

Abstract
Ischemic heart disease (IHD) is a common clinical disease and has a younger tendency in recent years. This study focused on the role of Ulinastatin (UTI) in the anti-oxidative stress and anti-inflammatory response of cardiomyocytes. H9c2 cells were divided into control group, ischemia-anoxic group (ischemic hypoxia group) and ischemia-anoxia + UTI group (UTI group). Cell morphology was observed by light microscopy, Cell staining, Western blotting, quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) and enzyme-linked immunosorbent assay (ELISA) were conducted to research the effect of UTI on the nuclear factor-κB (NF-κB) signaling pathway. H9c2 cells in the ischemic hypoxia group showed hypertrophy and irregular shape, while the cell morphology of the UTI group was close to the fusiform shape. The cell hypertrophy was lighter, and the number of irregular morphological cells decreased in UTI group than ischemic hypoxia group. The content of interleukin-1β (IL-1β) in the ischemic hypoxic group was significantly higher than that in the control group. In the UTI group, IL-1β was significantly lowly expressed than the ischemia hypoxia group. In addition, the expressions of SOD1, SOD2, GPX1, GPX3, Bcl-2 and Sirt1 in UTI group were higher than ischemic hypoxia group (P<0.05). The expressions of p65, Iκk-α kinase, Caspase3 and Bax in UTI group were lower than ischemic hypoxia group (P<0.05). UTI protects H9c2 cells from ischemia and hypoxia injuries by inhibiting the NF-κB pathway, thereby reducing inflammation, resisting oxidative stress, inhibiting apoptosis, and delaying cell senescence.
Keywords: Ischemic heart disease (IHD), heart failure (HF), Ulinastatin (UTI), nuclear factor-κB (NF-κB)
Introduction
Ischemic heart disease (IHD) is one of the serious health problems with extremely high morbidity and mortality in the world. In particular, myocardial infarction is a major disease that endangers human health [1]. Although significant progress has been made in controlling interventions such as risk factors, drug therapy, bypass surgery, and stenting, IHD often leads to heart failure, increases social burden, and increases mortality [2]. The current treatment of heart failure persists in delaying the progression of the disease without further repairing and regenerating damaged myocardium. Although heart transplantation is the only effective treatment for end-stage patients, donor heart supply is limited for the large demand for heart failure patients [3]. Under myocardial ischemia, the balance between coronary oxygen supply and myocardial aerobics is destroyed, resulting in severe persistent hypoxia. Eventually, imbalance of vascular compensation leads to irreversible damage to myocardial morphology and function, including oxidative stress (OS). OS is one of the major pathological changes [4]. OS is resulted by severe hypoxia stimulation, triggering the production of a large amount of reactive oxygen species (ROS) immediately. As a result, abundant harmful factors are released, including malondialdehyde (MDA), lactate dehydrogenase (LDH), and oxidative toxic intermediates. OS is capable of stimulating autophagy, Ca2+ overload, and endoplasmic reticulum stress, further aggravating myocardial hypoxia, myocardial dysfunction and eventually, the development of IHD [5].
Studies have shown that the occurrence and development of IHD is inseparable from the inflammatory response. The connection between inflammatory reactions and the development of IHD has become a hot issue in recent years, but the specific mechanism remains unclear [6].
The results of the study indicated that Ulinastatin (UTI) exerted anti-inflammatory and anti-oxidative effects, but its anti-oxidation and anti-inflammatory mechanisms have not been fully elucidated in ischemic IHD [7]. In this paper, we investigated the protective mechanism of UTI on H9c2 cells suffered from ischemic and hypoxia, and provided a reference for the development of new drugs for the treatment of myocardial ischemia and hypoxia injury.
Materials and methods
Cell culture and treatment
H9c2 cells (Cell Culture Center, Shanghai, China) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Life Technology, Wuhan, China) containing 10% fetal bovine serum (FBS) (Life Technology, Wuhan, China) and 1% penicillin/streptomycin (Life Technology, Wuhan, China). When the cells were grown to the appropriate density, they were induced with ischemic and hypoxia (no serum and oxygen-free environment for 12 h: We placed the cell culture bottle in a sealable plastic box. Then the iron powder bag was placed in the plastic box. At last the plastic box was put into an anoxic incubator for further culture) and UTI (UTI 500 μmol*l-1 pre-intervention for 6 h).
Drug preparation
UTI (Tianpu Biochemical Pharmaceutical, Guangzhou, China) were dissolved in phosphate-buffered saline (PBS), prepared into a stock solution, and stored in a refrigerator at -20°C. Before cell experiments, UTI was diluted in DMEM as a working solution.
Cell counting kit-8 (CCK8) assay
The optimal concentration and treatment time of UTI were determined by CCK-8 (Construction, Nanjing, China). H9c2 cells in logarithmic growth phase were inoculated into 96-well plates at a density of 3000/well, and cultured for 24 h. Cells were incubated with different concentrations of UTI, followed by applying CCK-8 solution for 6 h, 12 h, 24 h, and 48 h. The absorbance at 450 nm was measured by a microplate reader.
Determination of lactate dehydrogenase (LDH) and malondialdehyde (MDA) levels in cell supernatants
Cell supernatants were collected for measuring levels of LDH and MDA using commercial kits according to the manufacturer’s instructions (Construction, Nanjing, China).
Immunofluorescence
Cells were fixed with 4% paraformaldehyde and blocked in goat serum at room temperature for 1 h. Subsequently, cells were incubated with diluted primary antibody SOD1 (Abcam, Cambridge, MA, USA, Rabbit, 1:3000) and IL-1β (Abcam, Cambridge, MA, USA, Rabbit, 1:3000) overnight at 4°C. The fluorescent secondary antibody was added the next day and incubated for 1 hour in the dark. Cell nucleus was stained by 4’,6-diamidino-2-phenylindole (DAPI) (Construction, Nanjing, China) in the dark, and the film was sealed with a sealing liquid. Finally, the image was observed under a fluorescence microscope.
Western blotting technology
Cells were lysed to extract total protein using the total protein extraction kit (Camilo Biological, Nanjing, China). The protein-containing compound was centrifuged at a low temperature (4°C) using a high-speed centrifuge (13,000 rpm, 15 min), and the supernatant was taken. The concentration of the protein solution was determined by the bicinchoninic acid (BCA) (Construction, Nanjing, China) method. After normalizing the protein concentration of each sample, the protein was separated using a 10% sodium dodecyl sulfate-polyacrylamide gel. The dispersed protein was then transferred to a polyvinylidene difluoride (PVDF, Thermo Fisher Scientific, Waltham, MA, USA) membrane for 2 h at 4°C. 5% skim milk was prepared with Tris-buffered saline with Tween-20 (TBST) to block the specific antigen for 1 h. After rinsing 3 times with TBST, the membrane was incubated with primary antibodies (SOD1, Abcam, Cambridge, MA, USA, Rabbit, 1:3000; SOD2, Abcam, Cambridge, MA, USA, Rabbit, 1:3000; Bcl-2, Abcam, Cambridge, MA, USA, Mouse, 1:2000; Bax, Abcam, Cambridge, MA, USA, Mouse, 1:2000; Sirt1, Abcam, Cambridge, MA, USA, Rabbit, 1:3000; Caspase3, Abcam, Cambridge, MA, USA, Rabbit, 1:3000; IL-1β, Abcam, Cambridge, MA, USA, Rabbit, 1:5000, IL-6, Abcam, Cambridge, MA, USA, Rabbit, 1:5000; TNF-α, Abcam, Cambridge, MA, USA, Rabbit, 1:500; p65, Abcam, Cambridge, MA, USA, Rabbit, 1:500; p-p65, Abcam, Cambridge, MA, USA, Rabbit, 1:1000; IκKα, Abcam, Cambridge, MA, USA, Rabbit, 1:2000; IκB-α, Abcam, Cambridge, MA, USA, Rabbit, 1:2000; GAPDH, Proteintech, Rosemont, IL, USA, 1:5000) overnight at 4°C. After washing in TBST for 3 times, membranes were incubated with the second antibody (goat anti-rabbit IgG antibody, Yifei Xue Biotechnology, Nanjing, China, 1:3000) at room temperature for 2 h, and washed 3 times in TBST. The target protein was displayed on an exposure machine using enhanced chemiluminescence (ECL) technology.
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
0.5 mL of TRIzol (Thermo Fisher Scientific, Waltham, MA, USA) was added to each of the treated well of 6-well plates and shaken for 10 minutes. 0.1 mL of chloroform was added to each well, and the tube was shaken vigorously for 15 seconds and left at room temperature for 3 minutes. The mixture was centrifuged for 15 minutes, and the upper aqueous phase was aspirated and isopropanol was added. The mixture was shaken and left at room temperature for 10 minutes. After centrifugation 10 minutes, the precipitate was washed in 75% ethanol, followed by another 5-min centrifugation. The liquid was discarded and then dissolved by the addition of 30 μL of ribonuclease free water (Thermo Fisher Scientific, Waltham, MA, USA). The RNA concentration was determined on the nanodroplets and their absorbances at 260 nm, 230 nm and 280 nm were determined. If the A260/A280 was between 1.8 and 2.1, the RNA quality was considered to be standard and can be used in subsequent experiments.
QRT-PCR was performed using Prism 7300 Sequence Detection System 25 μL reaction system was prepared, including SYBR Green (12.5 μL, Thermo Fisher Scientific, Waltham, MA, USA), 10 mΜ primers (0.5 mL each from the stock, Thermo Fisher Scientific, Waltham, MA, USA), 10.5 μL of water and 0.5 μL of template. The PCR conditions were as follows: 10 min denaturation at 95°C; 40 cycles of denaturation at 95°C for 15 s; 60°C annealing for 30 s and 72°C extension for 30 s. The data were analyzed by SDS software and the results were then output to EXCEL for further analysis. Endogenous GAPDH (Thermo Fisher Scientific, Waltham, MA, USA) was used to standardize the data. The comparative threshold cycle (Ct) method, that was, the 2-ΔΔCt method was used to calculate fold amplification. Primers used were shown in Table 1.
Table 1.
RT-PCR primers
| Gene name | Forward (5’>3’) | Reverse (5’>3’) |
|---|---|---|
| Bax | CAGTTGAAGTTGCCATCAGC | CAGTTGAAGTTACCATCAGC |
| Bcl-2 | GACTGAGTACCTGAACCGGCATC | CTGAGCAGCGTCTTCAGAGACA |
| Caspase3 | TGGAACAAATGGACCTGTTGACC | AGGACTCAAATTCTGTTGCCACC |
| Sirt1 | CCAGATCCTCAAGCCATG | TTGGATTCCTGCAACCTG |
| SOD1 | GGTGAACCAGTTGTGTTGTC | CCGTCCTTTCCAGCAGTC |
| SOD2 | CAGACCTGCCTTACGACTATGG | CTCGGTGGCGTTGAGATTGTT |
| GPX1 | ATCATATGTGTGCTGCTCGGCTAGC | TACTCGAGGGCACAGCTGGGCCCTTGAG |
| GPX3 | AGAGCCGGGGACAAGAGAA | ATTTGCCAGCATACTGCTTGA |
| IL-1β | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| IL-6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| TNF-α | CCTCTCTCTAATCAGCCCTCTG | GAGGACCTGGGAGTAGATGAG |
| IκKα | GTCAGGACCGTGTTCTCAAGG | GCTTCTTTGATGTTACTGAGGGC |
| IκB-α | GGATCTAGCAGCTACGTACG | TTAGGACCTGACGTAACACG |
| P65 | ACTGCCGGGATGGCTACTAT | TCTGGATTCGCTGGCTAATGG |
| GAPDH | ACAACTTTGGTATCGTGGAAGG | GCCATCACGCCACAGTTTC |
RT-PCR, quantitative reverse-transcription polymerase chain reaction.
Enzyme linked immunosorbent assay (ELISA)
H9c2 cells were plated in a 6-well plate at 5000/well, and the cells were treated differently. The ELISA kit (Elabscience, Wuhan, China) was used to detect the concentrations of IL-1β, IL-6 in the cell supernatant according to the manufacturer’s instructions.
Intracellular reactive oxygen species (ROS) levels
Flow cytometry was used to detect intracellular ROS levels. After different treatments of the cells, H9c2 cells were collected and washed three times with cold PBS. Total ROS levels were measured by flow cytometry (Becton Dickinson, Heidelberg, Germany) at 37°C for 20 min using DCFH-DA (10 μM Kaiji, Nanjing, China).
Superoxide dismutase (SOD) activity assay
H9c2 cells were transferred to a 6-well plate. After treatment, cells were lysed, collected and centrifuged to remove the supernatant. Detection of SOD levels in cells was performed according to the SOD Assay Kit (Construction, Nanjing, China) manual.
Statistical analysis
Statistical Product and Service Solutions (SPSS) 21.0 (SPSS IBM, Armonk, NY USA) statistical software was used to analyze the experimental data. Measurement data were expressed as x ± s. The t-test was used for comparisons between the two groups. Comparison between multiple groups was done using One-way analysis of variance (ANOVA) test followed by Post-Hoc Test (Least Significant Difference). Least Significant Difference (LSD) test or Student-Newman-Keuls (SNK) test was used for pairwise comparison under the condition of homogeneity of variance. Test level α=0.05. All experiments were repeated 3 times.
Results
UTI inhibits ischemia-anoxic induced degeneration in H9c2 cardiomyocytes
We examined the optimal concentration and optimal culture time of H9c2 cells induced with UTI by CCK-8 assay. It was found that 24-h treatment of 500 μmol/l UTI achieved the highest survival in H9c2 cells (Figure 1A). The supernatant of H9c2 cells was detected by LDH and MDA kits. The results showed that UTI can effectively reduce the levels of LDH and MDA in the supernatant of H9c2 cells (Figure 1B, 1C). It is indicated that UTI can protect H9c2 cells from ischemia and hypoxia injuries.
Figure 1.
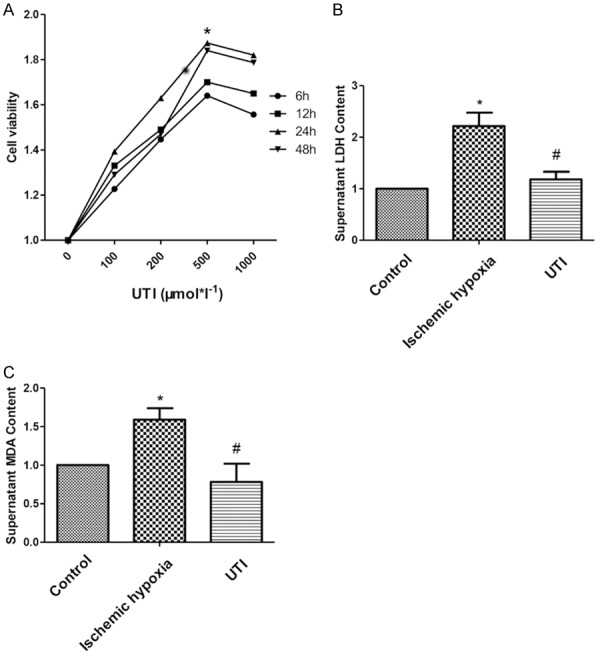
UTI promotes proliferation of H9c2 cells in vitro. A. Optimal concentration and time of UTI treatment in H9c2 cells are determine by CCK-8 assay. B, C. Levels of LDH and MDA in the supernatant are determine by LDH kit and MDA kit. (“*” indicates statistical difference from the control group P<0.05 and “#” indicates statistical difference from the ischemic hypoxia group P<0.05).
UTI inhibits ischemia-anoxic induced oxidative stress in H9c2 cardiomyocytes
Oxidative stress plays an important role in ischemic heart disease. During oxidative stress, SOD1 and SOD2 are important indicators of antioxidant stress. By Western blot and RT-PCR, we found that the anti-oxidative stress was significantly reduced in the hypoxia-ischemia group, and UTI treatment significantly increased the levels of SOD1, SOD2, GPX1 and GPX3 (Figure 2A-E). SOD Activity Assay showed that SOD level was significantly reduced in the ischemia hypoxia group, which was alleviated after UTI intervention (Figure 2F). Flow cytometry results showed that ROS level in the ischemic hypoxia group was significantly higher than that in the control group, and its increased level decreased after UTI treatment (Figure 2G). Immunofluorescence showed that SOD1 levels were significantly improved in the UTI group compared with the ischemic hypoxia group. These data indicated that UTI slowed down oxidative stress caused by ischemic and hypoxia in H9c2 cells by increasing the antioxidant levels (Figure 2H).
Figure 2.
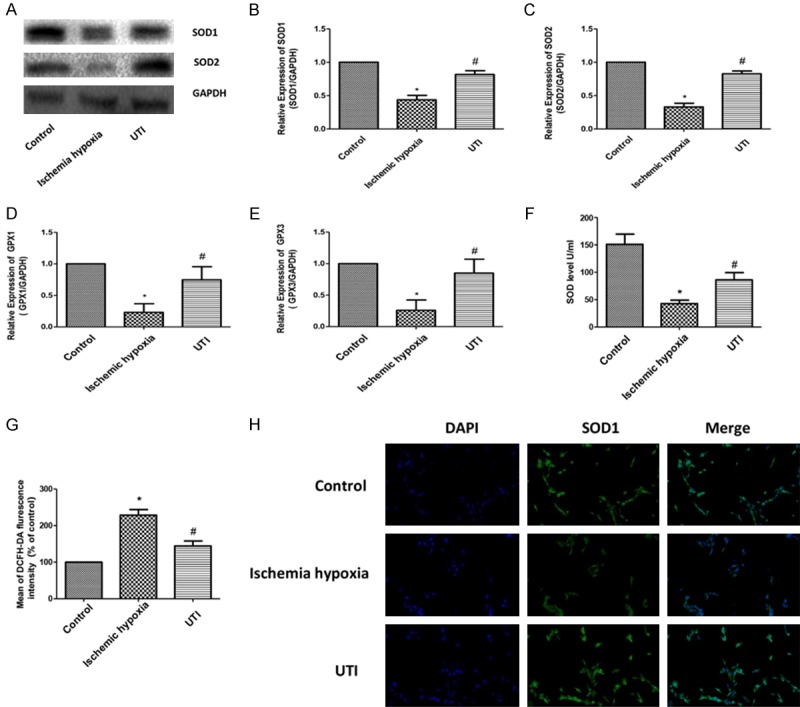
UTI inhibits oxidative stress induced by ischemia-anoxic in H9c2 cardiomyocytes. A. Protein expressions of SOD1 and SOD in three groups are determined by Western blotting. GAPDH is used as an internal control. B-E. The mRNA expressions of SOD1, SOD2, GPX1 and GPX3 in the three groups are determined by real-time PCR. F. SOD activity determined by the SOD activity assay. G. ROS level determined by flow cytometry. H. Positive expression of SOD1 determined by Immunofluorescence in the three groups (100×). (“*” indicates statistical difference from the control group P<0.05 and “#” indicates statistical difference from the ischemic hypoxia group P<0.05).
UTI inhibits ischemia-anoxic induced inflammation of H9c2 cardiomyocytes
The expressions of IL-1β, IL-6 and TNF-α were examined by Western blotting (Figure 3A). The results showed that the expressions of IL-1β, IL-6 and TNF-α inflammatory factors significantly increased in the ischemia hypoxia group, indicating that inflammatory factors were involved in IHD induced by ischemia and hypoxia. After UTI intervention, the expressions of IL-1β, IL-6 and TNF-α were significantly reduced. Similar results were obtained for their mRNA levels (Figure 3B-D). ELISA assay yielded the same results on expressions of IL-1β and IL-6 in three groups (Figure 3E, 3F). Immunofluorescence showed that the expression level of IL-1β in the UTI group was significantly lower than that in the ischemia hypoxia group (Figure 3G), suggesting that UTI may delay ischemic-hypoxia induced injuries in H9c2 cells by inhibiting inflammation.
Figure 3.
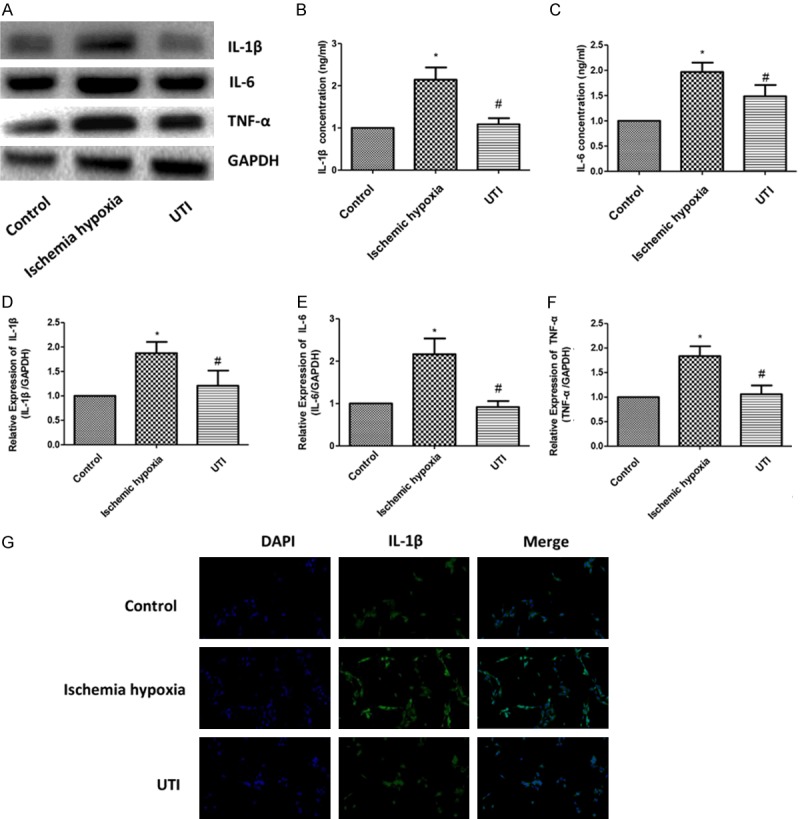
UTI inhibits inflammation induced by ischemia-anoxic in H9c2 cardiomyocytes. A. Protein expressions of IL-1β, IL-6 and TNF-α in the three groups are determined by Western blotting. GAPDH is used as an internal control. B-D. The mRNA expressions of IL-1β, IL-6 and TNF-α in the three groups are determined by real-time PCR. E, F. Protein expressions of IL-1β and IL-6 determined by ELISA in the three groups. G. Positive expression of IL-1β determined by Immunofluorescence in the three groups (100×). (“*” indicates statistical difference from the control group P<0.05 and “#” indicates statistical difference from the ischemic hypoxia group P<0.05).
UTI inhibits ischemia-anoxic induced senescence and apoptosis of H9c2 cardiomyocytes
The expression levels of Sirt1, Bcl-2, Bax and Caspase3 were detected by Western blot (Figure 4A). The results showed that the expressions of Sirt1 and Bcl-2 significantly decreased in the ischemia hypoxia group, while the expression levels of Bax and Caspase3 significantly increased compared with the control group, indicating that senescence and apoptotic factors were involved in ischemia-hypoxia induced IHD. After UTI intervention, the expressions of Sirt1 and Bcl-2 were significantly upregulated in the ischemia hypoxia group, while the expressions of Bax and Caspase3 were significantly downregulated. Bcl-2 to Bax ratio indicated that H9c2 cell apoptosis plays a major role after ischemia and hypoxia treatment (Figure 4B). Similar results were obtained for mRNA levels (Figure 4C-F).
Figure 4.
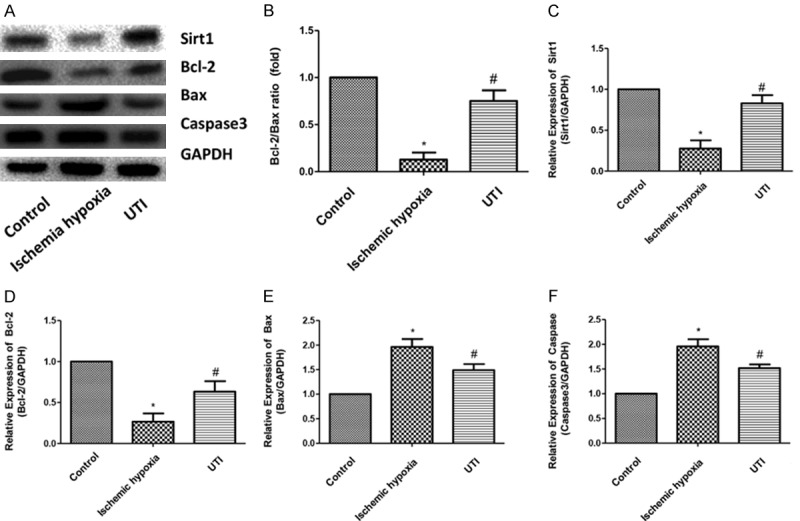
UTI inhibits senescence and apoptosis induced by ischemia-anoxic in H9c2 cardiomyocytes. A. Protein expressions of Sirt1, Bcl-2 and Bax in the three groups are determined by Western blotting. GAPDH is used as an internal control. B. Bcl-2 to Bax ratio. C-F. The mRNA expressions of Sirt1, Bcl-2, Bax and Caspase3 in the three groups are determined by real-time PCR. (“*” indicates statistical difference from the control group P<0.05 and “#” indicates statistical difference from the ischemic hypoxia group P<0.05).
UTI inhibits activation of the NF-κB pathway
The NF-κB pathway plays an important role in IHD. Herein, the expressions of key genes in the NF-κB pathway were detected in different treatment groups. Western blot results showed that in the ischemia hypoxia group, the expressions of p65 and IκB kinase α (IκKα) were significantly upregulated, and the expression of inflammatory inhibitor NF-κBα (IκBα) was significantly reduced compared with the control group. After stimulation with UTI, the expressions of p65 and IκKα were reduced, while the expression level of IκBα increased compared with the ischemic hypoxia group. We also detected the expression of p-p65 and found that p-p65 expression was significantly increased after ischemia and anoxic treatment, but significantly decreased after UTI treatment (Figure 5A). Similar results were also obtained for mRNA levels (Figure 5B-D). These data indicated that UTI inhibited the NF-κB pathway.
Figure 5.
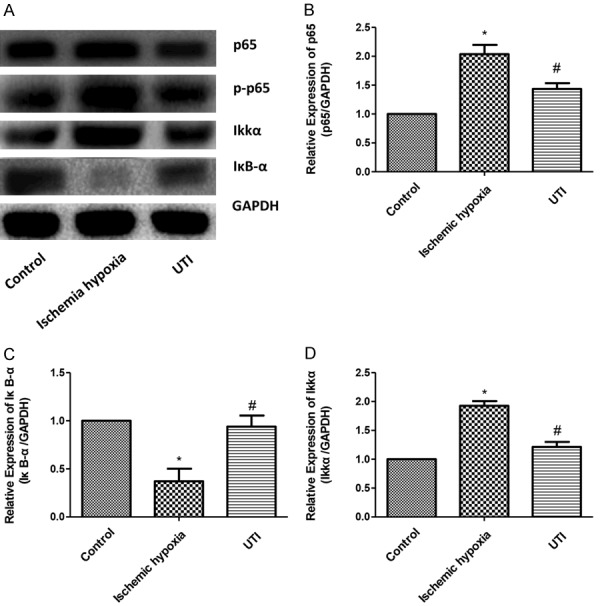
UTI inhibits the activation of the NF-κB pathway. A. Protein expressions of p65, p-P65, IκKα and IκB-α in three groups are detected by Western blotting. GAPDH is used as an internal control. B-D. The mRNA expressions of p65, IκKα and IκB-α in three groups are detected by real-time PCR. (“*” indicates statistical difference from the control group P<0.05 and “#” indicates statistical difference from the ischemic hypoxia group P<0.05).
Discussion
IHD concerns a health issue globally [8]. Previous studies have shown that ischemia-anoxia induced heart damage is thought to be associated with the production of highly reactive oxygen species, which directly damage myocardial cell membranes through membrane lipid peroxidation [9]. The antioxidant defense system is of significance in IHD, which triggers antioxidant depletion, increased lipid peroxidation and activation of inflammatory factors, leading to apoptosis and aging [10]. During the process of life oxidative metabolism, the body continuously produces various oxygen free radicals (OFR) and inflammatory factors. Under physiological conditions, the antioxidant and anti-inflammatory defense system in the body can eliminate a small amount of OFR and inflammatory factors, while the overproduction or clearance of OFR and inflammatory factors in the body is limited, leading to imbalanced ROS production and antioxidant defense [11]. OS can cause lipid peroxidation, damages on cell structure and functions, nucleobase modification and nucleic acid strand breakage, and thus activate inflammation [12]. Oxygen free radical accumulation leads to IL-17-induced monocyte migration and aggregation, promotes secretion of TNF-α and MCP-1 by endothelial cells and macrophages and promotes the expansion of inflammatory response. Myocardial ischemia and hypoxia, mitochondrial single electron reduction, neutrophil respiratory burst and other physiological and pathological changes result in a large number of OFR and ROS accumulated in the cardiomyocytes. Oxidative stress further leads to cells apoptosis, Ca2+ overload, inflammatory transmitters, damage to vascular endothelial cells and other mechanisms causing myocardial cell damage [13].
Studies [14] have found that UTI inhibits the production of oxygen free radicals and multiple inflammatory mediators. UTI is widely applied for the treatment of pancreatitis, sepsis, multiple organ dysfunction syndrome and other diseases [15]. In the study of craniocerebral injury, it was proved that UTI can inhibit neuronal apoptosis and death, improve nerve cell damage, and exert brain protection [16]. Studies [17] have found that UTI can prevent the effects of pulmonary edema and other clinical symptoms by inhibiting various inflammatory substances such as elastase and oxygen free radicals. In the rat model of hepatocyte injury, it is believed that UTI can enhance the antioxidant capacity of the body by down-regulating the expression of p65 in liver tissue [18]. UTI pretreatment can improve cardiac function in sepsis rats by regulating energy metabolism and immune response [19].
To discuss the protective effects of UTI on ischemia-anoxia induced injury in H9c2 cardiomyocytes, cell morphology, oxidative stress, inflammation and apoptosis in each treated group were assessed. The main findings were summarized as follows: 1) UTI inhibited inflammation of H9c2 cells induced by ischemia-anoxia through inhibiting levels of inflammatory factors. 2) UTI inhibited oxidative stress induced by ischemia-anoxia in H9c2 cells through improving cell antioxidant capacity. 3) UIT inhibited apoptosis and improved proliferation in H9c2 cells. 4) UTI protected ischemia-anoxia damage to H9c2 cardiomyocytes by inhibiting the NF-κB signaling pathway.
Studies [20] have shown that IHD involves multiple signaling pathways such as Rho/ROCK signaling pathway, MAPK signaling pathway, and NF-κB signaling pathway. Among them, NF-κB signaling pathway is a very classical signaling pathway. IHD is characterized by elevated levels of inflammatory factors and ROS accumulation, suggesting that the regulations on inflammatory response and oxidative stress play a key role in IHD disease. NF-κB can activate IHD through different exogenous and endogenous stimuli [21]. Previous studies have confirmed that inflammatory factors are closely related to the occurrence and development of intervertebral disc degeneration, including IL-1β, IL-6, IL-10 and NF-κB [22]. In addition, cytoplasmic NF-κB (an inactive state) binds to the inhibitory protein IκBα, preventing NF-κB from entering the nucleus. After being stimulated by many inductive factors or phosphorylation of IκBα, the subunit of NF-κB is transferred to the promoter region of the nuclear metalloprotein (MMP) where containing the binding site of NF-κB, thereby exerting transcriptional regulation. Activation of NF-κB induces MMP and inflammatory factors, which in turn can further activate and regulate MMPs, thereby forming a positive feedback cascade signaling pathway that aggravates inflammatory responses [23]. Therefore, the body’s inflammatory response greatly depends on NF-κB activated cells. In our study, the expressions of key genes in the NF-κB signaling pathway were downregulated by UTI treatment in H9c2 cardiomyocytes, suggesting that UTI can inhibit the NF-κB signaling pathway.
In summary, our study suggested that the protective effect of UTI on IHD relied on the inhibition of the NF-κB signaling pathway. This finding is of great help to the clinical treatment of IHD.
Conclusions
UTI inhibits NF-κB signaling pathway, reduces oxidative stress and inflammation levels of IHD, thereby delaying cell senescence and inhibiting apoptosis. Therefore, UTI is of great value in improving IHD.
Acknowledgements
This study was suppirted by Natural Science Foundatoin of Inner Mongolia (No: 2019MS08161).
Disclosure of conflict of interest
None.
References
- 1.Dalen JE, Alpert JS, Goldberg RJ, Weinstein RS. The epidemic of the 20(th) century: coronary heart disease. Am J Med. 2014;127:807–812. doi: 10.1016/j.amjmed.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 2.Lala A, Desai AS. The role of coronary artery disease in heart failure. Heart Fail Clin. 2014;10:353–365. doi: 10.1016/j.hfc.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Schmid JP. Cardiac rehabilitation and non-medical treatment of chronic heart failure (without devices) Ther Umsch. 2018;75:174–178. doi: 10.1024/0040-5930/a000985. [DOI] [PubMed] [Google Scholar]
- 4.Kurian GA, Rajagopal R, Vedantham S, Rajesh M. The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: revisited. Oxid Med Cell Longev. 2016;2016:1656450. doi: 10.1155/2016/1656450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stocker R, Keaney JJ. Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 6.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 7.He F, Song Y, Ying WJ, Jin XY, Wang ZX, Su JD, Wang GY. Effects of ulinastatin on myocardial oxidative stress and inflammation in severely burned rats. Eur Rev Med Pharmacol Sci. 2018;22:5719–5728. doi: 10.26355/eurrev_201809_15840. [DOI] [PubMed] [Google Scholar]
- 8.Chugh SS, Aro AL. Coronary ischemia: global trigger of sudden cardiac death. Heart Rhythm. 2017;14:88–89. doi: 10.1016/j.hrthm.2016.09.028. [DOI] [PubMed] [Google Scholar]
- 9.Namazi G, Jamshidi RS, Attar AM, Sarrafzadegan N, Sadeghi M, Naderi G, Pourfarzam M. Increased membrane lipid peroxidation and decreased Na+/K+-ATPase activity in erythrocytes of patients with stable coronary artery disease. Coron Artery Dis. 2015;26:239–244. doi: 10.1097/MCA.0000000000000196. [DOI] [PubMed] [Google Scholar]
- 10.Shahzad S, Hasan A, Faizy AF, Mateen S, Fatima N, Moin S. Elevated DNA damage, oxidative stress, and impaired response defense system inflicted in patients with myocardial infarction. Clin Appl Thromb Hemost. 2018;24:780–789. doi: 10.1177/1076029617725602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V, Squadrito F, Altavilla D, Bitto A. Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev. 2017;2017:8416763. doi: 10.1155/2017/8416763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dandekar A, Mendez R, Zhang K. Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol Biol. 2015;1292:205–214. doi: 10.1007/978-1-4939-2522-3_15. [DOI] [PubMed] [Google Scholar]
- 13.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao C, Yin C, Shou S, Wang J, Yu L, Li X, Chai Y. Ulinastatin protects against LPS-induced acute lung injury by attenuating TLR4/NF-kappaB pathway activation and reducing inflammatory mediators. Shock. 2018;50:595–605. doi: 10.1097/SHK.0000000000001104. [DOI] [PubMed] [Google Scholar]
- 15.Atal SS, Atal S. Ulinastatin - a newer potential therapeutic option for multiple organ dysfunction syndrome. J Basic Clin Physiol Pharmacol. 2016;27:91–99. doi: 10.1515/jbcpp-2015-0003. [DOI] [PubMed] [Google Scholar]
- 16.Zhou J, Lian J, Li HX, Hong GL, Zhao GJ, Zhi SC, Qiu QM, Li MF, Lu ZQ. Mechanism research and effect of ulinastatin in the brain tissue injury of acute hydrogen sulfide intoxicated rats. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2016;34:166–172. doi: 10.3760/cma.j.issn.1001-9391.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Tao Z, Hu FQ, Li CF, Zhang T, Cao BZ, Cui LQ. Effect of ulinastatin, a human urinary protease inhibitor, on heatstroke-induced apoptosis and inflammatory responses in rats. Exp Ther Med. 2017;13:335–341. doi: 10.3892/etm.2016.3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song Y, Miao S, Li Y, Fu H. Ulinastatin attenuates liver injury and inflammation in a cecal ligation and puncture induced sepsis mouse model. J Cell Biochem. 2019;120:417–424. doi: 10.1002/jcb.27396. [DOI] [PubMed] [Google Scholar]
- 19.Feng Z, Shi Q, Fan Y, Wang Q, Yin W. Ulinastatin and/or thymosin alpha1 for severe sepsis: a systematic review and meta-analysis. J Trauma Acute Care Surg. 2016;80:335–340. doi: 10.1097/TA.0000000000000909. [DOI] [PubMed] [Google Scholar]
- 20.Perrino C, Barabasi AL, Condorelli G, Davidson SM, De Windt L, Dimmeler S, Engel FB, Hausenloy DJ, Hill JA, Van Laake LW, Lecour S, Leor J, Madonna R, Mayr M, Prunier F, Sluijter JPG, Schulz R, Thum T, Ytrehus K, Ferdinandy P. Epigenomic and transcriptomic approaches in the post-genomic era: path to novel targets for diagnosis and therapy of the ischaemic heart? Position paper of the European society of cardiology working group on cellular biology of the heart. Cardiovasc Res. 2017;113:725–736. doi: 10.1093/cvr/cvx070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang JJ, Peng K, Zhang J, Meng XW, Ji FH. Dexmedetomidine preconditioning may attenuate myocardial ischemia/reperfusion injury by down-regulating the HMGB1-TLR4-MyD88-NF-small ka, CyrillicB signaling pathway. PLoS One. 2017;12:e172006. doi: 10.1371/journal.pone.0172006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma T, Guo CJ, Zhao X, Wu L, Sun SX, Jin QH. The effect of curcumin on NF-kappaB expression in rat with lumbar intervertebral disc degeneration. Eur Rev Med Pharmacol Sci. 2015;19:1305–1314. [PubMed] [Google Scholar]
- 23.Mitchell S, Vargas J, Hoffmann A. Signaling via the NFkappaB system. Wiley Interdiscip Rev Syst Biol Med. 2016;8:227–241. doi: 10.1002/wsbm.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]