

Abstract
The relationships between upadacitinib, an oral selective Janus kinase 1 inhibitor, plasma exposures, and its efficacy (assessed by the American College of Rheumatology 20%/50%/70% responses over time) in moderate‐to‐severe active rheumatoid arthritis (RA) were characterized using data from 574 patients, on background methotrexate and inadequate response to methotrexate or anti‐TNF therapy, from two phase II trials conducted with twice‐daily dosing of an immediate‐release formulation. The developed time‐continuous Markov models were used to simulate efficacy of once‐daily (q.d.). regimens of upadacitinib extended‐release incorporating sources of uncertainty. Upadacitinib plasma concentrations associated with 15 and 30 mg extended‐release q.d. doses were predicted to achieve that plateau of response across RA subpopulations. Results from these analyses provided the rationale that supported selection and de‐risked evaluation of upadacitinib extended‐release doses for the first time in >4,000 patients in five large phase III trials.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Upadacitinib is a selective Janus kinase 1 inhibitor evaluated in phase IIb trials in subjects with rheumatoid arthritis (RA) as twice‐daily regimens using the immediate‐release formulation.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The study characterized the relationships between upadacitinib plasma exposures and efficacy measures in subjects with RA using data from two phase IIb trials and provided predictions for the efficacy of once‐daily regimens using the extended‐release formulation.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The presented analyses represent the basis for selection of the 15 and 30 mg doses of upadacitinib extended‐release for evaluation in a subsequent large phase III development program that encompassed >4,000 patients with moderate‐to‐severe RA across five global clinical trials.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ These analyses demonstrate an example of model‐informed drug development, including leveraging exposure–response analyses to support dose selection decisions for phase III clinical trials and to de‐risk and instill efficiency in formulation and regimen changes late in clinical development.
Upadacitinib (ABT‐494) is a selective inhibitor of Janus kinase 1 (JAK1), an intracellular tyrosine kinase that mediates receptor signaling for several inflammatory cytokines.1 The JAKs are a family of four tyrosine kinases (JAK1, 2, and 3, and tyrosine kinase 2) that function as heterodimers or homodimers (for JAK2) to mediate receptor signaling for almost 40 cytokines involved in inflammatory diseases as well as in hematopoiesis and normal immune function.2 Selective inhibition of JAK1 has the potential to offer advantages over nonspecific inhibition of multiple JAKs by reducing inflammation while limiting the unwanted effects on hematopoiesis and normal immune responses.3, 4 Accordingly, upadacitinib has been or is being evaluated in several phase III studies in rheumatoid arthritis (RA),5, 6, 7, 8, 9, 10 psoriatic arthritis,11, 12 giant cell arteritis,13 and Crohn's disease,14, 15 and phase II studies in atopic dermatitis, ulcerative colitis, and ankylosing spondylitis.16, 17, 18
Upadacitinib was evaluated in two phase IIb studies in patients with RA who were inadequate responders to anti‐TNF (BALANCE‐I) or methotrexate (MTX; BALANCE‐II) therapies.19, 20 The immediate‐release formulation of upadacitinib was evaluated in these studies, with doses ranging from 3−18 mg twice‐daily (b.i.d.), in addition to a 24 mg once‐daily (q.d.) dose in BALANCE‐II. Treatment response, measured as the percentage of patients meeting American College of Rheumatology 20% improvement criteria (ACR20) at week 12, was higher for all doses of upadacitinib compared with placebo. The efficacy of upadacitinib was also demonstrated on more stringent end points, including ACR50, ACR70, low disease activity, and remission based on disease activity score 28 based on C‐reactive protein (DAS28‐CRP) criteria.
Upadacitinib effective half‐life is suited for twice‐daily administration with immediate‐release formulations.21, 22 In order to enhance patient compliance and provide a more convenient q.d. dosing regimen in phase III trials and beyond, an extended‐release tablet formulation of upadacitinib was developed.23 However, the efficacy of the extended‐release regimens was not evaluated prior to phase III. The exposure–response analyses reported herein for BALANCE‐I and BALANCE‐II were conducted to (i) characterize the exposure–response relationships for the effects of upadacitinib on ACR responses and (ii) to use these established relationships to predict the efficacy for q.d. regimens of upadacitinib extended‐release formulation in order to support phase III dose selection and de‐risk the formulation bridging.
Results
Upadacitinib plasma concentrations and efficacy data from 574 patients were analyzed. A summary of the demographics and baseline characteristics of the patients in each study is provided in Table 1.19, 20 In BALANCE I and II, baseline mean DAS28‐CRP was 5.77 and 5.66, baseline high sensitivity CRP (hsCRP) was 13.4 and 13.3 mg/L, and disease duration was 12 and 7 years, respectively.
Table 1.
Summary of patient demographics and baseline disease characteristics
| Study | BALANCE Ia N = 276 | BALANCE IIa N = 298b |
|---|---|---|
| Sex, n (%) | ||
| Male | 55 (20%) | 62 (21%) |
| Female | 221 (80%) | 236 (79%) |
| Race, n (%) | ||
| White | 247 (89%) | 292 (98%) |
| Black | 22 (8%) | 3 (1%) |
| Asian | 3 (1%) | 0 |
| Other | 4 (1%) | 3 (1%) |
| Age, year, mean ± SD (range) | 57 ± 12 (26–88) | 55 ± 12 (19–82) |
| Weight, kg, mean ± SD (range) | 78 ± 17 (42–134) | 75 ± 14 (44–122) |
| MTX use, n (%) | 276 (100%) | 298 (100%) |
| Baseline CRP, mg/L, mean ± SD (range) | 13.4 ± 18.6 (0.08–135) | 13.3 ± 18.2 (0.07–149) |
| Baseline DAS28‐CRP, mean ± SD (range) | 5.77 ± 0.92 (3.58–7.97) | 5.66 ± 0.99 (3–8.25) |
| Baseline RF status, n (%) | ||
| Positive | 230 (83%) | 260 (87%) |
| Negative | 46 (17%) | 38 (13%) |
| Baseline TJC, mean ± SD (range) | 15.8 ± 6.9 (2–28) | 15.8 ± 7.2 (0–28) |
| Baseline SJC, mean ± SD (range) | 12.2 ± 5.8 (0–28) | 11.9 ± 5.8 (2–28) |
| Baseline anti‐CCP status, n (%) | ||
| Positive | 230 (83%) | 251 (84%) |
| Negative | 46 (17%) | 47 (16%) |
| Prior biologics use, n (%) | ||
| Yes | 276 (100%) | 0 |
| No | 0 | 298 (100%) |
CCP, cyclic citrullinated peptide; CRP, C‐reactive protein; DAS28‐CRP, disease activity score 28 based on CRP; MTX, methotrexate; RA, rheumatoid arthritis; RF, rheumatoid factor; SJC, swollen joint count; TJC, tender joint count.
aIn BALANCE I, there were 55 patients in each upadacitinib dose group and 56 patients in the placebo group. In BALANCE II, there were 50 patients in the 3, 6, and 12 mg b.i.d. dose groups, 49 patients each in the 18 mg b.i.d. and 24 mg q.d. dose groups, and 50 patients in the placebo group. bA total of 300 patients were enrolled in the study and 298 were included in the exposure–response analyses. One patient in the 18 mg b.i.d. dose group and one patient in the 24 mg q.d. dose group were excluded from the analyses because their American College of Rheumatology data were insufficient to clearly define a Markov state for one or more study visits.
Upadacitinib exposure–response relationships for effects on ACR responses
Upadacitinib plasma exposures were dose proportional within the range of doses evaluated in the studies. A summary of upadacitinib model‐estimated average plasma concentrations over a dosing interval (Cave) is provided in Table 2. Overall, upadacitinib plasma exposures were consistent with prior pharmacokinetic evaluations of upadacitinib in subjects with RA.22
Table 2.
Upadacitinib estimated average plasma concentrations over a dosing interval for immediate‐release regimens in BALANCE I and BALANCE II
| Upadacitinib dose | Upadacitinib Cave, ng/mL, median (5th–95th percentile) | |
|---|---|---|
| BALANCE I (Anti‐TNF‐inadequate responders) | BALANCE II (MTX‐inadequate responders) | |
| 3 mg b.i.d. | 9.2 (5.3–15.1) | 9.7 (6.3–14.6) |
| 6 mg b.i.d. | 18.4 (11.9–34.6) | 19.7 (12.7–29.9) |
| 12 mg b.i.d. | 39 (24.4–51) | 40.5 (26.7–68.5) |
| 18 mg b.i.d. | 51.2 (33.1–92.3) | 59.8 (40.7–79.8) |
| 24 mg q.d. | – | 35.9 (23.9–53.2) |
b.i.d., twice daily; Cave, average plasma concentration; q.d., once daily.
The relationships between upadacitinib plasma concentrations and the ACR response were analyzed using time‐continuous Markov modeling approach within which a patient may transition between the different states of no‐response, ACR20, ACR50, and ACR70, and can drop out (i.e., prematurely discontinue participation in the trial) from any of the aforementioned response states (Figure 1). A separate model was developed for the anti‐TNF‐inadequate and MTX‐inadequate populations. In the models, upadacitinib enhanced transition to a higher response state with a maximum effect (Emax) relationship between upadacitinib plasma concentration and effect on the transition rates. Different measures of upadacitinib exposures (time course of plasma concentrations (Cp), average plasma concentrations over a dosing interval or trough plasma concentrations) were evaluated in the exposure–response models, and Cp was included in the final model based on the diagnostic and simulation‐based model‐selection criteria. Summary of the parameter estimates of the Markov models are presented in Tables S1 and S2 . The point estimate for upadacitinib plasma concentrations associated with 50% of the maximal effect (EC50) on the transition rates to higher ACR responses was numerically higher for anti‐TNF‐inadequate responders (point estimate of 12.2 ng/mL; % relative standard error (RSE) of 73%) compared with MTX‐inadequate responders (point estimate of 3 ng/mL; % RSE of 104%); however, the confidence intervals were highly overlapping. Baseline hsCRP was a statistically significant covariate for upadacitinib Emax in the anti‐TNF‐inadequate responder population, with higher estimated Emax in patients with higher baseline hsCRP. Significant covariates of the exposure‐ACR model for MTX‐inadequate responders were found to be sex, baseline anti–cyclic citrullinated peptide (CCP) status, and baseline DAS28‐CRP on the model parameters characterizing placebo response, but not on upadacitinib effect. None of the evaluated covariates had a statistically significant effect on upadacitinib effect parameters in MTX‐inadequate responders.
Figure 1.
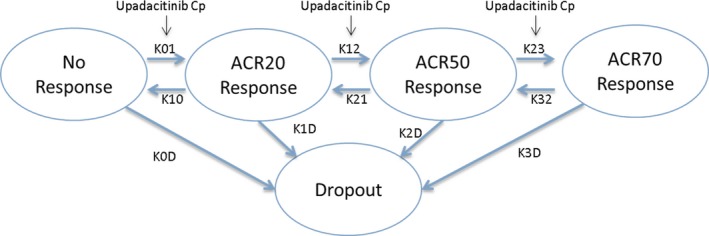
Schematic for Markov model analysis of the relationship between upadacitinib plasma exposures and American College of Rheumatology (ACR) responses. K01, K12, and K23 represent transition rates of the status of patients to higher levels of response. K10, K21, and K32 represent transition rates of the status of patients to lower levels of response. Model parameters K0D, K1D, K2D, and K3D represent transition of patients from different response states to dropout. Cp represents the upadacitinib plasma concentration. ACR20/50/70, American College of Rheumatology 20%/50%/70% improvement criteria. [Colour figure can be viewed at http://www.wileyonlinelibrary.com]
The time course of the observed and model‐simulated ACR20, ACR50, and ACR70 responses for placebo and each upadacitinib dose are shown in the plots in Figure 2. The proportions of anti‐TNF‐inadequate responders and MTX‐inadequate responders with ACR responses during the 12 weeks of treatment were well described by the Markov model. The percentage of patients achieving ACR20, ACR50, and ACR70 responses increased with increasing upadacitinib plasma exposures, with maximal efficacy estimated to be achieved at exposures corresponding to the 6 mg to 12 mg b.i.d. doses of the immediate‐release formulation (Table 2 ).
Figure 2.
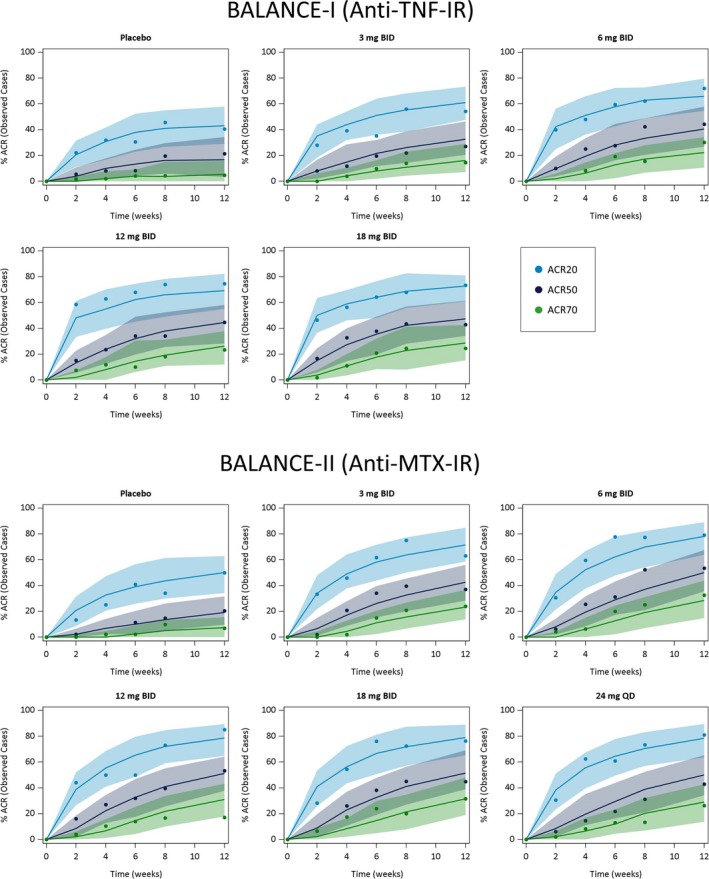
Observed and model‐predicted American College of Rheumatology 20%/50%/70% improvement criteria (ACR20/50/70) responses (observed cases) vs. time stratified by dose and patient population. Symbols represent the observed time course of the percentage of patients achieving ACR20, (blue) ACR50, (black) and ACR70 (green) responses. Solid lines and shaded areas represent the exposure–response model‐predicted median and 90% prediction intervals, respectively. IR, inadequate responder; MTX, methotrexate.
Simulations for the efficacy of the phase III extended‐release regimens compared with the efficacy of the phase II immediate‐release regimens
Simulated median and 90% prediction intervals for week 12 ACR responses for the 15 and 30 mg q.d. doses of the extended‐release formulation compared with b.i.d. doses of the immediate‐release formulation are shown in Figure 3. The model‐predicted median ACR 20/50/70 responses at week 12 for the extended‐release formulation were 57/34/18 and 69/43/25 for the 15 mg q.d. dose and 62/39/23 and 71/46/28 for the 30 mg q.d. dose in anti‐TNF and MTX‐inadequate responders, respectively. The corresponding predicted responses for placebo were 34/12/3 and 47/18/8, respectively, based on the phase II data.
Figure 3.
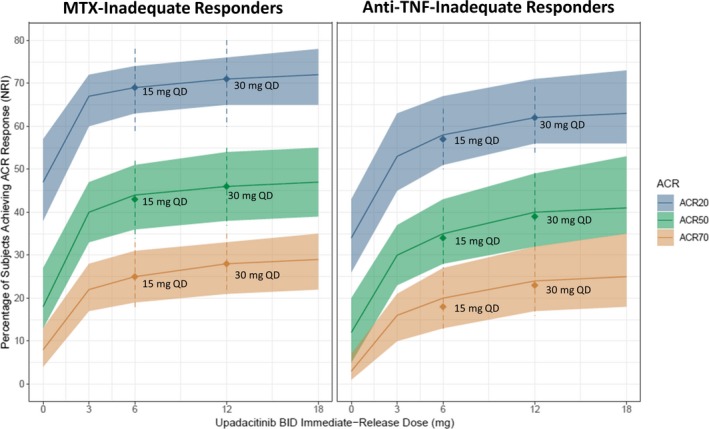
Simulated American College of Rheumatology (ACR) responses (nonresponder imputation (NRI)) at week 12 for the immediate‐release twice daily (b.i.d.) and extended‐release once daily (q.d.) dosing regimens. Lines and shaded areas represent the median and 90% prediction intervals for the immediate‐release 3, 6, 12, and 18 mg b.i.d. dosing regimens in the BALANCE I (anti‐TNF‐inadequate responders (IRs)) and II methotrexate (MTX‐IRs) studies. Symbols and dashed lines represent the simulated median and 90% prediction intervals for the extended‐release 15 and 30 mg q.d. dosing regimens.
The 15 mg q.d. dose of the extended‐release formulation was predicted to provide near maximal efficacy, similar to that achieved with the 6 mg b.i.d. dose of the immediate‐release formulation. The 30 mg q.d. dose of the extended‐release formulation was predicted to provide 4–6% (in anti‐TNF‐ inadequate responder) and 2–3% (in MTX‐inadequate responder) higher ACR responses than 15 mg q.d.
Comparison of the observed ACR responses in phase III trials for upadacitinib 15 mg and 30 mg q.d. (extended‐release) to the model‐simulated ACR responses prior to phase III
The model‐simulated ACR responses (difference from placebo) for 15 mg q.d. and 30 mg q.d. are presented in Figure 4 overlaid with the observed ACR responses in phase III trials SELECT‐NEXT (5) and SELECT‐COMPARE (8) in MTX‐inadequate responder/conventional synthetic disease‐modifying antirheumatic drugs (csDMARDs) patients and SELECT‐BEYOND (6) in anti‐TNF‐inadequate responder/biologics‐inadequate responder patients, which recently have become available. Overall, the efficacy observed in phase III trials for upadacitinib 15 mg q.d. and 30 mg q.d. is consistent with the predicted efficacy based on exposure–response analyses of phase II trials.
Figure 4.
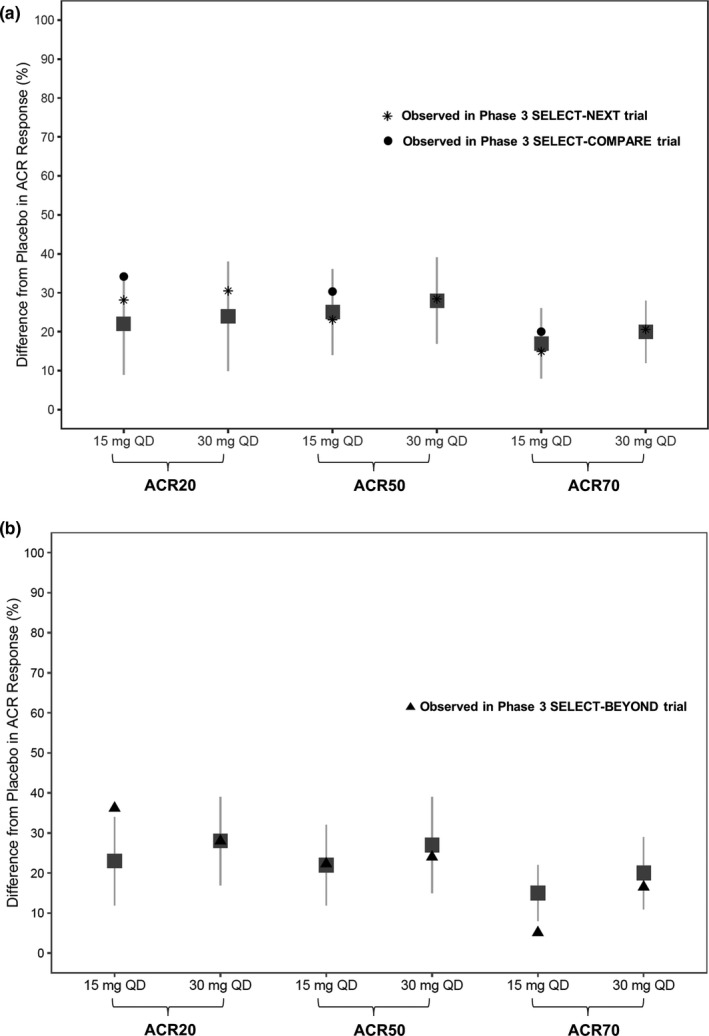
Simulated and observed American College of Rheumatology (ACR) responses (nonresponder imputation (NRI)) at week 12 in (a) methotrexate (MTX)‐inadequate responders (IRs)/conventional synthetic disease‐modifying antirheumatic drugs (csDMARD)‐IR and (b) anti‐TNF‐IR/biologics‐IR patients in phase III trials. Gray symbols and error bars represent the simulated median and 90% prediction intervals for 15 mg and 30 mg q.d. extended‐release regimens based on exposure–response analyses of the phase II studies BALANCE I and II. Black symbols represent the observed responses in phase III trials. q.d., once daily.
DISCUSSION
Exposure–response modeling is a powerful tool to characterize the relationship between drug exposures (rather than dose) and response, taking into account the different sources of variability (e.g., the time course of the plasma exposures, between‐subject differences in the pharmacokinetics, subject characteristics, between‐subject differences in response, and uncertainty in the estimates of pharmacokinetic and pharmacodynamic parameters).24 This enables use of data from certain evaluated trial designs or dosing regimens to predict efficacy of new dosing regimens or trial designs. In the present analyses, we characterized the exposure–response relationships for upadacitinib for effects on ACR20, ACR50, and ACR70 responses in patients with RA based on the phase IIb studies BALANCE I and II and utilized the exposure–response modeling to predict efficacy for a new formulation. These analyses enabled and de‐risked advancing upadacitinib q.d. regimens of the extended‐release formulation into six global phase III RA studies without the need for prior evaluation of these regimens in subjects with RA.
In the presented analyses, plasma concentrations of upadacitinib had a significant relationship with the rates of improvement in ACR response state for patients with RA; higher plasma concentrations were associated with faster transition from a lower to a higher ACR response state (e.g., from ACR20 to ACR50). Upadacitinib estimated EC50 in patients with an inadequate response to anti‐TNF treatment was numerically higher than the EC50 in patients with an inadequate response to MTX therapy; however, the RSE for the estimates indicated that the difference in the EC50 estimate between populations was not statistically significant with the available phase II data. Based on exposure–response analyses of BALANCE I and II studies, exposures corresponding to the 6 mg b.i.d. to 12 mg b.i.d. of the immediate‐release formulation of upadacitinib (corresponding to steady‐state Cave of 20–40 ng/mL) were predicted to maximize efficacy in patients with RA.
Upadacitinib extended‐release formulation was developed after the completion of the phase IIb studies with the goal of providing a more convenient dosing regimen for patients in phase III trials through q.d. administration. The oral bioavailability of the extended‐release formulation was estimated to be 7–80% compared with the same dose of the immediate‐release formulation used in BALANCE I and BALANCE II.23 Based on exposure–response analyses of the phase IIb studies (BALANCE I and II), doses of 15 and 30 mg q.d. of the upadacitinib extended‐release formulation were predicted to maximize efficacy of upadacitinib in RA and have similar ACR20, ACR50, and ACR70 responses to 6 mg b.i.d. and 12 mg b.i.d., respectively, of the immediate‐release formulation (Figure 3). Majority of upadacitinib efficacy was predicted to be achieved with the 15 mg q.d. dose; 30 mg q.d. dose group was included to demonstrate that the plateau of efficacy was achieved in patients with RA. As such, 15 mg and 30 mg q.d. doses of upadacitinib were advanced for evaluation in phase III trials. As shown in Figure 4, recent results from two phase III trials in inadequate responders to conventional synthetic DMARDs and anti‐TNF drugs demonstrate that the observed efficacy for upadacitinib 15 mg and 30 mg q.d. extended‐release are in close agreement with the model‐predicted efficacy based on phase II.19, 20 This confirms the robustness of the exposure–response analyses of the phase IIb trials and that the extended‐release formulation of upadacitinib performed as anticipated in the phase III trials.
The different ACR end points (ACR20, ACR50, and ACR70) and dropout state were analyzed simultaneously in the presented analyses using continuous‐time Markov analyses.25, 26 As previously noted, this approach is more informative than analyzing the different ACR end points separately as the model accounts for the correlation between different ACR states within each patient as well as for the dropout state over time. For example, a subject who is an ACR20 responder at a certain visit is more likely to become an ACR50 responder at the subsequent visit compared with a subject who has not achieved ACR20 response. This approach also utilizes more information than analysis of a single end point at a single time point of interest (e.g., at week 12 only). Additionally, by utilizing data from the full time course and across different ACR states, the analysis is less sensitive to the effect of potentially outlying observations at the time point of interest.25
The models utilized in these analyses differed in some aspects from the Markov model described by Lacroix et al.25 The approach from Lacroix et al.25 included a separate logistic model to describe dropout rather than using a dropout state in the Markov model and assumed an additional time dependency of the placebo transition rates. A model with time dependency of placebo transition rates was also tested in the current work but could not be estimated reliably. The exposure measure used by Lacroix et al.25 was the predicted plasma concentration at time of ACR assessment rather than the full individual plasma concentration time profile. Use of the plasma concentration time profiles as exposure metric was considered appropriate in the current work, as it takes into account subjects’ compliance to the study drug and enables use of the model to simulate efficacy of the extended‐release formulation regimens.
Initially, it was attempted to analyze data from both anti‐TNF and MTX‐inadequate responders simultaneously. However, due to the greater placebo response in MTX‐inadequate responders compared with anti‐TNF‐inadequate responders, different placebo‐response transition rates were needed in the model. This difference is due to anti‐TNF‐inadequate responder patients being more advanced in their disease with longer disease duration, making them less responsive to placebo, compared with MTX‐inadequate responders. Therefore, two separate analyses were conducted, one for anti‐TNF‐inadequate responders (BALANCE I) and another for MTX‐inadequate responders (BALANCE II).
The effects of different baseline characteristics (e.g., age, sex, total body weight, hsCRP, rheumatoid factor (RF) status, and anti‐CCP status) on upadacitinib exposure–response relationships for ACR responses were evaluated within the context of the analyses. Of the covariates evaluated, only baseline hsCRP showed a statistically significant correlation with the upadacitinib exposure–response relationship (higher upadacitinib Emax estimated with higher baseline hsCRP). In a previously reported population pharmacokinetic analysis, creatinine clearance and sex were identified as statistically significant covariates that affect upadacitinib oral clearance.21 Based on population pharmacokinetic analyses of phase I and RA phase II studies, patients with mild or moderate renal impairment were predicted to have 16% and 32% higher upadacitinib area under the curve values compared with patients with normal renal function, and female patients were predicted to have 16% higher upadacitinib area under the curve than male patients. Given that mild or moderate renal impairment results in a small increase (not decrease) in upadacitinib exposures, no decrease in efficacy is expected in patients with mild or moderate renal impairment. The slightly lower upadacitinib exposures in men compared with women is not clinically relevant either (ACR responses are predicted to be <2% different in men vs. women who receive 15 mg q.d. or 30 mg q.d. doses of upadacitinib extended–release formulation, data not shown). Therefore, the current analyses do not suggest that a certain subpopulation of patients with RA may require higher or lower upadacitinib doses compared with the rest of the patient population. Further assessments are warranted of the effects of different covariates on upadacitinib exposures as well as exposure–response relationships using the accumulating larger dataset from phase III trials.
Of note, all patients included in the presented exposure–response analyses received upadacitinib on a background MTX. Therefore, it was not possible to evaluate the effect of background MTX treatment on upadacitinib exposure–response relationships for ACR. Two of the phase III trials for upadacitinib are evaluating the efficacy of upadacitinib monotherapy (without background treatment with conventional synthetic DMARDs) in patients with RA.9, 10 Evaluation of the effect of MTX on the exposure–response relationships in patients with RA as part of phase III analyses is warranted.
In conclusion, exposure–response analyses of data from two phase IIb studies in patients with RA who were inadequate responders to MTX or anti‐TNF therapies demonstrated that extended‐release regimens of 15 mg q.d. and 30 mg q.d. were predicted to maximize efficacy in subjects with RA. The presented analyses were the basis for selection of the 15 and 30 mg doses of upadacitinib extended‐release for evaluation in a subsequent large phase III development program that encompassed >4,000 patients with moderate‐to‐severe RA across five global clinical trials.
METHODS
Participants and design of the studies
The studies were conducted in accordance with Good Clinical Practice Guidelines and the ethical principles that have their origin in the Declaration of Helsinki. The protocols were approved by the institutional review board or ethics committee at each site, and each patient provided written informed consent before any study‐related procedures were performed.
Details of the study designs for the 12‐week, randomized, double‐blind, parallel‐arm, placebo‐controlled, phase IIb BALANCE I (NCT01960855) and II (NCT02066389) studies have been previously described.19, 20 Briefly, men and women 18 years of age or older who were diagnosed with RA for at least 3 months were eligible to enroll into the studies. Eligible patients were required to have moderate‐to‐severe RA, which was defined as having at least six swollen joints (based on a 66‐joint count) and at least six tender joints (based on a 68‐joint count) and either an hsCRP concentration above 5 mg/L or seropositivity for RF and anti‐CCP. Patients must have been receiving a stable dose of oral or parenteral MTX (7.5–25 mg/week) for at least 4 weeks before receiving the first dose of study drug and must not have had prior exposure to a JAK inhibitor. Patients in BALANCE I were inadequate responders to anti‐TNF biologic therapies. Patients in BALANCE II were inadequate responders to MTX therapy and had not received any biologic treatment for RA prior to enrollment.
In BALANCE I, patients were randomized to receive b.i.d. doses of 3, 6, 12, or 18 mg of the immediate‐release formulation of upadacitinib or placebo for 12 weeks. In BALANCE II, patients were randomized to receive 3 mg b.i.d., 6 mg b.i.d., 12 mg b.i.d., 18 mg b.i.d., or 24 mg q.d. of upadacitinib immediate‐release formulation or placebo for 12 weeks. In both studies, patients continued to take their stable dose of background MTX therapy, an oral supplement of folic acid, and stable doses of any other non‐DMARDs throughout the study.
Pharmacokinetic and ACR assessments
Blood samples for determination of upadacitinib plasma concentrations were collected from each patient prior to dosing at weeks 2, 4, 6, 8, and 12 in each study. Additional blood samples were collected from ~ 30% of patients at 1, 2, and 3 hours after the morning dose on study day 1 at week 8. Plasma concentrations of upadacitinib were determined at AbbVie (North Chicago, IL) using a validated liquid chromatography method with mass spectrometric detection, as previously described.22
The proportions of patients with 20%, 50%, or 70% improvement in ACR criteria compared with baseline (i.e., ACR20, ACR50, and ACR70 responses) were determined at each study visit, as previously described19, 20 and were used as the efficacy measure for the exposure–response analyses described below.
Exposure–response analyses methodology
A population pharmacokinetic model was developed for upadacitinib using data from phase I and phase II studies, as previously described.21 This pharmacokinetic model was used to generate the full time course of the upadacitinib plasma concentration profile for each patient, which was used as an input for the exposure–response analyses in NONMEM version 7.3 (ICON Development Solutions, Hanover, MD).
The full time‐course data of individual ACR20, ACR50, and ACR70 responses as well as the dropout state for each patient from the BALANCE I and II studies were used to generate the analyses dataset for exposure–response modeling. All patients with at least a baseline and subsequent ACR assessment were included in the dataset. Exposure–response analyses were conducted separately for anti‐TNF and MTX‐inadequate responders due to differences in the placebo responses between the two populations and to allow for potential differences in treatment effect. The relationships between the time‐course of upadacitinib plasma concentrations and ACR20, ACR50, and ACR70 responses and patient dropouts were characterized using a Markov modeling approach similar to Lacroix et al.25 (Figure 1) and as previously utilized for analysis of other RA trials.26 In the Markov models, active treatment with upadacitinib was assumed to enhance the transition of the patients’ status to higher levels of response (e.g., no response to ACR20, ACR20 to ACR50, and ACR50 to ACR70). A transition from each response state to dropout was also allowed in the model.
Upadacitinib effect on the rate of transitions from a low to a high ACR response state was described in the models using Emax functions described as:
where C describes the exposure, Placebo describes the rate of placebo subjects, EC50 the concentration at half‐maximum effect on the transition rate, and Emax the maximum response in Emax model.
Different measures of upadacitinib exposures (Cp, average plasma concentrations over a dosing interval or trough plasma concentrations) were evaluated in the exposure–response models, and the optimal exposure measure was selected for inclusion in the model based on the diagnostic and simulation based model‐selection criteria. The predictive performance of the final exposure–response models was evaluated based on visual predictive checks27, 28 of 100 simulated replicates of the dataset generated using NONMEM and Perl Speaks NONMEM (PSN 4, https://uupharmacometrics.github.io/PsN/index.html, Uppsala University, Uppsala, Sweden). Simulated data were compared with the observed data by superimposing the median of the observed data binned by visit with the 90% prediction bands for these bins from the simulations.
After an appropriate base model was developed, the effect of potential covariates on upadacitinib exposure–response relationships was evaluated. The covariates tested in the Markov model were age, sex, body mass index, total body weight, race, baseline RF status, baseline anti‐CCP status, baseline DAS28‐CRP, baseline hsCRP, baseline MTX dose, both RF and anti‐CCP status being positive, number of prior DMARDs other than MTX (BALANCE II only), duration of RA, baseline tender joint count, baseline tender joint count category, baseline swollen joint count, and baseline swollen joint count category. Additional covariates for BALANCE I included the number of types of prior anti‐TNF use, use of non‐anti‐TNF biologics, and number of types of prior biologics use.
The final exposure–response models were used to predict the efficacy of different doses of upadacitinib in MTX and anti‐TNF‐inadequate responders assuming enrolling 300 patients for each dose. The pharmacokinetic parameters characterizing upadacitinib absorption for the extended‐release formulation were estimated using population pharmacokinetic analysis of data from a phase I study in healthy subjects.23 The estimated extended‐release absorption parameters were incorporated in the pharmacokinetic component of the exposure–response models (developed with immediate‐release formulation data) to predict the efficacy in patients with RA for a range of doses of upadacitinib immediate‐release and extended‐release formulations.
FUNDING
This work was supported by AbbVie.
CONFLICT OF INTEREST
M.‐E. Mohamed, B. Klünder, H. S. Camp, and A. A. Othman are employees of AbbVie and may hold AbbVie stock or stock options.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript. All authors designed the research; all authors performed the research.
DATA SHARING STATEMENT
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis datasets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Supporting information
Table S1. Summary of the Markov ACR model parameter estimates in anti TNF‐IR patients (BALANCE‐I study).
Table S2. Summary of the ACR Markov model parameter estimates in MTX‐IR patients (BALANCE II study).
ACKNOWLEDGMENTS
AbbVie funded this study (https://ClinicalTrials.gov, NCT01960855 and NCT02066389), contributed to the design, and was involved in the collection, analysis, and interpretation of the data and in the writing, review, and approval of the publication.
References
- 1. O'Shea, J.J. , Kontzias, A. , Yamaoka, K. , Tanaka, Y. & Laurence, A. Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 72 (suppl. 2), ii111–ii115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ghoreschi, K. , Laurence, A. & O'Shea, J.J. Janus kinases in immune cell signaling. Immunol. Rev. 228, 273–287 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Norman, P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin. Investig. Drugs 23, 1067–1077 (2014). [DOI] [PubMed] [Google Scholar]
- 4. Baker, K.F. & Isaacs, J.D. Novel therapies for immune‐mediated inflammatory diseases: what can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn's disease and ulcerative colitis? Ann. Rheum. Dis. (2017). 10.1136/annrheumdis-2017-211555. [DOI] [PubMed] [Google Scholar]
- 5. Burmester, G.R. et al Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease‐modifying anti‐rheumatic drugs (SELECT‐NEXT): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet 391, 2503–2512 (2018). [DOI] [PubMed] [Google Scholar]
- 6. Genovese, M.C. et al Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease‐modifying anti‐rheumatic drugs (SELECT‐BEYOND): a double‐blind, randomised controlled phase 3 trial. Lancet 391, 2513–2524 (2018). [DOI] [PubMed] [Google Scholar]
- 7. AbbVie . A phase 3 study to compare ABT‐494 to abatacept in subjects with rheumatoid arthritis on stable dose of conventional synthetic disease‐modifying antirheumatic drugs (csDMARDs) who have an inadequate response or intolerance to biologic DMARDs (SELECT‐CHOICE) [https://ClinicalTrials.gov Identifier NCT03086343]. Available from https://clinicaltrials.gov/ct2/show/NCT03086343. Accessed April 17, 2018.
- 8. Fleischmann, R. et al Upadacitinib versus Placebo or Adalimumab in Patients with Rheumatoid Arthritis and an Inadequate Response to Methotrexate: Results of a Phase 3, Double-Blind, Randomized Controlled Trial. Arthritis Rheumatol. In Press. doi: 10.1002/art.41032 (2019). [DOI] [PubMed] [Google Scholar]
- 9. van Vollenhoven, R. et al A phase 3, randomized, controlled trial comparing upadacitinib monotherapy to MTX monotherapy in MTX‐naïve patients with active rheumatoid arthritis [abstract]. Arthritis Rheum. 70 (suppl. 10). <https://acrabstracts.org/abstract/a-phase-3-randomized-controlled-trial-comparing-upadacitinib-monotherapy-to-mtx-monotherapy-in-mtx-naive-patients-with-active-rheumatoid-arthritis/> (2018). Accessed September 16, 2018. [Google Scholar]
- 10. Smolen, J. et al Upadacitinib versus Placebo or Adalimumab in Patients with Rheumatoid Arthritis and an Inadequate Response to Methotrexate: Results of a Phase 3, Double-Blind, Randomized Controlled Trial [abstract]. Arthritis Rheum. 70 (suppl. 10), (2018). [DOI] [PubMed] [Google Scholar]
- 11. AbbVie . A study comparing ABT‐494 to placebo and to adalimumab in participants with psoriatic arthritis who have an inadequate response to at least one non‐biologic disease modifying anti‐rheumatic drug (SELECT ‐ PsA 1) [https://ClinicalTrials.gov Identifier NCT03104400]. Available from https://clinicaltrials.gov/ct2/show/NCT03104400?term=NCT03104400&rank=1. Accessed January 3, 2018.
- 12. AbbVie . A study comparing ABT‐494 to placebo in participants with active psoriatic arthritis who have a history of inadequate response to at least one biologic disease modifying anti‐rheumatic drug (SELECT ‐ PsA 2) [https://ClinicalTrials.gov Identifier NCT03104374]. Available from https://clinicaltrials.gov/ct2/show/NCT03104374?term=NCT03104374&rank=1. Accessed January 3, 2018.
- 13. AbbVie . A study to evaluate the safety and efficacy of upadacitinib in participants with giant cell arteritis [https://ClinicalTrials.gov Identifier NCT03725202]. Available from https://clinicaltrials.gov/ct2/show/NCT03725202?term=NCT03725202&rank=1. Accessed April 4, 2019.
- 14. AbbVie . A study of the efficacy and safety of upadacitinib (ABT‐494) in subjects with moderately to severely active Crohn's disease who have inadequately responded to or are intolerant to biologic therapy. [https://ClinicalTrials.gov Identifier NCT03345836]. Available from https://clinicaltrials.gov/ct2/show/NCT03345836?term=NCT03345836&rank=1. Accessed April 20, 2018.
- 15. AbbVie . A study of the efficacy and safety of upadacitinib (ABT‐494) in subjects with moderately to severely active Crohn's disease who have inadequately responded to or are intolerant to conventional therapies but have not failed biologic therapy. [https://ClinicalTrials.gov Identifier NCT03345849]. Available from https://clinicaltrials.gov/ct2/show/NCT03345849?term=NCT03345849&rank=1Accessed April 20, 2018.
- 16. AbbVie . A study to evaluate ABT‐494 in adult subjects with moderate to severe atopic dermatitis [https://ClinicalTrials.gov Identifier NCT02925117]. Available from https://clinicaltrials.gov/ct2/show/NCT02925117?term=NCT02925117&rank=1. Accessed January 3, 2018.
- 17. AbbVie . A study to evaluate the safety and efficacy of ABT‐494 for induction and maintenance therapy in subjects with moderately to severely active ulcerative colitis [https://ClinicalTrials.gov Identifier NCT02819635]. Available from https://clinicaltrials.gov/ct2/show/NCT02819635?term=NCT02819635&rank=1. Accessed January 3, 2018.
- 18. AbbVie . A study evaluating the safety and efficacy of upadacitinib in subjects with active ankylosing spondylitis (SELECT Axis 1) [https://ClinicalTrials.gov Identifier NCT03178487]. Available from https://clinicaltrials.gov/ct2/show/NCT03178487?term=NCT03178487&rank=1. Accessed January 3, 2018.
- 19. Kremer, J.M. et al A phase IIb study of ABT‐494, a selective JAK‐1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti‐tumor necrosis factor therapy. Arthritis Rheum. 68, 2867–2877 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Genovese, M.C. et al Efficacy and safety of ABT‐494, a selective JAK‐1 inhibitor, in a phase IIb study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheum. 68, 2857–2866 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klunder, B. , Mohamed, M.F. & Othman, A.A. Population pharmacokinetics of upadacitinib in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I and II clinical trials. Clin. Pharmacokinet. 57, 977–988 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mohamed, M.F. , Camp, H.S. , Jiang, P. , Padley, R.J. , Asatryan, A. & Othman, A.A. Pharmacokinetics, safety and tolerability of ABT‐494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin. Pharmacokinet. 55, 1547–1558 (2016). [DOI] [PubMed] [Google Scholar]
- 23. Mohamed, M.F. , Zeng, J. , Marroum, P.J. , Song, I.H. & Othman, A.A. Pharmacokinetics of upadacitinib with the clinical regimens of the extended‐release formulation utilized in rheumatoid arthritis phase 3 trials. Clin. Pharmacol. Drug Dev. 8, 208–216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lovern, M. , Sargentini‐Maier, M.L. , Otoul, C. & Watelet, J.B. Population pharmacokinetic and pharmacodynamic analysis in allergic diseases. Drug Metab. Rev. 41, 475–485 (2009). [DOI] [PubMed] [Google Scholar]
- 25. Lacroix, B.D. , Karlsson, M.O. & Friberg, L.E. Simultaneous exposure‐response modeling of ACR25, ACR25, and ACR25 improvement scores in rheumatoid arthritis patients treated with certolizumab pegol. CPT Pharmacometrics Syst. Pharmacol. 3, e143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khatri, A. , Klunder, B. , Peloso, P.M. & Othman, A.A. Exposure‐response analyses demonstrate no evidence of interleukin 17A contribution to efficacy of ABT‐122 in rheumatoid or psoriatic arthritis. Rheumatology (Oxford) 58, 352–360 (2019). [DOI] [PubMed] [Google Scholar]
- 27. Karlsson, M.O. & Holford, N. A tutorial on visual predictive checks. Page 17 Abstract 1434. <https://www.page-meeting.org/default.asp?abstract=1434>. (2008).
- 28. Holford, N. The Visual Predictive Check – Superiority to Standard Diagnostic (Rorschach) Plots. Page 14 Abstract 738. <https://www.page-meeting.org/?abstract=738>. (2005).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of the Markov ACR model parameter estimates in anti TNF‐IR patients (BALANCE‐I study).
Table S2. Summary of the ACR Markov model parameter estimates in MTX‐IR patients (BALANCE II study).