

Summary
Primary tauopathies are characterized neuropathologically by inclusions containing abnormal forms of the microtubule-associated protein tau (MAPT) and clinically by diverse neuropsychiatric, cognitive, and motor impairments. Autosomal dominant mutations in the MAPT gene cause heterogeneous forms of frontotemporal lobar degeneration with tauopathy (FTLD-Tau). Common and rare variants in the MAPT gene increase the risk for sporadic FTLD-Tau, including progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD). We generated a collection of fibroblasts from 140 MAPT mutation/risk variant carriers, PSP, CBD, and cognitively normal controls; 31 induced pluripotent stem cell (iPSC) lines from MAPT mutation carriers, non-carrier family members, and autopsy-confirmed PSP patients; 33 genome engineered iPSCs that were corrected or mutagenized; and forebrain neural progenitor cells (NPCs). Here, we present a resource of fibroblasts, iPSCs, and NPCs with comprehensive clinical histories that can be accessed by the scientific community for disease modeling and development of novel therapeutics for tauopathies.
Keywords: tau, MAPT, fibroblasts, induced pluripotent stem cells, neural progenitor cells, tauopathy, frontotemporal dementia, progressive supranuclear palsy, corticobasal degeneration, CRISPR/Cas9
Graphical Abstract
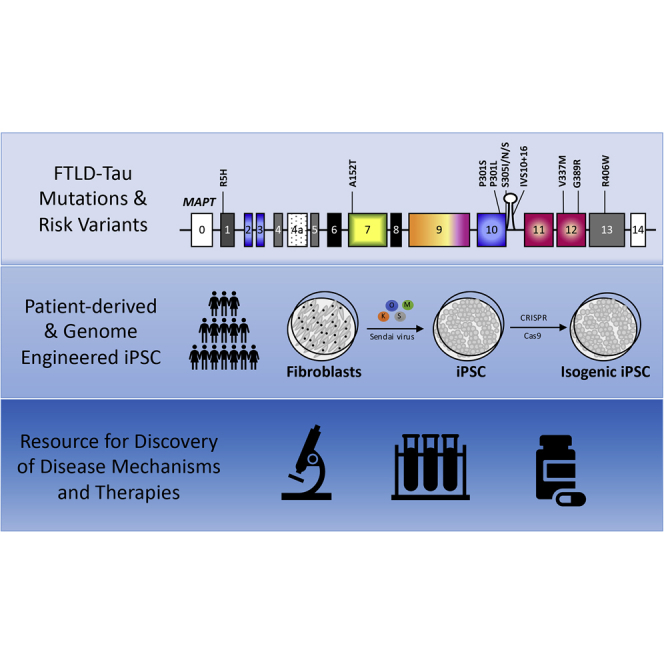
Highlights
-
•
A collection of fibroblasts from 140 MAPT mutation carriers, PSP, CBD, and controls
-
•
31 iPSC lines reprogrammed from MAPT mutation carriers, PSP patients, and controls
-
•
33 iPSC lines engineered with CRISPR/Cas9 or TALENs
-
•
Comprehensive resource for tauopathy modeling and discovery of novel therapeutics
In this article, Karch, Temple and colleagues describe a resource of fibroblasts, patient-derived induced pluripotent stem cells, and genome engineered stem cells with comprehensive clinical histories that can be accessed by the scientific community for disease modeling and development of novel therapeutics for primary tauopathies.
Introduction
Frontotemporal lobar degeneration (FTLD) with inclusions containing the microtubule-associated protein tau (FTLD-Tau) account for half of all cases of FTLD. This heterogeneous group of diseases includes progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick disease, and other rare forms of tauopathy. Patients with FTLD-Tau exhibit a broad range of neurological deficits including movement and motor neuron disease (e.g., gait and balance disturbances, impaired speech and swallowing, visual impairment), psychiatric impairment (e.g., mood and behavior), and cognitive impairment (e.g., memory, executive dysfunction, language and attention) (Perry et al., 2017). Due to significant overlap in the clinical syndromes, a definitive diagnosis can only be obtained by postmortem examination of brain tissue obtained at autopsy or more rarely by biopsy (Perry et al., 2017). Thus, to understand disease etiology, it is particularly valuable to generate a collection of induced pluripotent stem cells (iPSCs) from patients who have been followed clinically and from whom detailed neurological, neuroimaging, and neuropathological data and tissues are available. This requires a coordinated multidisciplinary effort and is the central impetus for the development of the resource described here.
While the majority of patients with primary tauopathy are sporadic, autosomal dominant FTLD-Tau families have been reported to carry mutations in the microtubule-associated protein tau (MAPT) gene. More than 50 MAPT mutations are reported to cause FTLD-Tau (Table 1; http://www.molgen.ua.ac.be/ADMutations/) (Cruts et al., 2012). The MAPT gene is alternatively spliced in the central nervous system (CNS) to produce six tau isoforms that differ based on the presence of the N-terminal insertion (0N, 1N, 2N) and the number of microtubule-binding repeats (MTBR; 3R, 4R; Figure 1). In normal adult human brains, the ratio of 3R/4R tau is 1:1 (Trabzuni et al., 2012). MAPT mutation carriers may bear 3-repeat (3R), 4-repeat (4R), or mixed 3R/4R tau inclusions (Table 1) (Cairns et al., 2007).
Table 1.
Neuropathology in FTLD-Tau Associated with MAPT Mutations
| Mutation | Clinical | Neuropathology |
Tau Isoforms | Referencesa | iPSCs Reported | ||
|---|---|---|---|---|---|---|---|
| Macroscopy | Microscopy | Tauopathy | |||||
| P301L | bvFTD, personality change, language abnormalities | atrophy of frontal and temporal lobes, basal ganglia, hippocampus, and depigmentation of substantia nigra | neuronal loss, ballooned neurons, and gliosis | neurons: tau-immunoreactive perinuclear, ring-like and dot-like cytoplasmic inclusions, fibrillary neuronal inclusions, and neuropil threads | 4R | Mirra et al., 1999 | Iovino et al., 2015, Paonessa et al., 2019, Silva et al., 2019 |
| astrocytes: thorn-shaped inclusions | |||||||
| oligodendrocytes: coiled bodies | |||||||
| S305I | bvFTD, personality change, language abnormalities, Parkinsonism | atrophy of medial temporal lobe, temporal pole, and hippocampus | neuronal loss, gliosis, and ballooned neurons | neurons: tau-immunoreactive fibrillary inclusions and diffuse cytoplasmic staining | 4R | Kovacs et al., 2008 | N/A |
| astrocytes: thorn-shaped inclusions | |||||||
| oligodendrocytes: coiled bodies | |||||||
| argyrophilic grains | |||||||
| S305N | bvFTD, personality change, memory loss | atrophy of frontal and temporal lobes | neuronal loss and gliosis | neurons: tau-immunoreactive Pick body-like and ring-like inclusions | 4R | Boeve et al., 2005, Iijima et al., 1999 | N/A |
| astrocytes: thorn-shaped inclusions | |||||||
| oligodendrocytes: coiled bodies | |||||||
| S305S | bvFTD, memory loss | atrophy of frontal and temporal lobes | neuronal loss, gliosis, and ballooned neurons | neurons: tau-immunoreactive neurofibrillary tangles and pretangles | 4R | Skoglund et al., 2008, Stanford et al., 2000 | N/A |
| astrocytes: tuft-shaped inclusions | |||||||
| oligodendrocytes: coiled bodies | |||||||
| IVS10+16 | bvFTD, personality change, executive dysfunction, memory loss, parkinsonism, non-fluent aphasia | atrophy of frontal and temporal lobes, cingulate and insular cortex, hippocampus, striatum, amygdala and brainstem | neuronal loss, gliosis, and ballooned neurons | neurons: tau-immunoreactive fibrillary inclusions and diffuse cytoplasmic staining | 4R | Janssen et al., 2002, Lantos et al., 2002 | Espuny-Camacho et al., 2017, Esteras et al., 2017, Paonessa et al., 2019, Sposito et al., 2015 |
| astrocytes: thorn-shaped inclusions | |||||||
| oligodendrocytes: coiled bodies | |||||||
| V337M | antisocial behavior, paranoia, executive dysfunction | atrophy of frontal and temporal lobes and hippocampus | neuronal loss and gliosis | neurons: tau-immunoreactive neurofibrillary tangles, pretangles, and neuropil threads | 3R & 4R | Spillantini et al., 1996, Spina et al., 2017 | Ehrlich et al., 2015, Sohn et al., 2019 |
| astrocytes: tuft-shaped inclusions | |||||||
| oligodendrocytes: none | |||||||
| G389R | progressive aphasia, apathy, rigidity | atrophy of frontal and temporal lobes, hippocampus, and amygdala | neuronal loss and gliosis | neurons: tau-immunoreactive Pick body-like and filamentous inclusions | 3R & 4R | Murrell et al., 1999 | N/A |
| astrocytes: none | |||||||
| oligodendrocytes: none | |||||||
| R406W | memory loss | severe atrophy of frontal and temporal lobes and hippocampus | neuronal loss, gliosis, and ballooned neurons | neurons: tau-immunoreactive neurofibrillary tangles, Pick body-like inclusions | 3R & 4R | Miyasaka et al., 2001, Reed et al., 1997 | Imamura et al., 2016, Jiang et al., 2018 |
| astrocytes: thorn-shaped inclusions | |||||||
| oligodendrocytes: coiled bodies | |||||||
| R406W/R406W | bvFTD | N/A | N/A | N/A | 3R & 4R | Behnam et al., 2015, Ng et al., 2015 | N/A |
N/A, not available.
See additional references at the AD/FTD Mutation Database (Cruts et al., 2012).
Figure 1.

MAPT Mutations Cause Primary Tauopathy
(A) Schematic of the location of MAPT mutations reported in this collection. MAPT A152T, V337M, G389R, and R406W occur in all tau isoforms expressed in the brain. MAPT P301L, P301S, and S305I/N/S occur exclusively in transcripts containing exon 10 (2N4R, 1N4R, and 0N4R). MAPT P301L/S, S305I/N/S and IVS10+16 alter splicing of tau such that more 4R-containing transcripts are expressed.
(B–I) Neuropathology in human brains with primary tauopathies. (B–E) MAPT R406W carrier. (B) Atrophy of the frontal lobe with dilatation of the lateral ventricle and prominent shrinkage of the medial temporal lobe. Scale bar, 0.5 cm. (C) Neuronal loss, gliosis, and microvacuolation of superficial laminae of the superior temporal gyrus. H&E. (D) Neuronal cytoplasmic PHF1-immunoreactive inclusions are seen in the hippocampal CA1 subfield. (E) Pick body-like, PHF1-immunoreactive inclusion bodies in the dentate fascia. Scale bar in (C), (D), and (E), 50 μm. (F and G) Anterior cingulate gyrus of a MAPT V337M carrier. (F) RD4-immunoreactive cytoplasmic inclusions in spindle, also called von Economo, neurons and surrounding layer V neurons. (G) R3 (RD3) tau-immunoreactive cytoplasmic inclusions in spindle and surrounding layer V neurons, and in the neuropil. (H) Dentate gyrus of MAPT P301L case showing typical pTAU (CP13) ring-like perinuclear deposit and Pick body-like inclusions. (I) PSP associated with a MAPT A152T variant. Tufted astrocyte (left; white arrow), neurofibrillary tangle (center; open arrow), and oligodendroglial coiled bodies (right; black arrow), stained with a phospho-tau antibody (CP13). Scale bar, 25 μm.
Several mechanisms have been proposed to explain how MAPT mutations cause disease: abnormal MAPT splicing, altered microtubule-binding kinetics, impaired degradation, or tau accumulation and aggregation, among others (van Swieten and Spillantini, 2007). We have focused our collection on mutations that represent these proposed mechanisms. A subset of MAPT mutations occur at sites that alter MAPT splicing, resulting in increased levels of exon 10-containing (4R) mRNA (e.g., IVS10+16, S305I, S305N, S305S) (Liu and Gong, 2008). In the case of intronic mutations such as IVS10+16, no mutant protein is produced. Instead, there is a shift in the levels of 4R tau, skewing the normally balanced 3R/4R tau ratio in human adult brain. Another set of mutations occurs in exon 10, which is exclusively present in 4R tau isoforms (e.g., P301L, P301S) (Hutton et al., 1998). Many of the mutations located in and around exon 10 have been implicated in disrupting microtubule-binding kinetics (Dayanandan et al., 1999, Fischer et al., 2007). Other MAPT mutations are located some distance from exon 10 and are expressed by all MAPT transcripts (e.g., R5H, V337M, G389R, R406W); thus, their mode of action may be linked to aspects of tau biology beyond microtubule binding, such as membrane association (Gauthier-Kemper et al., 2011). Additionally, all MAPT mutations increase the propensity for the tau protein to aggregate. Despite the clear association of MAPT mutations with FTLD-Tau, we have little understanding of the mechanisms by which these mutations lead to disease.
Rare and common variants in MAPT have been associated with increased risk for PSP, CBD, and frontotemporal dementia (FTD) (Coppola et al., 2012, Höglinger et al., 2011). MAPT A152T decreases binding of tau to microtubules and increases tau oligomer formation in vitro (Coppola et al., 2012). When expressed in Caenorhabditis elegans, MAPT A152T induces neuronal dysfunction, mislocalization of pre-synaptic proteins, and distorted mitochondrial distribution and trafficking, and reduces life span independently of protein aggregation (Butler et al., 2019, Pir et al., 2016). Mouse models expressing A152T demonstrate age-dependent neuronal loss and seizures, which occur both in the presence (Decker et al., 2016) or absence of tau aggregates (Maeda et al., 2016). iPSC-derived neurons from MAPT A152T carriers exhibit increased total tau levels and phosphorylation, detergent-insoluble tau, dysregulation of proteostasis pathways involving autophagy and lysosomal activity, and vulnerability to specific cellular stressors (Biswas et al., 2016, Fong et al., 2013, Silva et al., 2016). Our fibroblast and stem cell resource containing MAPT variants that are predicted to modify PSP and CBD risk will allow for the cellular and molecular dissection of disease phenotypes.
Patient-derived iPSCs have emerged as a powerful resource to study the molecular mechanisms underlying neurodegenerative diseases. These iPSCs can be differentiated into the neuronal and glial subtypes that are affected in primary tauopathies, giving us a tool toward understanding the biology of tau in a human cell model that may more faithfully reflect the endogenous condition. To date, iPSCs carrying the MAPT mutations N279K, P301L, V337M, R406W, and IVS10+16 and the risk variant A152T have been described, and their neural derivatives show phenotypes such as tau accumulation, tau hyperphosphorylation, tau insolubility, vulnerability to specific cellular stressors, and other phenotypes that begin to reveal possible disease mechanisms (Ehrlich et al., 2015, Hallmann et al., 2017, Imamura et al., 2016, Iovino et al., 2015, Jiang et al., 2018, Seo et al., 2017, Silva et al., 2016, Sposito et al., 2015, Wren et al., 2015). Most prior studies, however, have not included genome-edited isogenic controls, which increase the power to detect variant-specific phenotypes by decreasing inter-individual variability. These promising findings warrant the investment in a comprehensive, isogenically controlled collection of MAPT iPSC lines.
Here, we present a resource of fibroblast and iPSC lines that includes known disease-associated MAPT mutations and paired isogenic iPSCs, MAPT risk variant carriers, and PSP-syndrome (PSP-S) and corticobasal syndrome (CBS) lines from individuals where a MAPT mutation has not been detected. Most of the cell lines presented in this study were obtained from participants who underwent detailed clinical phenotyping, and for whom fluid biomarkers, imaging biomarkers, and genetic and neuropathological information are available that can be used for correlative analyses with cellular phenotypes. Together, this represents a comprehensive resource that can be accessed for tauopathy modeling and the discovery of novel therapeutics.
Results
Selection of Fibroblast Lines
Dermal fibroblasts were collected from: (1) families with pathogenic MAPT mutations; (2) individuals carrying MAPT missense variants that increase risk for PSP, CBD, and FTD; (3) sporadic PSP-S and CBS cases; and (4) cognitively normal (non-mutant) controls at the Memory and Aging Center at the University of California San Francisco, the Knight Alzheimer Disease Research Center at Washington University, and the National Institute of Neurological Disease and Stroke (NINDS) Cell Repository. A total of 36 fibroblast lines were generated from individuals with pathogenic MAPT mutations (P301L, S305I, IVS10+16, V337M, G389R, R406W; Figure 1 and Table 2). These mutations are representative of some of the most common MAPT mutations and capture the range of clinical and neuropathological phenotypes associated with FTLD-Tau (Tables 1 and 2). The collection also includes fibroblasts from eight MAPT A152T risk variant carriers, which has been reported to increase the risk for PSP, CBD, and FTD (Coppola et al., 2012). Additionally, we have banked fibroblast lines from research participants clinically diagnosed with PSP-S, CBS, or mixed dementias (Alzheimer's disease and Lewy body disease) and from cognitively normal controls (Table 2). These fibroblasts were obtained from subjects who are part of larger clinical programs that obtain a detailed clinical history, including physical and neurological examinations, cognitive testing, and neuroimaging (magnetic resonance imaging [MRI], β-amyloid positron emission tomography [PET], and tau PET). Many fibroblast lines also have corresponding plasma and cerebrospinal fluid (CSF) samples. Additional covariates for the fibroblast lines including age at biopsy, sex, and genotypic data are available upon request from http://neuralsci.org/tau.
Table 2.
Dermal Fibroblast Bank to Model Primary Tauopathies
| MAPT | Classification | Clinical Presentation | Tau Isoform | Mean AAOa | Mean Disease Durationa | Fibroblasts | Families |
|---|---|---|---|---|---|---|---|
| A152T | PSP, CBD, FTLD-Tau | bvFTD | 4R | 57.5 | N/A | 8 | N/A |
| P301L | FTLD-Tau | bvFTD | 4R | 52.6 | 6.7 | 13 | 3 |
| S305I | FTLD-Tau/AGD | bvFTD | 4R | 39 | 2 | 2 | 1 |
| IVS10+16 | FTLD-Tau | bvFTD/AD | 4R | 49.1 | 10.3 | 4 | 1 |
| V337M | FTLD-Tau | bvFTD | 3R & 4R | 51.5 | 15.4 | 4 | 2 |
| G389R | FTLD-Tau | bvFTD | 3R & 4R | 39.8 | 2.5 | 3 | 1 |
| R406W | FTLD-Tau | AD | 3R & 4R | 56.3 | 11.5 | 9 | 2 |
| R406W/R406W | FTLD-Tau | bvFTD | 3R & 4R | 34 | 7 | 1 | 1 |
| WT | PSP | PSP-S | 4R | N/A | N/A | 12 | N/A |
| WT | CBD | CBS | 4R | N/A | N/A | 5 | N/A |
| WT | PSP/CBD mixed | PSP-S/CBS/mixed | 4R | N/A | N/A | 10 | N/A |
| WT | normal | N/A | N/A | N/A | N/A | 69 | N/A |
AAO, age at onset; bvFTD, behavioral variant frontotemporal dementia; AGD, argyrophilic grain disease; AD, Alzheimer's disease; PSP, progressive supranuclear palsy; CBD, cortical basal degeneration; N/A, not available.
Data from the AD/FTD Mutation Database presented in years (Cruts et al., 2012).
The patient-specific fibroblasts in this collection capture classical aspects of clinical and neuropathology associated with primary tauopathies (Table 1). A MAPT R406W carrier presented with progressive memory loss and later developed the behavioral variant of FTD (bvFTD). Macroscopically, there was pronounced atrophy of the temporal lobe (cell line F11362; Figures 1B–1E). Microscopically, there was severe neuronal loss and gliosis and cortical neurofibrillary tangles similar to those seen in Alzheimer’s disease. Frequent Pick body-like, tau-immunoreactive inclusions were seen in affected areas including the hippocampus and dentate gyrus. Tau-immunoreactive glial inclusions were also present. A MAPT V337M carrier included in the collection exhibited bvFTD; neuropathological examination revealed inclusions containing both 3R and 4R tau (cell line GIH6; Figures 1F and 1G). MAPT P301L cases more commonly present clinically with bvFTD, and pathologically display neuronal cytoplasmic inclusions with perinuclear ring-like concentration and the presence of mini Pick body-like inclusions (Figure 1H). A MAPT A152T carrier (cell line FTD19; Figure 1I) included in the collection presented with symptoms characteristic of PSP-S, including motor slowing, falls, and cervical dystonia that progressed to dysarthria as well as supranuclear gaze palsy, and neuropathological examination revealed classical PSP neuropathology featuring 4R tau-immunoreactive tufted astrocytes (white arrow in Figure 1I), neurofibrillary tangles (open arrow in Figure 1I), and oligodendroglial coiled bodies (black arrow in Figure 1I).
Generation and Characterization of iPSCs
To establish cellular models that can inform on the pathophysiological mechanisms of MAPT mutations, MAPT risk variants, and sporadic PSP, including cell types affected by disease, we reprogrammed a subset of fibroblasts described in Table 2 into iPSCs. All iPSCs were generated using non-integrating Sendai virus carrying SOX2, OCT4, KLF4, and cMYC (Table 3, Figure 2A). Multiple clones are available for each line. iPSCs were grown in feeder-free conditions using Matrigel and maintained in mTeSR1. Resulting iPSCs have been characterized for pluripotency based on morphology and gene expression markers (Figures 2B and 2C). We confirmed the silencing of exogenous Sendai virus-driven pluripotent markers by qPCR (Figure 2C). Correct mutation propagation was verified by Sanger sequencing (Figure 2D), chromosomal stability was assessed by karyotyping (Figure 2E), and the capacity to form cell types from the three germ layers was also confirmed (Figures 2F and 2G). All iPSC lines reported in this study meet these quality-control criteria (Table S1 and Figure S1), are included in the cell bank, and are available upon request from http://neuralsci.org/tau.
Table 3.
Human iPSCs for Modeling Primary Tauopathies
| Donor ID | Alternative Donor ID | Mutation | Clinical Statusa | Autopsy | Corrected Line | Fibroblast Source | Neural Induction |
|---|---|---|---|---|---|---|---|
| FTD30 (FTD-FF) | 151209SBA1 | A152T/WT | S | pending | no | UCSF | yes |
| FTD19 (FTD-T) | 151209SBA2 | A152T/WT | S | PSP | no | UCSF | yes |
| GIH2 | 151209SBA3 | A152T/WT | A | N/A | no | UCSF | yes |
| FTD38 | 151209SBA4 | A152T/WT | A | N/A | no | UCSF | yes |
| GIH169 | 160311SBA5 | A152T/WT | S | N/A | no | UCSF | yes |
| GIH56 | 160311SBA6 | A152T/WT | S | N/A | no | UCSF | yes |
| TAU6 (Tau1225-7) | 160311SBA7 | A152T/WT | S | CBD | no | UCSF | yes |
| F0510 | F0510 | P301L/WT | A | N/A | yes | NINDS repository | yes |
| F13535 | F13535 | P301L/WT | A | N/A | no | WUSM | N/A |
| F14537 | F14537 | P301L/WT | S | FTLD-Tau | no | WUSM | N/A |
| F14536 | F14536 | WT/WTb | A | N/A | no | WUSM | N/A |
| MHF110 | 17524NCE1 | S305I/WT | S | N/A | no | UCSF | N/A |
| 75.11 | AG255075 | S305I/WT | N/A | N/A | yes | ARTFL/LEFFTDS | N/A |
| 300.12 | AG251300 | S305N/WT | N/A | N/A | no | ARTFL/LEFFTDS | N/A |
| GP1.1 | GP-1i | S305S/WT | N/A | FTLD-Tau | yes | NSWBB | N/A |
| GIH36 | 160311SBC1 | IVS10+16/WT | A | N/A | yes | UCSF | yes |
| GIH161 | I18XXYYNCG1 | WT/WTb | A | N/A | no | UCSF | N/A |
| GIH178 | I18XXYYNCC3 | IVS10+16/WT | N/A | N/A | no | UCSF | N/A |
| GIH6 | 160311SBB1 | V337M/WT | S | FTLD-Tau | yes | UCSF | yes |
| GIH7 | 160311SBB2 | V337M/WT | A | N/A | yes | UCSF | yes |
| GIH155 | 160311SBB3 | V337M/WT | S | N/A | no | UCSF | N/A |
| ND32951A | ND32951A | V337M/WT | A | N/A | yes | NINDS repository | yes |
| MHF100 | 171018NCD1 | G389R/WT | A | N/A | no | UCSF | N/A |
| MHF101 | 171018NCD2 | G389R/WT | A | N/A | no | UCSF | N/A |
| MHF102 | 171018NCD3 | G389R/WT | S | N/A | no | UCSF | N/A |
| F11374 | F11374 | R406W/WT | A | N/A | no | WUSM | N/A |
| F11362 | F11362 | R406W/WT | S | FTLD-Tau | yes | WUSM | yes |
| F11421 | F11421 | R406W/WT | A | N/A | yes | WUSM | yes |
| GIH143 | UCSF1 | R406W/R406W | S | N/A | no | UCSF | N/A |
| GIH131 | 170524NCF1 | WT/WT | S | PSP | no | UCSF | N/A |
| GIH92 | 171013NCF3 | WT/WT | S | PSP | no | UCSF | N/A |
UCSF, University of California San Francisco Memory and Aging Center; WUSM, Washington University, Knight Alzheimer's Disease Research Center; NINDS repository, National Institute of Neurologic Disorders and Stroke; ARTFL/LEFFTDS, Advancing Resource and Treatment for Frontotemporal Dementia/Longitudinal Evaluations of Familial Frontotemporal Dementia Subjects; NSWBB, New South Wales Brain Bank; N/A, not available.
At biopsy: A, asymptomatic; S, symptomatic.
Non-carrier, related to MAPT family.
Figure 2.

Generation and Characterization of iPSC Models of Tauopathy
Representative images of control (MAPT WT/WT), mutant (MAPT P301L/WT), and CRISPR/Cas9-edited, isogenic control (MAPT WT/WT-iso) iPSCs.
(A) Diagram of reprogramming and CRISPR/Cas9 editing.
(B and C) Immunostaining (B) and qPCR (C) for pluripotency markers. Graph represents mean ± SEM.
(D) Sanger sequencing.
(E) Karyotyping.
(F and G) Spontaneous differentiation into cells within the three germ layers evaluated by RT-PCR (F) and immunostaining (G). MAPT WT/WT (iPSC line: F11350); MAPT P301L/WT (iPSC line: F0510); MAPT WT-iso (iPSC line: F0510.2Δ2′H1). Scale bars, 50 μm.
See also Table S1.
Genome Editing and Characterization of iPSCs
Genetic background of individual donors is a large contributor to phenotypic variability in iPSCs (Kilpinen et al., 2017). To define phenotypes driven specifically by a mutant or risk allele, we used CRISPR/Cas9 genome editing to establish isogenic controls of donor iPSC lines (Figure 2A). For each set of edited lines, additional iPSC clones were selected that underwent the CRISPR/Cas9 editing pipeline but remained unmodified. These unmodified iPSC clones serve as important controls, in addition to the parental donor line, to account for selective pressures that may occur during the editing process (Budde et al., 2017). Donor iPSC lines carrying the MAPT mutations IVS10+16, P301L, S305I, R406W, and V337M have been corrected to wild type (WT; Tables 4 and S2).
Table 4.
CRISPR/Cas9-Edited iPSC Lines
| Donor ID | Donor Genotype | Isogenic Genotype | Ngn2 Integration | Engineering Methoda | Line Name | Neural Induction |
|---|---|---|---|---|---|---|
| F11362 | R406W/WT | WT/WT | no | CRISPR | F11362.1Δ1C11, F11362.1Δ1B6 | yes |
| F11421 | R406W/WT | WT/WT | no | CRISPR | F11421.12Δ2A07 | yes |
| F11374 | R406W/WT | N/A | yes | TALENs | NF11374.65 | yes |
| 160311SBB1 | V337M/WT | WT/WT | no | CRISPR | GIH6C1Δ1E11 | yes |
| 160311SBB2 | V337M/WT | WT/WT | no | CRISPR | GIH7C2Δ2B12, GIH7C2Δ2F02 | yes |
| ND32951A | V337M/WT | WT/WT | no | CRISPR | ND32951A.15Δ1B06, ND32951A.15Δ1C12 | yes |
| GIH36 | IVS10+16/WT | WT/WT | no | CRISPR | GIH36C2Δ1D01 | yes |
| F0510 | P301L/WT | WT/WT | no | CRISPR | F0510.2Δ2E7, F0510.2Δ2′H1 | yes |
| F0510 | P301L/WT | P301L/P301S | no | CRISPR | F0510.2Δ3A11, F0510.2Δ3A9 | yes |
| F0510 | P301L/WT | WT/P301S | no | CRISPR | F0510.2Δ3E10, F0510.2Δ4B3, F0510.2Δ4B4 | yes |
| F0510 | P301L/WT | P301S/P301S | no | CRISPR | F0510.2Δ3B5 | yes |
| F0510 | P301L/WT | N/A | yes | TALENs | NF0510.23, NF0510.12 | yes |
| 75.11 | S305I/WT | WT/WT | no | CRISPR | 75.11-IW1A12 | N/A |
| 75.11 | S305I/WT | S305I/S305I | no | CRISPR | 75.11-IH1B9 | N/A |
| GP1.1 | S305S/WT | S305S/S305S | no | CRISPR | GP1.1-SH1G8 | N/A |
| F13505 | WT/WT | S305I/WT | no | CRISPR | F13505.1-I1B10 | N/A |
| F13505 | WT/WT | S305S/WT | no | CRISPR | F13505.1-S3H5 | N/A |
| F11350 | WT/WT | WT/R5H | no | CRISPR | F11350.1.R5HΔ2F06 | N/A |
| F11350 | WT/WT | WT/G389R | no | CRISPR | F11350.1.G389R.1C05ΔE03 | N/A |
| F11350 | WT/WT | P301L/P301L | no | CRISPR | F11350.1.P301LΔ4A02, F11350.1.P301LΔ4A08 | N/A |
| F12468 | WT/WT | N/A | yes | TALENs | NF12468.131 | yes |
| WTC11 | WT/WT | N/A | yes | TALENs | NWTC11.G3 | yes |
| WTC11 | WT/WT | WT/WT | yes | TALENs/CRISPR | NWTC11.G3.0036 | yes |
| WTC11 | WT/WT | V337M/WT | yes | TALENs/CRISPR | NWTC11.G3.0212 | yes |
| WTC11 | WT/WT | V337M/V337M | yes | TALENs/CRISPR | NWTC11.G3.3917 | yes |
Ngn2 was engineered by TALENs; MAPT mutations/corrections were engineered by CRISPR/Cas9.
The most commonly used mouse model of tauopathy overexpresses MAPT P301S (Yoshiyama et al., 2007). Clinically, MAPT P301S carriers present with a more aggressive form of FTD than MAPT P301L carriers and have an earlier age at onset (P301S: mean age at onset 33.7 years and mean disease duration 4.2 years; P301L: mean age at onset 52.6 years and mean disease duration 6.7 years) (Cruts et al., 2012). However, fibroblast lines from MAPT P301S carriers, which would be useful to validate mouse studies, were not available for reprogramming at the time of this study. Thus, we used CRISPR/Cas9 to mutate the MAPT P301L donor iPSC line to MAPT P301S (Tables 4 and S2). For this series, we performed whole-genome sequencing and analysis of the mutational burden induced by genome editing and observed no modifications at computationally predicted off-target sites from CRISPR/Cas9 (Budde et al., 2017). The mutational burden that was observed in the edited iPSC lines was largely driven by selective pressures of culture (Bhutani et al., 2016, Budde et al., 2017, Merkle et al., 2017).
To understand the specific contribution of MAPT mutations and risk variants to disease phenotypes, we introduced MAPT mutations into an unaffected control line. In a control donor iPSC line (F11350) from a male individual carrying APOE 3/3 and MAPT H1/H1, we introduced MAPT R5H, P301L, or G389R (Tables 4 and S2). In a second control iPSC line (F13505) from a female individual carrying APOE 3/3 and MAPT H1/H1, we introduced MAPT S305I or S305S (Tables 4 and S2). All resulting edited or unmodified clones were characterized for pluripotency and chromosomal stability as described above (Table S2). We are continuing to build this collection on the same genetic background with additional MAPT mutations.
Differentiation of iPSCs into Neural Progenitor Cells and Differentiated Neural Cells
iPSCs have the capacity to form the diverse neural cell types affected by primary tauopathies. By exploiting our understanding of CNS development, several groups have established protocols to generate neuroectodermal neural progenitor cells (NPCs) that can be further patterned into specific neuronal subtypes (Doi et al., 2014, Elkabetz et al., 2008, Muratore et al., 2014). We adapted a neural aggregate-based method that allows for the efficient generation of a scalable pool of NPCs, which have the capacity to be patterned into cultures enriched for different types of neurons or glia (Figure 3). Production of cryopreserved banks of stable and expandable intermediate NPC populations will help to reduce time, effort, and variability across experiments.
Figure 3.

Differentiation of iPSCs into Neural Progenitor Cells
(A) Diagram for neural progenitor derivation protocol.
(B–E) Bright-field images. (B) iPSC. (C) Neural aggregates. (D) Neural rosettes. (E) NPCs.
(F) RT-PCR of neural progenitor cell markers, NESTIN, SOX2, PAX6, and the housekeeping gene, ACTIN.
(G) Immunostaining for neural progenitor cell marker, PAX6.
(H and I) Immunostaining of iPSC-derived neurons. (H) Tuj1. (I) MAP2.
(J) Immunostaining of iPSC-derived astrocytes with GFAP.
Scale bars, 50 μm.
We have applied this neural induction protocol to the donor lines in this collection across multiple laboratories (Figure S1; Tables S1 and S2). We have verified that the iPSC collection presented here has the capacity to form NPCs that can be expanded and cryopreserved (Figures 3A–3E). These NPCs express early neuroectodermal markers including PAX6, SOX2, and Nestin, and lack expression of SOX10, which marks the neural crest, indicating a CNS expression pattern (Figure 3F). The viability of these NPCs after thaw is high (mean 89% ± 1.2% live cells). These NPCs maintain their capacity to differentiate into neuronal subtypes when used at early passages (between passage 1 and passage 5), while astrocyte differentiation as measured by glial fibrillary acidic protein (GFAP) and S100β can be promoted using NPCs from early or later passages (beyond passage 5). By incorporating fluorescence-activated cell sorting for cell-surface markers CD133+, CD184+, and CD271−, the resulting selected pool of NPCs can be maintained with a high proportion of neuronal differentiation for at least 50 passages (Cheng et al., 2017). It is critical to culture NPCs at a high density for the successful maintenance of a stable and expandable population of progenitors (Cheng et al., 2017).
NPCs can be patterned into different neural CNS regions and differentiated into neuronal and glial subtypes to model primary tauopathies (Figures 3I and 3J) (Jiang et al., 2018, Silva et al., 2016, Tcw et al., 2017). By default, these NPCs tend to adopt anterior CNS characteristics, such as FOXG1 expression, but this fate can also be stimulated by the addition of patterning factors (Kirwan et al., 2015, Saurat et al., 2016). The resulting forebrain neurons produce tau that is physiologically similar to human CNS tau (Sato et al., 2018), with the exception of intracellular 4R tau levels. Achieving splicing of the six major tau isoforms expressed in adult brains remains a challenge in the iPSC system (Hefti et al., 2018, Sposito et al., 2015). Despite the low levels of 4R tau, iPSC-derived neural cells from MAPT mutation/risk variant carriers phenocopy aspects of primary tauopathies. This includes the accumulation of phosphorylated forms of tau (Ehrlich et al., 2015, Imamura et al., 2016, Iovino et al., 2015, Silva et al., 2016, Silva et al., 2019), mitochondrial defects (Esteras et al., 2017), and increased cell vulnerability (Hallmann et al., 2017, Silva et al., 2016, Wren et al., 2015). More recently, we have demonstrated that neurons expressing MAPT R406W (F11362) capture molecular signatures related to altered synaptic function that are also present in human brains from MAPT R406W carriers and in mouse models of primary tauopathies (Jiang et al., 2018). Cryopreserved NPCs from the iPSCs reported here (Tables 3 and 4) are available upon request from http://neuralsci.org/tau.
Generation of Integrated, Isogenic, and Inducible Neurogenin-2 iPSCs
Integrated, isogenic, and inducible neurogenin-2 (i3N) iPSCs engineered with a doxycycline-inducible mouse Neurogenin-2 (Ngn2) transgene in the AAVS1 safe-harbor locus can be scalably differentiated to homogeneous excitatory neurons, which enables the use of human neurons for high-throughput drug discovery (Wang et al., 2017). We have engineered two healthy control WT (F12468 and WTC11), as well as MAPT P301L (F0510) and MAPT R406W (F11374), to i3N iPSCs (Table 4) (Wang et al., 2017). We also mutagenized a control i3N line (WTC11) to be heterozygous or homozygous for MAPT V337M (Sohn et al., 2019). All modified lines were characterized for chromosomal stability, and confirmed for Ngn2 integration and neuronal differentiation (Figure S2 and Table S2), and are available upon request from http://neuralsci.org/tau.
Discussion
We present a comprehensive and valuable resource that can be used to model primary tauopathies and for drug discovery. Our patient-based cohort consists of a library of 140 dermal fibroblast lines and respective iPSC lines with multiple clones that are focused on primary tauopathies: 29 iPSC lines from patients carrying pathogenic MAPT mutations or risk variants; 2 iPSC lines from autopsy-confirmed PSP patients; 28 isogenic iPSCs; and 8 Ngn2-integrated iPSCs. Importantly, most of these cell lines were obtained from deeply clinically phenotyped individuals with detailed neurological and neuropsychological assessment and availability of fluid biomarkers (CSF and plasma), imaging biomarkers (MRI, β-amyloid PET, and tau PET), genetic data, and, for some, neuropathological data.
Phenotypic Diversity of MAPT Mutations
More than 50 mutations in MAPT have been reported to cause FTLD-Tau and are located primarily in exons 9–13; yet FTLD-Tau is both clinically and neuropathologically heterogeneous (Table 1). Broadly, FTLD-Tau is defined by neuronal loss, gliosis, and spongiform changes in layer II with predominant involvement of the frontal, temporal, cingulate, and insular cortices and variable involvement of subcortical nuclei. Clinically, patients with FTLD-Tau pathology can present with a broad range of phenotypes spanning behavioral, cognitive, and motor disturbances. Various different combinations of clinical symptoms may be seen in association with specific mutations and even among affected members of a single family (Spina et al., 2008). Hence, there is a need to understand the impact of specific MAPT mutations within the genetic background of individuals with known clinical and pathological manifestations. Our fibroblast and iPSC resources allow for the investigation of common and unique cellular phenotypes driven by these mutations.
The Clinical and Pathological Spectrum of the Most Common 4R Primary Tauopathies
FTLD-Tau, PSP, and CBD are neuropathologically defined as 4R tauopathies (Kovacs, 2015). PSP pathology occurs in neurons and glia (astrocytes and oligodendrocytes) and preferentially affects the tectum, tegmentum, globus pallidus, diencephalon, and superior cerebellar peduncle (Dickson et al., 2007). In CBD, neuronal and glial pathology occurs within gray and white mater regions of the cortex, basal ganglia, diencephalon, and rostral brainstem (Forman et al., 2002). While 4R-tau aggregation is characteristic of both PSP and CBD, differences in proteolytic processing of tau have been reported to distinguish the two diseases: detergent-insoluble tau occurs at a doublet around 37 kDa in CBD and as a single band at 33 kDa in PSP (Arai et al., 2001, Arai et al., 2004). The MAPT A152T risk variant has been associated with both clinical and pathological forms of PSP-S/PSP and CBS/CBD. Our fibroblast resources containing sporadic PSP-S and CBS will allow for the cellular and molecular dissection of disease phenotypes, providing a powerful system for understanding the cellular mechanisms that drive phenotypic differences between PSP-S/PSP and CBS/CBD.
Challenges in Modeling Primary Tauopathies in Traditional Cell and Mouse Models
Current cellular and animal models used to study primary tauopathies have several limitations. Critically, there is no natural animal model of tauopathy, as these diseases are largely restricted to Homo sapiens (Heuer et al., 2012, Holzer et al., 2004). While tau isoforms may share broad functional similarities, different isoforms likely play distinct physiological and pathological roles in the cell (Goedert and Jakes, 1990, Karch et al., 2012, Kosik et al., 1989, Panda et al., 2003). Notably, the expression of tau isoforms drastically differs between human and rodent brains. While the adult human brains have roughly equal levels of 3R and 4R tau, adult rodents express almost exclusively 4R tau, limiting the conclusions that can be drawn from mouse models (Trabzuni et al., 2012). Additionally, most cellular and transgenic models (e.g., C. elegans, Drosophila, and mice) rely on overexpression of a mutant transgene comprising a single tau isoform, which may produce effects that are a function of excessive protein expression, specific isoform expression, and possible off-target effects, rather than a disease-relevant phenotype. Finally, neuronal and glial cells are the primary cell types affected in tauopathies; therefore, studies in immortalized cell lines may fail to capture the phenotypes specific to neurons and glia. Thus, our understanding of how tau is metabolized in the human brain has been obtained from experimental paradigms that do not fully capture physiological conditions relevant to human tauopathies. Stem cell models begin to address these gaps; however, tau generated by stem cell-derived neurons remains in the fetal state (e.g., primarily 3R0N) (Hefti et al., 2018, Iovino et al., 2015, Sposito et al., 2015). Nevertheless, cells expressing 4R-containing MAPT mutations, such as P301L, exhibit altered tau phosphorylation, tau accumulation, and cell vulnerability (Iovino et al., 2015, Silva et al., 2019). These phenotypes can be reversed with novel tau degraders (Silva et al., 2019).
Basic Science, Clinical, and Translational Applications of Human Tauopathy Models
Beyond modeling the molecular and cellular pathophysiology of primary tauopathies, the derivation of patient-specific expandable NPCs enables large-scale functional genomics, proteomics, and small-molecule-based and CRISPR-based genetic modifier screens (Boselli et al., 2017, Cheng et al., 2017, Silva et al., 2016, Tian et al., 2019, Wang et al., 2017). This includes the use of high-content imaging methodologies with subcellular level resolution of molecular and morphological changes in defined neuronal subtypes with and without glial subtypes. We envision that using the framework of phenotypes from the autosomal dominant mutations and risk factors for tauopathy described here will also assist in the interpretation of genetic variants of unknown pathological significance that are being identified by exome and whole-genome sequencing projects.
One of the challenges in modeling diseases that typically present clinically in mid to late adulthood is to accelerate aging in iPSC-derived cells. This is particularly difficult given that during reprogramming to pluripotency, the features of aging present in originating somatic cells are reset. In contrast, fibroblasts directly reprogrammed into neurons retain their aging characteristics such as DNA methylation (Huh et al., 2016, Maherali et al., 2007, Mertens et al., 2015). Efforts to induce chronological aging in iPSC-derived neurons are under way (Miller et al., 2013). In the meantime, the ability to study both reprogrammed iPSCs and their source fibroblasts directly differentiated into neurons may help the field dissect how gene mutations contribute to the neurodegenerative process at both early and late age-dependent stages.
Our intention is to extend the existing resource, particularly by adding fibroblasts and iPSCs carrying novel mutations with clear pathogenicity and unique clinical features such as those associated with extremely early age of onset or rapid disease progression. Other variants in specific domains of tau or in non-coding genomic elements (e.g., 5′/3′ UTRs, enhancer sites) would also be of interest to the collection as well as additional genome engineering in existing lines.
Conclusions
In total, the resource presented here represents an opportunity to understand the mechanisms by which pathogenic mutations or risk variants in MAPT drive tauopathy. This resource will also be of interest to the broader community working on neurodegenerative disease. The collaborative efforts through which this resource has been generated can serve as a model for other neurodegenerative disease subtypes as well as other neurological and non-neurological diseases under genetic influence. Our intention is to broadly share the collection of fibroblasts, iPSCs, and NPCs and related data and information, which are available upon request. We are continuing to build this collection with additional reprogramming and genome editing, and updates will be available at http://neuralsci.org/tau (Figure S3).
Experimental Procedures
The Washington University and University of California San Francisco Institutional Review Boards reviewed the Neuropathology Cores (from whom the brains were obtained) operating protocols as well as this specific study and determined it was exempt from approval. Our participants provide this consent by signing the hospital's autopsy form. If the participant does not provide future consent before death the DPOA or next of kin provide it after death. All data were analyzed anonymously.
Skin punches were performed following written informed consent from the donor. The informed consent was approved by the Washington University School of Medicine and the University of California San Francisco Institutional Review Board and Ethics Committee (IRB 201104178, 201306108 and 10-03946). The consent allows for use of tissue by all parties, commercial and academic, for the purposes of research but not for use in human therapy.
Peripheral blood mononuclear cells or dermal fibroblasts were transduced with non-integrating Sendai virus carrying OCT3/4, SOX2, KLF4, and cMYC. iPSC lines were analyzed for pluripotency markers by immunocytochemistry (ICC) and qPCR; spontaneous differentiation into the three germ layers by ICC or qPCR; and chromosomal abnormalities by karyotyping. Human iPSCs were edited using CRISPR/Cas9 as previously reported (Budde et al., 2017). i3N iPSCs were generated as described previously (Wang et al., 2017). See Supplemental Experimental Procedures for additional details.
Consortia
Tau Consortium Stem Cell Group: Carolina Alquezar1, Kathryn R. Bowles2, David C. Butler3, John F. Crary4, Li Gan5, Alison M. Goate2, Stephen J. Haggarty6, Israel Hernandez7, Valerie Hennes8, Cindy Huang5, Justin K. Ichida8, Martin Kampmann9, Aimee W. Kao1, Celeste M. Karch10, Anna Karydas1, Kenneth S. Kosik7, Rita Martinez10, Khadijah Onanuga3, M. Catarina Silva6, Sally Temple3, Chao Wang5
1Division of Memory and Aging Center, Department of Neurology, University of California, San Francisco, San Francisco, CA 94158, USA
2Ronald M. Loeb Center for Alzheimer's Disease, Departments of Neuroscience, Neurology and Genetics & Genomic Sciences, Icahn School of Medicine, New York, NY 10029, USA
3Neural Stem Cell Institute, 1 Discovery Drive, Rensselaer, NY 12144, USA
4Department of Pathology, Fishberg Department of Neuroscience, Friedman Brain Institute, Ronald M. Loeb Center for Alzheimer's Disease, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA
5Gladstone Institutes of Neurological Disease, Department of Neurology, Neuroscience Graduate Program, University of California, San Francisco, CA 94158
6Chemical Neurobiology Laboratory, Center for Genomic Medicine, Departments of Neurology & Psychiatry, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA
7Department of Molecular Cellular and Developmental Biology, Neuroscience Research Institute, Biomolecular Science and Engineering Program, University of California, Santa Barbara, Santa Barbara, CA, USA
8Department of Stem Cell Biology and Regenerative Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
9Department of Biochemistry and Biophysics, Institute for Neurodegenerative Diseases and California Institute for Quantitative Biomedical Research, University of California, San Francisco, CA 94158, USA
10Department of Psychiatry, Washington University in St. Louis, St. Louis, MO 63110, USA
Author Contributions
Conceived and designed experiments: C.M.K., A.W.K., C.W., M.C.S., S.J.H., J.K.I., K.S.K., L.G., A.M.G., and S.T. Performed experiments: C.M.K., R.M., J.A.M., A.A., K.R.B., D.A.P., P.T., S.S., S.H., C.W., C.H., P.D.S., D.B., M.C.S., and N.J.C. Analyzed data: C.M.K., A.W.K., A.K., K.M., A.A., S.H., C.W., C.H., and P.D.S. Contributed reagents, materials, and analysis tools: C.M.K., A.W.K., A.K., K.R.B., K.M., Y.H., S.E.L., N.G., J.N., F.M., B.F.B., M.C.S., S.J.H., J.F.C., J.K.I., B.L.M., L.G., L.T.G., W.W.S., A.M.G., K.O., and S.T. Wrote the manuscript: C.M.K., A.W.K., S.S., and S.T. Edited the manuscript: C.M.K., C.W., L.T.G., M.C.S., S.J.H., J.K.I., K.S.K., L.G., A.M.G., S.T., N.G., M.K., N.J.C., and J.F.C.
Acknowledgments
We would like to thank the research subjects and their families who generously participated in this study. We thank Steven Lotz, Shawn Sutton, Brian Unruh, Isabel Tian, and Nicholas St. John at NeuraCell and Evan Y. Snyder and Yang M. Lui at Sanford Burnham Prebys Medical Discovery Institute for technical assistance. We thank the Icahn School of Medicine at Mount Sinai Pluripotent Stem Cell Core facility for technical assistance. The gRNAs were generated by the Genome Engineering and iPSC Center (GEiC) at the Washington University in St. Louis. We thank Amber Neilson at the GEiC for technical assistance. This work was supported by access to equipment made possible by the Hope Center for Neurological Disorders, and the Departments of Neurology and Psychiatry at Washington University School of Medicine. Data collection and dissemination of the data presented in this paper were supported by the LEFFTDS & ARTFL Consortium (LEFFTDS: U01AG045390; ARTFL: U54NS092089). The authors acknowledge the invaluable contributions of the participants in ARTFL & LEFFTDS as well as the assistance of the support staff at each of the participating sites. Funding: The Tau Consortium (C.M.K., A.W.K., Y.H., S.E.L., J.F.C., S.J.H., J.K.I., K.S.K., B.L.M., L.G., A.M.G., S.T., K.O., M.K.), NIH AG046374 (C.M.K.), CurePSP (A.W.K., K.R.B.), Brain Research Foundation (A.W.K.), MGH Research Scholars Program (S.J.H.), Association for Frontotemporal Degeneration, AFTD (M.C.S., K.R.B.), BrightFocus Foundation (K.R.B.), Farrell Family Alzheimer’s Disease Research Fund (C.M.K.), NIH/NIA (P50 AG023501, P01 AG019724, T32 AG023481-11S1, and P50 AG1657303 to B.L.M.), NIH (R01AG054008 and R01NS095252 to J.F.C.), NIH (P50 AG005681 to J.C.M.), NIH (K12 HD001459 to N.G.), NIH/NINDS (R35 NS097277 to S.T.), NIH/NIA (AG056293 to S.T.), NIH (K08 AG052648 to S.S.), NIH (U01 AG045390 to B.B.), NIH U54 NS092089, U24 AG21886. Neuracell received support from the Empire State Stem Cell Fund through New York State Department of Health contract #C029158. Opinions expressed here are solely those of the authors and do not necessarily reflect those of the Empire State Stem Cell Board, the New York State Department of Health, or the State of New York. J.K.I. is a New York Stem Cell Foundation-Robertson Investigator. A.W.K. and M.K. are Paul G. Allen Family Foundation Distinguished Investigators. M.K. is a Chan Zuckerberg Biohub Investigator. A.M.G. is a member of the Scientific Advisory Board for Denali Therapeutics and on the Genetic Scientific Advisory Panel for Pfizer. N.J.C. is a member of the Modeling Alliance of Systems Pharmacology in Tauopathies Scientific Advisory Board. S.J.H. is a member of the Scientific Advisory Board for Rodin Therapeutics, Frequency Therapeutics, and Psy Therapeutics and Souvien Therapeutics, none of whom had involvement in the present study. N.G. has participated or is currently participating in clinical trials of anti-dementia drugs sponsored by Bristol Myers Squibb, Eli Lilly/Avid Radiopharmaceuticals, Janssen Immunotherapy, Novartis, Pfizer, Wyeth, SNIFF (The Study of Nasal Insulin to Fight Forgetfulness) study, and A4 (The Anti-Amyloid Treatment in Asymptomatic Alzheimer's Disease) trial. S.T. is president of StemCultures, scientific co-founder of Luxa Biotech and has served on the scientific advisory boards of Sana Biotechnology and Blue Rock Therapeutics and as consultant to Merck. The remaining authors declare no competing interests.
Published: October 17, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.09.006.
Contributor Information
Celeste M. Karch, Email: karchc@wustl.edu.
Sally Temple, Email: sallytemple@neuralsci.org.
Tau Consortium Stem Cell Group:
Carolina Alquezar, Kathryn Bowles, David Butler, John F. Crary, Li Gan, Alison M. Goate, Stephen J. Haggarty, Israel Hernandez, Valerie Hennes, Cindy Huang, Justin K. Ichida, Martin Kampmann, Aimee W. Kao, Celeste M. Karch, Anna Karydas, Kenneth S. Kosik, Rita Martinez, Khadijah Onanuga, M. Catarina Silva, Sally Temple, and Chao Wang
Supplemental Information
References
- Arai T., Ikeda K., Akiyama H., Nonaka T., Hasegawa M., Ishiguro K., Iritani S., Tsuchiya K., Iseki E., Yagishita S. Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann. Neurol. 2004;55:72–79. doi: 10.1002/ana.10793. [DOI] [PubMed] [Google Scholar]
- Arai T., Ikeda K., Akiyama H., Tsuchiya K., Yagishita S., Takamatsu J. Intracellular processing of aggregated tau differs between corticobasal degeneration and progressive supranuclear palsy. Neuroreport. 2001;12:935–938. doi: 10.1097/00001756-200104170-00014. [DOI] [PubMed] [Google Scholar]
- Behnam M., Ghorbani F., Shin J.H., Kim D.S., Jang H., Nouri N., Sedghi M., Salehi M., Ansari B., Basiri K. Homozygous MAPT R406W mutation causing FTDP phenotype: a unique instance of a unique mutation. Gene. 2015;570:150–152. doi: 10.1016/j.gene.2015.06.033. [DOI] [PubMed] [Google Scholar]
- Bhutani K., Nazor K.L., Williams R., Tran H., Dai H., Dzakula Z., Cho E.H., Pang A.W., Rao M., Cao H. Whole-genome mutational burden analysis of three pluripotency induction methods. Nat. Commun. 2016;7:10536. doi: 10.1038/ncomms10536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas M.H.U., Almeida S., Lopez-Gonzalez R., Mao W., Zhang Z., Karydas A., Geschwind M.D., Biernat J., Mandelkow E.M., Futai K. MMP-9 and MMP-2 contribute to neuronal cell death in iPSC models of frontotemporal dementia with MAPT mutations. Stem Cell Reports. 2016;7:316–324. doi: 10.1016/j.stemcr.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve B.F., Tremont-Lukats I.W., Waclawik A.J., Murrell J.R., Hermann B., Jack C.R., Jr., Shiung M.M., Smith G.E., Nair A.R., Lindor N. Longitudinal characterization of two siblings with frontotemporal dementia and parkinsonism linked to chromosome 17 associated with the S305N tau mutation. Brain. 2005;128:752–772. doi: 10.1093/brain/awh356. [DOI] [PubMed] [Google Scholar]
- Boselli M., Lee B.H., Robert J., Prado M.A., Min S.W., Cheng C., Silva M.C., Seong C., Elsasser S., Hatle K.M. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J. Biol. Chem. 2017;292:19209–19225. doi: 10.1074/jbc.M117.815126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde J., Martinez R., Hsu S., Wen N., Chen J., Coppola G., Goate A.M., Cruchaga C., Karch C.M. Precision genome-editing with CRISPR/Cas9 in human induced pluripotent stem cells. bioRxiv. 2017 [Google Scholar]
- Butler V.J., Salazar D.A., Soriano-Castell D., Alves-Ferreira M., Dennissen F.J.A., Vohra M., Oses-Prieto J.A., Li K.H., Wang A.L., Jing B. Tau/MAPT disease-associated variant A152T alters tau function and toxicity via impaired retrograde axonal transport. Hum. Mol. Genet. 2019;28:1498–1514. doi: 10.1093/hmg/ddy442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns N.J., Bigio E.H., Mackenzie I.R.A., Neumann M., Lee V.M.-Y., Hatanpaa K.J., White C.L., Schneider J.A., Grinberg L.T., Halliday G. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C., Fass D.M., Folz-Donahue K., MacDonald M.E., Haggarty S.J. Highly expandable human iPS cell-derived neural progenitor cells (NPC) and neurons for central nervous system disease modeling and high-throughput screening. Curr. Protoc. Hum. Genet. 2017;92:21 28 21. doi: 10.1002/cphg.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G., Chinnathambi S., Lee J.J., Dombroski B.A., Baker M.C., Soto-Ortolaza A.I., Lee S.E., Klein E., Huang A.Y., Sears R. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer's diseases. Hum. Mol. Genet. 2012;21:3500–3512. doi: 10.1093/hmg/dds161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M., Theuns J., Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 2012;33:1340–1344. doi: 10.1002/humu.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayanandan R., Van Slegtenhorst M., Mack T.G., Ko L., Yen S.H., Leroy K., Brion J.P., Anderton B.H., Hutton M., Lovestone S. Mutations in tau reduce its microtubule binding properties in intact cells and affect its phosphorylation. FEBS Lett. 1999;446:228–232. doi: 10.1016/s0014-5793(99)00222-7. [DOI] [PubMed] [Google Scholar]
- Decker J.M., Kruger L., Sydow A., Dennissen F.J., Siskova Z., Mandelkow E., Mandelkow E.M. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. 2016;17:552–569. doi: 10.15252/embr.201541439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson D.W., Rademakers R., Hutton M.L. Progressive supranuclear palsy: pathology and genetics. Brain Pathol. 2007;17:74–82. doi: 10.1111/j.1750-3639.2007.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi D., Samata B., Katsukawa M., Kikuchi T., Morizane A., Ono Y., Sekiguchi K., Nakagawa M., Parmar M., Takahashi J. Isolation of human induced pluripotent stem cell-derived dopaminergic progenitors by cell sorting for successful transplantation. Stem Cell Reports. 2014;2:337–350. doi: 10.1016/j.stemcr.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M., Hallmann A.L., Reinhardt P., Arauzo-Bravo M.J., Korr S., Ropke A., Psathaki O.E., Ehling P., Meuth S.G., Oblak A.L. Distinct neurodegenerative changes in an induced pluripotent stem cell model of frontotemporal dementia linked to mutant TAU protein. Stem Cell Reports. 2015;5:83–96. doi: 10.1016/j.stemcr.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkabetz Y., Panagiotakos G., Al Shamy G., Socci N.D., Tabar V., Studer L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008;22:152–165. doi: 10.1101/gad.1616208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espuny-Camacho I., Arranz A.M., Fiers M., Snellinx A., Ando K., Munck S., Bonnefont J., Lambot L., Corthout N., Omodho L. Hallmarks of Alzheimer's disease in stem-cell-derived human neurons transplanted into mouse brain. Neuron. 2017;93:1066–1081.e8. doi: 10.1016/j.neuron.2017.02.001. [DOI] [PubMed] [Google Scholar]
- Esteras N., Rohrer J.D., Hardy J., Wray S., Abramov A.Y. Mitochondrial hyperpolarization in iPSC-derived neurons from patients of FTDP-17 with 10+16 MAPT mutation leads to oxidative stress and neurodegeneration. Redox. Biol. 2017;12:410–422. doi: 10.1016/j.redox.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D., Mukrasch M.D., Von Bergen M., Klos-Witkowska A., Biernat J., Griesinger C., Mandelkow E., Zweckstetter M. Structural and microtubule binding properties of tau mutants of frontotemporal dementias. Biochemistry. 2007;46:2574–2582. doi: 10.1021/bi061318s. [DOI] [PubMed] [Google Scholar]
- Fong H., Wang C., Knoferle J., Walker D., Balestra M.E., Tong L.M., Leung L., Ring K.L., Seeley W.W., Karydas A. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Reports. 2013;1:226–234. doi: 10.1016/j.stemcr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman M.S., Zhukareva V., Bergeron C., Chin S.S., Grossman M., Clark C., Lee V.M., Trojanowski J.Q. Signature tau neuropathology in gray and white matter of corticobasal degeneration. Am. J. Pathol. 2002;160:2045–2053. doi: 10.1016/S0002-9440(10)61154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier-Kemper A., Weissmann C., Golovyashkina N., Sebo-Lemke Z., Drewes G., Gerke V., Heinisch J.J., Brandt R. The frontotemporal dementia mutation R406W blocks tau's interaction with the membrane in an annexin A2-dependent manner. J. Cell Biol. 2011;192:647–661. doi: 10.1083/jcb.201007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M., Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmann A.L., Arauzo-Bravo M.J., Mavrommatis L., Ehrlich M., Ropke A., Brockhaus J., Missler M., Sterneckert J., Scholer H.R., Kuhlmann T. Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci. Rep. 2017;7:42991. doi: 10.1038/srep42991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefti M.M., Farrell K., Kim S., Bowles K.R., Fowkes M.E., Raj T., Crary J.F. High-resolution temporal and regional mapping of MAPT expression and splicing in human brain development. PLoS One. 2018;13:e0195771. doi: 10.1371/journal.pone.0195771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer E., Rosen R.F., Cintron A., Walker L.C. Nonhuman primate models of Alzheimer-like cerebral proteopathy. Curr. Pharm. Des. 2012;18:1159–1169. doi: 10.2174/138161212799315885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglinger G.U., Melhem N.M., Dickson D.W., Sleiman P.M.A., Wang L.-S., Klei L., Rademakers R., de Silva R., Litvan I., Riley D.E. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 2011;43:699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer M., Craxton M., Jakes R., Arendt T., Goedert M. Tau gene (MAPT) sequence variation among primates. Gene. 2004;341:313–322. doi: 10.1016/j.gene.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Huh C.J., Zhang B., Victor M.B., Dahiya S., Batista L.F., Horvath S., Yoo A.S. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. eLife. 2016;5 doi: 10.7554/eLife.18648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M., Lendon C.L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Iijima M., Tabira T., Poorkaj P., Schellenberg G.D., Trojanowski J.Q., Lee V.M., Schmidt M.L., Takahashi K., Nabika T., Matsumoto T. A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport. 1999;10:497–501. doi: 10.1097/00001756-199902250-00010. [DOI] [PubMed] [Google Scholar]
- Imamura K., Sahara N., Kanaan N.M., Tsukita K., Kondo T., Kutoku Y., Ohsawa Y., Sunada Y., Kawakami K., Hotta A. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci. Rep. 2016;6:34904. doi: 10.1038/srep34904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovino M., Agathou S., Gonzalez-Rueda A., Del Castillo Velasco-Herrera M., Borroni B., Alberici A., Lynch T., O'Dowd S., Geti I., Gaffney D. Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain. 2015;138:3345–3359. doi: 10.1093/brain/awv222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen J.C., Warrington E.K., Morris H.R., Lantos P., Brown J., Revesz T., Wood N., Khan M.N., Cipolotti L., Fox N.C. Clinical features of frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology. 2002;58:1161–1168. doi: 10.1212/wnl.58.8.1161. [DOI] [PubMed] [Google Scholar]
- Jiang S., Wen N., Li Z., Dube U., Del Aguila J., Budde J., Martinez R., Hsu S., Fernandez M.V., Cairns N.J. Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl. Psychiatry. 2018;8:265. doi: 10.1038/s41398-018-0319-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch C.M., Jeng A.T., Goate A.M. Extracellular tau levels are influenced by variability in tau that is associated with tauopathies. J. Biol. Chem. 2012;287:42751–42762. doi: 10.1074/jbc.M112.380642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen H., Goncalves A., Leha A., Afzal V., Alasoo K., Ashford S., Bala S., Bensaddek D., Casale F.P., Culley O.J. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature. 2017;546:370–375. doi: 10.1038/nature22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirwan P., Turner-Bridger B., Peter M., Momoh A., Arambepola D., Robinson H.P., Livesey F.J. Development and function of human cerebral cortex neural networks from pluripotent stem cells in vitro. Development. 2015;142:3178–3187. doi: 10.1242/dev.123851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik K.S., Orecchio L.D., Bakalis S., Neve R.L. Developmentally regulated expression of specific tau sequences. Neuron. 1989;2:1389–1397. doi: 10.1016/0896-6273(89)90077-9. [DOI] [PubMed] [Google Scholar]
- Kovacs G.G. Invited review: neuropathology of tauopathies: principles and practice. Neuropathol. Appl. Neurobiol. 2015;41:3–23. doi: 10.1111/nan.12208. [DOI] [PubMed] [Google Scholar]
- Kovacs G.G., Pittman A., Revesz T., Luk C., Lees A., Kiss E., Tariska P., Laszlo L., Molnar K., Molnar M.J. MAPT S305I mutation: implications for argyrophilic grain disease. Acta Neuropathol. 2008;116:103–118. doi: 10.1007/s00401-007-0322-6. [DOI] [PubMed] [Google Scholar]
- Lantos P.L., Cairns N.J., Khan M.N., King A., Revesz T., Janssen J.C., Morris H., Rossor M.N. Neuropathologic variation in frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology. 2002;58:1169–1175. doi: 10.1212/wnl.58.8.1169. [DOI] [PubMed] [Google Scholar]
- Liu F., Gong C.-X. Tau exon 10 alternative splicing and tauopathies. Mol. Neurodegener. 2008;3:8. doi: 10.1186/1750-1326-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S., Djukic B., Taneja P., Yu G.Q., Lo I., Davis A., Craft R., Guo W., Wang X., Kim D. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 2016;17:530–551. doi: 10.15252/embr.201541438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N., Sridharan R., Xie W., Utikal J., Eminli S., Arnold K., Stadtfeld M., Yachechko R., Tchieu J., Jaenisch R. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Merkle F.T., Ghosh S., Kamitaki N., Mitchell J., Avior Y., Mello C., Kashin S., Mekhoubad S., Ilic D., Charlton M. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. 2017;545:229–233. doi: 10.1038/nature22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J., Paquola A.C.M., Ku M., Hatch E., Bohnke L., Ladjevardi S., McGrath S., Campbell B., Lee H., Herdy J.R. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell. 2015;17:705–718. doi: 10.1016/j.stem.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.D., Ganat Y.M., Kishinevsky S., Bowman R.L., Liu B., Tu E.Y., Mandal P.K., Vera E., Shim J.W., Kriks S. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13:691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra S.S., Murrell J.R., Gearing M., Spillantini M.G., Goedert M., Crowther R.A., Levey A.I., Jones R., Green J., Shoffner J.M. Tau pathology in a family with dementia and a P301L mutation in tau. J. Neuropathol. Exp. Neurol. 1999;58:335–345. doi: 10.1097/00005072-199904000-00004. [DOI] [PubMed] [Google Scholar]
- Miyasaka T., Morishima-Kawashima M., Ravid R., Heutink P., van Swieten J.C., Nagashima K., Ihara Y. Molecular analysis of mutant and wild-type tau deposited in the brain affected by the FTDP-17 R406W mutation. Am. J. Pathol. 2001;158:373–379. doi: 10.1016/S0002-9440(10)63979-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratore C.R., Srikanth P., Callahan D.G., Young-Pearse T.L. Comparison and optimization of hiPSC forebrain cortical differentiation protocols. PLoS One. 2014;9:e105807. doi: 10.1371/journal.pone.0105807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell J.R., Spillantini M.G., Zolo P., Guazzelli M., Smith M.J., Hasegawa M., Redi F., Crowther R.A., Pietrini P., Ghetti B. Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J. Neuropathol. Exp. Neurol. 1999;58:1207–1226. doi: 10.1097/00005072-199912000-00002. [DOI] [PubMed] [Google Scholar]
- Ng A.S., Sias A.C., Pressman P.S., Fong J.C., Karydas A.M., Zanto T.P., De May M., Coppola G., Geschwind D.H., Miller B.L. Young-onset frontotemporal dementia in a homozygous tau R406W mutation carrier. Ann. Clin. Transl Neurol. 2015;2:1124–1128. doi: 10.1002/acn3.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda D., Samuel J.C., Massie M., Feinstein S.C., Wilson L. Differential regulation of microtubule dynamics by three- and four-repeat tau: implications for the onset of neurodegenerative disease. Proc. Natl. Acad. Sci. U S A. 2003;100:9548–9553. doi: 10.1073/pnas.1633508100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paonessa F., Evans L.D., Solanki R., Larrieu D., Wray S., Hardy J., Jackson S.P., Livesey F.J. microtubules deform the nuclear membrane and disrupt nucleocytoplasmic transport in tau-mediated frontotemporal dementia. Cell Rep. 2019;26:582–593.e5. doi: 10.1016/j.celrep.2018.12.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry D.C., Brown J.A., Possin K.L., Datta S., Trujillo A., Radke A., Karydas A., Kornak J., Sias A.C., Rabinovici G.D. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. 2017;140:3329–3345. doi: 10.1093/brain/awx254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pir G.J., Choudhary B., Mandelkow E., Mandelkow E.M. Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol. Neurodegener. 2016;11:33. doi: 10.1186/s13024-016-0096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L.A., Grabowski T.J., Schmidt M.L., Morris J.C., Goate A., Solodkin A., Van Hoesen G.W., Schelper R.L., Talbot C.J., Wragg M.A. Autosomal dominant dementia with widespread neurofibrillary tangles. Ann. Neurol. 1997;42:564–572. doi: 10.1002/ana.410420406. [DOI] [PubMed] [Google Scholar]
- Sato C., Barthelemy N.R., Mawuenyega K.G., Patterson B.W., Gordon B.A., Jockel-Balsarotti J., Sullivan M., Crisp M.J., Kasten T., Kirmess K.M. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;97:1284–1298.e7. doi: 10.1016/j.neuron.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saurat N.G., Livesey F.J., Moore S. Cortical differentiation of human pluripotent cells for in vitro modeling of Alzheimer's disease. Methods Mol. Biol. 2016;1303:267–278. doi: 10.1007/978-1-4939-2627-5_16. [DOI] [PubMed] [Google Scholar]
- Seo J., Kritskiy O., Watson L.A., Barker S.J., Dey D., Raja W.K., Lin Y.T., Ko T., Cho S., Penney J. Inhibition of p25/Cdk5 attenuates tauopathy in mouse and iPSC models of frontotemporal dementia. J. Neurosci. 2017;37:9917–9924. doi: 10.1523/JNEUROSCI.0621-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva M.C., Cheng C., Mair W., Almeida S., Fong H., Biswas M.H.U., Zhang Z., Huang Y., Temple S., Coppola G. Human iPSC-derived neuronal model of tau-A152T frontotemporal dementia reveals tau-mediated mechanisms of neuronal vulnerability. Stem Cell Reports. 2016;7:325–340. doi: 10.1016/j.stemcr.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva M.C., Ferguson F.M., Cai Q., Donovan K.A., Nandi G., Patnaik D., Zhang T., Huang H.T., Lucente D.E., Dickerson B.C. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. eLife. 2019;8 doi: 10.7554/eLife.45457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund L., Viitanen M., Kalimo H., Lannfelt L., Jonhagen M.E., Ingelsson M., Glaser A., Herva R. The tau S305S mutation causes frontotemporal dementia with parkinsonism. Eur. J. Neurol. 2008;15:156–161. doi: 10.1111/j.1468-1331.2007.02017.x. [DOI] [PubMed] [Google Scholar]
- Sohn P., Tracy T., Huang C., Yan R., Camargo C., Mok S., Freilich R., Roberson E., Karch C., Gestwicki J. Pathogenic Tau impairs axon initial segment plasticity and excitability homeostasis. Neuron. 2019 doi: 10.1016/j.neuron.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M.G., Crowther R.A., Goedert M. Comparison of the neurofibrillary pathology in Alzheimer's disease and familial presenile dementia with tangles. Acta Neuropathol. 1996;92:42–48. doi: 10.1007/s004010050487. [DOI] [PubMed] [Google Scholar]
- Spina S., Farlow M.R., Unverzagt F.W., Kareken D.A., Murrell J.R., Fraser G., Epperson F., Crowther R.A., Spillantini M.G., Goedert M. The tauopathy associated with mutation +3 in intron 10 of Tau: characterization of the MSTD family. Brain. 2008;131:72–89. doi: 10.1093/brain/awm280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina S., Schonhaut D.R., Boeve B.F., Seeley W.W., Ossenkoppele R., O'Neil J.P., Lazaris A., Rosen H.J., Boxer A.L., Perry D.C. Frontotemporal dementia with the V337M MAPT mutation: tau-PET and pathology correlations. Neurology. 2017;88:758–766. doi: 10.1212/WNL.0000000000003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sposito T., Preza E., Mahoney C.J., Seto-Salvia N., Ryan N.S., Morris H.R., Arber C., Devine M.J., Houlden H., Warner T.T. Developmental regulation of tau splicing is disrupted in stem cell-derived neurons from frontotemporal dementia patients with the 10 + 16 splice-site mutation in MAPT. Hum. Mol. Genet. 2015;24:5260–5269. doi: 10.1093/hmg/ddv246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford P.M., Halliday G.M., Brooks W.S., Kwok J.B., Storey C.E., Creasey H., Morris J.G., Fulham M.J., Schofield P.R. Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain. 2000;123(Pt 5):880–893. doi: 10.1093/brain/123.5.880. [DOI] [PubMed] [Google Scholar]
- Tcw J., Wang M., Pimenova A.A., Bowles K.R., Hartley B.J., Lacin E., Machlovi S.I., Abdelaal R., Karch C.M., Phatnani H. An efficient platform for astrocyte differentiation from human induced pluripotent stem cells. Stem Cell Reports. 2017;9:600–614. doi: 10.1016/j.stemcr.2017.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R., Gachechiladze M.A., Ludwig C.H., Laurie M.T., Hong J.Y., Nathaniel D., Prabhu A.V., Fernandopulle M.S., Patel R., Abshari M. CRISPR interference-based platform for multimodal genetic screens in human iPSC-derived neurons. Neuron. 2019 doi: 10.1016/j.neuron.2019.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabzuni D., Wray S., Vandrovcova J., Ramasamy A., Walker R., Smith C., Luk C., Gibbs J.R., Dillman A., Hernandez D.G. MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum. Mol. Genet. 2012;21:4094–4103. doi: 10.1093/hmg/dds238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Swieten J., Spillantini M.G. Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol. 2007;17:63–73. doi: 10.1111/j.1750-3639.2007.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Ward M.E., Chen R., Liu K., Tracy T.E., Chen X., Xie M., Sohn P.D., Ludwig C., Meyer-Franke A. Scalable production of iPSC-derived human neurons to identify Tau-lowering compounds by high-content screening. Stem Cell Reports. 2017;9:1221–1233. doi: 10.1016/j.stemcr.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wren M.C., Zhao J., Liu C.C., Murray M.E., Atagi Y., Davis M.D., Fu Y., Okano H.J., Ogaki K., Strongosky A.J. Frontotemporal dementia-associated N279K tau mutant disrupts subcellular vesicle trafficking and induces cellular stress in iPSC-derived neural stem cells. Mol. Neurodegener. 2015;10:46. doi: 10.1186/s13024-015-0042-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y., Higuchi M., Zhang B., Huang S.-M., Iwata N., Saido T.C., Maeda J., Suhara T., Trojanowski J.Q., Lee V.M.-Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.