

A benzoquinone compound was unintentionally synthesized by photocyclization and subsequent oxidation in air while attempting to transform the E isomer of a nicotinamide derivative to its Z isomer. The chemical and molecular structures of the product was established crystallographically. The surprising synthesis, chromatographic purification and comprehensive characterization of this compound including single crystal structural analysis are reported and its structure including crystal packing is discussed in detail.
Keywords: crystal structure, benzoquinoline, nicotinamide derivative, photocyclization
Abstract
The title compound, C14H10N2O, crystallizes in the monoclinic space group P21/c with four molecules in the unit cell. All 17 non-H atoms of one molecule lie essentially in one plane. In the unit cell, two pairs of molecules are exactly coplanar, while the angle between these two orientations is close to perfectly perpendicular at 87.64 (6)°. In the crystal, molecules adopt a 50:50 crisscross arrangement, which is held together by two nonclassical and two classical intermolecular hydrogen bonds. The hydrogen-bonding network together with off-centre π–π stacking interactions between the pyridine and outermost benzene rings, stack the molecules along the b-axis direction.
Chemical context
Quinoline and benzoquinoline scaffolds are common structural motifs in artificial, as well as natural products, and many of these compounds are of enormous value for pharmacotherapy. Their multifaceted biological efficacy is outstanding and ranges from cardiovascular (Ferlin et al., 2002 ▸; Abouzid et al., 2008 ▸) and anti-inflammatory effects (Kumar et al., 2009 ▸; Hussaini, 2016 ▸) to antimicrobial (El Shehry et al., 2018 ▸), as well as anticancer activity (Abdelsalam et al., 2019 ▸; Haiba et al., 2019 ▸; Jafari et al., 2019 ▸; Musiol, 2017 ▸; Marzaro et al., 2016 ▸). In a report on 3-(tetrazol-5-yl)quinolines with antiallergic potential, benzo[h]quinoline-3-carboxamide was mentioned as a synthetic intermediate, though its biological activity was not determined in that work (Erickson et al., 1979 ▸). In our recent studies on photoswitchable sirtuin inhibitors, we obtained benzo[h]quinoline-3-carboxamide as a side product of azastilbene photoisomerization (Grathwol et al., 2019 ▸). By UV radiation, (E)-5-styrylnicotinamide was transformed to its Z isomer as envisioned, but underwent photocyclization and successive oxidation, yielding two isomeric benzoquinoline derivatives; the identity of one of these was determined to be the benzo[h]quinoline derivative and its crystal structure is reported here.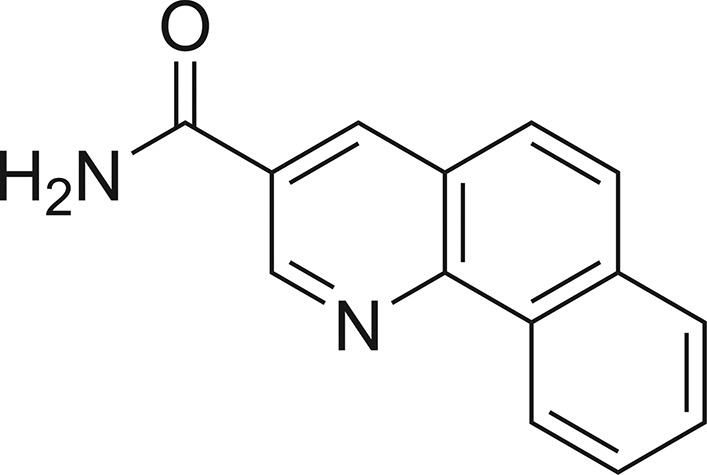
Structural commentary
The title compound, benzo[h]quinoline-3-carboxamide, crystallizes in the monoclinic space group P21/c. Four molecules are present in the unit cell (Z = 4) and there is one molecule in the asymmetric unit. Benzo[h]quinoline-3-carboxamide consists of a nicotinamide unit being fused with a benzo[h]quinoline moiety, while the pyridine ring is shared between these two common structural building blocks (Fig. 1 ▸). The molecule is essentially flat, with a largest deviation from the plane through all 17 non-H atoms of 0.050 (2) Å (O1) and an r.m.s. deviation of 0.020 (2) Å. In the unit cell, the four molecules are arranged in two perfectly coplanar pairs, with a nearly perpendicular angle between the respective planes of the two pairs of 87.64 (6)° (Fig. 2 ▸). A plethora of crystal structures are known for compounds with one or other of the two building blocks that make up this molecule [for the nicotinamide scaffold, ConQuest finds over 2000 hits in the Cambridge Structural Database (CSD), while for benzoquinoline, there are over 500; Groom et al., 2016 ▸]. However, the specific combination in the title compound is unprecedented. Comparing the title compound to the known structures of unsubstituted nicotinamides, its pronounced planarity is most notable. In the six published structures in the space groups P21/c or P21/a, the angles between the aromatic plane (here C2/C3/N2/C4/C13/C14) and the amide substituent (here O1/N1/C1) range from 22.1 to 23.3° (general CSD refcode NICOAM; Wright & King, 1954 ▸; Miwa et al., 1999 ▸; Fábián et al., 2011 ▸; Jarzembska et al., 2014 ▸), i.e. this angle is quite consistent. In the only distinct polymorph of a nicotinamide in the space group P2/a, four distinct molecules were refined with this angle ranging from 8.1 to 22.4° (Li et al., 2011 ▸), i.e. they are not very consistent but still considerably larger than the corresponding angle found in the title compound, which is a mere 3.3 (4)°. This points toward an extension of the aromatic resonance systems to include the amide substituent. In the parent nicotinamide scaffolds, this does not occur. Similarly, the comparatively long C1=O1 distance of 1.238 (3) Å (average 1.23 Å) and the comparatively short C1—C2 distance of 1.491 (3) Å (average 1.50 Å in other nicotinamide structures) indicate some involvement of these atoms in resonance effects. In support of this extended resonance, in the nicotinamide structures, the aromatic C—C bonds are much less diverse (range 1.38–1.39 Å, indicating very strong aromaticity in the pyridine ring) than in the structure reported here. In fact, the C—C [range 1.376 (3)–1.414 (3) Å] and C—N [1.321 (3) and 1.360 (3) Å] bond lengths here are much more similar to the two known structures of 2-unsubstituted and 3-substituted benzo[h]quinolines (refcodes JAFVEU and SUDVES), with ranges of average C—C and C—N bond lengths of 1.38–1.42 and 1.32–1.36 Å, respectively (Martínez et al., 1992 ▸; Luo et al., 2015 ▸). The benzo[h]quinoline structural motif therefore dominates the observed metrical parameters of the molecule reported here, representing a fusion between a nicotinamide and a benzo[h]quinoline, with a partial extension of the aromaticity beyond the ring system and extending towards the amide substituent.
Figure 1.

The molecular structure of benzo[h]quinoline-3-carboxamide. Displacement ellipsoids are shown at the 50% probability level.
Figure 2.

The unit cell of benzo[h]quinoline-3-carboxamide in P21/c, with its four molecules in a coplanar and perpendicular arrangement, viewed along the ac diagonal.
Supramolecular features
In the crystal, the planar molecules are all arranged in planes in two distinct orientations, which are nearly perpendicular to each other [angle 87.64 (6)°]. This forms a crisscross pattern when viewed along the ac diagonal (Fig. 2 ▸). Classical inversion-related N1—H1P⋯O1 hydrogen bonds form dimers and generate  (8) ring motifs (Fig. 3 ▸). Each molecule forms two classical (N—H⋯O and N—H⋯N) and two nonclassical (C—H⋯N and C—H⋯O) hydrogen bonds (Table 1 ▸), and these contacts link adjacent dimers into zigzag chains along the c-axis direction (Fig. 4 ▸). The observed packing is further stabilized by off-centre π–π stacking between the pyridine and outermost benzene rings of each of the coplanar layers [centroid-to-centroid distance = 3.610 (1) Å] (Fig. 5 ▸). These contacts combine to stack the molecules along the b-axis direction (Fig. 6 ▸).
(8) ring motifs (Fig. 3 ▸). Each molecule forms two classical (N—H⋯O and N—H⋯N) and two nonclassical (C—H⋯N and C—H⋯O) hydrogen bonds (Table 1 ▸), and these contacts link adjacent dimers into zigzag chains along the c-axis direction (Fig. 4 ▸). The observed packing is further stabilized by off-centre π–π stacking between the pyridine and outermost benzene rings of each of the coplanar layers [centroid-to-centroid distance = 3.610 (1) Å] (Fig. 5 ▸). These contacts combine to stack the molecules along the b-axis direction (Fig. 6 ▸).
Figure 3.

Dimers formed by N—H⋯O hydrogen bonds.
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1N⋯N2i | 0.97 (3) | 2.17 (3) | 3.133 (3) | 173 (2) |
| N1—H1P⋯O1ii | 0.93 (3) | 1.96 (3) | 2.895 (3) | 175 (3) |
| C3—H3⋯N2i | 0.95 | 2.41 | 3.361 (3) | 174 |
| C7—H7⋯O1iii | 0.95 | 2.45 | 3.140 (3) | 129 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  .
.
Figure 4.

Chains of dimers along the c-axis.
Figure 5.

π–π stacking interactions, with centroids shown as coloured spheres. Cg1 and Cg2 are the centroids of the C5–C10 and C2/C3/N2/C4/C13/C14 rings, respectively.
Figure 6.

The overall packing of the title compound, viewed along the b-axis direction.
Synthesis and crystallization
A solution of (E)-5-styrylnicotinamide (673 mg, 3.00 mmol, 1.00 equiv.) in methanol (350 ml) was treated with a solution of iodine (38 mg, 0.15 mmol, 0.05 equiv.) in methanol (50 ml). A slow stream of compressed air was bubbled through the reaction mixture while it was irradiated with UV light (six Vilber-Lourmat T8-C lamps, 8 W, 254 nm). After complete consumption of the starting material (24 h), the solvent was removed under reduced pressure. Purification of the residue by silica-gel column chromatography (n-hexane/THF, 1:1 v/v) gave pure benzo[h]quinoline-3-carboxamide as a colourless solid (yield 80 mg, 0.36 mmol, 12%). Crystallization was accomplished by slow evaporation of a solution in THF (5 mg ml−1) and yielded the title compound as colourless needles: R F = 0.32 (n-hexane/THF, 1:1 v/v); m.p. 549.8 K (decomposition); 1H NMR, H,H-COSY (400 MHz, DMSO-d 6): δ (ppm) 9.48 (d, J = 2.2 Hz, 1H, C3-H), 9.26–9.19 (m, 1H, C6-H), 8.89 (d, J = 2.1 Hz, 1H, C14-H), 8.36 (s, br, 1H, N1-H), 8.12–8.07 (m, 1H, C9-H), 8.03 (d, J = 8.9 Hz, 1H, C11-H), 7.95 (d, J = 8.9 Hz, 1H, C12-H), 7.85–7.78 (m, 2H, C7-H, C8-H), 7.74 (s, br, 1H, N1-H); 13C NMR, DEPT135, HSQC, HMBC (101 MHz, DMSO-d 6): δ (ppm) 166.4 (C1), 147.8 (C3), 146.7 (C4), 135.5 (C14), 133.8 (C13), 130.3 (C5), 129.0 (C8), 128.1 (C9/C11), 128.0 (C9/C11), 127.8 (C2), 127.3 (C7), 125.8 (C12), 124.9 (C10), 124.1 (C6); IR (ATR): ν (cm−1) 3336, 3136, 1686, 1482, 1395, 1295, 801, 691, 539, 489; ESI–HRMS calculated for [C14H10N2O + H]+ 222.0793, found 222.0796; compound purity (220 nm): 100%.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. All C-bound H atoms constitute aromatic protons, which were attached in calculated positions and treated as riding with U iso(H) = 1.2U eq(C). The two amine H atoms were found and refined without any constraints or restraints.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C14H10N2O |
| M r | 222.24 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 170 |
| a, b, c (Å) | 12.634 (3), 4.9426 (10), 16.778 (3) |
| β (°) | 100.53 (3) |
| V (Å3) | 1030.0 (4) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.09 |
| Crystal size (mm) | 0.37 × 0.07 × 0.04 |
| Data collection | |
| Diffractometer | Stoe IPDS-2T |
| Absorption correction | Numerical face indexed |
| T min, T max | 0.727, 0.997 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 10053, 2551, 1320 |
| R int | 0.087 |
| (sin θ/λ)max (Å−1) | 0.667 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.055, 0.167, 0.98 |
| No. of reflections | 2551 |
| No. of parameters | 163 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.23, −0.27 |
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989019014440/sj5580sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989019014440/sj5580Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989019014440/sj5580Isup3.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors acknowledge support for the Article Processing Charge from the DFG (German Research Foundation) and the Open Access Publication Fund of the University of Greifswald.
supplementary crystallographic information
Crystal data
| C14H10N2O | F(000) = 464 |
| Mr = 222.24 | Dx = 1.433 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 12.634 (3) Å | Cell parameters from 11960 reflections |
| b = 4.9426 (10) Å | θ = 6.4–59.0° |
| c = 16.778 (3) Å | µ = 0.09 mm−1 |
| β = 100.53 (3)° | T = 170 K |
| V = 1030.0 (4) Å3 | Needle, colourless |
| Z = 4 | 0.37 × 0.07 × 0.04 mm |
Data collection
| Stoe IPDS2T diffractometer | 2551 independent reflections |
| Radiation source: fine-focus sealed tube | 1320 reflections with I > 2σ(I) |
| Detector resolution: 6.67 pixels mm-1 | Rint = 0.087 |
| ω scans | θmax = 28.3°, θmin = 3.2° |
| Absorption correction: numerical face indexed | h = −16→16 |
| Tmin = 0.727, Tmax = 0.997 | k = −6→6 |
| 10053 measured reflections | l = −22→22 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.055 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.167 | w = 1/[σ2(Fo2) + (0.086P)2] where P = (Fo2 + 2Fc2)/3 |
| S = 0.98 | (Δ/σ)max < 0.001 |
| 2551 reflections | Δρmax = 0.23 e Å−3 |
| 163 parameters | Δρmin = −0.27 e Å−3 |
| 0 restraints | Extinction correction: SHELXL2018 (Sheldrick, 2015b), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: dual | Extinction coefficient: 0.019 (4) |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.60103 (14) | −0.2438 (3) | 0.51392 (9) | 0.0364 (4) | |
| N2 | 0.63400 (15) | 0.2758 (4) | 0.28016 (11) | 0.0304 (5) | |
| N1 | 0.48278 (17) | −0.3250 (4) | 0.39872 (12) | 0.0331 (5) | |
| C1 | 0.56558 (19) | −0.1907 (4) | 0.44191 (13) | 0.0312 (5) | |
| C2 | 0.61839 (18) | 0.0252 (4) | 0.40098 (12) | 0.0287 (5) | |
| C3 | 0.58725 (19) | 0.0909 (4) | 0.31880 (13) | 0.0311 (5) | |
| H3 | 0.528010 | −0.004871 | 0.288573 | 0.037* | |
| C4 | 0.71867 (17) | 0.4149 (4) | 0.32278 (12) | 0.0274 (5) | |
| C5 | 0.77008 (18) | 0.6182 (4) | 0.28123 (13) | 0.0292 (5) | |
| C6 | 0.7362 (2) | 0.6735 (5) | 0.19826 (13) | 0.0332 (5) | |
| H6 | 0.678537 | 0.573337 | 0.167687 | 0.040* | |
| C7 | 0.7858 (2) | 0.8702 (5) | 0.16151 (14) | 0.0360 (6) | |
| H7 | 0.762331 | 0.905703 | 0.105414 | 0.043* | |
| C8 | 0.8707 (2) | 1.0200 (5) | 0.20521 (15) | 0.0375 (6) | |
| H8 | 0.904089 | 1.157365 | 0.178843 | 0.045* | |
| C9 | 0.90571 (19) | 0.9702 (5) | 0.28530 (14) | 0.0346 (6) | |
| H9 | 0.963477 | 1.073076 | 0.314661 | 0.041* | |
| C10 | 0.85689 (18) | 0.7665 (4) | 0.32529 (13) | 0.0305 (5) | |
| C11 | 0.8935 (2) | 0.7074 (5) | 0.40961 (14) | 0.0350 (6) | |
| H11 | 0.952662 | 0.805664 | 0.438992 | 0.042* | |
| C12 | 0.84594 (19) | 0.5161 (5) | 0.44794 (13) | 0.0329 (5) | |
| H12 | 0.872211 | 0.481242 | 0.503736 | 0.039* | |
| C13 | 0.75657 (17) | 0.3646 (4) | 0.40605 (12) | 0.0291 (5) | |
| C14 | 0.70365 (19) | 0.1668 (4) | 0.44428 (13) | 0.0307 (5) | |
| H14 | 0.726853 | 0.130640 | 0.500358 | 0.037* | |
| H1N | 0.449 (2) | −0.279 (6) | 0.344 (2) | 0.059 (9)* | |
| H1P | 0.452 (2) | −0.464 (6) | 0.4246 (16) | 0.054 (8)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0445 (10) | 0.0389 (9) | 0.0247 (8) | −0.0018 (8) | 0.0036 (7) | 0.0056 (7) |
| N2 | 0.0311 (11) | 0.0302 (10) | 0.0293 (9) | −0.0018 (8) | 0.0037 (8) | 0.0005 (8) |
| N1 | 0.0373 (12) | 0.0331 (11) | 0.0280 (10) | −0.0018 (9) | 0.0036 (9) | 0.0020 (8) |
| C1 | 0.0351 (13) | 0.0302 (11) | 0.0285 (11) | 0.0031 (10) | 0.0064 (10) | 0.0001 (9) |
| C2 | 0.0310 (12) | 0.0281 (11) | 0.0275 (10) | 0.0048 (9) | 0.0068 (9) | 0.0004 (9) |
| C3 | 0.0333 (12) | 0.0306 (11) | 0.0286 (11) | −0.0012 (10) | 0.0033 (9) | 0.0002 (9) |
| C4 | 0.0278 (11) | 0.0261 (11) | 0.0279 (11) | 0.0029 (9) | 0.0036 (9) | −0.0021 (9) |
| C5 | 0.0299 (12) | 0.0276 (11) | 0.0307 (11) | 0.0034 (10) | 0.0068 (9) | −0.0007 (9) |
| C6 | 0.0345 (13) | 0.0355 (13) | 0.0295 (11) | −0.0024 (10) | 0.0058 (10) | 0.0009 (9) |
| C7 | 0.0372 (14) | 0.0387 (13) | 0.0327 (11) | −0.0005 (11) | 0.0082 (10) | 0.0042 (10) |
| C8 | 0.0367 (13) | 0.0348 (13) | 0.0428 (13) | −0.0015 (11) | 0.0124 (11) | 0.0025 (11) |
| C9 | 0.0325 (13) | 0.0324 (12) | 0.0393 (13) | 0.0003 (10) | 0.0079 (10) | −0.0006 (10) |
| C10 | 0.0291 (12) | 0.0280 (11) | 0.0351 (11) | 0.0030 (9) | 0.0079 (9) | −0.0021 (9) |
| C11 | 0.0319 (13) | 0.0379 (13) | 0.0342 (12) | −0.0024 (10) | 0.0037 (10) | −0.0063 (10) |
| C12 | 0.0324 (12) | 0.0372 (12) | 0.0274 (11) | 0.0009 (10) | 0.0009 (9) | −0.0027 (9) |
| C13 | 0.0303 (12) | 0.0302 (11) | 0.0260 (10) | 0.0045 (10) | 0.0033 (9) | 0.0006 (9) |
| C14 | 0.0352 (13) | 0.0312 (12) | 0.0248 (10) | 0.0053 (10) | 0.0034 (9) | 0.0009 (9) |
Geometric parameters (Å, º)
| O1—C1 | 1.238 (3) | C6—H6 | 0.9500 |
| N2—C3 | 1.321 (3) | C7—C8 | 1.396 (3) |
| N2—C4 | 1.360 (3) | C7—H7 | 0.9500 |
| N1—C1 | 1.335 (3) | C8—C9 | 1.358 (3) |
| N1—H1N | 0.97 (3) | C8—H8 | 0.9500 |
| N1—H1P | 0.93 (3) | C9—C10 | 1.413 (3) |
| C1—C2 | 1.491 (3) | C9—H9 | 0.9500 |
| C2—C14 | 1.376 (3) | C10—C11 | 1.436 (3) |
| C2—C3 | 1.401 (3) | C11—C12 | 1.346 (3) |
| C3—H3 | 0.9500 | C11—H11 | 0.9500 |
| C4—C13 | 1.414 (3) | C12—C13 | 1.428 (3) |
| C4—C5 | 1.443 (3) | C12—H12 | 0.9500 |
| C5—C6 | 1.406 (3) | C13—C14 | 1.404 (3) |
| C5—C10 | 1.411 (3) | C14—H14 | 0.9500 |
| C6—C7 | 1.364 (3) | ||
| C3—N2—C4 | 118.11 (19) | C6—C7—H7 | 119.6 |
| C1—N1—H1N | 124.6 (17) | C8—C7—H7 | 119.6 |
| C1—N1—H1P | 117.5 (17) | C9—C8—C7 | 120.2 (2) |
| H1N—N1—H1P | 118 (2) | C9—C8—H8 | 119.9 |
| O1—C1—N1 | 122.2 (2) | C7—C8—H8 | 119.9 |
| O1—C1—C2 | 119.3 (2) | C8—C9—C10 | 120.5 (2) |
| N1—C1—C2 | 118.55 (19) | C8—C9—H9 | 119.7 |
| C14—C2—C3 | 117.0 (2) | C10—C9—H9 | 119.7 |
| C14—C2—C1 | 119.62 (19) | C5—C10—C9 | 119.1 (2) |
| C3—C2—C1 | 123.4 (2) | C5—C10—C11 | 119.4 (2) |
| N2—C3—C2 | 125.0 (2) | C9—C10—C11 | 121.5 (2) |
| N2—C3—H3 | 117.5 | C12—C11—C10 | 121.5 (2) |
| C2—C3—H3 | 117.5 | C12—C11—H11 | 119.3 |
| N2—C4—C13 | 121.5 (2) | C10—C11—H11 | 119.3 |
| N2—C4—C5 | 118.57 (19) | C11—C12—C13 | 121.0 (2) |
| C13—C4—C5 | 119.92 (19) | C11—C12—H12 | 119.5 |
| C6—C5—C10 | 119.0 (2) | C13—C12—H12 | 119.5 |
| C6—C5—C4 | 122.1 (2) | C14—C13—C4 | 118.1 (2) |
| C10—C5—C4 | 118.95 (19) | C14—C13—C12 | 122.68 (19) |
| C7—C6—C5 | 120.3 (2) | C4—C13—C12 | 119.3 (2) |
| C7—C6—H6 | 119.9 | C2—C14—C13 | 120.37 (19) |
| C5—C6—H6 | 119.9 | C2—C14—H14 | 119.8 |
| C6—C7—C8 | 120.9 (2) | C13—C14—H14 | 119.8 |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N···N2i | 0.97 (3) | 2.17 (3) | 3.133 (3) | 173 (2) |
| N1—H1P···O1ii | 0.93 (3) | 1.96 (3) | 2.895 (3) | 175 (3) |
| C3—H3···N2i | 0.95 | 2.41 | 3.361 (3) | 174 |
| C7—H7···O1iii | 0.95 | 2.45 | 3.140 (3) | 129 |
Symmetry codes: (i) −x+1, y−1/2, −z+1/2; (ii) −x+1, −y−1, −z+1; (iii) x, −y+1/2, z−1/2.
Funding Statement
This work was funded by Deutsche Forschungsgemeinschaft grant 393148499.
References
- Abdelsalam, E. A., Zaghary, W. A., Amin, K. M., Abou Taleb, N. A., Mekawey, A. A. I., Eldehna, W. M., Abdel-Aziz, H. A. & Hammad, S. F. (2019). Bioorg. Chem. 89, 102985. [DOI] [PubMed]
- Abouzid, K., Abdel Hakeem, M., Khalil, O. & Maklad, Y. (2008). Bioorg. Med. Chem. 16, 382–389. [DOI] [PubMed]
- El Shehry, M. F., Ghorab, M. M., Abbas, S. Y., Fayed, E. A., Shedid, S. A. & Ammar, Y. A. (2018). Eur. J. Med. Chem. 143, 1463–1473. [DOI] [PubMed]
- Erickson, H. E., Hainline, C. F., Lenon, L. S., Matson, C. J., Rice, T. K., Swingle, K. F. & Van Winkle, M. (1979). J. Med. Chem. 22, 816–823. [DOI] [PubMed]
- Fábián, L., Hamill, N., Eccles, K. S., Moynihan, H. A., Maguire, A. R., McCausland, L. & Lawrence, S. E. (2011). Cryst. Growth Des. 11, 3522–3528.
- Ferlin, M. G., Chiarelotto, G., Antonucci, F., Caparrotta, L. & Froldi, G. (2002). Eur. J. Med. Chem. 37, 427–434. [DOI] [PubMed]
- Grathwol, C. W., Wössner, N., Swyter, S., Smith, A. C., Tapavicza, E., Hofstetter, R. K., Bodtke, A., Jung, M. & Link, A. (2019). Beilstein J. Org. Chem. 15, 2170–2183. [DOI] [PMC free article] [PubMed]
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Haiba, M. E., Al-Abdullah, E. S., Ahmed, N. S., Ghabbour, H. A. & Awad, H. M. (2019). J. Mol. Struct. 1195, 702–711.
- Hussaini, S. M. A. (2016). Expert Opin. Ther. Pat. 26, 1201–1221. [DOI] [PubMed]
- Jafari, F., Baghayi, H., Lavaee, P., Hadizadeh, F., Soltani, F., Moallemzadeh, H., Mirzaei, S., Aboutorabzadeh, S. M. & Ghodsi, R. (2019). Eur. J. Med. Chem. 164, 292–303. [DOI] [PubMed]
- Jarzembska, K. N., Hoser, A. A., Kamiński, R., Madsen, A., Durka, K. & Woźniak, K. (2014). Cryst. Growth Des. 14, 3453–3465.
- Kumar, S., Bawa, S. & Gupta, H. (2009). Mini Rev. Med. Chem. 9, 1648–1654. [DOI] [PubMed]
- Li, J., Bourne, S. A. & Caira, M. R. (2011). Chem. Commun. 47, 1530–1532. [DOI] [PubMed]
- Luo, C.-Z., Gandeepan, P., Wu, Y.-C., Chen, W.-C. & Cheng, C.-H. (2015). RSC Adv. 5, 106012–106018.
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- Martínez, R., Toscano, R. A., Linzaga, I. E. & Sánchez, H. (1992). J. Heterocycl. Chem. 29, 1385–1388.
- Marzaro, G., Dalla Via, L., García-Argáez, A. N., Dalla Via, M. & Chilin, A. (2016). Bioorg. Med. Chem. Lett. 26, 4875–4878. [DOI] [PubMed]
- Miwa, Y., Mizuno, T., Tsuchida, K., Taga, T. & Iwata, Y. (1999). Acta Cryst. B55, 78–84. [DOI] [PubMed]
- Musiol, R. (2017). Exp. Opin. Drug Discov. 12, 583–597. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Stoe & Cie (2010). X-AREA. Stoe & Cie GmbH, Darmstadt, Germany.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Wright, W. B. & King, G. S. D. (1954). Acta Cryst. 7, 283–288.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989019014440/sj5580sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989019014440/sj5580Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989019014440/sj5580Isup3.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report