

Abstract
Background: Ulcerative colitis (UC) is a chronic inflammatory intestinal disease, and its morbidity is rising worldwide. Previous study indicated that astragaloside II (AS II), a monomeric compound, was used to treat bowel disease. However, the effects of AS II on UC remains unclear. Thus, this study aimed to investigate the therapeutic effects of AS II on experimental UC in vitro and in vivo. Methods: CCD-18Co cells were stimulated by 1 μg/mL LPS to mimic UC in vitro. In addition, dextran sulfate sodium (DSS)-induced UC mouse model was established in vivo. CCK-8 assay was used to detect cell proliferation in vitro. Moreover, the concentrations of inflammatory factors interleukin 6 (IL-6), tumor necrosis factor-α (TNF-α), interleukin 1β (IL-1β), nitric oxide (NO), superoxide dismutase (SOD) and malondialdehyde (MDA) in CCD-18Co cells and colon tissues were determined by ELISA, respectively. Meanwhile, the expressions of hypoxia-inducible factor 1α (HIF-α), phospho-inhibitor of NF-κB (p-IκB) and phospho-NF-κB p65 (p-p65) were detected by western blotting in vitro and in vivo, respectively. Results: In this study, the levels of pro-inflammatory cytokines TNF-α, IL-1β and IL-6 were significantly increased in lipopolysaccharide (LPS)-stimulated CCD-18Co cells. However, LPS-induced inflammatory response was markedly alleviated by AS II. In addition, LPS-induced HIF-α, p-IκB and p-p65 proteins increases were markedly ameliorated by AS II treatment. Moreover, AS II reduced disease activity index (DAI) scores and increased the colon lengths in DSS-treated mice. Meanwhile, AS II decreased the levels of IL-6, TNF-α, IL-1β, NO, MPO and MDA, and increased the level of SOD in colon of DSS-treated mice. Furthermore, AS II downregulated the expressions of HIF-α, p-IκB and p-p65 in DSS-induced UC in mice. Conclusion: Our findings indicated that AS II could alleviate inflammatory response in LPS-induced CCD-18Co cells and in DSS-induced UC in mice. In conclusion, AS II may serve as a potential agent for the treatment of UC.
Keywords: Astragaloside II, lipopolysaccharide, dextran sulfate sodium, ulcerative colitis
Introduction
Ulcerative colitis (UC) is a chronic and inflammatory disease of the rectum and colon [1]. Patients with UC in adolescence or young adulthood were diagnosed with symptoms of diarrhea, abdominal pain, and rectal bleeding [2]. The morbidity of UC was increasing with globalization [3]. 15% of the patients had been hospitalized during the exacerbations [4]. Conventional colonoscopy is an important detection method to evaluate the disease severity degree in patients with UC [5,6]. However, conventional colonoscopy has some limitations, including adverse events and low patient compliance when patients receive treatment [7,8]. Therefore, it is necessary to explore better therapeutic strategies for treating UC.
Astragaloside II (AS II) is a Chinese monomeric compound, which extracted from the Chinese traditional herb Astragalus membranaceu [9]. Previous reports demonstrated that AS II exhibited multiple pharmacological activities, such as anti-tumor activity, anti-bacteria activity, anti-inflammation activity and so on [10-12]. In addition, AS II could reduce cell proliferation and induce cell apoptosis in cancer cells [13]. Moreover, AS II could repair irritable bowel disease by activating the mTOR pathway [9].
It has been shown that inflammatory cell infiltration could lead to decreased mucosal perfusion in inflamed mucosal tissues, and thus could activate hypoxia-inducible factor 1-alpha (HIF-α) [14]. HIF-α is transcription factor, which could link inflammatory pathways, including nuclear factor-kappa B (NF-κB) pathway [15]. NF-κB, a pro-inflammatory cytokine, which was associated to the development and pathogenesis of UC [16]. Previous study indicated that activation of NF-κB could increase the release of proinflammatory cytokines TNF-α, IL-1β and IL-6 in DSS-induced UC mice [17]. In addition, colon length shortening and colonic pathological damage were observed in a mouse model of DSS-induced UC [17]. However, the anti-inflammatory effect of ASII on DSS-induced experimental UC in mice remains unclear. Therefore, this study aimed to explore the effect of ASII on experimental UC in vitro and in vivo.
Materials and methods
Cell line and cell culture
Human colon fibroblast CCD-18Co was purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were incubated in Eagle’s Minimum Essential Medium (ATCC), supplemented with 10% fetal bovine serum (FBS, Gibco, Carlsbad, CA, USA), penicillin and streptomycin (100 U/ml) in a humidified 5% CO2 incubator at 37°C.
Cell viability detection
Cell viability was detected using the Cell Counting Kit-8 (CCK-8, Beyotime Biotechnology, Beijing, China) according to the manufacturer’s protocols. CCD-18Co cells (5×103 cells/well) were plated into a 96-well plate and cultured overnight. Then, AS II (0, 0.1, 0.33, 1, 3 μM) were added into CCD-18Co cells at 37°C for 48 h. After that, 10 μL of CCK-8 reagent was added into each well. The absorbance at a wavelength of 450 nm was measured using a microplate reader (Bio-Rad Laboratories, Benicia, California, USA). LPS was provided by sigma (Sigma, St. Louis, MO, USA). AS II was obtained from MCE (Med Chem Express, Monmouth Junction, NJ, USA).
Enzyme-linked immunosorbent assay (ELISA) for detection of pro-inflammatory factors
CCD-18Co cells were treated with LPS (1 μg/mL) for 0, 12, 24 and 48 h to produce inflammatory mediators. In addition, CCD-18Co cells were treated with 1 μM AS II and 1 μg/mL LPS for 48 h. Samples of the supernatant were obtained from CCD-18Co cells. ELISA was used to detect the levels of IL-6, TNF-α and IL-1β in cell culture supernatant. IL-6, TNF-α and IL-1β ELISA kits were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
qRT-PCR
The total RNA from CCD-18Co cells were extracted by TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. After that, cDNA was synthesized using the cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Later on, real-time PCR was performed using a SYBR premix Ex Taq II kit (TaKaRa, Dalian, China) on an ABI 7900HT instrument (ABI, NY, USA). HIF-α, F: 5’-CCAGTGGACTTGGGGCCTTG-3’; R: 5’-AAAGGCAAAGACACCCGCGA-3’. GAPDH, F: 5’-ATGGCCTTCCGTGTTCCTAC-3’; R: 5’-CTTTACAAAGTTGTCGTTGA-3’. Relative quantification of gene expression was performed using the 2-ΔΔCT as folder changes. The primers for p-p65 was obtained from GenePharma (Shanghai, China).
Western blotting
CCD-18Co cells and colonic samples were homogenized using RIPA buffer in ice-cold homogenization. Bradford Protein Assay Kit (Beyotime, Biotechnology) was used to assess the quantity of protein. Equal quantity of proteins (40 μg) in lysate were then separated on 10% SDS polyacrylamide gel electrophoresis. After that, the proteins were transferred onto a polyvinylidene fluoride membrane (PVDF, Thermo Fisher Scientific). The membranes were blocked with TBST containing 5% nonfat milk for 2 h, and then membranes were applied with primary antibodies overnight at 4°C as follows: anti-HIF-α (1:1000), anti-p-p65 (1:1000), anti-p65 (1:1000), anti-p-IκB (1:1000), anti-IκB (1:1000) and anti-β-actin (1:1000). Then, the PVDF membranes were incubated with secondary antibody anti-rabbit (1:5000). The protein was detected by enhanced chemiluminescence (ECL) system (Bio-Rad, Hercules, CA, USA). The density of blots for targets were normalized to β-actin. All antibodies were purchased from Abcam (Cambridge, UK).
DSS-induced ulcerative colitis in mice
Male BABL/c mice (20-22 g) were provided by Kay Biological Technology (Shanghai, China). The animals were housed according to the guideline of National Institutes Health for the Care and Use of Laboratory Animals. 48 mice were randomly separated into 8 experimental groups (n = 6): group I, control; group II, AS II_10 mg/kg; group III, AS II_30 mg/kg; group IV, AS II_50 mg/kg; group V, AS II_80 mg/kg; group VI, model group (DSS); group VII, DSS + 30 mg/kg AS II; group VIII, DSS + 50 mg/kg AS II.
The mice were treated with AS II (dissolved in 2% Tween 20 in PBS) via oral gavage once per day for 10 days in the morning. On the day 3, UC were induced by 3% (w/v) DSS added to drinking water for 5 consecutive days. The body weights of mice were monitored every day. The DAI score and was determined as previously reported [18]. In the termination of the study, the mice were euthanized using CO2, and the colon was removed from animals. The length of colon was measured. All experimental procedures were approved by the Ethical Committee of Nanjing University of Chinese Medicine. DSS was obtained from Sigma. National Institutes of Health guide for the care and use of laboratory animals was followed.
Histological evaluation
The colonic samples were fixed in 10% formalin, and then embedded in paraffin for histological evaluation. The colonic specimens (5 μm) were stained with hematoxylin & eosin (H&E), and then observed by a light microscope. The grading of histological damage was assessed as described previously [19].
Cytokine analysis by ELISA
The colonic samples were weighed, and homogenized for 3 min on ice using a homogenizer. The levels of IL-6, TNF-α, IL-1β and NO in the colon homogenates were measured with ELISA kits (Nanjing Jiancheng Co., Nanjing, China) in accordance with the manufacturer’s instructions.
Measurement of MPO, MDA and SOD
The distribution number of neutrophils in colonic samples was detected by (myeloperoxidase) MPO activity. Colonic samples were weighed, and homogenized using reaction buffer. Then, MPO activity assay kit (Nanjing Jiancheng Co., China) was applied to assess the MPO activity. The activity of SOD and the content of MDA were determined using SOD or MDA activity assay kits according to manufacturer’s protocols (Nanjing Jiancheng Co., China).
Immunohistochemistry (IHC) analysis
p-p65 was detected by IHC staining according to the methods reported before. The samples were incubated with the primary antibodies overnight at 4°C. Later on, biotinylated immunoglobulin cocktail of goat anti-rabbit IgG was applied at room temperature. The colon specimens were observed using a fluorescence microscope.
Statistical analysis
Each group were performed at least three independent experiments and all data were presented as the mean ± standard deviation (SD). Statistical analysis was analyzed by GraphPad Prism v7.0 (GraphPad Software, Inc., La Jolla, CA, USA). Protein expressions of HIF-α, p-p65 and p-IκB and the levels of proinflammatory cytokines IL-6, TNF-α, IL-1β were analyzed with one-way analysis of variance (ANOVA) followed by Tukey’s test. In addition, histological evaluation and IHC assays were analyzed with one-way analysis of variance (ANOVA) followed by Tukey’s test. P<0.05 was considered statistically significant.
Results
AS II attenuated oxidative stress damage in LPS-stimulated CCD-18Co cells via decreasing the production of inflammatory factors
The chemical structure of AS II was indicated in Figure 1A. CCK-8 assay was used to assess the potential cytotoxic effect of AS II on CCD-18Co cells. As indicated in Figure 1B, 3 μM AS II significantly decreased the viability of CCD-18Co cells, compared with control group. Meanwhile, 1 μM AS II had very limited on the cytotoxicity of CCD-18Co cells. Therefore, 1 μM AS II was utilized in following in vitro experiments. As shown in Figure 1C-E, LPS significantly increased the levels of IL-6, TNF-α and IL-β in CCD-18Co cells, which were obviously reversed by AS II. In addition, the results of NO, SOD and MDA activity assays indicated that LPS significantly decreased the level of SOD, and increased the levels of NO and MDA in CCD-18Co cells. However, the effect of LPS on the productions of NO, SOD and MDA were obviously reversed in the presence of AS II (Figure 1F-H). These data indicated that AS II could decrease the production of inflammatory factors and attenuate oxidative stress damage in LPS-stimulated CCD-18Co cells.
Figure 1.

AS II attenuated oxidative stress damage in LPS-stimulated CCD-18Co cells via decreasing the production of inflammatory factors. A. The chemical structure of AS II. B. CCD-18Co cells were treated with AS II (0, 0.1, 0.33, 1, 3 μM) for 48 h. CCK-8 assay was used to detect the viability of CCD-18Co cells. C. CCD-18Co cells were treated with LPS (1 μg/mL) for 0, 12, 24 and 48 h. In addition, CCD-18Co cells were treated with 1 μM AS II and 1 μg/mL LPS for 48 h. The level of IL-6 in the culture media was measured with ELISA. D. The level of TNF-α in the culture media was measured with ELISA. E. The level of IL-β in the culture media was measured with ELISA. F. CCD-18Co cells were treated with 1 μg/mL LPS or/and 1 μM AS II for 48 h. The level of NO in cells was measured with ELISA. G. The level of SOD in cells was measured with ELISA. H. The level of MDA in cells was measured with ELISA. *P<0.05, **P<0.01 compared with 0 h group; ##P<0.01 compared with 48 h group.
AS II inhibited the levels of HIF-α, p-p65 and p-IκB in LPS-treated CCD-18Co cells in vitro
We next investigated whether AS II exhibited anti-inflammation effect in LPS-stimulated CCD-18Co cells via inhibition of inflammatory proteins. QRT-PCR and western blotting data showed that LPS significantly increased the level of HIF-α, which was notably decreased in the presence of AS II (Figure 2A-C). In addition, the expressions of pro-inflammatory proteins p-p65 and p-IκB were detected by western blotting. As indicated in Figure 2B, 2D and 2E, the levels of p-p65 and p-IκB were markedly increased in LPS-stimulated CCD-18Co cells, compared with control group. However, LPS-induced p-p65 and p-IκB proteins upregulation were notably alleviated by AS II treatment. All these results suggested that AS II exhibited anti-inflammation effect in LPS-stimulated CCD-18Co cells via inhibition of inflammatory proteins.
Figure 2.
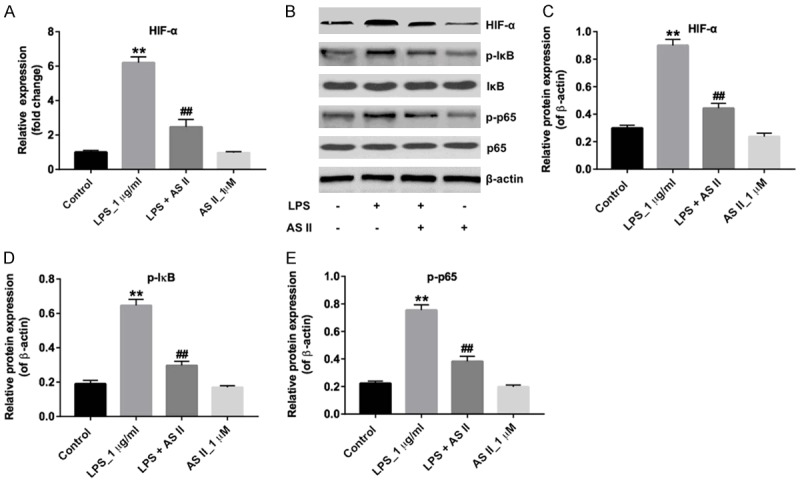
AS II inhibited the levels of HIF-α, p-p65 and p-IκB in LPS-treated CCD-18Co cells in vitro. CCD-18Co cells were treated with 1 μg/mL LPS or/and 1 μM AS II for 48 h. A. Relative expression of HIF-α in CCD-18Co cells was detected by qRT-PCR. B. Expression levels of HIF-α, p-IκB and p-p65 in CCD-18Co cells were detected with western blotting. β-actin was used as an internal control. C. The relative expression of HIF-α was quantified via normalization to β-actin. D. The relative expression of p-IκB was quantified via normalization to β-actin. E. The relative expression of p-p65 was quantified via normalization to β-actin. **P<0.01 compared with control group; ##P<0.01 compared with LPS_1 μg/ml group.
AS II alleviated the symptom of DSS-induced UC in mice
A mouse model of DSS-induced UC was established to further investigate the anti-inflammation effect of AS II in vivo. As illustrated in Figure 3A and 3B, the body weights of mice were notably decreased at 10 days in AS II_80 mg/kg group, compared with control group. Meanwhile, 30 or 50 mg/kg AS II had very limited system toxicity on mice. Therefore, AS II (30 and 50 mg/kg) was utilized in following in vivo experiments.
Figure 3.
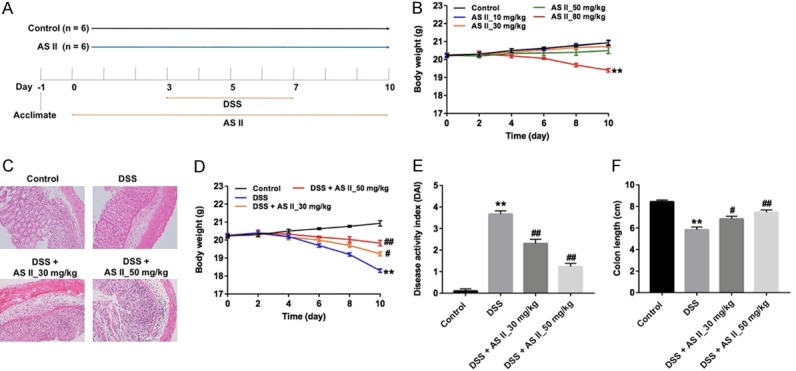
AS II attenuated DSS-induced UC in mice. A. Drug administration in a mouse model of DSS-induced UC. B. Mice were treated with AS II (0, 10, 30, 50 or 80 mg/kg) for 0, 2, 4, 6, 8 and 10 day and the body weight of mice were monitored. C. The mice were treated with DSS, DSS plus AS II (30 or 50 mg/kg) for 0, 2, 4, 6, 8 and 10 day. HE staining was performed by photomicrography (magnification at 400×). D. Body weights of mice in each group were monitored. E. Disease activity index (DAI) of mice in each group was measured. F. The colon lengths of mice in each group were evaluated. **P<0.01 compared with control group; #P<0.05, ##P<0.01 compared with DSS group.
H&E staining data showed that the infiltration of inflammatory cells and damage to the surface epithelium were observed in the DSS group. However, preventive effect of AS II on DSS-induced UC was observed in DSS + AS II_50 mg/kg group (Figure 3C). In addition, 50 mg/kg AS II markedly prevented the body weight loss in DSS-treated mice (Figure 3D). As we know, the severity of weight loss, stool consistency and blood in stool were reflected by DAI. As indicated in Figure 3E, 30 or 50 mg/kg AS II significantly reduced the DAI caused by DSS in mice, compared with the DSS group. Meanwhile, 50 mg/kg AS II significantly increased colon length during the progression of experimental UC in mice (Figure 3F). These data suggested that AS II could alleviate the symptom of DSS-induced UC in mice.
AS II attenuated oxidative stress damage in DSS-induced UC mice via decreasing the levels of inflammatory factors
Next, the levels of inflammatory cytokines in DSS-induced UC mice was detected by ELISA. As illustrated in Figure 4A-C, the levels of pro-inflammatory cytokines IL-6, TNF-α and IL-1β in colon tissues were markedly increased in the DSS group, compared with control group. However, DSS-induced IL-6, TNF-α and IL-β upregulations were significantly alleviated by AS II treatment. In addition, the contents of NO, MDA and MPO were increased, and SOD activity was markedly reduced in colon tissues of DSS treated mice, which were markedly reversed by AS II treatment (Figure 4D-G). These data indicated that AS II could decrease the levels of inflammatory factors and attenuate oxidative stress damage in DSS-treated mice.
Figure 4.

AS II attenuated oxidative stress damage in DSS-induced UC mice via decreasing the levels of inflammatory factors. The mice were treated with DSS, DSS and 30 mg/kg AS II or DSS plus 50 mg/kg AS II for 0, 2, 4, 6, 8 and 10 day. A-C. The levels of IL-6, TNF-α or IL-β in the colon tissues were measured with ELISA. D-G. The levels of NO, MPO, SOD and MDA in colon tissues were measured with ELISA. **P<0.01 compared with control group; #P<0.05, ##P<0.01 compared with DSS group.
AS II decreased the expression of inflammatory proteins in DSS-treated mice
It is reported that NF-κB plays an important role in the mediation of inflammation in UC [20]. We next explored the effects of AS II on NF-κB pathway. IHC results indicated that the level of p-p65 was significantly increased in DSS-treated mice, which was markedly reduced by AS II treatment (Figure 5A). In addition, western blotting results showed the expressions of HIF-α, p-p65 and p-IκB were markedly upregulated in the DSS group. However, DSS-induced upregulations of HIF-α, p-p65 and p-IκB were significantly reversed by AS II treatment (Figure 5B-E). All these data illustrated that AS II could decrease the expressions of inflammatory proteins in DSS-treated mice.
Figure 5.
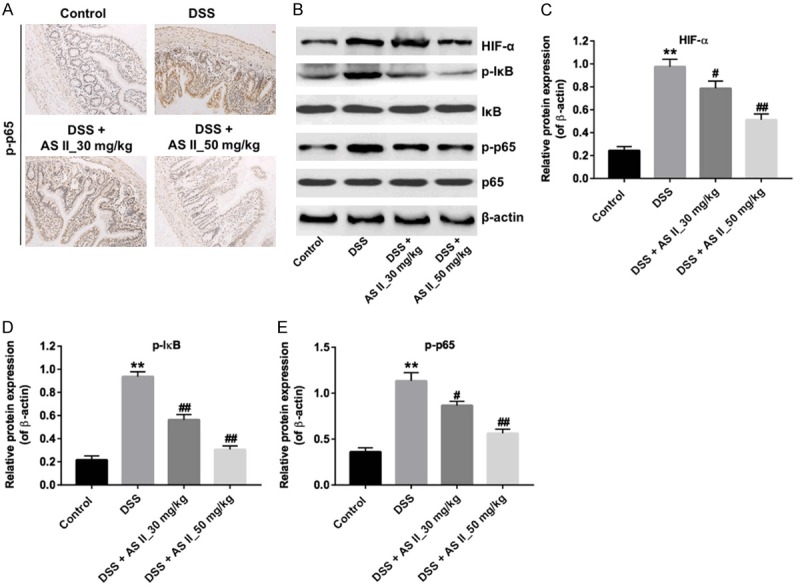
AS II inhibited the expression of inflammatory proteins in DSS-induced UC mice. The mice were treated with DSS, DSS and 30 mg/kg AS II or DSS and 50 mg/kg AS II for 0, 2, 4, 6, 8 and 10 day. A. The level of p-p65 in colon tissues was detected by immunohistochemical staining assay (magnification at 400×). B. Expression levels of HIF-α, p-IκB and p-p65 in colon tissues were detected with western blotting. β-actin was used as an internal control. C-E. The relative expressions of HIF-α, p-IκB and p-p65 were quantified via normalization to β-actin. **P<0.01 compared with control group; #P<0.05, ##P<0.01 compared with DSS group.
Discussion
In this study, we investigated the mechanisms by which AS II repaired ulcerative colitis barrier function in vitro and in vivo. Based on in vitro and in vivo results, we found AS II significantly alleviated LPS-induced inflammatory response on CCD-18Co cells and markedly reduced the production of inflammatory proteins in DSS-treated mice.
AS II is cycloartane-type triterpene glycosides, which has been reported to promote intestinal epithelial repair [9]. In present study, 3 μM AS II significantly inhibited the proliferation of CCD-18Co cells, while the effect of 1 μM AS II on cell proliferation was extremely limited. These data illustrated that 1 μM was an appropriate concentration. In order to investigate the mechanisms responsible for the anti-inflammatory activities of AS II, LPS was added into CCD-18Co cells to create an in vitro model of UC. LPS increased the production of pro-inflammatory cytokines IL-6, TNF-α and IL-β, while AS II markedly decreased the production of inflammatory factors in LPS-stimulated CCD-18Co cells. Ke et al found that a traditional Chinese formulation (Qing Hua Chang Yin) markedly suppressed the secretion of IL-6 in LPS-stimulated Caco-2 cells [21]. These data suggested that AS II could decrease the secretion of inflammatory factors in LPS-stimulated CCD-18Co cells.
In addition, dextran sulfate sodium (DSS)-induced UC mice model was used to further investigate the anti-UC effects of AS II. In this study, higher levels of pro-inflammatory factors TNF-α, IL-1β, IL-6 were observed in the DSS group. However, AS II markedly alleviated DSS-induced production of pro-inflammatory factors. Moreover, MPO and NO also play important role in local intestinal damage [22,23]. Meanwhile, SOD is an antioxidant enzyme that can reduce harmful substances produced during metabolism [24]. However, MDA is the product of lipid peroxidation, which reflect the severity of membrane damage in vivo [25]. In this study, we found that DSS-induced MPO, NO and MDA activity increases and SOD activity decreases in colon tissues were significantly reversed by AS II treatment. These data suggested that AS II could alleviate DSS-induced colonic injury via decreasing the production of inflammatory factors and attenuating oxidative stress damage. The above results strongly suggested that AS II alleviated the symptoms of UC in vitro and in vivo.
Hypoxia inducible factor-α (HIF-α) is a master regulatory transcription factor, which plays a vital role in regulating the inflammatory response [26]. When inflammation was appeared in cells or tissues, it would lead to inflammation-associated tissue hypoxia. Then, the level of HIF-α was increased in hypoxic cells or tissues [27]. Cristina et al found that HIF-α could induce the production of NO [28]. In addition, previous study indicated that HIF-α could induce inflammatory response via phosphorylation of IκB and NF-κB p-65 [29]. Meanwhile, large amount of NO could induce the activation of NF-κB [30]. NF-κB is a transcription factor, which could regulate the intestinal inflammatory response [31]. Various inflammatory factors, such as TNF-α, IL-6 and IL-12, have been shown to be regulated by NF-κB p-65 [32]. In this study, the levels of HIF-α, NO, p65 and IκB were markedly increased in colon tissues in DSS-induced UC mice, which were markedly decreased in the presence of AS II. Zhou et al indicated that Brusatol could alleviates the symptoms of UC via suppression of NF-κB-mediated inflammatory responses [33]. In addition, ke et al found that Qing Hua Chang Yin attenuated LPS-induced inflammatory response in Caco-2 cells via suppressing the level of NF-κB [34]. These evidences illustrated that AS II could alleviate the symptoms of UC via inhibiting the NF-κB signaling. Hypoxia could occur under inflammation situation, which could activate HIF-α [35]. Meanwhile, HIF-α could activate NF-κB and proinflammatory cytokines in hypoxia, then hypoxia and inflammation are linked [36]. For the first time, we found that AS II could attenuate LPS-induced inflammatory response in CCD-18Co cells via suppressing the level of HIF-α, and then inactivating NF-κB. Therefore, our evidence suggests that AS II could alleviate the symptoms of UC via inhibiting the HIF-α/NF-κB pathway.
Conclusion
In summary, AS II could alleviate the symptoms of experimental UC in vitro and in vivo via inhibition of HIF-α/NF-κB pathway. The evidences suggested that AS II may act as a potential agent for the treatment of UC.
Acknowledgements
This work was supported by the Science and Technology Projects of Shandong Provincial of Traditional Chinese Medicine (2017-190) and National Natural Science Foundation of China (81673982).
Disclosure of conflict of interest
None.
References
- 1.Antonelli E, Villanacci V, Bassotti G. Novel oral-targeted therapies for mucosal healing in ulcerative colitis. World J Gastroenterol. 2018;24:5322–5330. doi: 10.3748/wjg.v24.i47.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ranson N, Veldhuis M, Mitchell B, Fanning S, Cook AL, Kunde D, Eri R. NLRP3-dependent and -independent processing of interleukin (IL)-1beta in active ulcerative colitis. Int J Mol Sci. 2018;20 doi: 10.3390/ijms20010057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplan GG, Ng SC. Understanding and preventing the global increase of inflammatory bowel disease. Gastroenterology. 2017;152:313–321. e2. doi: 10.1053/j.gastro.2016.10.020. [DOI] [PubMed] [Google Scholar]
- 4.Akpinar MY, Ozin YO, Kaplan M, Ates I, Kalkan IH, Kilic ZMY, Yuksel M, Kayacetin E. Platelet-to-lymphocyte ratio and neutrophil-to-lymphocyte ratio predict mucosal disease severity in ulcerative colitis. J Med Biochem. 2018;37:155–162. doi: 10.1515/jomb-2017-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hata K, Kishikawa J, Anzai H, Shinagawa T, Kazama S, Ishii H, Nozawa H, Kawai K, Kiyomatsu T, Tanaka J, Tanaka T, Nishikawa T, Otani K, Yasuda K, Yamaguchi H, Ishihara S, Sunami E, Kitayama J, Watanabe T. Surveillance colonoscopy for colitis-associated dysplasia and cancer in ulcerative colitis patients. Dig Endosc. 2016;28:260–265. doi: 10.1111/den.12505. [DOI] [PubMed] [Google Scholar]
- 6.Dignass A, Lindsay JO, Sturm A, Windsor A, Colombel JF, Allez M, D’Haens G, D’Hoore A, Mantzaris G, Novacek G, Oresland T, Reinisch W, Sans M, Stange E, Vermeire S, Travis S, Van Assche G. Second European evidence-based consensus on the diagnosis and management of ulcerative colitis part 2: current management. J Crohns Colitis. 2012;6:991–1030. doi: 10.1016/j.crohns.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Verschuren EC, Ong DE, Kamm MA, Desmond PV, Lust M. Inflammatory bowel disease cancer surveillance in a tertiary referral hospital: attitudes and practice. Intern Med J. 2014;44:40–49. doi: 10.1111/imj.12285. [DOI] [PubMed] [Google Scholar]
- 8.Takano R, Osawa S, Uotani T, Tani S, Ishida N, Tamura S, Yamade M, Iwaizumi M, Hamaya Y, Furuta T, Miyajima H, Sugimoto K. Evaluating mucosal healing using colon capsule endoscopy predicts outcome in patients with ulcerative colitis in clinical remission. World J Clin Cases. 2018;6:952–960. doi: 10.12998/wjcc.v6.i15.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee SY, Tsai WC, Lin JC, Ahmetaj-Shala B, Huang SF, Chang WL, Chang TC. Astragaloside II promotes intestinal epithelial repair by enhancing L-arginine uptake and activating the mTOR pathway. Sci Rep. 2017;7:12302. doi: 10.1038/s41598-017-12435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M, Huang C, Su Y, Yang C, Xia Q, Xu DJ. Astragaloside II sensitizes human hepatocellular carcinoma cells to 5-fluorouracil via suppression of autophagy. J Pharm Pharmacol. 2017;69:743–752. doi: 10.1111/jphp.12706. [DOI] [PubMed] [Google Scholar]
- 11.Li X, Pi Z, Liu S, Wang W, Liu Z, Song F. Online monitoring of astragaloside II metabolism using a homemade cultural device coupled with microdialysis and ultra-performance liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1063:141–148. doi: 10.1016/j.jchromb.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Wan CP, Gao LX, Hou LF, Yang XQ, He PL, Yang YF, Tang W, Yue JM, Li J, Zuo JP. Astragaloside II triggers T cell activation through regulation of CD45 protein tyrosine phosphatase activity. Acta Pharmacol Sin. 2013;34:522–530. doi: 10.1038/aps.2012.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang C, Wu C, Xu D, Wang M, Xia Q. AstragalosideII inhibits autophagic flux and enhance chemosensitivity of cisplatin in human cancer cells. Biomed Pharmacother. 2016;81:166–175. doi: 10.1016/j.biopha.2016.03.025. [DOI] [PubMed] [Google Scholar]
- 14.Kim DS, Ko JH, Jeon YD, Han YH, Kim HJ, Poudel A, Jung HJ, Ku SK, Kim SJ, Park SH, Park JH, Choi BM, Park SJ, Um JY, Hong SH. Ixeris dentata NAKAI reduces clinical score and HIF-1 expression in experimental colitis in mice. Evid Based Complement Alternat Med. 2013;2013:671281. doi: 10.1155/2013/671281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003;17:2115–2117. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- 16.Karrasch T, Jobin C. NF-kappaB and the intestine: friend or foe? Inflamm Bowel Dis. 2008;14:114–124. doi: 10.1002/ibd.20243. [DOI] [PubMed] [Google Scholar]
- 17.Shen P, Zhang Z, Zhu K, Cao H, Liu J, Lu X, Li Y, Jing Y, Yuan X, Fu Y, Cao Y, Zhang N. Evodiamine prevents dextran sulfate sodium-induced murine experimental colitis via the regulation of NF-kappaB and NLRP3 inflammasome. Biomed Pharmacother. 2018;110:786–795. doi: 10.1016/j.biopha.2018.12.033. [DOI] [PubMed] [Google Scholar]
- 18.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, Conklin LS, Centola M, Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 20.Zhang H, Li W. microRNA-15 activates NF-kappaB pathway via down regulating expression of adenosine A2 receptor in ulcerative colitis. Cell Physiol Biochem. 2018;51:1932–1944. doi: 10.1159/000495718. [DOI] [PubMed] [Google Scholar]
- 21.Ke X, Hu G, Fang W, Chen J, Zhang X, Yang C, Peng J, Chen Y, Sferra TJ. Qing Hua Chang Yin inhibits the LPS-induced activation of the IL-6/STAT3 signaling pathway in human intestinal Caco-2 cells. Int J Mol Med. 2015;35:1133–1137. doi: 10.3892/ijmm.2015.2083. [DOI] [PubMed] [Google Scholar]
- 22.Shimada S, Tanigawa T, Watanabe T, Nakata A, Sugimura N, Itani S, Higashimori A, Nadatani Y, Otani K, Taira K, Hosomi S, Nagami Y, Tanaka F, Kamata N, Yamagami H, Shiba M, Fujiwara Y. Involvement of gliadin, a component of wheat gluten, in increased intestinal permeability leading to non-steroidal anti-inflammatory drug-induced small-intestinal damage. PLoS One. 2019;14:e0211436. doi: 10.1371/journal.pone.0211436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turan I, Sayan Ozacmak H, Ozacmak VH, Barut F, Ozacmak ID. The effects of S-nitrosoglutathione on intestinal ischemia reperfusion injury and acute lung injury in rats: roles of oxidative stress and NF-kappaB. Tissue Cell. 2018;52:35–41. doi: 10.1016/j.tice.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 24.Zhu MM, Wang L, Yang D, Li C, Pang ST, Li XH, Li R, Yang B, Lian YP, Ma L, Lv QL, Jia XB, Feng L. Wedelolactone alleviates doxorubicin-induced inflammation and oxidative stress damage of podocytes by IkappaK/IkappaB/NF-kappaB pathway. Biomed Pharmacother. 2019;117:109088. doi: 10.1016/j.biopha.2019.109088. [DOI] [PubMed] [Google Scholar]
- 25.Wei LF, Zhang HM, Wang SS, Jing JJ, Zheng ZC, Gao JX, Liu Z, Tian J. Changes of MDA and SOD in brain tissue after secondary brain injury with seawater immersion in rats. Turk Neurosurg. 2016;26:384–288. doi: 10.5137/1019-5149.JTN.8265-13.1. [DOI] [PubMed] [Google Scholar]
- 26.Ehrentraut SF, Kominsky DJ, Glover LE, Campbell EL, Kelly CJ, Bowers BE, Bayless AJ, Colgan SP. Central role for endothelial human deneddylase-1/SENP8 in fine-tuning the vascular inflammatory response. J Immunol. 2013;190:392–400. doi: 10.4049/jimmunol.1202041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elks PM, Renshaw SA, Meijer AH, Walmsley SR, van Eeden FJ. Exploring the HIFs, buts and maybes of hypoxia signalling in disease: lessons from zebrafish models. Dis Model Mech. 2015;8:1349–1360. doi: 10.1242/dmm.021865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Branco-Price C, Evans CE, Johnson RS. Endothelial hypoxic metabolism in carcinogenesis and dissemination: HIF-A isoforms are a NO metastatic phenomenon. Oncotarget. 2013;4:2567–2576. doi: 10.18632/oncotarget.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scortegagna M, Cataisson C, Martin RJ, Hicklin DJ, Schreiber RD, Yuspa SH, Arbeit JM. HIF-1alpha regulates epithelial inflammation by cell autonomous NFkappaB activation and paracrine stromal remodeling. Blood. 2008;111:3343–3354. doi: 10.1182/blood-2007-10-115758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seo JY, Yu JH, Lim JW, Mukaida N, Kim H. Nitric oxide-induced IL-8 expression is mediated by NF-kappaB and AP-1 in gastric epithelial AGS cells. J Physiol Pharmacol. 2009;60(Suppl 7):101–106. [PubMed] [Google Scholar]
- 31.Wang KZ, Feng L, Jiang WD, Wu P, Liu Y, Jiang J, Kuang SY, Tang L, Zhang YA, Zhou XQ. Dietary gossypol reduced intestinal immunity and aggravated inflammation in on-growing grass carp (Ctenopharyngodon idella) Fish Shellfish Immunol. 2019;86:814–831. doi: 10.1016/j.fsi.2018.12.014. [DOI] [PubMed] [Google Scholar]
- 32.Xie Z, Wang Y, Huang J, Qian N, Shen G, Chen L. Anti-inflammatory activity of polysaccharides from Phellinus linteus by regulating the NF-kappaB translocation in LPS-stimulated RAW264.7 macrophages. Int J Biol Macromol. 2019;129:61–67. doi: 10.1016/j.ijbiomac.2019.02.023. [DOI] [PubMed] [Google Scholar]
- 33.Zhou J, Wang T, Dou Y, Huang Y, Qu C, Gao J, Huang Z, Xie Y, Huang P, Lin Z, Su Z. Brusatol ameliorates 2, 4, 6-trinitrobenzenesulfonic acid-induced experimental colitis in rats: Involvement of NF-kappaB pathway and NLRP3 inflammasome. Int Immunopharmacol. 2018;64:264–274. doi: 10.1016/j.intimp.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 34.Ke X, Chen J, Zhang X, Fang W, Yang C, Peng J, Chen Y, Sferra TJ. Qing Hua Chang Yin attenuates lipopolysaccharide-induced inflammatory response in human intestinal cells by inhibiting NF-kappaB activation. Exp Ther Med. 2013;6:189–193. doi: 10.3892/etm.2013.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Ignazio L, Bandarra D, Rocha S. NF-kappaB and HIF crosstalk in immune responses. FEBS J. 2016;283:413–424. doi: 10.1111/febs.13578. [DOI] [PMC free article] [PubMed] [Google Scholar]