

Abstract
Certain mutations in mitochondrial DNA (mtDNA) are associated with Leber's hereditary optic neuropathy (LHON). In particular, the well-known NADH dehydrogenase 4 (ND4) m.11778G>A mutation is one of the most common LHON-associated primary mutations worldwide. However, how specific mtDNA mutations, or variants, affect LHON penetrance is not fully understood. The aim of the current study was to explore the relationship between mtDNA mutations and LHON, and to provide useful information for early detection and prevention of this disease. Following the molecular characterization of a Han Chinese family with maternally inherited LHON, four out of eight matrilineal relatives demonstrated varying degrees of both visual impairment and age of onset. Through PCR amplification of mitochondrial genomes and direct Sanger sequencing analysis, a homoplasmic mitochondrial-encoded ND4 m.11778G>A mutation, alongside a set of genetic variations belonging to human mtDNA haplogroup B5b1 were identified. Among these sequence variants, alanine transfer RNA (tRNA)Ala m.5601C>T was of particular interest. This variant occurred at position 59 in the TψC loop and altered the base pairing, which led to mitochondrial RNA (mt-RNA) metabolism failure and defects in mitochondrial protein synthesis. Bioinformatics analysis suggested that the m.5601C>T variant altered tRNAAla structure. Therefore, impaired mitochondrial functions caused by the ND4 m.11778G>A mutation may be enhanced by the mt-tRNAAla m.5601C>T variant. These findings suggested that the tRNAAla m.5601C>T variant might modulate the clinical manifestation of the LHON-associated primary mutation.
Keywords: mitochondrial DNA, Leber's hereditary optic neuropathy, m.11778G>A, m.5601C>T, alanine transfer RNA
Introduction
Leber's hereditary optic neuropathy (LHON) is a maternally inherited disease that affects 1 in 31,000-50,000 people and culminates in the bilateral loss of central vision (1–3). In the North East of England, it has been reported that 1:8,500 individuals harbor a primary LHON-causing mutation and 1:31,000 experience visual loss as a result of LHON (4). Patients with LHON may exhibit abnormal symptoms, including movement disorders, dystonia or multiplesclerosislike symptoms, which pose a significant challenge for clinicians (5,6). Few significant improvements in visual acuity are reported following atrophy of the optic discs. LHON demonstrates an incomplete penetrance for both vision loss and gender bias; LHON affects males more frequently than females (7,8). Three primary mutations including the NADH dehydrogenase (ND) 4 m.11778G>A, ND6 m.14484T>C and ND1 m.3460G>A have been identified in 90% of patients with LHON (9–11). Yet the molecular mechanisms of these mtDNA mutations in the phenotypic manifestation of LHON have not been elucidated.
To understand the role of mitochondrial dysfunction in LHON, an extended genetic screen for mtDNA variants was performed in a Han Chinese family with a high prevalence of LHON. Sequence analysis of the complete mitochondrial genome identified the occurrence of an ND4 m.11778G>A mutation and an alanine transfer RNA (tRNAAla) m.5601C>T variant within matrilineal relatives of the proband. In addition, bioinformatics analysis was performed in order to explore whether the m.5601C>T affected the tRNAAla secondary structure.
Patients and methods
Patients and genetic screening
To identify mtDNA variations in Chinese patients with LHON, a Han Chinese family was recruited from Hangzhou First People's Hospital (Hangzhou, China) in January 2018, and blood samples (5 ml) were collected from each matrilineal relative of the proband. Blood samples (5 ml) from unrelated control subjects (n=300) from the same geographical region recruited at the Hangzhou First People's Hospital were also used in the present study. These healthy subjects consisted of 200 males and 100 females, aged 11–48 years, and were enrolled from January 2018 to January 2019. The present study was approved by the Ethics Committee of Hangzhou First People's Hospital. Written informed consent was obtained from all participants, or their parent/guardian, prior to enrollment in the study.
Clinical examinations
The proband (II-12) and other affected matrilineal relatives (II-5, II-7 and III-8; Fig. 1) underwent comprehensive ophthalmic examinations, including visual field tests, examination of visual acuity, fundus photography, visual evoked potentials and determination of the degree of visual impairment, performed as previously described (12,13). The degree of visual impairment was classified based on the following criteria (12,13): healthy, ≥0.300; mild, 0.100–0.299; moderate, 0.050–0.099; severe, 0.020–0.049; and profound, <0.020.
Figure 1.

A three-generation Han Chinese family pedigree chart of Leber's hereditary optic neuropathy. The arrow indicates the proband (II-12) and filled symbols represent the visually impaired individuals.
PCR and genetic sequencing to identify mtDNA variants
Genomic DNA from LHON patients and control subjects was extracted using a DNA extraction kit (QIAamp® DNA Blood Mini kit; Qiagen GmbH), according to the manufacturer's protocol. The complete mitochondrial genomes of II-5, II-7, II-12 and III-8 were amplified in 24 overlapping fragments using 200 µM dNTP, 10X buffer, Taq DNA polymerase and 15 mmol/l Mg2 (cat. no. R004A; Takara Biotechnology, Co., Ltd.). The 24 sequences of light-strand and heavy-strands oligonucleotide primers for amplification of mtDNA genes were used according to a previous report (14). The following thermocycling conditions were used for PCR: 95°C for 5 min; 30 cycles of 94°C for 10 sec, 60°C for 30 sec and 72°C for 1 min; and a final extension at 72°C for 5 min. After confirmation of band of interest, the PCR products were purified using the PureLink Gel Extraction kit (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's recommendations. DNA samples with concentrations >1.0 ng/µl were sequenced using the BigDye™ Terminator Cycle Sequencing reaction kit (Applied Biosystems; Thermo Fisher Scientific, Inc.) and an ABI PRISM® 3700 DNA Analyzer. Sequencing data were compared with the updated Cambridge consensus human mitochondrial genome sequence (accession no. NC_012920) using DNASTAR version 5.01 (DNASTAR Inc.) (15).
Phylogenetic conservation analysis
Phylogenetic analysis was performed to determine the potential pathogenic role of the identified mtDNA mutations. Briefly, 17 different species were selected for phylogenetic analysis (Table I). The conservation index (CI) was measured by comparing the human nucleotide alternations with the nucleotide sequences of other species. CI≥70% was implicated to have functional significance (16).
Table I.
Mitochondrial DNA sequence accession number of 17 vertebrate species used in the phylogenetic analyses.
| Species | GenBank accession no. |
|---|---|
| Homo sapiens | NC_012920 |
| Cebus albifron | NC_002763 |
| Gorilla gorilla | NC_011120 |
| Hylobates lar | NC_002082 |
| Lemur catta | NC_004025 |
| Macaca mulatta | NC_005943 |
| Macaca sylvanus | NC_002764 |
| Nycticebus coucang | NC_002765 |
| Pan paniscus | NC_001644 |
| Pan troglodytes | NC_001643 |
| Papio hamadryas | NC_001992 |
| Pongo pygmaeus | NC_001646 |
| Pongo pygmaeus abelii | NC_002083 |
| Tarsius bancanus | NC_002811 |
| Mus musculus | NC_006914.1 |
| Bos taurus | HM045018.1 |
| Xenopus laevis | NC_001573.1 |
Bioinformatics analysis
To determine whether the m.5601C>T variant affected tRNAAla secondary structure, the RNAfold web server program was used (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi), as previously described (17).
Determining the pathogenicity of the variant
The role of the tRNAAla m.5601C>T variant was determined using the pathogenicity scoring system, as described by Yarham et al (18). In brief, mutations were classified as: ‘neutral polymorphism’, ≤6; ‘possible pathogenic’, 7–10; ‘definitely pathogenic’, ≥11.
Statistical analysis
SPSS 17.0 software (SPSS Inc.) was used for statistical analysis. Fisher's exact test was used to assess the differences between groups. P<0.05 was considered to indicate a statistically significant difference.
Results
Clinical presentation of a Han Chinese family with LHON
A pedigree chart from a Han Chinese family with a history of LHON is presented in Fig. 1. There were four LHON patients presented in the pedigree (three males and one female), aged 7–39 years old. Medical history analysis of the proband (II-12) confirmed that no other clinical disorders, such as deafness, diabetes mellitus, cardiovascular diseases, cancer or neurological disorders, were present. Following comprehensive genetic counseling at the Department of Ophthalmology in Hangzhou First People's Hospital, the proband (age, 39), was found to have begun suffering from painless and progressive bilateral loss of vision at the age of 19, manifesting as a dark cloud in the central vision and difficulty differentiating different colors. Ophthalmic examination revealed large centrocecal scotoma in both eyes, a typical clinical feature of LHON (13). A total of three out of seven matrilineal relatives (II-5, II-7 and III-8), in addition to the proband (II-12), suffered from moderate to profound visual impairment (Table II).
Table II.
Summary of the clinical data for the proband (II-12) and matrilineal relatives (II-5, II-7 and III-8) in the Han Chinese family with maternally inherited Leber's hereditary optic neuropathy.
| Visual impairment score | ||||||
|---|---|---|---|---|---|---|
| Subject | Sex | Age at onset (years) | Age at test (years) | Right eye | Left eye | Degree of visual impairmenta |
| II-5 | Male | 11 | 35 | 0.03 | 0.05 | Severe |
| II-7 | Male | 16 | 33 | 0.1 | 0.2 | Moderate |
| II-12 | Female | 19 | 39 | 0.01 | 0.01 | Profound |
| III-8 | Male | 3 | 7 | 0.02 | 0.01 | Profound |
The degree of visual impairment was classified based on criteria stated in the clinical examinations section of the Methods.
Screening for mtDNA mutations
To investigate the molecular basis of LHON, II-5, II-7, II-12 and III-8 were screened for mutations following PCR amplification of the mtDNA genomes. Sequence analysis of the mtDNA PCR products revealed 32 genetic polymorphisms (Table III), all of which belonged to the human mtDNA B5b1 haplogroup (19). Of these, there were nine variants in the D-loop gene, two variants in the 12S rRNA gene, one variant in the 16S rRNA gene and one variant in a tRNA gene (m.5601C>T). The other variants were mainly localized within oxidative phosphorylation encoding genes. Notably, five missense mutations were identified: Mitochondrial encoded NADH dehydrogenase 1 (ND1) m.3593T>C (p.V96A), ND2 m.5442T>C (p.F325L), ATP 6 m.9103T>C (p.F193L), ND3 m.10398A>G (p.T114A) and ND4 m.11778G>A (p.R340H). The CIs of these variants were investigated between different species, including mouse, bovine and Xenopus laevis (20–22). Of all identified variants, only tRNAAla m.5601C>T and ND4 m.11778G>A were conserved. Notably, the m.5601C>T and m.11778G>A mutations were absent in the 300 control subjects compared with the mtDNA genomes of the matrilineal relatives (P<0.05). Taken together, these results indicated that tRNAAla m.5601C>T and ND4 m.11778G>A may have active roles in the pathogenesis of LHON.
Table III.
Sequence analysis of mitochondrial DNA mutations in a Han Chinese family with maternally inherited Leber's hereditary optic neuropathy.
| Gene | Position | Base change | Conservation (H/B/M/X)a | CI (%) | Previously reportedb |
|---|---|---|---|---|---|
| D-loop | 73 | A>G | Yes | ||
| 152 | T>C | Yes | |||
| 189 | A>C | Yes | |||
| 263 | A>G | Yes | |||
| 489 | T>C | Yes | |||
| 16117 | T>C | Yes | |||
| 16172 | T>C | Yes | |||
| 16223 | T>C | Yes | |||
| 16519 | T>C | Yes | |||
| 12S rRNA | 709 | G>A | G/A/A/- | Yes | |
| 1438 | A>G | A/A/A/G | Yes | ||
| 16S rRNA | 2706 | A>G | A/G/A/A | Yes | |
| ND1 | 3593 | T>C (p.V96A) | V/I/I/A | 25 | Yes |
| 4102 | Yes | ||||
| ND2 | 4769 | A>G | Yes | ||
| 4833 | A>G | Yes | |||
| 5108 | T>C | Yes | |||
| 5442 | T>C (p. F325L) | F/F/M/L | 23 | Yes | |
| tRNAAla | 5601 | C>T | C/C/C/C | 100 | Yes |
| CO1 | 7028 | C>T | Yes | ||
| 7600 | G>A | Yes | |||
| CO2 | 8167 | C>T | Yes | ||
| ATP6 | 8547 | C>T | Yes | ||
| 8748 | C>T | Yes | |||
| 9103 | T>C (p. F193L) | F/F/F/S | 52 | Yes | |
| ND3 | 10398 | A>G (p. T114A) | T/T/T/A | 36 | Yes |
| ND4 | 11719 | G>A | Yes | ||
| 11778 | G>A (p. R340H) | R/R/R/R | 100 | Yes | |
| ND5 | 12705 | C>T | Yes | ||
| ND6 | 14668 | C>T | Yes | ||
| Cyt b | 15043 | G>A | Yes | ||
| 15301 | G>A | Yes |
Conservation of amino acid for polypeptide.
From Mitomap database (www.mitomap.org). Conserved nucleotide residues are shown in bold font. B, bovine; CO, cytochrome c oxidase; cyt b, cytochrome b; H, human; M, mouse; ND, mitochondrial encoded NADH dehydrogenase; rRNA, ribosomal RNA; tRNA, transfer RNA; X, Xenopus laevis.
In addition, the results revealed that the ratio between affected males and females carrying ND4 m.11778G>A mutations in this case study was 3:1, which was similar to previous studies on families with LHON carrying ND4 m.11778G>A mutations (Table IV) (23–28). These findings suggested that the m.11778G>A mutation may be the molecular basis for the LHON phenotype.
Table IV.
Clinical and molecular data for eight Han Chinese pedigrees carrying the ND4 11778G>A primary mutation in LHON.
| Author, year | Pedigree number | Affected ratio (male:female) | Penetrance of LHON (%) | Secondary variants | MthNA haplogroup | (Refs.) |
|---|---|---|---|---|---|---|
| Ding et al, 2019 | 1 | 3:1 | 40 | tRNAAla m.5601C>T | B5b1 | – |
| Qu et al, 2006 | 2 | 3:1 | 61.5 | tRNAMet m.4435A>G | D5 | (23) |
| Li et al, 2006 | 3 | 2:1 | 60 | tRNAThr m.15951A>G | D4 | (24) |
| Zhang et al, 2010 | 4 | 3:0 | 37.5 | ND1 m.3394T>C | M9a | (25) |
| Qu et al, 2007 | 5 | 3.5:1 | 33 | ND4 m.11696G>A | D4 | (26) |
| Zhang et al, 2010 | 6 | 1:1 | 57.1 | ND6 m.14502T>C | M10a | (27) |
| Qu et al, 2009 | 7 | 1:0 | 14.2 | None | M8a2 | (28) |
| Qu et al, 2009 | 8 | 2:0 | 8 | None | D4g2 | (28) |
LHON, Leber's hereditary optic neuropathy; ND, mitochondrial encoded NADH dehydrogenase; tRNA, transfer RNA.
m.5601C>T variant alters the tRNAAla structure
The m.5601C>T variant is located at a highly conserved position in the TψC loop within the tRNAAla (29); thus, the point mutation results in a missense mutation that creates a novel base pairing (55T-59C) (Figs. 2 and 3). Subsequent bioinformatics analysis revealed that the m.5601C>T variant caused a structural alteration of tRNAAla (Fig. 4), which indicated that m.5601C>T may have an impact on tRNAAla function.
Figure 2.

Identification of the (A) ND4 m.11778G>A mutation and (B) tRNAAla m.5601C>T variant in the mitochondrial genome. Partial sequence chromatograms of ND4 and tRNAAla genes from the proband (II-12; mutant) and a control subject (wild-type). Arrows indicate the location of the base change. ND4, mitochondrial encoded NADH dehydrogenase 4; tRNAAla, alanine transfer RNA.
Figure 3.

Location of the tRNAAla m.5601C>T variant. The secondary structure of tRNAAla protein (wild-type) was derived from the Mitomap database (www.mitomap.org). Arrow indicates the m.5601C>T variant. tRNAAla, alanine-transfer RNA.
Figure 4.
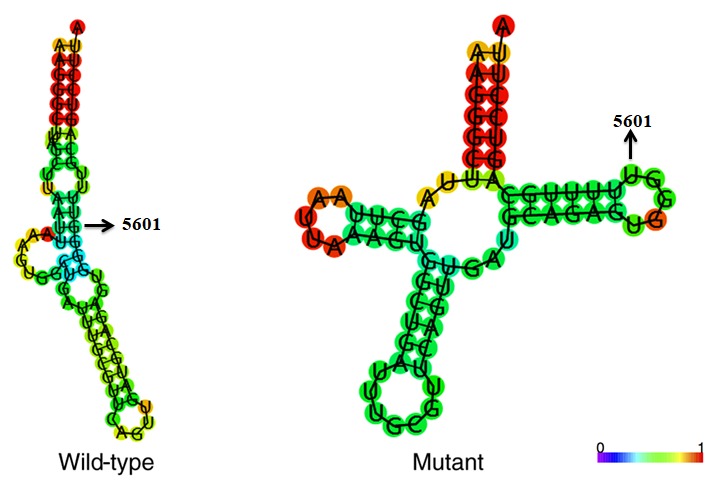
Predicted secondary protein structure of tRNAAla with (mutant) and without (wild-type) the m.5601C>T variant (indicated by arrow). The RNA Fold Webserver program (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) was used to predict the structure. The structure is colored according to the base-pairing probability (0-1, as denoted in the color scale bar).
m.5601C>T variant is ‘possibly pathogenic’ for LHON
The pathogenicity scoring system described by Yarham et al (18) was used to determine the role of the tRNAAla m.5601C>T variant. As presented in Table V, the total pathogenicity score for the m.5601C>T variant was 8, placing it within the ‘possibly pathogenic’ category for LHON.
Table V.
Pathogenicity scoring system for the m.5601C>T mutation.
| Scoring criteria | m.5601C>T mutation | Score | Classification |
|---|---|---|---|
| More than one independent report | Yes | 2 | |
| Evolutionary conservation of the base pair | No changes | 2 | |
| Variant heteroplasmy | No | 0 | |
| Segregation of the mutation with disease | Yes | 2 | |
| Histochemical evidence of mitochondrial disease | Strong evidence | 2 | |
| Biochemical defect in complex I, III or IV | No | 0 | |
| Evidence of mutation segregation with biochemical defect from single-fiber studies | No | 0 | |
| Mutant mt-tRNA steady-state level or evidence of pathogenicity in trans-mitochondrial cybrid studies | No | 0 | |
| Total score | 8 | Possibly pathogenic |
Discussion
In the present study, a Han Chinese family with maternally inherited LHON was clinically and molecularly characterized. One of the most common features of LHON is bilateral loss of vision in the matrilineal relatives of the proband (9); this preferential effect on vision has facilitated the positive association between mtDNA mutations and LHON (30). Clinical evaluation of this family revealed that the age of onset for visual impairment between 3 and 19 years. The association between m. 11778G>A and LHON was reported as early as 1988 (31). In the present study, patients harboring the m.11778G>A mutation had different mtDNA haplogroups, suggesting that the m.11778G>A mutation occurred sporadically and multiplied through evolution of the mtDNA in China. The varying degree of visual impairment in this Chinese family suggested that modifying factors, such as nuclear genes, environmental factors and mitochondrial genetic polymorphisms, may also contribute to LHON penetrance (32). In particular, secondary LHON-associated variants, such as ND1 m.4216T>C and ND5 m.13708G>A mutations in the mtDNA haplogroup J, may increase the penetrance and severity of LHON, in combination with the primary mutations, in European populations (33). mtDNA haplogroups M7b1′2 and M8a have been implicated in the clinical expression of the LHON-associated ND4 m.11778G>A mutation (33).
In the present study, sequencing of the complete mitochondrial genomes of the matrilineal relatives (II-5, II-7, II-12 and III-8) revealed a set of genetic polymorphisms from the Asian mtDNA haplogroup B5b1 (19). Of these variants, tRNAAla m.5601C>T was of most interest because this variant is located at a highly conserved nucleotide in the TψC loop of tRNAAla (position 59), which is thought to be involved in tertiary interactions between the TψC loop and the truncated D-arm (34). Bioinformatics analysis revealed that the m.5601C>T variant created a novel Watson-Crick base-pairing (55T-59C). The tRNAAla m.5601C>T variant has previously been associated with maternally inherited hypertension and mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (35,36). Therefore, the m.5601C>T may alter the secondary structure of tRNAAla and impair the mt-tRNA metabolism and protein translation, and contribute to the LHON phenotype. A previous study has demonstrated that the m.12192G>A mutation, occurring at a similar position on tRNAHis, modulates the clinical expression of deafness in a Chinese pedigree (37), whereas the m.5601C>T variant may increase the penetrance of the hypertension-associated tRNAMet m.4435A>G mutation (35). Zhou et al have previously described the association between the tRNAAla m.5601C>T variant and LHON in seven Han Chinese families (38). However, these families only carried the m.5601C>T variant, and did not harbor the three LHON-associated primary mutations (ND4 m.11778G>A, ND6 m.14484T>C and ND1 m.3460G>A), thus exhibiting very low penetrance and severity of visual impairment (4.5–25.0%) (38). In the present study, the penetrance of LHON-induced visual impairment was 40%, which suggested that the combination of the ND4 m.11778G>A mutation and the tRNAAla m.5601C>T variant may be responsible for the higher prevalence of LHON in this family.
Results from the present study suggested that the tRNAAla m.5601C>T variant could increase both the prevalence and the expression of the LHON-associated ND4 m.11778G>A mutation. Evidence to support this includes the fact that the mutation occurs at a highly conserved nucleotide of tRNAAla, which is critical for basal tRNA activity and normal function (29). The present data demonstrated that the m.5601C>T variant alters the secondary structure of the tRNAAla gene. Finally, the pathogenicity scoring system generated indicated that the m.5601C>T variant was ‘possibly pathogenic’ (18). Therefore, the mitochondrial dysfunction, caused by the ND4 m. 11778G>A mutation, may be worsened by the m. 5601C>T variant. In conclusion, the m. 5601C>T variant may have a modified role in clinical expression of LHON-associated m. 11778G>A mutation in this family.
Nevertheless, the incomplete penetrance of visual impairment in this family (as evidenced by family members harboring these mutations but exhibiting normal vision) indicated that the ND4 m.11778G>A and tRNAAla m.5601C>T variants are insufficient alone to produce the observed clinical phenotypes. Therefore, it is likely that other risk factors, including environmental factors, nuclear genes and epigenetic modifications, may contribute to the clinical manifestation of LHON in this pedigree. The main limitation of this study is the lack of functional analysis of the tRNAAla m.5601C>T variant. Further studies, such as the use of cytoplasmic hybrid cells carrying the tRNAAla m.5601C>T variant are required to confirm our conclusions and to identify additional contributing risk factors.
Acknowledgements
Not applicable.
Funding
The present study was supported by The Hangzhou Health and Family Planning Commission (grant no. 2015A04), The Hangzhou Bureau of Science and Technology (grant no. 20150633B16), The Zhejiang Provincial Administration of Traditional Chinese Medicine (grant no. 2018ZB082) and The Ministry of Public Health from Zhejiang Province (grant nos. 2013KYA158 and 2018ZH019).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
YD and YFY designed the study. MYL and BHX performed the molecular analysis. YFY collected the samples and performed the clinical examinations. JHL analyzed the datasets and carried out the phylogenetic analysis. JHL and YD wrote the paper. All authors discussed the results and implications and commented on the manuscript at all stages. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by The Ethics Committee of Hangzhou First People's Hospital (Hangzhou, China). Written informed consent was obtained from all participants, or their parent/guardian, prior to enrollment in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Yu-Wai-Man P, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. J Med Genet. 2002;39:162–169. doi: 10.1136/jmg.39.3.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carelli V, Rugolo M, Sgarbi G, Ghelli A, Zanna C, Baracca A, Lenaz G, Napoli E, Martinuzzi A, Solaini G. Bioenergetics shapes cellular death pathways in Leber's hereditary optic neuropathy: A model of mitochondrial neurodegeneration. Biochim Biophys Acta. 2004;1658:172–179. doi: 10.1016/j.bbabio.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Puomila A, Hämäläinen P, Kivioja S, Savontaus ML, Koivumäki S, Huoponen K, Nikoskelainen E. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet. 2007;15:1079–1089. doi: 10.1038/sj.ejhg.5201828. [DOI] [PubMed] [Google Scholar]
- 4.Yu-Wai-Man P, Griffiths PG, Brown DT, Howell N, Turnbull DM, Chinnery PF. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am J Hum Genet. 2003;72:333–339. doi: 10.1086/346066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies-disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30:81–114. doi: 10.1016/j.preteyeres.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia X, Li S, Xiao X, Guo X, Zhang Q. Molecular epidemiology of mtDNA mutations in 903 Chinese families suspected with Leber hereditary optic neuropathy. J Hum Genet. 2006;51:851–856. doi: 10.1007/s10038-006-0032-2. [DOI] [PubMed] [Google Scholar]
- 7.Fraser JA, Biousse V, Newman NJ. The neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol. 2010;55:299–334. doi: 10.1016/j.survophthal.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackey DA, Oostra RJ, Rosenberg T, Nikoskelainen E, Bronte-Stewart J, Poulton J, Harding AE, Govan G, Bolhuis PA, Norby S. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet. 1996;59:481–485. [PMC free article] [PubMed] [Google Scholar]
- 9.Catarino CB, Ahting U, Gusic M, Iuso A, Repp B, Peters K, Biskup S, von Livonius B, Prokisch H, Klopstock T. Characterization of a Leber's hereditary optic neuropathy (LHON) family harboring two primary LHON mutations m.11778G>A and m.14484T>C of the mitochondrial DNA. Mitochondrion. 2017;36:15–20. doi: 10.1016/j.mito.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Yu D, Jia X, Zhang AM, Guo X, Zhang YP, Zhang Q, Yao YG. Molecular characterization of six Chinese families with m.3460G>A and Leber hereditary optic neuropathy. Neurogenetics. 2010;11:349–356. doi: 10.1007/s10048-010-0236-7. [DOI] [PubMed] [Google Scholar]
- 11.Asanad S, Meer E, Tian JJ, Fantini M, Nassisi M, Sadun AA. Leber's hereditary optic neuropathy: Severe vascular pathology in a severe primary mutation. Intractable Rare Dis Res. 2019;8:52–55. doi: 10.5582/irdr.2018.01126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang M, Jiang P, Li F, Zhang J, Ji Y, He Y, Xu M, Zhu J, Meng X, Zhao F, et al. Frequency and spectrum of mitochondrial ND6 mutations in 1218 Han Chinese subjects with leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2014;55:1321–1331. doi: 10.1167/iovs.13-13011. [DOI] [PubMed] [Google Scholar]
- 13.Jiang P, Liang M, Zhang J, Gao Y, He Z, Yu H, Zhao F, Ji Y, Liu X, Zhang M, et al. Prevalence of mitochondrial ND4 mutations in 1281 Han Chinese subjects with leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2015;56:4778–4788. doi: 10.1167/iovs.14-16158. [DOI] [PubMed] [Google Scholar]
- 14.Rieder MJ, Taylor SL, Tobe VO, Nickerson DA. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: Analysis of the human mitochondrial genome. Nucleic Acids Res. 1998;26:967–973. doi: 10.1093/nar/26.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 16.Ruiz-Pesini E, Wallace DC. Evidence for adaptive selection acting on the tRNA and rRNA genes of human mitochondrial DNA. Hum Mutat. 2006;27:1072–1081. doi: 10.1002/humu.20378. [DOI] [PubMed] [Google Scholar]
- 17.Ding Y, Xia BH, Zhang CJ, Zhuo GC. Mitochondrial tRNALeu(UUR) C3275T, tRNAGln T4363C and tRNALys A8343G mutations may be associated with PCOS and metabolic syndrome. Gene. 2018;642:299–306. doi: 10.1016/j.gene.2017.11.049. [DOI] [PubMed] [Google Scholar]
- 18.Yarham JW, Al-Dosary M, Blakely EL, Alston CL, Taylor RW, Elson JL, McFarland R. A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum Mutat. 2011;32:1319–1325. doi: 10.1002/humu.21575. [DOI] [PubMed] [Google Scholar]
- 19.Kong QP, Bandelt HJ, Sun C, Yao YG, Salas A, Achilli A, Wang CY, Zhong L, Zhu CL, Wu SF, et al. Updating the East Asian mtDNA phylogeny: A prerequisite for the identification of pathogenic mutations. Hum Mol Genet. 2006;15:2076–2086. doi: 10.1093/hmg/ddl130. [DOI] [PubMed] [Google Scholar]
- 20.Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981;26:167–180. doi: 10.1016/0092-8674(81)90300-7. [DOI] [PubMed] [Google Scholar]
- 21.Gadaleta G, Pepe G, De Candia G, Quagliariello C, Sbisa E, Saccone C. The complete nucleotide sequence of the rattus norvegicus mitochondrial genome: Cryptic signals revealed by comparative analysis between vertebrates. J Mol Evol. 1989;28:497–516. doi: 10.1007/BF02602930. [DOI] [PubMed] [Google Scholar]
- 22.Roe BA, Ma DP, Wilson RK, Wong JF. The complete nucleotide sequence of the xenopus laevis mitochondrial genome. J Biol Chem. 1985;260:9759–9774. [PubMed] [Google Scholar]
- 23.Qu J, Li R, Zhou X, Tong Y, Lu F, Qian Y, Hu Y, Mo JQ, West CE, Guan MX. The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation. Invest Ophthalmol Vis Sci. 2006;47:475–483. doi: 10.1167/iovs.05-0665. [DOI] [PubMed] [Google Scholar]
- 24.Li R, Qu J, Zhou X, Tong Y, Hu Y, Qian Y, Lu F, Mo JQ, West CE, Guan MX. The mitochondrial tRNA(Thr) A15951G mutation may influence the phenotypic expression of the LHON-associated ND4 G11778A mutation in a Chinese family. Gene. 2006;376:79–86. doi: 10.1016/j.gene.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 25.Zhang M, Zhou X, Li C, Zhao F, Zhang J, Yuan M, Sun YH, Wang J, Tong Y, Liang M, et al. Mitochondrial haplogroup M9a specific variant ND1 T3394C may have a modifying role in the phenotypic expression of the LHON-associated ND4 G11778A mutation. Mol Genet Metab. 2010;101:192–199. doi: 10.1016/j.ymgme.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 26.Qu J, Li R, Zhou X, Tong Y, Yang L, Chen J, Zhao F, Lu C, Qian Y, Lu F, Guan MX. Cosegregation of the ND4 G11696A mutation with the LHON-associated ND4 G11778A mutation in a four generation Chinese family. Mitochondrion. 2007;7:140–146. doi: 10.1016/j.mito.2006.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Zhou X, Zhou J, Li C, Zhao F, Wang Y, Meng Y, Wang J, Yuan M, Cai W, et al. Mitochondrial ND6 T14502C variant may modulate the phenotypic expression of LHON-associated G11778A mutation in four Chinese families. Biochem Biophys Res Commun. 2010;399:647–653. doi: 10.1016/j.bbrc.2010.07.135. [DOI] [PubMed] [Google Scholar]
- 28.Qu J, Zhou X, Zhang J, Zhao F, Sun YH, Tong Y, Wei QP, Cai W, Yang L, West CE, Guan MX. Extremely low penetrance of Leber's hereditary optic neuropathy in 8 Han Chinese families carrying the ND4 G11778A mutation. Ophthalmology. 2009;116:558–564. doi: 10.1016/j.ophtha.2008.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Florentz C, Sohm B, Tryoen-Toth P, Putz J, Sissler M. Human mitochondrial tRNAs in health and disease. Cell Mol Life Sci. 2003;60:1356–1375. doi: 10.1007/s00018-003-2343-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallace DC, Lott MT. Leber hereditary optic neuropathy: Exemplar of an mtDNA disease. Handb Exp Pharmacol. 2017;240:339–376. doi: 10.1007/164_2017_2. [DOI] [PubMed] [Google Scholar]
- 31.Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, II, Nikoskelainen EK. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Ji Y, Lu Y, Fu R, Xu M, Liu X, Guan MX. Leber's hereditary optic neuropathy (LHON)-associated ND5 12338T > C mutation altered the assembly and function of complex I, apoptosis and mitophagy. Hum Mol Genet. 2018;27:1999–2011. doi: 10.1093/hmg/ddy107. [DOI] [PubMed] [Google Scholar]
- 33.Ji Y, Zhang AM, Jia X, Zhang YP, Xiao X, Li S, Guo X, Bandelt HJ, Zhang Q, Yao YG. Mitochondrial DNA haplogroups M7b1′2 and M8a affect clinical expression of leber hereditary optic neuropathy in Chinese families with the m.11778G->a mutation. Am J Hum Genet. 2008;83:760–768. doi: 10.1016/j.ajhg.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueda T, Yotsumoto Y, Ikeda K, Watanabe K. The T-loop region of animal mitochondrial tRNA(Ser)(AGY) is a main recognition site for homologous seryl-tRNA synthetase. Nucleic Acids Res. 1992;20:2217–2222. doi: 10.1093/nar/20.9.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng P, Li S, Liu C, Zha Z, Wei X, Yuan Y. Mitochondrial tRNAAla C5601T mutation may modulate the clinical expression of tRNAMet A4435G mutation in a Han Chinese family with hypertension. Clin Exp Hypertens. 2018;40:595–600. doi: 10.1080/10641963.2017.1411497. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka M, Ino H, Ohno K, Ohbayashi T, Ikebe S, Sano T, Ichiki T, Kobayashi M, Wada Y, Ozawa T. Mitochondrial DNA mutations in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) Biochem Biophys Res Commun. 1991;174:861–868. doi: 10.1016/0006-291X(91)91497-Z. [DOI] [PubMed] [Google Scholar]
- 37.Ding Y, Teng YS, Zhuo GC, Xia BH, Leng JH. The mitochondrial tRNAHis G12192A mutation may modulate the clinical expression of deafness-associated tRNAThr G15927A mutation in a Chinese pedigree. Curr Mol Med. 2019;19:136–146. doi: 10.2174/1566524019666190308121552. [DOI] [PubMed] [Google Scholar]
- 38.Zhou HH, Dai XN, Lin B, Mi H, Liu XL, Zhao FX, Zhang JJ, Zhou XT, Sun YH, Wei QP, et al. The analysis of Leber's hereditary optic neuropathy associated with mitochondrial tRNAAla C5601T mutation in seven Han Chinese families. Yi Chuan. 2012;34:1031–1042. doi: 10.3724/SP.J.1005.2012.01031. (In Chinese) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.