (Figure 2A) showed that the increased apoptosis rate could be observed in the IL-1β group, compared with the control group; while the IL-1β+ETX group exhibited the reducing trend of apoptosis rate. However, there was no statistical difference between groups in both early apoptosis rate (Figure 2B) and late apoptosis rate (Figure 2C).
Figure 2.

Effect of etoricoxib on apoptosis of human osteoarthritic chondrocytes. Flow cytometry was used to detect the effect of etoricoxib on chondrocyte apoptosis. A. Representative images of the apoptosis analysis. B. Quantified early apoptosis rate of chondrocytes. C. Quantified late apoptosis rate of chondrocytes.
Effects of etoricoxib on inflammatory factor expression and ECM degradation in IL-1β-stimulated chondrocytes
Inflammation and ECM degradation are factors that clearly enhance the progression of OA. Here we examined the influence of etoricoxib on the production of inflammatory mediators (COX-2, iNOS, PGE2, and NO) and matrix degrading enzymes (MMP-13 and ADAMTS-5). We found that IL-1β markedly increased the mRNA expression of COX2, iNOS, MMP-13 and ADAMT-5 compared with control group (Figure 3A-D), while IL-1β and etoricoxib co-treatment (IL-1β+ETX) downregulated the expression of iNOS (Figure 3B) and MMP-13 (Figure 3C). Consistent with the RT-PCR results, the protein levels of COX-2, MMP-13, and ADAMTS-5 in IL-1β+ETX group were significantly lower than those in IL-1β group (Figure 3E, 3F). Moreover, the concentration of PGE2 and NO in IL-1β+ETX group was significantly lower than IL-1β group (Figure 3G, 3H).
Figure 3.
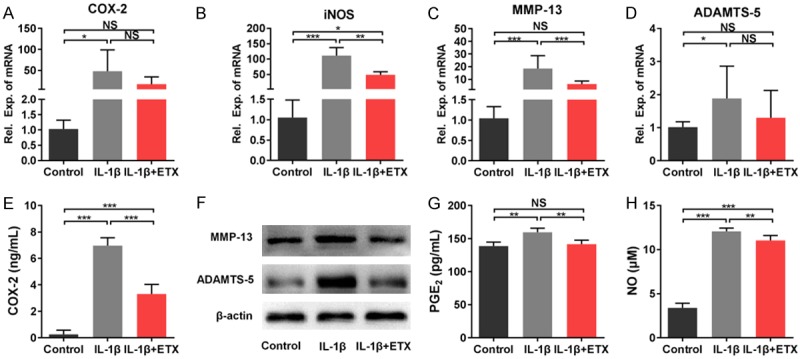
Effects of etoricoxib on inflammatory factor expression and ECM degradation in IL-1β-stimulated chondrocytes. A-D. Gene expression of COX-2, iNOS, MMP-13, and ADAMTS-5 detected by qPCR analysis. E. Concentration of COX-2 measured by ELISA. F. Protein expression of MMP-13 and ADAMTS-5 detected by western blot analysis. G. Concentration of PGE2 measured by ELISA. H. NO levels measured by Griess reaction. (NS: not significant, *P<0.05, **P<0.01, ***P<0.001).
Effects of etoricoxib on the chondrogenic phenotype of IL-1β-stimulated chondrocytes
The chondrogenic markers type II collagen, aggrecan, and SOX9 were measured by immunofluorescence staining, RT-PCR, and western blot. We found that IL-1β stimulation induced a weaker staining of these marker proteins, whereas the staining intensity of IL-1β+ETX group was comparable to that of control group (Figure 4A). PCR results showed that the mRNA expression of SOX9 (Figure 4B), ACAN (Figure 4C), and COL2A1 (Figure 4D) in IL-1β+ETX group was higher than that in control group and IL-1β group. The western blot results were consistent with the immunofluorescence staining, with IL-1β group having lower expression, and IL-1β+ETX group having comparable expression of type II collagen, aggrecan, and SOX9 compared to control group (Figure 4E).
Figure 4.
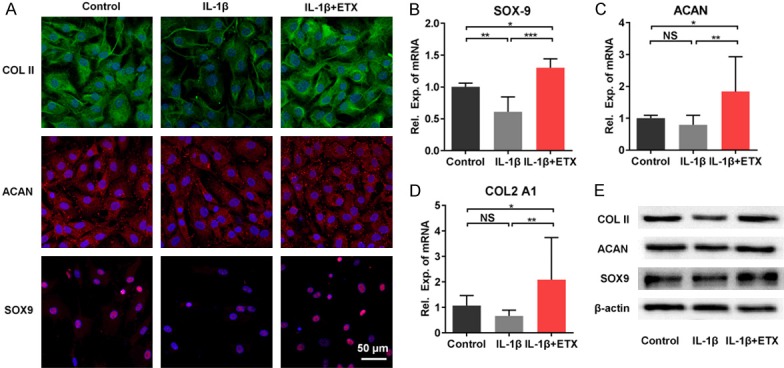
Effects of etoricoxib on the chondrogenic phenotype of IL-1β-stimulated chondrocytes. A. Immunofluorescent staining of type II collagen, aggrecan, and SOX9. B-D. Gene expression of SOX-9, ACAN, and COL2A1. (NS: not significant, *P<0.05, **P<0.01, ***P<0.001). E. Protein expression of type II collagen, aggrecan, and SOX9.
Characterization of etoricoxib-loaded PLGA-PEG-PLGA NPs
FTIR analyses (Figure 5A) were used to find any changes in the chemical structure of PLGA-PEG-PLGA and etoricoxib during the formation of NPs. The FTIR spectra of pure etoricoxib showed that the stretching vibration of C=N group appeared at 1597.7 cm-1. Pure PLGA-PEG-PLGA copolymer showed that a peak at 1750.3 cm-1 belonged to the carbonyl group. The absorption band of pure etoricoxib and PLGA-PEG-PLGA both appeared on the FTIR spectra of etoricoxib-loaded PLGA-PEG-PLGA NPs; there were no new peaks, indicating no chemical interaction between etoricoxib and the NPs. The surface morphology of etoricoxib-loaded PLGA-PEG-PLGA NPs was studied by SEM and TEM (Figure 5B, 5C). From the micrographs, it was clearly shown that the NPs were spherical and regular in shape. The size distribution of the NPs was characterized by DLS (Figure 5D), the average diameter of the NPs was 339 nm (PDI=0.207) with a uniform size distribution. These nanoparticles had a slight positive charge (1.68±0.85 mV) as determined by zeta potential measurements.
Figure 5.
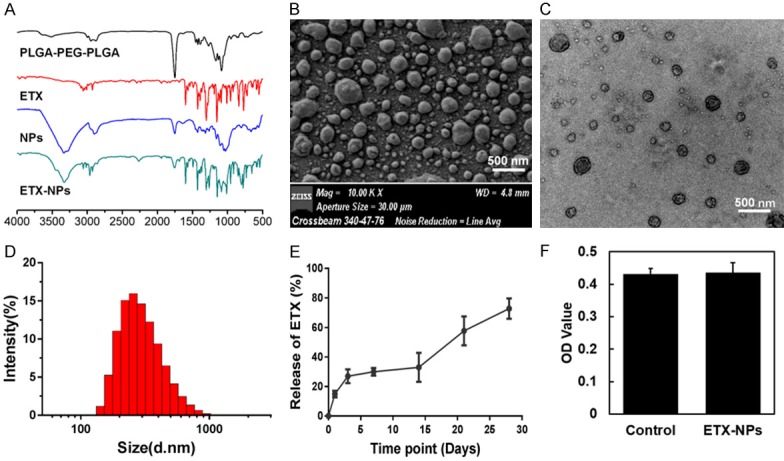
Characterization of etoricoxib-loaded PLGA-PEG-PLGA NPs. (A) FTIR analyses were used to find any changes in the chemical structure of PLGA-PEG-PLGA and etoricoxib during the formation of NPs. SEM (B) and TEM (C) images showed that the NPs were spherical and regular in shape. The size distribution of the NPs was characterized by DLS (D), the average diameter was 339 nm (PDI=0.207). (E) In vitro release of etoricoxib-loaded NPs in PBS buffer showed an initial burst release at the third day. (F) Cytotoxicity of the NPs to human chondrocytes.
In vitro release of etoricoxib from the NPs
To investigate whether etoricoxib-loaded PLGA-PEG-PLGA NPs were able to slowly release etoricoxib, in vitro release performance was tested. Following a short initial burst release of ~26.92% of total drug load in the first three days, etoricoxib was slowly released throughout time (Figure 5E). Cell viability assay (Figure 5F) showed that the etoricoxib-loaded NPs did not affect the viability of chondrocytes in vitro.
In vivo performance of etoricoxib-loaded NPs in rat OA model
To examine the changes of subchondral bone after IA injection of etoricoxib-loaded NPs, the tibial subchondral bone volume was determined by three-dimensional µCT (Figure 6A). Quantitative analysis of the µCT data including bone volume/total volume (BV/TV), trabecular number, trabecular thickness, and trabecular separation were performed. The ETX-NPs group showed an increase in both BV/TV (Figure 6B) and trabecular number (Figure 6C) compared with control group and NPs group. However, there was no significant difference in trabecular thickness (Figure 6D) and trabecular separation (Figure 6E) between groups.
Figure 6.
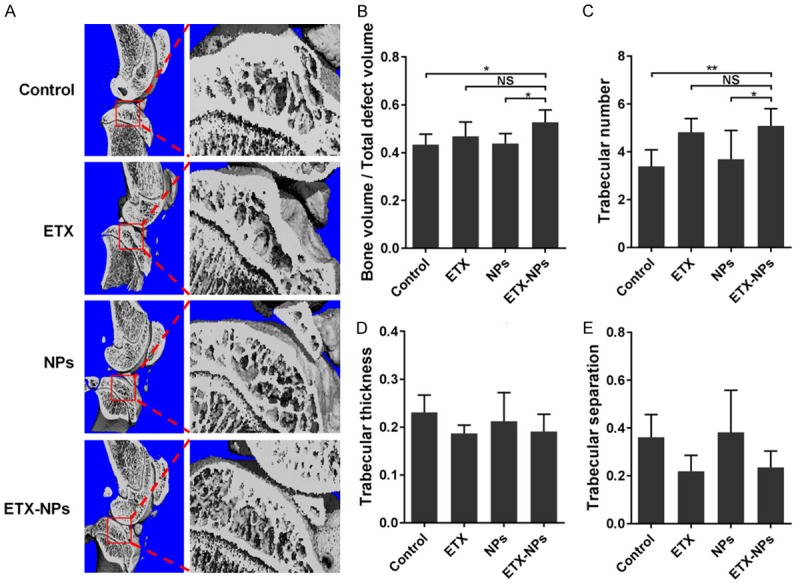
The changes of subchondral bone after IA injection of etoricoxib-loaded NPs. μCT scanning was performed to measure subchondral bone of knee joints at 12 weeks after OA induction (A). (B-E) Quantified bone volume/total volume, trabecular number, trabecular thickness, and trabecular separation, respectively. (NS: not significant, *P<0.05, **P<0.01).
After three IA injections (with 3 weeks interval between each injection), the rats were sacrificed at 12 weeks post OA induction (Figure 7A). There was no inflammation reaction or tissue necrosis at the surgery site. Gross images of the femoral condyle showed that, ETX group and ETX-NPs group had a smoother articular surface than control group (Figure 7B). Safranin-O/Fast green staining further showed that the ETX+NPs group had the best osteochondral interface among all groups (Figure 7C). Consistently, the OARSI score of ETX+NPs group was 8.67±1.56, lowest of all groups (Figure 7D). Hematoxylin and eosin staining showed that, the synovial lining cell layer slightly enlarged, and the density of the synovial stroma slightly increased in the ETX-NPs group (Figure 7E). Similarly, the synovitis score of ETX+NPs group was 1.45±0.69, lowest of all groups (Figure 7F).
Figure 7.

Histology evaluation after IA injection of the etoricoxib-loaded NPs. A. OA was induced by ACLT and IA injections were performed at 3, 6, and 9 weeks. Rats were sacrificed for analysis at 12 weeks after OA induction. B. Gross images of the femoral condyle. C. Safranin-O/Fast green staining of the joints. D. OARSI scores of the four groups at indicated time points. (**P<0.01, ***P<0.001). E. Hematoxylin and eosin staining of the synovium. In addition to ETX NPs group, the other groups showed a slight increase in the synovial lining cell layer, a slight increase in the density of synovial matrix, and inflammatory infiltration of small follicular like lymphocytes. F. Synovitis score. (**P<0.01, ***P<0.001).
The metabolism of cartilage matrix was evaluated by immunohistochemistry of matrix protein type II collagen and aggrecan, and catabolic markers MMP-13 and ADAMTS-5 (Figure 8). We found that the expression of type II collagen and aggrecan in the ETX-NPs group was strengthened compared with other groups. Correspondingly, the expression of MMP-13 and ADAMTS-5 in ETX-NPs group was lower than that in other groups, indicating that IA injection of ETX-NPs could inhibit the activity of matrix degrading enzymes to maintain the cartilaginous matrix.
Figure 8.
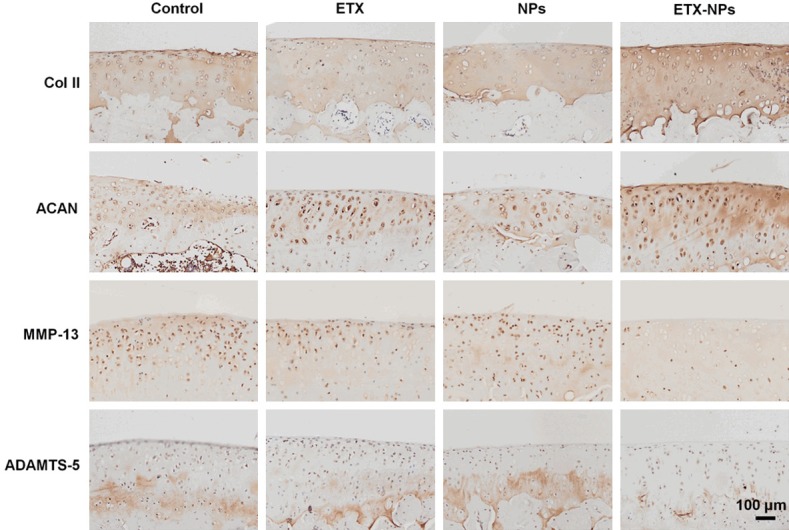
Immunohistochemistry of type II collagen, aggrecan, MMP-13, and ADAMTS-5 after IA injection of the etoricoxib-loaded NPs in rats at 12 weeks.
Discussion
In the last decade, tremendous research work on the NSAIDs treatment for OA has been done, and the understanding of NSAIDs has changed dramatically. The COX-2 inhibitors not only act as analgesics, but also show a great potential in delaying OA progression. For example, the selective COX-2 inhibitor celecoxib has shown a protective effect on OA cartilage [22,23]. Etoricoxib is a highly selective COX-2 inhibitor. Compared with celecoxib, etoricoxib has a higher selectivity for COX-2 and a long half-life (22 h). However, it is unclear whether etoricoxib has a similar chondro-protective effect to celecoxib.
In this study, we for the first time investigated the possible chondro-protective effect of etoricoxib. In cellular experiments, we used articular chondrocytes from OA patients, and stimulated the cells with IL-1β. After IL-1β stimulation, we found an upregulation of MMP-13, ADAMTS-5, COX-2, and PGE2, and a downregulation of chondrogenic markers SOX9, type II collagen, and aggrecan. These changes could be mostly inhibited when the IL-1β-treated chondrocytes were administered with etoricoxib, indicating a chondro-protective role of etoricoxib in vitro. Etoricoxib offset the impact of IL-1β treatment on COX-2 and PGE2 expression, because etoricoxib targets COX-2, and reduces the production of PGE2 by inhibiting COX-2. As for MMP-13 and ADAMTS-5, the former is mainly responsible for degrading type II collagen [24], and the latter is mainly responsible for degrading proteoglycan [25]. Therefore, the expression of MMP-13 and ADAMTS-5 is the core link of cartilage matrix degeneration in OA. There are at least two isoforms of COX. COX-1 is expressed constitutively for physiologic tissue homeostasis, while COX-2 expression is induced during inflammatory state. Among the COX metabolites, PGE2 is considered one of the major mediators of inflammation. It is reported that the spontaneous release of PGE2 is at least 50-fold higher than that in normal cartilage [26]. The therapeutic effect of NSAIDs is mainly attributed to their ability to inhibit COX activity [22]. It has been shown that celecoxib can hinder MMPs and aggrecanase from digesting ECM, and restore matrix production by inhibiting COX-2, PGE2, and NF-kB. Here we speculated that COX-2 and PGE2 could activate matrix degrading enzymes (Figure 9); although the detailed mechanism is not yet clear, it is certain that the effect of etoricoxib was partially achieved by inhibiting the production of MMP-13 and ADAMTS-5 (Figure 8). Whether there are other pathways involved in the protection of chondrocytes from IL-1β-induced matrix degradation requires further investigation. In addition, we found that the level of IL-1β-induced iNOS and NO expression was lowered by etoricoxib administration, but was still much higher than control (Figure 3B, 3H). This result was consistent with a previous study showing that IL-1β induced NO is involved in inhibition of proteoglycan synthesis, independent of PGE2, and thus is insensitive to regulation of COX-2 inhibitors [27].
Figure 9.
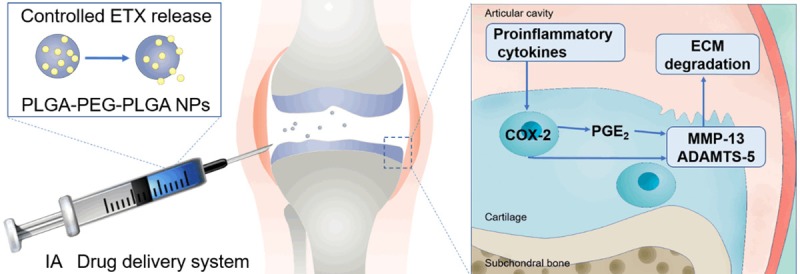
Controlled release of etoricoxib delayed OA progression. Catabolic enzymes, such as MMP-13 and ADAMTS-5 released by chondrocytes, degrade the cartilage matrix. The cartilage degradation products along with the pro-inflammatory cytokines, act on the synovium to induce joint inflammation, which further stimulates the release of catabolic enzymes by chondrocytes. In this study we showed that the controlled release of etoricoxib effectively inhibited the production of MMP-13 and ADAMTS-5, therefore delaying the OA progression of ACLT rats.
Drug administration by IA injection becomes an emerging strategy for OA treatment as it minimizes the adverse effects of drugs administered systemically or orally, and maximizes local effect [28]. However, traditional oral drugs delivered via IA injection are limited by the lack of sustained release [29], in this case an IA DDS might be a better option. At present, the use of PLGA-based DDS for OA treatment has been approved by FDA [30]. Although many benefits have been achieved, some shortcomings also limit the further application of this DDS. In particular, PLGA is a hydrophobic macromolecule likely being scavenged by macrophages [31], meanwhile, the low loading capacity and the high initial release of PLGA is not favorable in drug delivery [32]. To overcome these drawbacks, PLGA copolymers have attracted increasing attention. The amphiphilic triblock copolymer PLGA-PEG-PLGA can self-assemble into NPs due to the hydrophobic interaction between water insoluble segments [33]. The PLGA-PEG-PLGA copolymer NPs have good biocompatibility, biodegradability, high stability, and high loading capacity for hydrophobic drugs, therefore they are ideal candidates for IA DDS.
Drug release from a polymer DDS can be divided into three stages: (1) initial burst release of drug; (2) diffusion release of drug due to dissolution of polymer; (3) second burst release of drug due to polymer degradation [34]. In this study, the in vitro release of etoricoxib from PLGA-PEG-PLGA NPs was done through dialysis in PBS. The initial burst release for the etoricoxib-loaded NPs was 26.92%, and second burst release was observed in 21 days, indicating a well-controlled release of etoricoxib. Similarly, Kamali et al. also used PLGA-PEG-PLGA triblock copolymer to successfully decrease the initial burst release of buprenorphine from an in situ forming gel [32]. The charge of particles is a factor affecting the penetration and retention in cartilage matrix [35]. The negatively charged extracellular matrix inside cartilage provides a unique opportunity to use electrostatic interactions to augment transport, uptake and binding of drug carriers. Positively charged drug-carriers showed faster penetration and higher uptake than their neutral same-sized counterpart [36].
Biocompatibility is a prerequisite for the successful use of DDS in vivo. It has been reported that IA injection of NPs could induce acute inflammatory reactions in mouse joints [37]. In this study, the biocompatibility of PLGA-PEG-PLGA NPs were confirmed both in vitro and in vivo: at cellular levels, the NPs had no significant effect on chondrocyte viability; in animal experiments, after IA injection of NPs, no inflammatory infiltration, hyperplasia, or necrosis of synovium was observed. It seems that the biocompatibility of a delivery vector is related directly to the degree of the hydrophilicity of material surface [38]. Here, the hydrophilic component PEG might account for the excellent biocompatibility of PLGA-PEG-PLGA NPs.
Traditionally, OA was considered to be a disease affected by articular cartilage; nowadays OA has been recognized as a disease of the whole joint affecting articular cartilage, subchondral bone, and synovium [39]. Here we evaluated the changes of these tissues after IA injection of etoricoxib-loaded NPs. Our μCT data showed that, in ACLT-induced OA model the subchondral bone resorption increased, after etoricoxib treatment (both direct IA injection and IA DDS), the remodeling of subchondral bone was delayed as compared to untreated control (Figure 6A-C). This is crucial because in early OA the delayed bone remodeling would slow down the degeneration of the overlying articular cartilage [40,41]. Increasing evidence has shown that chronic inflammation plays an important role in the pathogenesis of OA [42,43], and the synovium is a major source of inflammation [44]. Here we used histological sections to evaluate the inflammatory condition of synovium. Inflammatory cell infiltration and synovial tissue thickening were observed in the rat OA model; after IA injection of etoricoxib-loaded NPs, the inflammatory infiltration of synovium was reduced (Figure 7E). The degeneration of the articular cartilage is another hallmark of OA. Here immunohistochemistry was used to observe cartilage matrix. The light staining of type II collagen and aggrecan in control group indicated the loss of cartilage matrix after OA induction, while the enhanced staining in ETX-NPs group indicated that sustained release of etoricoxib prevented the further loss of cartilage matrix possibly through inhibiting the activity of matrix degrading enzymes (MMP-13, ADAMTS-5) and controlling the production of local inflammatory mediators.
Poly caprolactone (PCL) microparticles (MPs) [45] and PCL MPs loaded chitosan thermogels [46] as IA DDS for etoricoxib have been studied by Arunkumer et al. However, their studies only showed a prolonged drug retention, and lacked the actual therapeutic effect. Our study for the first time verified the chondro-protective effect of etoricoxib-loaded NPs in vivo. Besides, the manufacturing process of PLGA-PEG-PLGA triblock copolymeric NPs is relatively simple, which is conducive to future translational application.
Conclusion
In this study, we confirmed that etoricoxib had a chondro-protective effect in addition to its anti-inflammatory role in OA. The PLGA-PEG-PLGA NPs as an IA DDS displayed a 28-days sustained release of etoricoxib in vitro. When IA delivered in rat OA model, etoricoxib-loaded NPs relieved the symptoms of subchondral bone, synovium, and in particular, cartilage. Therefore, etoricoxib might have a similar disease modifying potential to celecoxib in OA, and IA delivery of etoricoxib via PLGA-PEG-PLGA triblock NPs might be an effective modality for OA treatment.
Acknowledgements
This work was supported by Science Foundation of Guizhou Province (grant numbers: [2019]1428 and [2019]1429).
Disclosure of conflict of interest
None.
References
- 1.Glyn-Jones S, Palmer AJ, Agricola R, Price AJ, Vincent TL, Weinans H, Carr AJ. Osteoarthritis. Lancet. 2015;386:376–87. doi: 10.1016/S0140-6736(14)60802-3. [DOI] [PubMed] [Google Scholar]
- 2.Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, Goldring SR, Jones G, Teichtahl AJ, Pelletier JP. Osteoarthritis. Nat Rev Dis Primers. 2016;2:16072. doi: 10.1038/nrdp.2016.72. [DOI] [PubMed] [Google Scholar]
- 3.Hochberg MC, Altman RD, April KT, Benkhalti M, Guyatt G, McGowan J, Towheed T, Welch V, Wells G, Tugwell P American College of Rheumatology. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken) 2012;64:465–474. doi: 10.1002/acr.21596. [DOI] [PubMed] [Google Scholar]
- 4.Schnitzer TJ American College of Rheumatology. Update of ACR guidelines for osteoarthritis: role of the coxibs. J Pain Symptom Manage. 2002;23(Suppl):S24–30. doi: 10.1016/s0885-3924(02)00372-x. discussion S31-4. [DOI] [PubMed] [Google Scholar]
- 5.Harirforoosh S, Asghar W, Jamali F. Adverse effects of nonsteroidal antiinflammatory drugs: an update of gastrointestinal, cardiovascular and renal complications. J Pharm Pharm Sci. 2013;16:821–847. doi: 10.18433/j3vw2f. [DOI] [PubMed] [Google Scholar]
- 6.Coxib and traditional NSAID Trialists’ (CNT) Collaboration. Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, Bombardier C, Cannon C, Farkouh ME, FitzGerald GA, Goss P, Halls H, Hawk E, Hawkey C, Hennekens C, Hochberg M, Holland LE, Kearney PM, Laine L, Lanas A, Lance P, Laupacis A, Oates J, Patrono C, Schnitzer TJ, Solomon S, Tugwell P, Wilson K, Wittes J, Baigent C. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382:769–779. doi: 10.1016/S0140-6736(13)60900-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takemoto JK, Reynolds JK, Remsberg CM, Vega-Villa KR, Davies NM. Clinical pharmacokinetic and pharmacodynamic profile of etoricoxib. Clin Pharmacokinet. 2008;47:703–720. doi: 10.2165/00003088-200847110-00002. [DOI] [PubMed] [Google Scholar]
- 8.Chen YF, Jobanputra P, Barton P, Bryan S, Fry-Smith A, Harris G, Taylor RS. Cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs (etodolac, meloxicam, celecoxib, rofecoxib, etoricoxib, valdecoxib and lumiracoxib) for osteoarthritis and rheumatoid arthritis: a systematic review and economic evaluation. Health Technol Assess. 2008;12:1–278. iii. doi: 10.3310/hta12110. [DOI] [PubMed] [Google Scholar]
- 9.Song GG, Seo YH, Kim JH, Choi SJ, Ji JD, Lee YH. Relative efficacy and tolerability of etoricoxib, celecoxib, and naproxen in the treatment of osteoarthritis: a Bayesian network meta-analysis of randomized controlled trials based on patient withdrawal. Z Rheumatol. 2016;75:508–516. doi: 10.1007/s00393-015-0023-9. [DOI] [PubMed] [Google Scholar]
- 10.Martin Arias LH, Martin Gonzalez A, Sanz Fadrique R, Vazquez ES. Cardiovascular risk of nonsteroidal anti-inflammatory drugs and classical and selective cyclooxygenase-2 inhibitors: a meta-analysis of observational studies. J Clin Pharmacol. 2019;59:55–73. doi: 10.1002/jcph.1302. [DOI] [PubMed] [Google Scholar]
- 11.Zingler G, Hermann B, Fischer T, Herdegen T. Cardiovascular adverse events by non-steroidal anti-inflammatory drugs: when the benefits outweigh the risks. Expert Rev Clin Pharmacol. 2016;9:1479–1492. doi: 10.1080/17512433.2016.1230495. [DOI] [PubMed] [Google Scholar]
- 12.Maudens P, Jordan O, Allemann E. Recent advances in intra-articular drug delivery systems for osteoarthritis therapy. Drug Discov Today. 2018;23:1761–1775. doi: 10.1016/j.drudis.2018.05.023. [DOI] [PubMed] [Google Scholar]
- 13.Rai MF, Pham CT. Intra-articular drug delivery systems for joint diseases. Curr Opin Pharmacol. 2018;40:67–73. doi: 10.1016/j.coph.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zentner GM, Rathi R, Shih C, McRea JC, Seo MH, Oh H, Rhee BG, Mestecky J, Moldoveanu Z, Morgan M, Weitman S. Biodegradable block copolymers for delivery of proteins and water-insoluble drugs. J Control Release. 2001;72:203–215. doi: 10.1016/s0168-3659(01)00276-0. [DOI] [PubMed] [Google Scholar]
- 15.Zentner GM. Biodegradable block copolymers for delivery of proteins and water-insoluble drugs: reflections and commentary a decade later: original research article: Biodegradable block copolymers for delivery and proteins water-insoluble drugs, 2001. J Control Release. 2014;190:63–64. doi: 10.1016/j.jconrel.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 16.Huang S, Song X, Li T, Xiao J, Chen Y, Gong X, Zeng W, Yang L, Chen C. Pellet coculture of osteoarthritic chondrocytes and infrapatellar fat pad-derived mesenchymal stem cells with chitosan/hyaluronic acid nanoparticles promotes chondrogenic differentiation. Stem Cell Res Ther. 2017;8:264. doi: 10.1186/s13287-017-0719-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gajendiran M, Divakar S, Raaman N, Balasubramanian S. In vitro drug release behavior, mechanism and antimicrobial activity of rifampicin loaded low molecular weight PLGA-PEG-PLGA triblock copolymeric nanospheres. Curr Drug Deliv. 2013;10:722–731. doi: 10.2174/15672018113109990002. [DOI] [PubMed] [Google Scholar]
- 18.Chen ZP, Liu W, Liu D, Xiao YY, Chen HX, Chen J, Li W, Cai H, Li W, Cai BC, Pan J. Development of brucine-loaded microsphere/thermally responsive hydrogel combination system for intra-articular administration. J Control Release. 2012;162:628–635. doi: 10.1016/j.jconrel.2012.07.037. [DOI] [PubMed] [Google Scholar]
- 19.Galois L, Etienne S, Grossin L, Watrin-Pinzano A, Cournil-Henrionnet C, Loeuille D, Netter P, Mainard D, Gillet P. Dose-response relationship for exercise on severity of experimental osteoarthritis in rats: a pilot study. Osteoarthritis Cartilage. 2004;12:779–786. doi: 10.1016/j.joca.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, Salter D, van den Berg WB. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage. 2006;14:13–29. doi: 10.1016/j.joca.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Cochrane DJ, Jarvis B, Keating GM. Etoricoxib. Drugs. 2002;62:2637–2651. doi: 10.2165/00003495-200262180-00006. discussion 2652-3. [DOI] [PubMed] [Google Scholar]
- 22.Zweers MC, de Boer TN, van Roon J, Bijlsma JW, Lafeber FP, Mastbergen SC. Celecoxib: considerations regarding its potential disease-modifying properties in osteoarthritis. Arthritis Res Ther. 2011;13:239. doi: 10.1186/ar3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Boer TN, Huisman AM, Polak AA, Niehoff AG, van Rinsum AC, Saris D, Bijlsma JW, Lafeber FJ, Mastbergen SC. The chondroprotective effect of selective COX-2 inhibition in osteoarthritis: ex vivo evaluation of human cartilage tissue after in vivo treatment. Osteoarthritis Cartilage. 2009;17:482–488. doi: 10.1016/j.joca.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Tetlow LC, Adlam DJ, Woolley DE. Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 2001;44:585–594. doi: 10.1002/1529-0131(200103)44:3<585::AID-ANR107>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 25.Stanton H, Rogerson FM, East CJ, Golub SB, Lawlor KE, Meeker CT, Little CB, Last K, Farmer PJ, Campbell IK, Fourie AM, Fosang AJ. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;434:648–652. doi: 10.1038/nature03417. [DOI] [PubMed] [Google Scholar]
- 26.Honda K, Ohno S, Tanimoto K, Ijuin C, Tanaka N, Doi T, Kato Y, Tanne K. The effects of high magnitude cyclic tensile load on cartilage matrix metabolism in cultured chondrocytes. Eur J Cell Biol. 2000;79:601–609. doi: 10.1078/0171-9335-00089. [DOI] [PubMed] [Google Scholar]
- 27.Mastbergen SC, Bijlsma JW, Lafeber FP. Synthesis and release of human cartilage matrix proteoglycans are differently regulated by nitric oxide and prostaglandin-E2. Ann Rheum Dis. 2008;67:52–58. doi: 10.1136/ard.2006.065946. [DOI] [PubMed] [Google Scholar]
- 28.Jones IA, Togashi R, Wilson ML, Heckmann N, Vangsness CT Jr. Intra-articular treatment options for knee osteoarthritis. Nat Rev Rheumatol. 2019;15:77–90. doi: 10.1038/s41584-018-0123-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larsen C, Ostergaard J, Larsen SW, Jensen H, Jacobsen S, Lindegaard C, Andersen PH. Intra-articular depot formulation principles: role in the management of postoperative pain and arthritic disorders. J Pharm Sci. 2008;97:4622–4654. doi: 10.1002/jps.21346. [DOI] [PubMed] [Google Scholar]
- 30.McAlindon TE, Bannuru RR. Osteoarthritis in 2017: latest advances in the management of knee OA. Nat Rev Rheumatol. 2018;14:73–74. doi: 10.1038/nrrheum.2017.219. [DOI] [PubMed] [Google Scholar]
- 31.Feczkó T, Fodor-Kardos A, Sivakumaran M, Haque Shubhra QT. In vitro IFN-alpha release from IFN-alpha- and pegylated IFN-alpha-loaded poly(lactic-co-glycolic acid) and pegylated poly(lactic-co-glycolic acid) nanoparticles. Nanomedicine (Lond) 2016;11:2029–2034. doi: 10.2217/nnm-2016-0058. [DOI] [PubMed] [Google Scholar]
- 32.Kamali H, Khodaverdi E, Hadizadeh F, Mohajeri SA. In-vitro, ex-vivo, and in-vivo evaluation of buprenorphine HCl release from an in situ forming gel of PLGA-PEG-PLGA using Nmethyl2pyrrolidone as solvent. Mater Sci Eng C Mater Biol Appl. 2019;96:561–575. doi: 10.1016/j.msec.2018.11.058. [DOI] [PubMed] [Google Scholar]
- 33.Hong W, Chen D, Jia L, Gu J, Hu H, Zhao X, Qiao M. Thermo- and pH-responsive copolymers based on PLGA-PEG-PLGA and poly(L-histidine): synthesis and in vitro characterization of copolymer micelles. Acta Biomater. 2014;10:1259–1271. doi: 10.1016/j.actbio.2013.12.033. [DOI] [PubMed] [Google Scholar]
- 34.Zolnik BS, Leary PE, Burgess DJ. Elevated temperature accelerated release testing of PLGA microspheres. J Control Release. 2006;112:293–300. doi: 10.1016/j.jconrel.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 35.Bajpayee AG, Grodzinsky AJ. Cartilage-targeting drug delivery: can electrostatic interactions help? Nat Rev Rheumatol. 2017;13:183–193. doi: 10.1038/nrrheum.2016.210. [DOI] [PubMed] [Google Scholar]
- 36.Bajpayee AG, Wong CR, Bawendi MG, Frank EH, Grodzinsky AJ. Avidin as a model for charge driven transport into cartilage and drug delivery for treating early stage post-traumatic osteoarthritis. Biomaterials. 2014;35:538–549. doi: 10.1016/j.biomaterials.2013.09.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vermeij EA, Koenders MI, Bennink MB, Crowe LA, Maurizi L, Vallee JP, Hofmann H, van den Berg WB, van Lent PL, van de Loo FA. The in-vivo use of superparamagnetic iron oxide nanoparticles to detect inflammation elicits a cytokine response but does not aggravate experimental arthritis. PLoS One. 2015;10:e0126687. doi: 10.1371/journal.pone.0126687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hezi-Yamit A, Sullivan C, Wong J, David L, Chen M, Cheng P, Shumaker D, Wilcox JN, Udipi K. Impact of polymer hydrophilicity on biocompatibility: implication for DES polymer design. J Biomed Mater Res A. 2009;90:133–141. doi: 10.1002/jbm.a.32057. [DOI] [PubMed] [Google Scholar]
- 39.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–1707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calvo E, Castañeda S, Largo R, Fernández-Valle ME, Rodríguez-Salvanés F, Herrero-Beaumont G. Osteoporosis increases the severity of cartilage damage in an experimental model of osteoarthritis in rabbits. Osteoarthritis Cartilage. 2007;15:69–77. doi: 10.1016/j.joca.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 41.Goldring MB, Goldring SR. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann N Y Acad Sci. 2010;1192:230–237. doi: 10.1111/j.1749-6632.2009.05240.x. [DOI] [PubMed] [Google Scholar]
- 42.Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!) Osteoarthritis Cartilage. 2013;21:16–21. doi: 10.1016/j.joca.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 43.Sokolove J, Lepus CM. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis. 2013;5:77–94. doi: 10.1177/1759720X12467868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19:18. doi: 10.1186/s13075-017-1229-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arunkumar P, Indulekha S, Vijayalakshmi S, Srivastava R. Synthesis, characterizations, in vitro and in vivo evaluation of Etoricoxib-loaded Poly (Caprolactone) microparticles--a potential Intra-articular drug delivery system for the treatment of Osteoarthritis. J Biomater Sci Polym Ed. 2016;27:303–316. doi: 10.1080/09205063.2015.1125564. [DOI] [PubMed] [Google Scholar]
- 46.Arunkumar P, Indulekha S, Vijayalakshmi S, Srivastava R. Poly (caprolactone) microparticles and chitosan thermogels based injectable formulation of etoricoxib for the potential treatment of osteoarthritis. Mater Sci Eng C Mater Biol Appl. 2016;61:534–544. doi: 10.1016/j.msec.2015.12.039. [DOI] [PubMed] [Google Scholar]