

Abstract
Esophageal cancer (EC) is the seventh most common cancer worldwide and the sixth leading cause of death, according to Globocan 2018. Despite efforts made for therapeutic advances, EC remains highly lethal, portending a five-year overall survival of just 15–20%. Hence, the discovery of new molecular targets that might improve therapeutic efficacy is urgently needed. Due to high proliferative rates and also the limited oxygen and nutrient diffusion in tumors, the development of hypoxic regions and consequent activation of hypoxia-inducible factors (HIFs) are a common characteristic of solid tumors, including EC. Accordingly, HIF-1α, involved in cell cycle deregulation, apoptosis, angiogenesis induction and proliferation in cancer, constitutes a predictive marker of resistance to radiotherapy (RT). Deregulation of epigenetic mechanisms, including aberrant DNA methylation and histone modifications, have emerged as critical factors in cancer development and progression. Recently, interactions between epigenetic enzymes and HIF-1α transcription factors have been reported. Thus, further insight into hypoxia-induced epigenetic alterations in EC may allow the identification of novel therapeutic targets and predictive biomarkers, impacting on patient survival and quality of life.
Keywords: Epigenetic, esophageal cancer, hypoxia, microenvironment, radiobiology, radioresistance
1. Introduction
Esophageal cancer (EC) incidence has increased in recent decades, representing the seventh most common cancer worldwide and the sixth cause of cancer-related death, according to Globocan 2018 [1], with an overall five-year survival ranging from 15–20% [2,3]. There are two major EC subtypes—Adenocarcinoma (EAC) and squamous cell carcinoma (ESCC)—Which disclose unlike geographical distributions as a consequence of differentially associated risk factors and ethnicity [4,5,6]. Indeed, tobacco, alcohol consumption and dietary habits constitute the main risk factors for ESCC, whereas, Barrett’s esophagus (BE), gastroesophageal reflux disease (GERD) and obesity are associated with EAC [7].
Despite major advances in patients’ management and therapeutic strategies, including multimodality neoadjuvant approaches, the prognosis of advanced stage EC remains poor [8,9]. Currently, localized tumors are primarily treated with radical surgery, with an expected five-year survival rate of 10–40% [8]. Nonetheless, most ECs are diagnosed at advanced stages, in which primary surgery is not an effective therapy [9], entailing the need for multimodal treatments. In this setting, neoadjuvant chemoradiotherapy (CRT) achieves the highest complete pathological response rates, significantly improving EC patients’ overall outcomes [8,9,10]. Moreover, a synergistic effect is achieved, combining radio- and chemotherapy with surgical resection, i.e., trimodal therapy [8].
Discovery of cancer-specific molecular targets has been a major focus of research to improve cancer survival rates [11]. Taking into account tumor biology and microenvironment heterogeneity, tumors respond differently to radiotherapy (RT), presenting several forms of therapy resistance [12]. Since RT acts through a fundamentally biological, rather than physical, mechanism both the understanding of how radiation influences tumor cells’ behavior, and its surrounding microenvironment is required to maximize therapeutic efficacy.
A common feature of solid tumors, including EC, is the presence of hypoxic regions, and its extent has been previously correlated with poor prognosis [13].
It is well known that molecular oxygen local availability permanently fixes radiation induces DNA lesions produced by free radicals, leading to unrepairable damage, and thus, enhancing RT efficacy. Moreover, one of the cancer cells’ goal consists in the maintenance of a pool of reductive molecules that protect against redox stress [14]. RT causes a redox imbalance in favor of excessive oxidation [14], and must overcome the redox defenses of tumor cells in order to be effective. In this context, hypoxia is highly detrimental as it interferes with the fixation of DNA damage causing resistance to RT.
A more detailed characterization of the molecular alterations associated with hypoxia could, not only, serve as a surrogate marker of this phenomenon, but also as relevant therapeutic targets, eventually impacting in EC response to CRT. Furthermore, epigenetic alterations are a recognized cancer hallmark owing to its role in altered chromatin organization and consequent transcriptional gene deregulation [15,16]. Moreover, it has been suggested that hypoxic tumor microenvironment might unleash several epigenetic changes resulting in increased tumor aggressiveness and RT resistance [17]. In radioresistant tumor cells, a phenotypic switch occurs during the course of radiation delivery, associated with both genetic and epigenetic changes [18]. Nonetheless, there is only limited evidence regarding the usefulness of epigenetic alterations as a potential predictor of response to radiotherapy [19]. Thus, the aim of this review is to summarize published evidence on the role of hypoxia-induced epigenetic alterations which might impact on RT resistance in EC.
2. Biological Basis of Cancer Radiation Therapy
RT plays an important role in improving the survival of EC patients who are not eligible for surgery [9]. The standard dose recommended by European and North American guidelines is 50.4 Gy as total dose [20], in 28 fractions of 1.8 Gy/day. Regarding Asian countries, the standard dose recommended is a total treatment of 60 Gy [20]. Although RT efficacy has developed considerably in recent years, due to advances in precision techniques, such as intensity-modulated RT (IMRT) or volumetric-modulated arc therapy (VMAT), inter-individual differences are observed, and significant response is not achieved in some patient subgroups. Cancer cells exposed to radiation, depending on the surrounding tumor microenvironment, may then follow multiple pathways that culminate in cell death [21].
The patients’ outcome to standard radiation treatment is determined by the 5R´s of radiobiology: Repair, redistribution, repopulation, reoxygenation and radiosensitivity [22]. Repair of sublethal DNA damage is more effective in normal than tumor cells, as observed from cell recovery after exposure to ionizing radiation (IR) [22]. Radiation randomly interacts with several cellular molecules, with DNA being the main target for the biological effects of radiation, including cell killing, carcinogenesis and mutation [21]. Overall, the main goal of RT is clonogenic capacity loss [22]. Thus, radiation causes a wide range of lesions in DNA and double-strand breaks (DSB) which represents the most deleterious lesion [23]. DSBs repair might be accomplished by several mechanisms, emphasizing the importance and difficulty of repairing this type of DNA injury.
Redistribution refers to different cellular radiosensitivity depending on cell cycle phase: S phase cells are more radioresistant than cells in late G2 and M phases [24]. Thus, small fractionated doses allow greater damage to tumor cells, due to reassortment of cells into more sensitive stages. Repopulation by increased cell division after irradiation depends on normal tissues capability to compensate killed cell populations [25].
Furthermore, tumor oxygenation level is a major determinant of RT effectiveness. Due to abnormal vasculature development during tumor angiogenesis, the tumor microenvironment developed areas with a decreased pH, lack of nutrients and hypoxia [26]. As a result of prolonged exposure to hypoxia, cells may acquire apoptosis resistance, which also favors clonal selection towards a more aggressive phenotype [27]. Indeed, during reoxygenation, surviving hypoxic tumor cells may increase the oxygen supply and become more radiosensitive [22].
During RT treatment, fractionation spares normal tissue, due to sublethal damage repair and cell repopulation occurring between treatment fractions [21]. Concomitantly, it allows increased tumor damage as a result of reoxygenation, as well as redistribution into more radiosensitive stages of the cell cycle [19]. Therefore, while DNA repair and cell repopulation induce normal tissue resistance to RT [21,28], redistribution and reoxygenation have the opposite effect increasing the radiosensitivity of tumor cells [19,22,28].
Tumor-Associated Microenvironment: Hypoxia, HIF-1α, and Radioresistance
Uncontrolled cell proliferation leads to oxygen demand exceeding oxygen supply. Tumor growth surpasses the capacity of the existing vasculature to provide nutrients and oxygen, leading to hypoxia. The permanent changes in blood flow and oxygen availability resulting from the tumor abnormal vasculature associate with poor prognosis and therapy resistance. In particular, RT that kills tumor cells by generating reactive oxygen species (ROS), has the ability to chemically modify radiation-induced DSB leading to permanent damage fixing [29] (Figure 1).
Figure 1.

Oxygen effect on radiotherapy response. During tumor development and progression, abnormal vasculature provides a hypoxic microenvironment. Conventional radiation therapy interacts by an indirect effect, due to water radiolysis’ free radicals. Tumor reoxygenation constitutes a major determinant of radiotherapy (RT) effectiveness. Indeed, the oxygen present in the cell is a potent radiosensitizer once it participates in these free radical reactions. Thus, in the absence of oxygen, ionizing radiation effectiveness is compromised, contributing to radioresistance. Abbreviations: DSB, double strand breaks; ROS, reactive oxygen species.
Briefly, hypoxia is mediated by key transcription factors, including the hypoxia-inducible factors —HIFs that promote oxygen delivery or adaptation to oxygen deprivation, regulating the expression of over 100 genes involved in processes, such as the promotion of angiogenesis, cell proliferation, metastasis and metabolic adaptation [30]. Ubiquitously expressed oxygen-labile α subunit, HIF-1α, is known as the main hypoxia-inducible gene expression regulator [27].
Under normoxic conditions, HIF-1α is rapidly hydroxylated by prolyl-hydroxylases (PHDs), which are oxygen sensor proteins [31]. Then, these hydroxylated molecules are ubiquitinated by Von Hippel-Lindau protein (pVHL) containing E3 ubiquitin ligase, leading to HIF-1α degradation in the proteasome complex [31]. Conversely, in hypoxic conditions, HIF-1α becomes constitutively expressed owing to PHDs inhibition [31].
In chronic hypoxia, HIF-1α, through hypoxia-responsive elements (HRE), binds to functional promoter regions of several genes inducing transcriptional activation. Hypoxia is, therefore, associated with key cancer hallmarks, namely, angiogenesis, metastasis and invasion, evasion of apoptosis, metabolic reprogramming and genomic instability [26,31,32]. Overall, hypoxia and HIF-1α pathway activation are both frequently associated with therapy resistance and poor clinical outcomes [26,33].
In EC, high HIF-1α levels have been correlated with tumor depth of invasion, differentiation, lymphatic permeation and metastization [34]. Accordingly, HIF-1α and hypoxic-associated biomarkers, such as carbonic anhydrase IX (CAIX) and glucose transporter 1 (GLUT-1) were strongly associated with worse prognosis and poor treatment outcome of EC patient’s [13,35,36]. Furthermore, higher HIF-1α expression was reported to be associated with shorter patients’ survival and poor CRT response [33,35], and it was also extensively demonstrated that higher radiation doses are required to kill hypoxic cancer cells compared to their well-oxygenated counterparts [37].
Previous experiments in ESCC have suggested that HIF-1α signaling pathway’s suppression with berberine, led to tumor radiosensitization [38]. Specifically, Kato, Y et al., indicated that hypoxia leads to downregulation of homologous recombinant proteins BRCA1 and BRCA2 in EC cells, promoting G0-G1 cell cycle arrest and consequently lower radiotherapy response [39]. Indeed, cell cycle kinetics and DNA damage play a critical role in tumor radiosensitivity evaluation at hypoxia conditions. Accordingly, experimental studies are required to unveil molecular mechanisms in tumor-surrounding microenvironment that allow hypoxic cell selection, oxygenation and sensitization to allow to improve therapeutic efficacy in EC patients [33,40].
3. Cancer Epigenetics: A Brief Overview
Epigenetics consists of a set of heterodynamic and heritable changes which pass from cell generation to cell generation, affecting chromatin organization and transcriptional gene regulation, without altering the DNA sequence [16]. Although certain genomic mutations have an important influence on the malignant phenotype, tumor heterogeneity goes beyond somatic mutations and can often be explained by epigenetic changes. Thus, the epigenome landscape is currently recognized as having an important role in the process of neoplastic transformation and progression [41]. Furthermore, several changes in the epigenetic landscape mediate gene expression regulation and play key roles in signaling pathways which contributes to tumor development and progression [41].
The major epigenetic mechanisms currently known are histone post-translational modifications (PTMs), histone variants, DNA methylation and chromatin remodeling (Figure 2). The basic structure of human chromatin comprises the DNA chain coupled with histone proteins, including the histone octamer composed by H2A, H2B, H3 and H4. These specific proteins may acquire post-translational covalent alterations in C and N-terminal tails [41,42]. The most studied histone modifications are arginine’s and lysine’s methylation, acetylation and phosphorylation [41,43]. In normal cells, epigenetic mechanisms regulate constitutive gene expression, which maintains cellular variability and identity.
Figure 2.

Epigenetic mechanisms. Epigenetic machinery and overall gene transcriptional deregulation. (a) Chromatin remodeler complexes catalyze chromatin changes. (b) Histone post-translational modifications are covalent dynamic changes of histones that affect chromosomal structure and function. (c) DNA methylation: The addition of a methyl group to CpG islands of cancer related genes’ promotor regions induce transcriptional repression. (d) Histone variants, proteins that replace the core canonical histone in the nucleosome, are often characteristic of specific structural and functional features in the cell.
Several epigenetic alterations, including aberrant DNA methylation and histone onco-modifications, found in several cancers and associated with oncogene activation and tumor suppressor gene silencing [15,41], have emerged as potential cancer biomarkers [44]. Concerning PTMs, histone acetylation, which is often associated with transcriptional activation, is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDAC), subdivided into four classes [45,46]. Histone methylation has a more pleiotropic effect, depending on the residue involved: Whereas, H3K4me3 and H3K36me2/3 are mostly associated with transcriptional activation, H3K9me3 and H3K27me3 are frequently associated with transcriptional repression [45,47]. Remarkably, histone methylation is a frequent event in cancer, and its dynamic transition plays a critical role in transcriptional regulation and subsequently in signaling pathways related with cell cycle, tumor metabolism and overall tumor aggressiveness capacities [48]. Accordingly, lysine methyltransferases (KMTs) and histone lysine demethylases (KDMs) regulate the methylation status of histones, and also protein substracts. KMTs, which added methyl groups in histones, comprise two major classes—those with or without SET [Su(var)3-9, E(z) (enhancer of zeste) and trithorax] domain [49]. Conversely, these methyl groups are specifically removed by several KDMs, which also comprise two major subgroups—lysine-specific demethylase (LSD1)/KDM1A containing a FAD (flavin adenine dinucleotide) dependent domain and the 2-oxoglutarate-dependent dioxygenase superfamily which encompasses most enzymes, composed by 7 KDMs subfamilies (KDM2-KDM8), with a Jumonji C catalytic domain with O2, Fe (II) and α-ketoglutarate as cofactor to catalyze the demethylation process [49,50].
Importantly, aberrant histone PTMs represents an essential contribution to EC progression and development. Briefly, HDAC6 overexpression induced higher proliferation and migration in ESCC cancer cell lines [51]. Accordingly, higher HDAC1 expression in ESCC patients with advanced stages, associated with aggressiveness [52]. Also, a significant H4 hyperacetylation in the early stage ESCC tissues was also observed [52]. However, when tumors progress and are invasive, global hypoacetylation might occur, indicating a dynamic interaction and equilibrium between HATs and HDACs in the tumor cells [52]. Particularly, in EC, several research teams suggest that histone methylation also plays a critical role in the maintenance of cells balance state, depending on the lysine residue involved. Moreover, high KMTs expression, such as SMYD3/KMT3E promotes ESCC cell proliferation. This KMT enzyme negatively interacts with RIZ1 (retinoblastoma protein-interacting zinc finger gene 1), leading to decreased H3K9 methylation levels [53]. Concerning histone demethylases, Jumonji C domain KDMs members of family GASC1/KDM4C and JMJD1C/KDM3C demethylating agents of repressive histone marker H3K9 and KDM7B/PHF8 which specifically demethylate H3K27me1/2, play a critical role in ESCC cancer development [54,55,56]. Moreover, LDS1 also contributes to EC malignant progression, leading to a patient’s poor prognosis [57,58]. Overall, high expression levels of these enzymes induce changes in several pathways, including, proliferation, migration and invasion.
Another extensively studied epigenetic alteration occurring in cancer cells is aberrant DNA methylation [59]. This process mostly interferes with gene promoter regions to induce their downregulation [59]. Briefly, DNA methylation consists in the addition of methyl groups (-CH3) at the 5’ position of the cytosines of the DNA chain, mainly at so-called CpG islands, which are regions enriched with cytosine and guanine nucleotides [59]. Similarly, to PTMs, DNA methylation is also mediated by DNA methyltransferases (DNMTs) [59]. Particularly, DNMT1, DNMT3a and DNMT3b are the main enzymes implicated in cytosines methylation during DNA replication [59]. Importantly, DNA hypermethylation is associated with onco-suppressor genes silencing [59], whereas, promoter hypomethylation leads to oncogene overexpression and global genomic instability [59]. Specifically, CDKN2A, MGMT, APC, RUNX3, TIMP-3 and CRBP1 hypermethylation have been frequently reported in EC. Aberrant DNA methylation is the most studied epigenetic mechanism in EC [60,61]. Epigenetic modifications, primarily in the form of DNA hypermethylation of cancer-related genes frequently occur in both ESCC and EAC, as well as in BE (the EAC precursor lesion), with some of these methylated tumor suppressor genes preceding the progression of BE to EAC or dysplasia to ESCC, thus, being good cancer biomarker candidates’ [61].
3.1. Epigenetic Remodeling and Hypoxic Microenvironment
Considering tumor heterogeneity and microenvironment, hypoxia was suggested to play a critical role in epigenetic regulation. Consequently, this interaction fosters tumor progression and increased aggressiveness [62]. Indeed, activation of HIF-1α induces several cellular adaptations through oxygen-dependent manner [62]. Remarkably, hypoxia-induced aberrant DNA methylation, altered histone PTMs patterns and modifications in their respective chromatin remodelers have been reported in several neoplasms [62] (Figure 3). Furthermore, HIF-1α plays a critical role in expression regulation, through HRE binding sites at promoter regions of the genes encoding for those enzymes. Interestingly, Skowronski and co-workers demonstrated that DNMT1 and DNMT3a proteins were significantly downregulated under hypoxia, in human colorectal cancer cell lines, entailing oncogene transcriptional activation and overall genetic instability [63,64].
Figure 3.

Epigenetic remodeling through hypoxic-dependent pathways. The accumulation of epigenetic alterations is characteristic of a hypoxic microenvironment in tumors. (A) Epigenetic changes. Briefly, is has been intensively reported that after HIF-1α stabilization, decreased DNA methyltransferases (DNMTs) activity correlates with oncogene activation. Conversely, HCADs and KDMs increased activity induces overall genomic instability. (B) Histone modifications. Histone methylation, depending on the residue, induces gene transcription activation or repression. Specifically, H3K4me3 and H3K36me2/3 are often correlated with transcriptional activation, whereas H3K9me3 and H3k27me3 are associated with transcriptional repression, both implicated tumor development and progression. Abbreviations: Ac—acetylation; DNMTs—DNA methyltransferases; HDACs—Histone deacetylases; HIF—Hypoxia-inducible factors; KDMs—Histone lysine demethylases; Me—methylation; PHD—Prolyl hydroxylases; TS—Tumor suppressor.
Conversely, ten-eleven translocation (TETs) dioxygenases proteins family, which mediate DNA methylation, particularly TET1 and TET3, have been reported to be overexpressed in tumor cells in association with hypoxia-induced pathways [65]. TET1 was shown to be a transcriptional co-activator associated with HIF-1α enhancement regulation in several human cancer cell lines from kidney, lung, liver and head and neck [66]. Moreover, TET proteins’ overexpression was closely associated with tumor angiogenesis, invasion and migration, since it allows for transcriptional activation of epithelial mesenchymal transition (EMT) promoting genes [66].
In addition, PTMs specifically associated with aggressive malignant phenotypes have been correlated with hypoxia and HIF-1α activation [62]. Accordingly, hypoxia was implicated in decreased histone acetylation levels, which are involved in overall gene transcriptional repression [67]. In accordance, global HDACs activity is modulated in hypoxic condition [67]. For instance, HDAC1 was associated with HIF-1α signaling pathways, being significantly overexpressed in the Lewis lung carcinoma model, increasing angiogenesis [67]. In accordance, Kim et al. have demonstrated the critical role of HDACs-induced hypoxia in angiogenesis and tumor progression [67]. Indeed, HDAC1, HDAC2 and HDAC3 were upregulated under hypoxic microenvironment in human kidney and lung carcinoma cell lines [67], and thus, HDAC inhibitors (HDACi) were suggested as a potential therapeutic agent in tumors with significant hypoxic regions [67].
Furthermore, several studies have identified genome binding sites for other key regulators, namely, JmjC-KDMs, and some of them were proposed as novel therapeutic targets in several cancer models [68]. Hypoxia-induced KDM3A, KDM4B, KDM4C (specifically in EC [54]), KDM6A, KDM6B, KDM2B, KDM5B and KDM5C overexpression [69,70]. Accordingly, Ikeda et al. demonstrated that KDM3A/JMJD1A was tightly regulated by HIF-1α, under hypoxic conditions, in multiple myeloma cell lines [71]. These dynamic alterations led to anti-apoptotic phenomena, invasive and pro-angiogenic properties [72,73]. Moreover, the KDM4 subfamily, particularly KDM4C/JMJD2C, which target methylated H3K9 and H3K36 histone marks, contributes to cancer initiation and development, and is associated with hypoxic status in breast cancer [74,75], whereas, in renal cell carcinoma cell lines, KDM3A, KDM4B and KDM5B were shown to be regulated by HIF family. Also, KDM6B/JMJD3, which demethylate H3K27me3, was previously demonstrated to play a critical role in the hypoxic signaling pathway, in the human liver and kidney cell lines [76,77]. The involvement of these main JmjC-KDMs highlight the importance of HIF-1α-dependent epigenetic regulation in hypoxia, leading to several molecular changes, like cell cycle arrest and DNA repair, which contribute to cancer progression.
In EC, the interplay between the hypoxia and epigenetic mechanism regulation remains poorly understood. Considering that both hypoxia and epigenetic alterations are common features in EC, however, its relevance in this particular cancer type is very likely [35]. HDAC1/2 were previously found to be deregulated in EC, and HDAC activity was associated with in vitro anti-neoplastic activity, in human EC cell lines [78]. Conversely, KDM4C/JMJD2C/GASC1 overexpression induced EC initiation and development [54]. Although this epigenetic mechanism is intensively studied in EC, the association of these alterations with tumor hypoxic microenvironment has not been reported, yet. Nonetheless, KDM3A/JMJD1A and KDM4C/JMJD2C were suggested to be hypoxia-inducible genes in kidney and prostate cancer cell lines [79]. Although it is acknowledged that hypoxia confers resistance to RT and that some epigenetic alterations are regulated by HIF-1α, the interaction between the hypoxia and epigenetic mechanism remains poorly understood, particularly in EC.
3.2. Epigenetic Modifications and Radiosensitivity
The role of epigenetic modifications, especially DNA methylation, has been widely implicated in radiation resistance, playing a critical role in cell cycle progression and checkpoint regulation, DNA repair and apoptosis, all of which decisive in determining radiation cell response (Figure 4). Recently, dysregulated epigenetic control through DNA methylation was associated with tumors’ radiation resistance [80]. Exposure to IR results in DNMTs decreased levels.
Figure 4.

Radiation-induced epigenetic alterations. A phenotypic switch during radiation treatment, leads several epigenetic modifications in the tumor cells. In fact, DNA methylation has been the most studied mechanism in RT resistance. Changes in DNA methylation did not occur uniformly into the cell genome. It has been described IR-induced specific methylated genes as TP53, CDKN1C and RUNX3, among others, in EC, resulting in uncontrolled cell cycle checkpoints and tumor growth. Additionally, PTMs, such as histone acetylation and/or methylation are also deregulated after IR treatment to promote cancer progression and development. Accordingly, there is a dynamic balance between tumor suppressor genes and oncogenes, resulting in lower apoptosis and higher cell proliferation and consequently, poor RT response. Abbreviations: Ac—acetylation; CDKN1C—cyclin dependent kinase inhibitor 1C; DNMTs—DNA methyltransferases; HDAC—histone deacetylases; IR—ionizing radiation; RT—radiotherapy; RUNX3—runt related transcription factor 3; TP53—tumor protein p53.
Indeed, decreased global DNA methylation was reported in tumor cells after IR. Moreover, in the course of DNA repair induced IR, polymerases are not able to incorporate methylcytosine during DNA synthesis, but rather cytosine [81]. Thus, a global DNA hypomethylation has been observed after IR and was associated with oncogene transcription activation and consequently carcinogenesis [81].
In EC, increased methylation levels of specific genes, such as TP73 (tumor protein p53), CDKN1C (cyclin dependent kinase inhibitor 1C) and RUNX3 (runt related transcription factor 3), among others, were significantly correlated with poor radiation response, resulting in uncontrolled cell growth [82]. RUNX3, a tumor suppressor mediating TGF-β-dependent apoptosis pathway was reported to be hypermethylated and downregulated in radioresistant EC cells. Thus, the assessment of respective expression and methylation levels in pretreatment samples might be useful for predicting ESCC radiosensitivity [83].
Furthermore, some histone PTMs may also affect cellular response to external IR stimuli. Indeed, global histone hypoacetylation, through increased HDAC activity and decreased H3K4me3 transcriptional activation-associated marker, were already associated with radioresistance [84,85]. Conversely, increased H3K9me3, a transcriptional repressive-associated marker [85], paralleling DNA methylation studies, was also associated with therapy resistance and tumor progression.
Despite increasing evidence of an association between epigenetic changes and radiosensitivity, there are no epigenetic biomarkers used in clinical practice for determining RT response. Some epigenetic drugs that might be used as radiosensitizers (such as DNMTi or HDACi, among others), are still being evaluated, although recent studies suggest their increased efficacy under hypoxic conditions.
4. Emerging Novel Therapeutic Targets through Epigenetic Chromatin Modulation under Hypoxia
Interestingly, the development of new therapeutic strategies targeting epigenetic effectors is progressively contributing to the discovery of novel molecular biomarkers [86]. Although several epigenetic targeting drugs, which reverse aberrant DNA methylation and histone onco-modifications, have been approved for clinical use [86] (Table 1), new epigenetic-based biomarkers, predictive of response to RT, and consequently, of radiosensitization of hypoxic cells, remain mostly unexplored, particularly in EC.
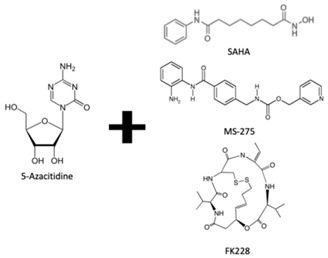
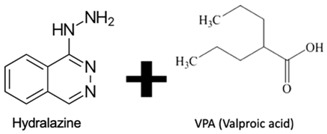
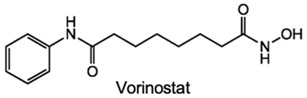
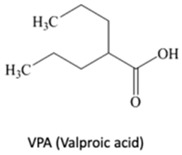
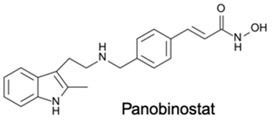
DNA methylation inhibition, using DNMTi, has been emerged as a potential therapeutic strategy in combination with radio- and chemotherapy [87]. Indeed, after IR exposure, significant alterations in DNA methylation status occur [87]. Thus, well established DNMTi, like Azacytidine (AZA) and Decitabine (DAC), might improve therapy response in several cancers through transcriptional reactivation of tumor suppressor genes [88]. Currently, those drugs, which were approved by Food and Drug Administration (FDA) and European medicine agency (EMA) for treatment of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) [86], are being tested in EC. Indeed, Ahrens et al. showed, in vitro, that combination of AZA with HDACi, such as SAHA, MS-275 and FK228, leads to a significant decrease in tumor cell viability [78] (Table 1). Accordingly, clinical trials testing the combination of DNMTi and HDACi to improve chemoradiation patient’s outcome are currently ongoing. With this purpose, Hydralazine plus Valproate or Valproic acid (VPA), an anti-epileptic drug, were used in clinical trials phase II for cervical cancer (NCT00404326) [89]. Herein, patients obtained a complete response to external radiation, thus, supporting further investigation [89]. Moreover, HDAC class I and II inhibitors, such as Vorinostat and VPA in combination with RT showed to increased tumor radiosensitization [90,91]. In particular, a phase I clinical trial with Panobinostat, an epidrug that function as HDAC inhibitor, sensitized prostate, head and neck and Esophageal tumors for external RT (NCT00670553) [90] (Table 1).
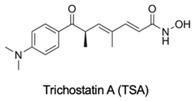
Additionally, Jonsson et al. showed that HDACi treatment-induced radioresistant-associated genes deregulation, including HIF-1α downregulation, in in vitro studies [90]. Also, Trichostatin A (TSA) treatment was found to decrease HIF-1α levels in tumor cells [92]. Nevertheless, the molecular mechanism subjacent to hypoxia and HDACs activity is still lacking [90,93].
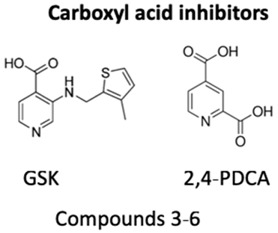
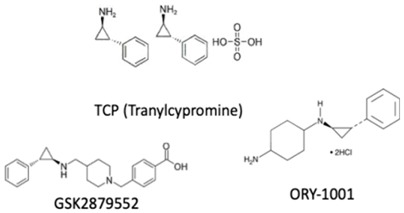
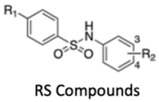
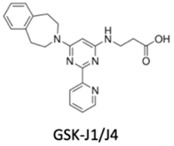
Likewise, histone methylation remodelers upregulated during hypoxia stabilization, were also suggested as targets for cancer therapy [94]. Specifically, hypoxia-induced JmjC-KDMs, which are HIF-1α direct targets, were pinpointed as potential candidates for therapeutic intervention [62,94]. Glioblastoma, characterized by extensive hypoxia were reported to display KDM5 subfamily upregulation, which specific inhibition induced cell cycle arrest and apoptosis associated with improved overall survival rates [95]. KDM4 family members are attractive therapeutic targets for EC, because KDM4C has been strongly implicated in tumor initiation and progression [96]. Remarkably, KDM4 inhibitors, such as carboxylic acid inhibitors, including GSK, compound 3–6 and 2,4-PDCA, demonstrated substantial antiproliferative effects in EC cell lines [96] (Table 1). Therefore, advances in the development of new KDMs inhibitors have been reported to improve cancer survival [97]. In particular, inhibitors targeting demethylating remodelers belonging to LSD1 subgroup, including KDM1A, are currently tested in Phase I clinical trials [97]. In accordance, tranylcypromine (TCP), structurally similar to chemicals previously used in the treatment of other disorders, including Parkinson’s disease, has been tested as KDM1 inhibitor [97] (Table 1). Furthermore, a broad-spectrum of pan KDM inhibitors, most of them 2OG mimic, like NOG, SAHA, IOX1, 2,4-PDCA and JIB-04, have demonstrated significant anti-neoplastic potential [97]. JIB-04 was shown to radiosensitize lung squamous cell carcinoma cell lines by decreasing DNA repair activity after IR therapy [98]. Moreover, specific inhibitory drugs have been developed and are currently in pre-clinical trials, including EPT103182, a KDM5 inhibitor in hematological cancers, such as multiple myeloma, and GSK-J1, a KDM6 inhibitor, in glioma, both with high efficacy against cancer cells [97,99] (Table 1). RS compounds were suggested to increase breast cancer cells sensitivity to RT, affecting DNA repair mechanisms [100] (Table 1). Additionally, KDM6 subfamily inhibitors, such as GSK-J1/J4, also enhanced glioblastoma cells radiosensitivity, as well as, in breast and lung adenocarcinoma cell lines [99] (Table 1).
Table 1.
Epigenetic targeting drugs for clinical use. Current link between hypoxic-dependent modulation and radiotherapeutic improvement.
| Epigenetic Drug | Target Function | Role in EC Treatment (Yes/No) | RT Response Improvement (Yes/No) | Study with Hypoxia Linked (Yes/No) | Other Models | References |
|---|---|---|---|---|---|---|
|
DNMTi combined with HDACi | Yes | Yes | No | - | [78] |
|
DNMTi combined with HDACi | No | Yes | No | Cervical cancer | [89] |
|
HDACi | No | Yes | Yes | Prostate cancer | [90] |
|
HDACi | Yes | Yes | No | - | [91] |
|
HDACi | Yes | Yes | No | Prostate cancer and head and neck tumors | [90] |
|
HDACi | No | No | Yes | - | [92] |
|
KDM4 inhibitors | Yes | No | No | - | [96] |
|
LSD inhibitors | No | No | No | Acute myeloid leukemia (AML) and small cell lung carcinoma (SCLC) | [97] |
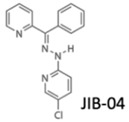
|
KDM5B inhibition (JmjC-KDM PAN inhibitor) | No | Yes | No | Lung cancer | [98] |
| EPT103182 | KDM5 subfamily inhibitor | No | No | No | Multiple myeloma | [97] |
|
KDM5 subfamily inhibitors | No | Yes | No | Breast cancer | [100] |
|
KDM6 subfamily inhibitor | No | Yes | No | Glioblastoma, breast and lung adenocarcinoma | [99] |
1 Abbreviations: AML—Acute myeloid leukemia; DNMTi—DNA methyltransferase inhibitor; EC—Esophageal cancer; GSK—Glycogen synthase; HDACi—Histone deacetylase inhibitor; JmjC-KDM—Jumonji contain c domain histone lysine demethylase; KDM—Histone lysine demethylase; kinase; LSD—Lysine-specific demethylase; PDCA—Pyridinedicarboxylic acid; RT—radiotherapy; SAHA – suberoylanilide hydroxamic acid; TCP—tranylcypromine; TSA—Trichostatin A; VPA—Valproic acid. table
Overall, according to the premise that epigenetic modulation promotes radiation resistance under hypoxic microenvironment, additional pre-clinical and clinical trials are required to confirm the efficacy and safety of new epigenetic drugs that may increase cancer cells’ radiosensitivity, particularly in hypoxic microenvironment.
5. Conclusions
For many years, research in Oncology has been focused on the identification of key targetable molecules among the most relevant signaling pathways, which might allow for the implementation of Precision Medicine. Because RT is the standard of care for treatment of many cancer types, including EC, new complementary therapeutic strategies are needed to overcome radioresistance. Indeed, hypoxic microenvironment, mainly driven by HIF-1α activation, drastically reduces cell radiosensitivity. Furthermore, epigenetic mechanisms involved in the dynamic regulation of chromatin induce a hypoxic microenvironment, triggering a series of cellular processes, including cell cycle alterations, increased migration and invasion, angiogenesis, DNA repair adaptations and cell survival. Therefore, the discovery of new molecular epigenetic targets implicated in hypoxia-responsive pathways seems to be key for improving the management and survival of EC patients.
Author Contributions
C.M.-S., V.M.-G., and C.J. conceptualized the paper. C.M.-S. and V.M.-G. collected, analyzed the information and elaborated the figures. C.M.-S. and V.M.-G. drafted the manuscript, R.H., I.B. and C.J. revised the paper. All authors read and approved the final manuscript.
Funding
This study was partially supported by Research Center IPO-Porto: PI 112-CI-IPOP-92-2018 and ESTIMA-NORTE-01-0145-740 FEDER-000027. VMG received a fellowship from ESTIMA-NORTE-01-0145-740 FEDER-000027: Early-stage cancer treatment, driven by the context of molecular Imaging 741.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Miller K.D., Siegel R.L., Lin C.C., Mariotto A.B., Kramer J.L., Rowland J.H., Stein K.D., Alteri R., Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016;66:271–289. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 3.Napier K.J., Scheerer M., Misra S. Esophageal cancer: A Review of epidemiology, pathogenesis, staging workup and treatment modalities. World. J. Gastrointest. Oncol. 2014;6:112–120. doi: 10.4251/wjgo.v6.i5.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnold M., Soerjomataram I., Ferlay J., Forman D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut. 2015;64:381–387. doi: 10.1136/gutjnl-2014-308124. [DOI] [PubMed] [Google Scholar]
- 5.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D., Bray F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 6.Pickens A., Orringer M.B. Geographical distribution and racial disparity in esophageal cancer. Ann. Thorac. Surg. 2003;76:S1367–S1369. doi: 10.1016/S0003-4975(03)01202-5. [DOI] [PubMed] [Google Scholar]
- 7.Engel L.S., Chow W.H., Vaughan T.L., Gammon M.D., Risch H.A., Stanford J.L., Schoenberg J.B., Mayne S.T., Dubrow R., Rotterdam H., et al. Population attributable risks of esophageal and gastric cancers. J. Natl. Cancer Inst. 2003;95:1404–1413. doi: 10.1093/jnci/djg047. [DOI] [PubMed] [Google Scholar]
- 8.Shah R.D., Cassano A.D., Neifeld J.P. Neoadjuvant therapy for esophageal cancer. World J. Gastrointest. Oncol. 2014;6:403–406. doi: 10.4251/wjgo.v6.i10.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell N.P., Villaflor V.M. Neoadjuvant treatment of esophageal cancer. World J. Gastroenterol. 2010;16:3793–3803. doi: 10.3748/wjg.v16.i30.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koshy M., Greenwald B.D., Hausner P., Krasna M.J., Horiba N., Battafarano R.J., Burrows W., Suntharalingam M. Outcomes after trimodality therapy for esophageal cancer: The impact of histology on failure patterns. Am. J. Clin. Oncol. 2011;34:259–264. doi: 10.1097/COC.0b013e3181e841ce. [DOI] [PubMed] [Google Scholar]
- 11.Wald O., Smaglo B., Mok H., Groth S.S. Future directions in esophageal cancer therapy. Ann. Cardiothorac. Surg. 2017;6:159–166. doi: 10.21037/acs.2017.02.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lovly C.M., Salama A.K., Salgia R. Tumor Heterogeneity and Therapeutic Resistance. Am. Soc. Clin. Oncol. Educ. Book. 2016;35:e585–e593. doi: 10.1200/EDBK_158808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peerlings J., Van De Voorde L., Mitea C., Larue R., Yaromina A., Sandeleanu S., Spiegelberg L., Dubois L., Lambin P., Mottaghy F.M. Hypoxia and hypoxia response-associated molecular markers in esophageal cancer: A systematic review. Methods. 2017;130:51–62. doi: 10.1016/j.ymeth.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Danhier P., Banski P., Payen V.L., Grasso D., Ippolito L., Sonveaux P., Porporato P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017;1858:556–572. doi: 10.1016/j.bbabio.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Biswas S., Rao C.M. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol. Therapeutics. 2017;173:118–134. doi: 10.1016/j.pharmthera.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 16.Jaenisch R., Bird A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33(Suppl. S3):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen M.P., Lee S., Lee Y.M. Epigenetic regulation of hypoxia inducible factor in diseases and therapeutics. Arch. Pharmacal Res. 2013;36:252–263. doi: 10.1007/s12272-013-0058-x. [DOI] [PubMed] [Google Scholar]
- 18.Chi H.C., Tsai C.Y., Tsai M.M., Lin K.H. Impact of DNA and RNA Methylation on Radiobiology and Cancer Progression. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19020555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smits K.M., Melotte V., Niessen H.E., Dubois L., Oberije C., Troost E.G., Starmans M.H., Boutros P.C., Vooijs M., van Engeland M., et al. Epigenetics in radiotherapy: Where are we heading? Radiolther. Oncol. 2014;111:168–177. doi: 10.1016/j.radonc.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Luo Y., Mao Q., Wang X., Yu J., Li M. Radiotherapy for esophageal carcinoma: Dose, response and survival. Cancer Manag. Res. 2018;10:13–21. doi: 10.2147/CMAR.S144687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baskar R., Dai J., Wenlong N., Yeo R., Yeoh K.W. Biological response of cancer cells to radiation treatment. Front. Mol. Biosci. 2014;1:24. doi: 10.3389/fmolb.2014.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrington K., Jankowska P., Hingorani M. Molecular biology for the radiation oncologist: The 5Rs of radiobiology meet the hallmarks of cancer. Clin. Oncol. 2007;19:561–571. doi: 10.1016/j.clon.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Lomax M.E., Folkes L.K., O′Neill P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. 2013;25:578–585. doi: 10.1016/j.clon.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 24.Pawlik T.M., Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004;59:928–942. doi: 10.1016/j.ijrobp.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Yang J., Yue J.B., Liu J., Yu J.M. Repopulation of tumor cells during fractionated radiotherapy and detection methods (Review) Oncol. Lett. 2014;7:1755–1760. doi: 10.3892/ol.2014.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muz B., de la Puente P., Azab F., Azab A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia. 2015;3:83–92. doi: 10.2147/HP.S93413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hall E.J., Giaccia A.J. Radiobiology for the Radiologist. 7th ed. Volume II. Wolters Kluwer Health/Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2012. pp. 432–447. [Google Scholar]
- 28.Kuwahara Y., Roudkenar M.H., Urushihara Y., Saito Y., Tomita K., Roushandeh A.M., Sato T., Kurimasa A., Fukumoto M. Clinically relevant radioresistant cell line: A simple model to understand cancer radioresistance. Med. Mol. Morphol. 2017;50:195–204. doi: 10.1007/s00795-017-0171-x. [DOI] [PubMed] [Google Scholar]
- 29.Lv X., Li J., Zhang C., Hu T., Li S., He S., Yan H., Tan Y., Lei M., Wen M., et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes. Dis. 2017;4:19–24. doi: 10.1016/j.gendis.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al Tameemi W., Dale T.P., Al-Jumaily R.M.K., Forsyth N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019;7:4. doi: 10.3389/fcell.2019.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan D.A., Giaccia A.J. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev. 2007;26:333–339. doi: 10.1007/s10555-007-9063-1. [DOI] [PubMed] [Google Scholar]
- 32.Martin J.D., Fukumura D., Duda D.G., Boucher Y., Jain R.K. Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb. Perspect. Med. 2016:6. doi: 10.1101/cshperspect.a027094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koukourakis M.I., Giatromanolaki A., Skarlatos J., Corti L., Blandamura S., Piazza M., Gatter K.C., Harris A.L. Hypoxia inducible factor (HIF-1a and HIF-2a) expression in early esophageal cancer and response to photodynamic therapy and radiotherapy. Cancer Res. 2001;61:1830–1832. [PubMed] [Google Scholar]
- 34.Jing S.W., Wang J., Xu Q. Expression of hypoxia inducible factor 1 alpha and its clinical significance in esophageal carcinoma: A meta-analysis. Tumour Biol. 2017;39:1010428317717983. doi: 10.1177/1010428317717983. [DOI] [PubMed] [Google Scholar]
- 35.Ogawa K., Chiba I., Morioka T., Shimoji H., Tamaki W., Takamatsu R., Nishimaki T., Yoshimi N., Murayama S. Clinical significance of HIF-1alpha expression in patients with esophageal cancer treated with concurrent chemoradiotherapy. Anticancer Res. 2011;31:2351–2359. [PubMed] [Google Scholar]
- 36.Matsuyama T., Nakanishi K., Hayashi T., Yoshizumi Y., Aiko S., Sugiura Y., Tanimoto T., Uenoyama M., Ozeki Y., Maehara T. Expression of hypoxia-inducible factor-1alpha in esophageal squamous cell carcinoma. Cancer Sci. 2005;96:176–182. doi: 10.1111/j.1349-7006.2005.00025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown J.M., Wilson W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 38.Yang X., Yang B., Cai J., Zhang C., Zhang Q., Xu L., Qin Q., Zhu H., Ma J., Tao G., et al. Berberine enhances radiosensitivity of esophageal squamous cancer by targeting HIF-1alpha in vitro and in vivo. Cancer Biol. 2013;14:1068–1073. doi: 10.4161/cbt.26426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato Y., Yashiro M., Fuyuhiro Y., Kashiwagi S., Matsuoka J., Hirakawa T., Noda S., Aomatsu N., Hasegawa T., Matsuzaki T., et al. Effects of acute and chronic hypoxia on the radiosensitivity of gastric and esophageal cancer cells. Anticancer Res. 2011;31:3369–3375. [PubMed] [Google Scholar]
- 40.Rockwell S., Dobrucki I.T., Kim E.Y., Marrison S.T., Vu V.T. Hypoxia and radiation therapy: Past history, ongoing research, and future promise. Curr. Mol. Med. 2009;9:442–458. doi: 10.2174/156652409788167087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma S., Kelly T.K., Jones P.A. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chew Y.C., Camporeale G., Kothapalli N., Sarath G., Zempleni J. Lysine residues in N-terminal and C-terminal regions of human histone H2A are targets for biotinylation by biotinidase. J. Nutr. Biochem. 2006;17:225–233. doi: 10.1016/j.jnutbio.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 44.Fullgrabe J., Kavanagh E., Joseph B. Histone onco-modifications. Oncogene. 2011;30:3391–3403. doi: 10.1038/onc.2011.121. [DOI] [PubMed] [Google Scholar]
- 45.Peterson C.L., Laniel M.A. Histones and histone modifications. Curr. Biol. 2004;14:R546–R551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 46.Seto E., Yoshida M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014;6:a018713. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thinnes C.C., England K.S., Kawamura A., Chowdhury R., Schofield C.J., Hopkinson R.J. Targeting histone lysine demethylases - progress, challenges, and the future. Biochim. Biophys. Acta. 2014;1839:1416–1432. doi: 10.1016/j.bbagrm.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi Y. Histone lysine demethylases: Emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007;8:829–833. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- 49.Black J.C., Van Rechem C., Whetstine J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell. 2012;48:491–507. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.D′Oto A., Tian Q.W., Davidoff A.M., Yang J. Histone demethylases and their roles in cancer epigenetics. J. Med. Oncol. Ther. 2016;1:34–40. doi: 10.35841/medical-oncology.1.2.34-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tao H., Chen Y.Y., Sun Z.W., Chen H.L., Chen M. Silence of HDAC6 suppressed esophageal squamous cell carcinoma proliferation and migration by disrupting chaperone function of HSP90. J Cell. Biochem. 2018;119:6623–6632. doi: 10.1002/jcb.26841. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Toh Y., Yamamoto M., Endo K., Ikeda Y., Baba H., Kohnoe S., Yonemasu H., Hachitanda Y., Okamura T., Sugimachi K. Histone H4 acetylation and histone deacetylase 1 expression in esophageal squamous cell carcinoma. Oncol. Rep. 2003;10:333–338. doi: 10.3892/or.10.2.333. [DOI] [PubMed] [Google Scholar]
- 53.Dong S.W., Zhang H., Wang B.L., Sun P., Wang Y.G., Zhang P. Effect of the downregulation of SMYD3 expression by RNAi on RIZ1 expression and proliferation of esophageal squamous cell carcinoma. Oncol. Rep. 2014;32:1064–1070. doi: 10.3892/or.2014.3307. [DOI] [PubMed] [Google Scholar]
- 54.Sun L.L., Holowatyj A., Xu X.E., Wu J.Y., Wu Z.Y., Shen J.H., Wang S.H., Li E.M., Yang Z.Q., Xu L.Y. Histone demethylase GASC1, a potential prognostic and predictive marker in esophageal squamous cell carcinoma. Am. J. Cancer Res. 2013;3:509–517. [PMC free article] [PubMed] [Google Scholar]
- 55.Sun X., Qiu J.J., Zhu S., Cao B., Sun L., Li S., Li P., Zhang S., Dong S. Oncogenic features of PHF8 histone demethylase in esophageal squamous cell carcinoma. PLoS ONE. 2013;8:e77353. doi: 10.1371/journal.pone.0077353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai Y., Fu X., Deng Y. Histone demethylase JMJD1C regulates esophageal cancer proliferation Via YAP1 signaling. Am. J. Cancer Res. 2017;7:115–124. [PMC free article] [PubMed] [Google Scholar]
- 57.Kosumi K., Baba Y., Sakamoto A., Ishimoto T., Harada K., Nakamura K., Kurashige J., Hiyoshi Y., Iwatsuki M., Iwagami S., et al. Lysine-specific demethylase-1 contributes to malignant behavior by regulation of invasive activity and metabolic shift in esophageal cancer. Int. J. Cancer. 2016;138:428–439. doi: 10.1002/ijc.29714. [DOI] [PubMed] [Google Scholar]
- 58.Yu Y., Wang B., Zhang K., Lei Z., Guo Y., Xiao H., Wang J., Fan L., Lan C., Wei Y., et al. High expression of lysine-specific demethylase 1 correlates with poor prognosis of patients with esophageal squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2013;437:192–198. doi: 10.1016/j.bbrc.2013.05.123. [DOI] [PubMed] [Google Scholar]
- 59.Kulis M., Esteller M. DNA methylation and cancer. Adv. Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 60.Gaur P., Hunt C.R., Pandita T.K. Emerging therapeutic targets in esophageal adenocarcinoma. Oncotarget. 2016;7:48644–48655. doi: 10.18632/oncotarget.8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaz A.M., Grady W.M. Epigenetic biomarkers in esophageal cancer. Cancer Lett. 2014;342:193–199. doi: 10.1016/j.canlet.2012.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perez-Perri J.I., Acevedo J.M., Wappner P. Epigenetics: New questions on the response to hypoxia. Int. J. Mol. Sci. 2011;12:4705–4721. doi: 10.3390/ijms12074705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsai Y.P., Wu K.J. Epigenetic regulation of hypoxia-responsive gene expression: Focusing on chromatin and DNA modifications. Int. J. Cancer. 2014;134:249–256. doi: 10.1002/ijc.28190. [DOI] [PubMed] [Google Scholar]
- 64.Skowronski K., Dubey S., Rodenhiser D., Coomber B. Ischemia dysregulates DNA methyltransferases and p16INK4a methylation in human colorectal cancer cells. Epigenetics. 2010;5:547–556. doi: 10.4161/epi.5.6.12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen H.F., Wu K.J. Epigenetics, TET proteins, and hypoxia in epithelial-mesenchymal transition and tumorigenesis. Biomedicine. 2016;6:1–8. doi: 10.7603/s40681-016-0001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsai Y.P., Chen H.F., Chen S.Y., Cheng W.C., Wang H.W., Shen Z.J., Song C., Teng S.C., He C., Wu K.J. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 2014;15:13. doi: 10.1186/s13059-014-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim M.S., Kwon H.J., Lee Y.M., Baek J.H., Jang J.E., Lee S.W., Moon E.J., Kim H.S., Lee S.K., Chung H.Y., et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001;7:437–443. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 68.Vavilala D.T., Ponnaluri V.K., Vadlapatla R.K., Pal D., Mitra A.K., Mukherji M. Honokiol inhibits HIF pathway and hypoxia-induced expression of histone lysine demethylases. Biochem. Biophys. Res. Commun. 2012;422:369–374. doi: 10.1016/j.bbrc.2012.04.143. [DOI] [PubMed] [Google Scholar]
- 69.Salminen A., Kaarniranta K., Kauppinen A. Hypoxia-Inducible Histone Lysine Demethylases: Impact on the Aging Process and Age-Related Diseases. Aging Dis. 2016;7:180–200. doi: 10.14336/AD.2015.0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hancock R.L., Dunne K., Walport L.J., Flashman E., Kawamura A. Epigenetic regulation by histone demethylases in hypoxia. Epigenomics. 2015;7:791–811. doi: 10.2217/epi.15.24. [DOI] [PubMed] [Google Scholar]
- 71.Ikeda S., Kitadate A., Abe F., Takahashi N., Tagawa H. Hypoxia-inducible KDM3A addiction in multiple myeloma. Blood Adv. 2018;2:323–334. doi: 10.1182/bloodadvances.2017008847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chakraborty D., Cui W., Rosario G.X., Scott R.L., Dhakal P., Renaud S.J., Tachibana M., Rumi M.A., Mason C.W., Krieg A.J., et al. HIF-KDM3A-MMP12 regulatory circuit ensures trophoblast plasticity and placental adaptations to hypoxia. Proc. Natl. Acad. Sci. USA. 2016;113:E7212–E7221. doi: 10.1073/pnas.1612626113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Osawa T., Tsuchida R., Muramatsu M., Shimamura T., Wang F., Suehiro J., Kanki Y., Wada Y., Yuasa Y., Aburatani H., et al. Inhibition of histone demethylase JMJD1A improves anti-angiogenic therapy and reduces tumor-associated macrophages. Cancer Res. 2013;73:3019–3028. doi: 10.1158/0008-5472.CAN-12-3231. [DOI] [PubMed] [Google Scholar]
- 74.Luo W., Chang R., Zhong J., Pandey A., Semenza G.L. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc. Natl. Acad. Sci. USA. 2012;109:E3367–E3376. doi: 10.1073/pnas.1217394109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Berry W.L., Janknecht R. KDM4/JMJD2 histone demethylases: Epigenetic regulators in cancer cells. Cancer Res. 2013;73:2936–2942. doi: 10.1158/0008-5472.CAN-12-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee H.Y., Choi K., Oh H., Park Y.K., Park H. HIF-1-dependent induction of Jumonji domain-containing protein (JMJD) 3 under hypoxic conditions. Mol. Cells. 2014;37:43–50. doi: 10.14348/molcells.2014.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo X., Tian Z., Wang X., Pan S., Huang W., Shen Y., Gui Y., Duan X., Cai Z. Regulation of histone demethylase KDM6B by hypoxia-inducible factor-2alpha. Acta Biochim. Biophys. Sin. 2015;47:106–113. doi: 10.1093/abbs/gmu122. [DOI] [PubMed] [Google Scholar]
- 78.Ahrens T.D., Timme S., Hoeppner J., Ostendorp J., Hembach S., Follo M., Hopt U.T., Werner M., Busch H., Boerries M., et al. Selective inhibition of esophageal cancer cells by combination of HDAC inhibitors and Azacytidine. Epigenetics. 2015;10:431–445. doi: 10.1080/15592294.2015.1039216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beyer S., Kristensen M.M., Jensen K.S., Johansen J.V., Staller P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 2008;283:36542–36552. doi: 10.1074/jbc.M804578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu X., Wang Y., Tan L., Fu X. The pivotal role of DNA methylation in the radio-sensitivity of tumor radiotherapy. Cancer Med. 2018;7:3812–3819. doi: 10.1002/cam4.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miousse I.R., Kutanzi K.R., Koturbash I. Effects of ionizing radiation on DNA methylation: From experimental biology to clinical applications. Int. J. Radiat. Biol. 2017;93:457–469. doi: 10.1080/09553002.2017.1287454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hamilton J.P., Sato F., Greenwald B.D., Suntharalingam M., Krasna M.J., Edelman M.J., Doyle A., Berki A.T., Abraham J.M., Mori Y., et al. Promoter methylation and response to chemotherapy and radiation in esophageal cancer. Clin. Gastroenterol. Hepatol. 2006;4:701–708. doi: 10.1016/j.cgh.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 83.Sakakura C., Miyagawa K., Fukuda K.I., Nakashima S., Yoshikawa T., Kin S., Nakase Y., Ida H., Yazumi S., Yamagishi H., et al. Frequent silencing of RUNX3 in esophageal squamous cell carcinomas is associated with radioresistance and poor prognosis. Oncogene. 2007;26:5927–5938. doi: 10.1038/sj.onc.1210403. [DOI] [PubMed] [Google Scholar]
- 84.Maroschik B., Gurtler A., Kramer A., Rossler U., Gomolka M., Hornhardt S., Mortl S., Friedl A.A. Radiation-induced alterations of histone post-translational modification levels in lymphoblastoid cell lines. Radiat. Oncol. 2014;9:15. doi: 10.1186/1748-717X-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang P., Yuan D., Guo F., Chen X., Zhu L., Zhang H., Wang C., Shao C. Chromatin remodeling modulates radiosensitivity of the daughter cells derived from cell population exposed to low- and high-LET irradiation. Oncotarget. 2017;8:52823–52836. doi: 10.18632/oncotarget.17275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ahuja N., Sharma A.R., Baylin S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016;67:73–89. doi: 10.1146/annurev-med-111314-035900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim J.G., Park M.T., Heo K., Yang K.M., Yi J.M. Epigenetics meets radiation biology as a new approach in cancer treatment. Int. J. Mol. Sci. 2013;14:15059–15073. doi: 10.3390/ijms140715059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Subramaniam D., Thombre R., Dhar A., Anant S. DNA methyltransferases: A novel target for prevention and therapy. Front. Oncol. 2014;4:80. doi: 10.3389/fonc.2014.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Oronsky B., Scicinski J., Kim M.M., Cabrales P., Salacz M.E., Carter C.A., Oronsky N., Lybeck H., Lybeck M., Larson C., et al. Turning on the Radio: Epigenetic Inhibitors as Potential Radiopriming Agents. Biomolecules. 2016;6 doi: 10.3390/biom6030032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jonsson M., Ragnum H.B., Julin C.H., Yeramian A., Clancy T., Frikstad K.M., Seierstad T., Stokke T., Matias-Guiu X., Ree A.H., et al. Hypoxia-independent gene expression signature associated with radiosensitisation of prostate cancer cell lines by histone deacetylase inhibition. Br. J. Cancer. 2016;115:929–939. doi: 10.1038/bjc.2016.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Makita N., Ninomiya I., Tsukada T., Okamoto K., Harada S., Nakanuma S., Sakai S., Makino I., Kinoshita J., Hayashi H., et al. Inhibitory effects of valproic acid in DNA double-strand break repair after irradiation in esophageal squamous carcinoma cells. Oncol. Rep. 2015;34:1185–1192. doi: 10.3892/or.2015.4089. [DOI] [PubMed] [Google Scholar]
- 92.Fath D.M., Kong X., Liang D., Lin Z., Chou A., Jiang Y., Fang J., Caro J., Sang N. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. J. Biol. Chem. 2006;281:13612–13619. doi: 10.1074/jbc.M600456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen S.Y., Sang N.L. Histone Deacetylase Inhibitors: The Epigenetic Therapeutics That Repress Hypoxia-Inducible Factors. J. Biomed. Biotechnol. 2011;2011 doi: 10.1155/2011/197946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Martinez E.D., Gazdar A.F. Inhibiting the Jumonji family: A potential new clinical approach to targeting aberrant epigenetic mechanisms. Epigenomics. 2016;8:313–316. doi: 10.2217/epi.15.115. [DOI] [PubMed] [Google Scholar]
- 95.Banelli B., Daga A., Forlani A., Allemanni G., Marubbi D., Pistillo M.P., Profumo A., Romani M. Small molecules targeting histone demethylase genes (KDMs) inhibit growth of temozolomide-resistant glioblastoma cells. Oncotarget. 2017;8:34896–34910. doi: 10.18632/oncotarget.16820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen Y.K., Bonaldi T., Cuomo A., Del Rosario J.R., Hosfield D.J., Kanouni T., Kao S.C., Lai C., Lobo N.A., Matuszkiewicz J., et al. Design of KDM4 Inhibitors with Antiproliferative Effects in Cancer Models. ACS Med. Chem. Lett. 2017;8:869–874. doi: 10.1021/acsmedchemlett.7b00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maes T., Carceller E., Salas J., Ortega A., Buesa C. Advances in the development of histone lysine demethylase inhibitors. Curr. Opin. Pharmacol. 2015;23:52–60. doi: 10.1016/j.coph.2015.05.009. [DOI] [PubMed] [Google Scholar]
- 98.Bayo J., Tran T.A., Wang L., Pena-Llopis S., Das A.K., Martinez E.D. Jumonji Inhibitors Overcome Radioresistance in Cancer through Changes in H3K4 Methylation at Double-Strand Breaks. Cell Rep. 2018;25:1040–1050. doi: 10.1016/j.celrep.2018.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rath B.H., Waung I., Camphausen K., Tofilon P.J. Inhibition of the Histone H3K27 Demethylase UTX Enhances Tumor Cell Radiosensitivity. Mol. Cancer Ther. 2018;17:1070–1078. doi: 10.1158/1535-7163.MCT-17-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pippa S., Mannironi C., Licursi V., Bombardi L., Colotti G., Cundari E., Mollica A., Coluccia A., Naccarato V., La Regina G. Small Molecule Inhibitors of KDM5 Histone Demethylases Increase Radio-Sensitivity of Breast Cancer Cells Over-Expressing JARID1B. Molecules. 2019;24:1739. doi: 10.3390/molecules24091739. [DOI] [PMC free article] [PubMed] [Google Scholar]