

SUMMARY
Id helix-loop-helix (HLH) proteins (Id1–4) bind E protein bHLH transcription factors, preventing them from forming active transcription complexes that drive changes in cell states. Id proteins are primarily expressed during development to inhibit differentiation, but they become re-expressed in adult tissues in diseases of the vasculature and cancer. We show that the genetic loss of Id1/Id3 reduces ocular neovascularization in mouse models of wet age-related macular degeneration (AMD) and retinopathy of prematurity (ROP). An in silico screen identifies AGX51, a small-molecule Id antagonist. AGX51 inhibits the Id1-E47 interaction, leading to ubiquitin-mediated degradation of Ids, cell growth arrest, and reduced viability. AGX51 is well-tolerated in mice and phenocopies the genetic loss of Id expression in AMD and ROP models by inhibiting retinal neovascularization. Thus, AGX51 is a first-in-class compound that antagonizes an interaction formerly considered undruggable and that may have utility in the management of multiple diseases.
Graphical Abstract
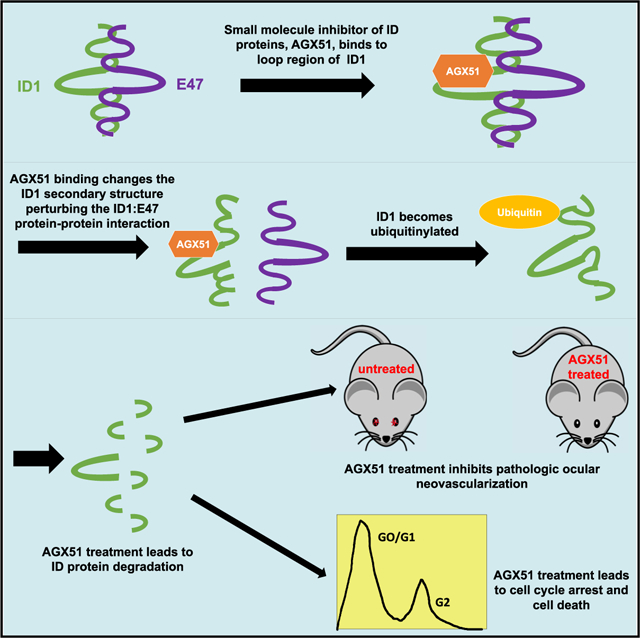
In Brief
Wojnarowicz et al., describe the identification, by an in silico screen, and characterization of a small molecule, AGX51, that targets Id proteins. AGX51 treatment of cells lead to Id protein degradation, cell cycle arrest, and reduced cell viability. AGX51 inhibited pathologic ocular neovascularization in mouse models, phenocopying genetic Id loss.
INTRODUCTION
Id proteins are dominant-negative antagonists of the basic HLH family of transcription factors that function in a variety of cellular processes. In normal embryonic development, high-level expression of all four members of the Id family (Id1–4) is required to inhibit differentiation and maintain stem cell self-renewal capacity. Id protein expression is silenced in many adult tissues but is re-activated in diverse disease processes such as cancer (Lasorella et al., 2014; Ling et al., 2014), diabetes (Kjørholt et al., 2005), Diamond Blackfan anemia (Zhang et al., 1997), and Rett syndrome (Gao et al., 2015; Peddada et al., 2006) (also reviewed in Wang and Baker, 2015). Initial data associating Id1 and Id3 with cancer emerged from xenograft studies and spontaneous tumors in genetically engineered mouse models that typically showed decreased tumor growth and impaired angiogenesis in Id1- and/or Id3-deficient backgrounds (Lyden et al., 1999; Ruzinova et al., 2003; Lasorella et al., 2014). ID1 and ID3 are highly expressed in virtually all human cancers in the vasculature and/or the tumor cells. Importantly, Id1 and Id3 are required in the neovessels for proper vascularization at the primary and metastatic site in mouse models (Gao et al., 2008; Lyden et al., 1999). Given these findings, we hypothesized that reducing Id protein levels would be protective in ocular neovascularization-related pathologies. The contribution of Id proteins in this context has not yet been explored.
Wet age-related macular degeneration (referred to as AMD in this manuscript) and retinopathy of prematurity (ROP) are two examples of pathologic angiogenesis in the posterior segment of the eye, which interfere with retinal function and are the leading causes of vision loss in adults over 60 and premature babies, respectively (Klein et al., 1992) (for a review see Liu et al., 2017). AMD is associated with choroidal neovascularization (CNV), which refers to the growth of new blood vessels through a degraded Bruch’s membrane into the subretinal space. Vascular endothelial growth factor (VEGF) from retinal pigment epithelial cells is thought to be a main stimulant of endothelial cell proliferation and antibodies, and related biologics that target VEGF have proven effective in treating patients with AMD (Heier et al., 2016; Amin et al., 1994). Unfortunately, some patients become resistant to anti-VEGF therapies (Yang et al., 2016). Thus, identifying alternative targets and routes of administration would be clinically valuable. Laser-induced disruption of the Bruch’s membrane in mouse eyes is a widely used model of AMD that recapitulates pathologic angiogenesis seen in patients (Lambert et al., 2013). ROP results from incomplete retinal vasculature development exacerbated by the suppression of neovascularization by the relative hyperoxia of the extrauterine space and supplemental oxygen, often needed for respiratory support. Subsequent exposure to normoxic conditions leads to a rebound effect and overgrowth of blood vessels mediated, at least in part, by increased VEGF production. Murine models in which animals are exposed to cycling hyperoxia recapitulate two phases of the disease: vaso-obliteration followed by hypoxia-induced neovascularization. Importantly, Id1 is known to be a downstream target of VEGF-mediating pro-angiogenic effects and is expressed at very low levels in resting vasculature. Thus, Id1 could be a therapeutic target in AMD and ROP (Ding et al., 2010; Lyden et al., 2001; Lasorella et al., 2014).
The primary mechanism of action of the Id proteins is to sequester other proteins by protein-protein interactions (PPIs). A wide array of PPIs involving Ids have been reported and reviewed extensively (Kee, 2009). We have focused on the Id-E protein PPI because it is found in all cell types examined, is extremely avid, and accounts for many of the transcriptional effects seen in loss- and gain-of-function experiments, i.e., overexpression of Id proteins inhibit the ability of E proteins to act as transcriptional activators or co-activators (with proteins such as Twist, MyoD, and NeuroD) and loss of Id function leads to ectopic or enhanced E-protein-mediated transcription.
Id protein expression in normal adult tissues is often limited to stem and progenitor cell populations. Remarkably, phenotypes of single- and double-knockout mice are rarely manifest until cells are challenged with stress or injury, requiring cell cycle entry. Id1 knockout mice, for example, have no obvious colon phenotype but are defective in mobilizing stem cells in response to chemical injury (Zhang et al., 2014). Id1 loss alone has a modest effect on steady-state hematopoietic stem cell numbers but upon serial bone marrow transplantation becomes activated and is required for maximal proliferative response, escape from senescence, and prevention of exhaustion (Jankovic et al., 2007; Suh et al., 2009). Id1 loss does not lead to a vascular phenotype in adults, which is consistent with low-level expression in resting vessels, but causes severe defects in primary and metastatic tumors, due to the support of local angiogenesis and systemic vasculogenesis of endothelial progenitor cells (Gao et al., 2008; Ruzinova et al., 2003). Thus, the reactivation of the Id proteins in many disease states makes them attractive targets for intervention, with wide therapeutic windows and minimal effects on normal tissues predicted. However, these potential benefits are tempered by the difficulty in developing viable therapeutic approaches to inhibit PPIs. A variety of strategies have been attempted to interfere with Id activity, including the use of targeted antisense delivery, cell-permeable peptides, antagonists of Id expression, and inhibitors of stability-inducing deubiquitinases (Anido et al., 2010; Henke et al., 2008; Mern et al., 2010a, 2010b; Mistry et al., 2013; Murase et al., 2014; Soroceanu et al., 2013). However, some of these approaches need to overcome pharmacological hurdles of delivery in humans and/or lack target specificity. Interestingly, bHLH proteins Myc and Hif2α with second-adjacent dimerization motifs (leucine zipper and PAS domains, respectively) have been targeted with small molecules (Fletcher and Prochownik, 2015; Chen et al., 2016; Cho et al., 2016), but successful targeting of pure bHLH proteins has not yet been reported.
Here, we set out to find a small-molecule antagonist of Id proteins based on X-ray crystal structure coordinates presented in this study. We identified a hydrophobic crevice adjacent to the HLH dimerization motif that was targeted in a 2,234,000-compound in silico screen, followed by electrophoretic mobility shift assays (EMSAs). The strongest hit that emerged from this screen, AGX51, inhibited Id activity and led to the destabilization of the protein by ubiquitin-mediated proteolysis. We report here the finding that the genetic loss of Id1 and Id3 is protective against ocular neovascular disease and demonstrate the ability of AGX51 to phenocopy this effect through intravitreal or systemic delivery.
RESULTS
Genetic Loss of Id1 or Id3 Reduces Ocular Neovascularization
Because Id proteins are known to participate in neovascularization within tumors (Lasorella et al., 2014), we hypothesized that they also participate in CNV, which occurs in AMD (Hamdy et al., 1994). The mouse model of laser-induced CNV (Tobe et al., 1998) used here has previously provided results predictive of outcomes in clinical trials investigating AMD treatments (Saishin et al., 2003; Heier et al., 2012). The laser-induced rupture of Bruch’s membrane was carried out on Id1−/−or Id3−/− mice and littermate control Id1+/+ or Id3+/+ mice. After 14 days, mice were euthanized and choroidal flat mounts were stained with fluorescein isothiocyanate (FITC)-labeled Griffonia simplicifolia lectin, which selectively stains vascular cells, and the area of CNV was measured. As shown in Figures 1A and 1B, genetic deletion of Id1 or Id3 significantly suppressed CNV (p < 0.03).
Figure 1. Genetic Deletion of Id1 or Id3 Suppresses Neovascularization in Mouse Models of AMD.

(A and B) Id1−/− (A) or Id3−/− (B) mice and littermate control Id1+/+ or Id3+/+ mice (n = 12 for each group) had laser-induced rupture of Bruch’s membrane at three locations in one eye, and after 14 days, the mean area of choroidal neovascularization (NV) was significantly less in Id1−/− or Id3−/− than in their corresponding controls (*p < 0.03 by unpaired t test; error bars represent SEM).
(C) Id1−/− or Id3−/− mice and littermate control Id1+/+ or Id3+/+ mice were placed in a 75% O2 chamber at P7 for 5 days to induce ROP. At P17, the animals were euthanized, and the area of retinal neovascularization was assessed by Griffonia simplicifolia lectin staining (selective for vascular cells) of retinal flat mounts (representative images of staining are shown) (white scale bar represents 500 μm). Quantification of the mean total area of NV per group is plotted with n = number of pups and *p < 0.0001 by unpaired t test; error bars represent SEM.
Retinal neovascularization pathology can also be studied in mouse models of ROP. In this model, post-natal day (P) 7 mice are placed in a 75% O2 chamber, which causes retinal capillary depletion. At P12, the mice are returned to room air, which leads to the development of retinal ischemia and proliferative vascular disease in the retinal vasculature. When this model was applied to Id1 or Id3 knockout mice, a significant decrease in retinal neovascularization was observed (p < 0.0001) (Figure 1C). Together, these studies suggest that antagonizing Id proteins pharmacologically may be a useful approach to treating ocular neovascularization diseases.
In Silico Screening Identifies AGX51, an Id1 Antagonist
To facilitate the search for small molecules that may antagonize Id proteins, we solved the crystal structures of two fragments of Id1 encompassing the HLH domain, residues 51–104 (PDB: 6MGM) and 59–104 (PDB: 6U2U) (identical in mouse and human) and an E47-Id1 complex, Id1 (59–104)-E47 (558–609) (PDB: 6MGN) (Figure 2A; Table S1). Similar to the other members of HLH superfamily (Jones, 2004), the structures comprise two α helices connected by a loop consisting of ten residues in Id1 and seven residues in E47. The HLH domain of Id1 is a homodimer and the α helices from both monomers form a four-helix bundle. The interface area is 1,045Å2 and is formed by leucine zipper-like regions of hydrophobic interactions and seven hydrogen bonds. The Id1-E47 interface is formed by the same region of Id1 as the homodimeric interface. The residues of E47 interacting with Id1 are structurally equivalent to the interface area of Id1, resulting in a four-helix bundle, which is similar to the structure of the Id1 homodimer with a buried area of 1,131Å2. In addition to hydrophobic interactions and six hydrogen bonds (Id1-L59: E47-Q590, Id1-Q89: E47-R558, Id1-Q89: E47-V559, Id1-Y94: E47-E600, Id1-L102: E47-R606, and Id1-S104: E47-R606), two salt bridges (Id1-R99: E47-E568 and E47-E568: E47-R571) are formed (Figure 2A). The loop region between the two helices of Id1 is flexible, resulting in different conformations observed in the three structures and high B-factors. Crystal structures of the HLH region of E47 alone (Ahmadpour et al., 2012) and in complex with other proteins and DNA (El Omari et al., 2013; Longo et al., 2008) were published previously. All structures superimpose on the E47 structure reported here with a root-mean-square deviation (RMSD) of 0.4 to 0.8Å between the Cα atoms. The dimerization interface both in E47 homodimers (Ahmadpour et al., 2012) and heterodimers with T-cell acute lymphocytic leukemia protein 1 (El Omari et al., 2013) and neurogenic differentiation factor 1 (Longo et al., 2008) is the same as in the Id1-E47 complex. However, the loop region of E47 is more rigid than that of Id1, as the conformations of the loop are similar in all structures of E47.
Figure 2. Identification of AGX51.

(A) Crystal structure of Id1 and E47, with hydrogen bonds depicted with black dashed lines and salt bridges depicted by red dashed lines with the participating residues labeled.
(B) Hydrophobic crevice analysis of Id1. Black arrow and gray region indicate the identified cleft.
(C) In vitro electrophoretic mobility shift assay (EMSA) of compounds A, B, and C. Wedges indicate increasing concentrations of small molecule from 1–100 μM: lanes 3–5: 20, 50, and 100μM compound A; lanes 6–8: 20, 50, and 100 μM compound B; lanes 10–16: 1, 5, 10, 20, 30, 50, and 100 μM compound C.
(D) Structures of compounds A, B, and C from Figure 2C, where C is AGX51, with red asterisk indicating the stereocenter.
(E) Prediction of AGX51 (depicted by blue stick figure) docking site (depicted in gray) at the Id1 HLH domain (depicted in green).
(F) Prediction of Id1 residues interacting with AGX51.
(G) Circular dichroism (CD) of Id1 with DMSO (left plot) or AGX51 (0, 10, 20, and 50 μM) (middle plot) and E47 with AGX51 (0, 10, 20, and 50 μM) (right plot).
A hydrophobic crevice analysis revealed a cleft adjacent to the loop region of Id1 present in the Id1-E47 heterodimer (Figure 2B), a region of Id1 that is highly conserved between members of the family and across species and is critical in maintaining Id activity (Pesce and Benezra, 1993). An in silico screen of 2,234,000 compounds for crevice binding was performed (see STAR Methods), yielding 3,000 hits that were pared for drug-like properties. A total of 364 candidates emerged and were tested for their capacity to inhibit the ability of Id1 to antagonize E47 binding to DNA by EMSAs, as previously described (Benezra, 1994). A representative EMSA showing three of the compounds tested is shown in Figure 2C, where compounds B and C show a dose-dependent increase in E protein binding (lanes 6 to 8 and 9 to 16, respectively). Compound A showed no such recovery. The strong recovery of E protein binding activity relative to E protein alone (compare lanes 1 to 16) suggests that compound C antagonizes Id1 activity and has little effect on E47-DNA binding. In summary, our hit rate was 2/364, or 0.55%, as only two compounds showed strong activity in the EMSA assay. This anti-Id1 activity is most likely due to the perturbation of the Id1-E47 interaction, but other explanations, such as the binding of an Id1-E47 complex to DNA in the presence of the compound, are possible. We note that, previously, EMSAs were carried out in the presence of reticulocyte lysate (Benezra et al., 1990), and here, also, HeLa nuclear extract was added to facilitate the observed interactions. Compound C, being more potent than B, was chosen for further analyses as an Id1 antagonist and is referred to as AGX51 (the structures of A, B, and C from Figure 2C are shown in Figure 2D).
AGX51 possesses a single stereocenter (Figure 2D). SiteMap (Halgren, 2009; Schrödinger, LLC, 2016a) was used to predict a binding pocket on Id1, which was the same cleft that was identified in Figure 2B (Figure 2E), whereas no AGX51-binding pocket on E47 was found. An analysis of the AGX51 binding pose (predicted by a subsequent high resolution docking calculation using Glide XP) shows close proximity of AGX51 to seven residues in the loop domain and four residues in helix 1 of Id1, with Lys70 forming a hydrogen bond with AGX51 (Figure 2F). Importantly, as shown in Figure S1, the majority of these residues (7/11) are highly conserved among the four Id family members and 9/11 generate a consensus amino acid sequence that is found in the Drosophila melanogaster Id ortholog.
To demonstrate a physical interaction between Id1 and AGX51, we performed circular dichroism (CD) measurements. An analysis of the CD spectra showed that Id1 interacts with AGX51, resulting in a significant alteration in the 2° structure of Id1 (Figure 2G). The CD changes were saturated at 20 μM, implying that the dissociation constant is in this range. Importantly, there was no evidence of an interaction between AGX51 and E47, nor were the changes observed attributable to DMSO or buffer used (Figure 2G). DMSO has strong absorbance in the wavelengths used in these assays, resulting in spikes in the spectra. Such noise unfortunately was unavoidable because AGX51 is water insoluble and the inclusion of DMSO and the range of wavelengths used in these assays were required. We were unable to purify sufficient quantities of Id1-bearing mutations in the pocket region, their inactivity (Pesce and Benezra, 1993) possibly a reflection of the instability of these proteins. The CD assay was also performed with purified Id3, and there was a small but reproducible effect of AGX51 on the Id3 secondary structure following the trend observed with Id1 (Figure S2). Although aggregation of AGX51 might affect the CD spectrum of proteins, this is unlikely here because such an effect would not be expected to be specific to the Id proteins over the highly related E47 bHLH protein. We also attempted to co-crystalize AGX51 and Id1 but were unsuccessful despite trying over 1,000 conditions, including all commonly used sparse matrix screens and soaking the Id1 crystals. Interestingly, when Id1 crystals were exposed to AGX51, but not DMSO alone, the crystals melted. The dissolution of the Id1 crystals after AGX51 exposure is consistent with our CD data, suggesting a conformational change that is incompatible with lattice formation.
To demonstrate on-target engagement in cells, we developed a NanoBRET assay for ID1 and AGX51. This assay is based on the NanoBRET Target Engagement Intracellular BET BRD Assay (Promega). We generated a construct expressing a NanoLuc luciferase ID1 fusion protein and an AGX51-fluorescent tracer (Figure S3A). Upon target engagement, bioluminescence resonance energy transfer (BRET) occurs by the transfer of the luminescent energy from the NanoLuc luciferase to the fluorescent tracer that is bound to the target protein portion of the fusion protein. We found a dose-dependent increase in the BRET ratio when permeabilized 293T cells, transfected with the fusion protein, were treated with the AGX51 tracer (0–4 μM) (Figure S3B), indicating target engagement. Furthermore, we were able to compete the AGX51 tracer with AGX51 and more efficiently with an AGX51 derivative, AGXA, which has greater activity in our biological assays (described below) (Figure S3C). The effective compound concentrations identified in these assays may vary from those found in biochemical and cell-based assays (see below), as this assay uses the AGX51 tracer, a distinct entity from AGX51 with its own properties, and a NanoLuc luciferase ID1 fusion protein rather than endogenous ID1. Furthermore, the assay is carried out in digitonin-permeabilized cells due to the size of the AGX51 tracer, which can alter endogenous cellular conditions and, hence, affect binding properties. These results support ID1 being a direct target of AGX51 in a cellular milieu.
To further validate our physical interaction data and investigate which Id1 residues interact with AGX51, we used an AGX51 analog, AGX51-XL2, which incorporates a benzophenone moiety that upon UV irradiation forms a covalent bond with residues in close proximity (Figure S1B). AGX51-XL2 is predicted to bind the same pocket in Id1 as AGX51 (data not shown). We mixed AGX51-XL2 and purified Id1 (59–104), exposed it to UV light and analyzed the samples by mass spectrometry. We found evidence of AGX51-XL2 covalently binding to Id1 at six residues (V73, P74, T75, P77, Q78, and R80) (Figure S1C). These residues overlap the four helix 1 residues and seven loop residues predicted to be in close proximity to AGX51 (Figure S1; Table S2). No evidence of covalent binding was observed in the sample not exposed to UV light, nor was there evidence of AGX51-XL2 binding to E47 (data not shown). To support the notion that AGX51 binds the same region as AGX51-XL2, we added excess amounts of AGX51 to compete for the Id1 binding site. Mass spectrometry analysis found a significant decrease in AGX51-XL2 binding to Id1 following the addition of a 10-fold excess of AGX51 (Table S3). These data support a direct interaction between AGX51 and Id1 that is consistent with the in silico screen and modeling described above. We were unable to identify a cell-permeable, UV-reactive form of AGX51 to carry out this analysis in living cells.
Effects of AGX51 on Id Proteins in Cells
We tested the activity of AGX51 on primary human umbilical vein endothelial cells (HUVECs) and the HCT116 colorectal cancer cell line, two cell types with different ID1–4 expression profiles. Unbound Id proteins are short-lived with half-lives on the order of 10–20 min but are significantly stabilized when complexed to E proteins (Bounpheng et al., 1999; Deed et al., 1996). If Id-E interactions are disrupted by AGX51 in cells in culture as seen in vitro, this would lead to an increase in unbound Id proteins that would then be predicted to be degraded rapidly. HUVECs were treated with increasing concentrations of AGX51 (0–40 μM) for 24 h, and a significant decrease in ID1 protein levels was observed at 10 μM (Figure 3A). A similar pattern of protein loss was observed for ID3 (Figure 3A); ID2 and ID4 proteins were undetectable in this cell line (data not shown). We note the effects of AGX51 on ID3 loss in HUVECs was diminished in the 20–40 μM range compared with ID1, consistent with the weaker perturbation observed in the CD spectra. A similarly reduced effect on Id3 is also observed in 4T1 breast cancer cells (R.B., P.W. unpublished). In HCT116 cells, AGX51 treatment resulted in reduced levels of ID1, ID2, ID3, and ID4, suggesting that AGX51 antagonizes all four members of the protein family (Figure S4A). Although ID protein levels were reduced by AGX51 treatment, we observed, paradoxically, an increase in ID1 mRNA levels (Figure S4B) perhaps due to activation of the ID1 promoter by liberated E proteins (Bhattacharya and Baker, 2011). These results demonstrate that the reduction in ID1 steady-state protein levels by AGX51 is strong enough to overcome a significant increase in ID1 mRNA production.
Figure 3. Effects of AGX51 on ID Protein Levels, ID1 Ubiquitylation, and E Protein Interactions.

(A) Western blot for ID1 and ID3 on whole cell lysates from HUVECs treated with 0–40 μM AGX51 for 24 h.
(B) Western blot for FLAG on whole-cell lysates from HCT116 cells (expressing FLAG-tagged ID1) treated with 60 μM AGX51 for 0–24 h.
(C) Ubiquitylation assay on U87MG and HCT116 cells treated with MG132 and AGX51.
(D) Immunoprecipitation (IP) for endogenous ID1 and E47 in HCT116 cells treated with 60 μM AGX1 for 1 h, with corresponding immunoblots on whole-cell lysates to the right of the IP blots.
See also Figure S4.
Id proteins are degraded by the ubiquitin 26S proteasome system (Lasorella et al., 2014). We sought to determine if this degradation pathway mediated the effects of AGX51 on Id protein levels. HUVECs are difficult to transfect with expression constructs, so HCT116 cells were utilized for this assay. HCT116 cells were transfected with a construct expressing Flag-ID1 and then treated with 60 μM AGX51 for 2–24 h (Figure 3B). We then co-transfected HCT116 and U87 glioma cells with FLAG-ID1 and hemagglutinin (HA)-ubiquitin and treated them either with vehicle or AGX51 for 2 h in an attempt to visualize ubiquitylation prior to degradation. The proteasomal inhibitor MG132 was added for an additional 6 h prior to immunoprecipitation with an anti-FLAG antibody and immunoblotted using an anti-HA antibody to visualize ubiquitylated ID1. As expected, ID1 ubiquitylation was observed in lysates from cells treated with MG132 (Figure 3C). Treatment with AGX51 further increased ID1 polyubiquitylation in both cell types.
Loss of Id proteins in response to AGX51 should result in an increase in E protein binding activity, assuming little interference of the compound with the E proteins themselves. After treating HCT116 cells with AGX51 for 24 h, we observed a small increase in E protein binding in cell lysates relative to controls, as expected (Figure S4C). To determine if the loss of Id activity precedes Id protein loss, we carried out EMSAs on cell lysates from HCT116 cells treated with AGX51 for 1 h. We saw a similar increase in E protein binding in response to AGX51 at a time when ID1 protein levels are not detectably reduced (Figure S4C), suggesting that the observed increase in E protein binding is due to disruption of the ID1-E protein heterodimer, as opposed to decreased overall ID protein levels. A similar result is observed in 4T1 breast cancer cells treated with AGX51 (R.B., P.W., unpublished).
To confirm that the increase in E47 binding to DNA in the presence of AGX51 was caused by AGX51-induced dissociation of the endogenous E47-ID1 cellular complex, we performed immunoprecipitation using ID1 or E47 antibodies and western blots for endogenous E47 or ID1, respectively. In both assays, treatment with AGX51 markedly reduced the levels of the co-precipitated proteins prior to any detectable loss of the ID1 protein. Thus, AGX51 is able to block the ID1-E47 PPI in cells (Figure 3D).
Together, these results suggest that AGX51 treatment disrupts the ID1-E47 complex, leading to proteasomal-mediated degradation of ID1 and the liberation of E proteins to drive transcription. We note that it is formally possible that the ubiquitinylation event precedes and possibly enhances the complete dissociation of the complex, followed by Id degradation.
Effects of AGX51 on Cell Growth
AGX51 treatment resulted in reduced cell viability, G0/G1 growth arrest, and a reduction in Cyclin D1 levels in both HUVECs (Figures 4A–4C) and HCT116 cells (Figures S5A–S5C). Other protein changes were also observed by whole-proteome stable isotope labeling with amino acids in cell culture (SILAC) analysis after AGX51 treatment (R.B., P.W., unpublished). These data are consistent with genetic experiments in which a threshold level of Id protein expression is essential for proliferation and/or viability of essentially all cell types examined in culture but not in most adult tissues in which these proteins are silenced (see toxicity studies below).
Figure 4. Effects of AGX51 on HUVEC Growth.

(A) Cell growth of HUVECs treated with DMSO or 20 μM AGX51 for 5 days.
(B) Cell cycle analysis of HUVECs treated with DMSO or 20 μM AGX51 for 24 h.
(C) Western blot for Cyclin D1 on whole-cell lysates from HUVECs treated with 0–40 μM AGX51 for 24 h. Tubulin is used as a protein loading control. See also Figure S5.
(D) HUVEC branching was observed after 18–20 h of culturing on matrigel in the absence or presence of 0–40 μM AGX51; images were taken at 10× magnification.
(E) Quantification of the number of nodes, junctions, meshes, and total branching length (n = 4 replicates per concentration tested); *p < 0.05 by Wilcoxon test.
(F) HUVEC monolayers were scratched, then media were replaced with media containing 0–40 μM AGX51, and migration was observed after 24 h, with images taken at 20× magnification.
To characterize the effects of AGX51 on HUVEC vascular branching, we measured the number of nodes, junctions, and meshes as well as branch length following AGX51 treatment. When HUVECs were cultured on matrigel in the presence of AGX51 for 18–20 h, vascular branching was significantly impaired in a dose-dependent manner across all parameters tested, relative to a vehicle control (p < 0.05) (Figures 4D and 4E). AGX51 also significantly impaired HUVEC migration after monolayers were scratched and then cultured in AGX51-containing media for 24 h (Figure 4F). Thus, AGX51 treatment impairs normal growth properties of human endothelial cells in culture.
Pharmacokinetics and Toxicity of AGX51 after Intraperitoneal Injection
We sought to determine the feasibility of administering AGX51 systemically for the treatment of ocular retinopathies. To determine the half-life of AGX51 in serum, mice were treated by intraperitoneal (i.p.) injection with a single dose of 30 mg/kg or 50 mg/kg AGX51 in 70% DMSO, and blood was collected over a 24 h period. A time-dependent decrease in AGX51 serum levels was observed, with a half-life of about 3 h. The mean maximum serum concentration of AGX51 achieved following the 30 mg/kg or 50 mg/kg dose was 1.1 and 1.6 μg/mL (2.7 and 4 μM), respectively, and was not increased further if mice were treated with a 100 mg/kg dose. Of note, significant Id loss was seen in HUVECs at 10 μM. Higher mean serum concentrations could be achieved in 100% DMSO (~12 μM at 100 mg/kg), but animals displayed injection site toxicity with DMSO alone and, thus, 70% formulations were used in all future studies.
Following a 14-day treatment period where mice were dosed i.p. with either vehicle or AGX51 at 60 mg/kg bis in die (bid), no mortality or morbidity was observed; in general, all mice looked healthy and displayed normal behavior throughout; no significant weight loss was evident in either group and clinical chemistry parameters and hematology were all within normal limits (Table S4). No abnormal findings were detected during gross necropsy, nor following complete histopathological evaluation of all major organs.
Effect of AGX51 Treatment on Ocular Neovascularization
To determine whether AGX51 treatment would phenocopy the effects seen in the genetic models of Id1 and Id3 loss described above, we again used the AMD mouse model (Tobe et al., 1998). We found that two intravitreal injections of 10 μg of AGX51 1 week apart (immediately and 7 days post laser treatment: analyses carried out on day 14) significantly suppressed CNV relative to vehicle alone (p < 0.05) (Figures 5A and 5B). Similar results were seen with a single dose of AGX51 immediately post laser treatment and day 14 analysis (data not shown). A total of 5 μg of AGX51 was also effective at significantly reducing CNV, whereas 1 μg was not (p < 0.05) (Figure S6A). Twice-daily i.p. injections of AGX51 (~30 mg/kg) also significantly reduced CNV relative to vehicle-treated mice (p < 0.05) (Figures 5C and 5D). Id1 protein was readily detected in control eyes in CNV regions (arrow heads) and co-localized with lectin-stained endothelial cells (Figure 5E, top row, arrows). Treatment with AGX51 yielded no Id1-positive cells in regions devoid of CNV (Figure 5E, middle row), and in rare sections where CNV was observed, there was no Id1 staining (Figure 5E, bottom row).
Figure 5. AGX51 Treatment Suppresses Ocular Neovascularization in Mouse Models of AMD and ROP.

(A) Wild-type mice had rupture of Bruch’s membrane at three locations in each eye, followed by intravitreal injection of 10 μg of AGX51 or vehicle in one eye immediately and after 7 days (n = 10 per group). Fourteen days after laser-induced rupture, the mean area of CNV was significantly less in AGX51-injected eyes than control eyes (*p < 0.05 by ANOVA with Bonferroni correction for multiple comparisons; error bars represent SEM).
(B) Representative Griffonia-simplicifolia-lectin (marking vascular cells)-stained choroidal flat mounts from a control-injected eye and an AGX51-injected eye (bar represents 100 μm).
(C) Twice-daily i.p. injection of 500 μg of AGX51 also significantly suppressed CNV (n = 10 mice per group; *p < 0.05 by unpaired t test; error bars represent SEM).
(D) Representative Griffonia-simplicifolia-lectin-stained choroidal flat mounts of eyes from mice treated with AGX51 (500 μg) or vehicle by twice-daily i.p. injection (bar represents 100 μm).
(E) Immunofluorescence for Id1 of CNV regions from mice treated with AGX51 or DMSO by intravitreal injection (bar represents 50 μm).
(F) Pups (n = 15) were placed in a 75% O2 chamber at P7 to induce ROP. At P12, the mice were returned to room air and received intravitreal injection of 10 μg of AGX51 in one eye or DMSO in the FE. On P17, the mice were euthanized, and the area of retinal neovascularization (RNV) was assessed. FE refers to “fellow eye” and is defined as the untreated eye in an animal in which both eyes received the laser treatment (*p < 0.01 by unpaired t test; error bars represent SEM).
The efficacy observed by i.p. administration suggests that AGX51 can reach the eye after systemic injection. We, thus, measured the concentration of AGX51 in the eyes of mice dosed i.p. with 30 mg/kg AGX51 by mass spectrometry over 24 h. The maximum concentration of AGX51 after 30 min was ~4 ng/eye, with a 3.7-h half-life. The amount of AGX51 reaching the eye is well below the amount required by intraocular injection to show efficacy, which could be due to incomplete recovery from the extraction of dissected eyes or that effective dose ranges vary considerably with delivery route.
We also assessed the effects of AGX51 in the ROP mouse model. We treated mice exposed to hyperoxic and then normoxic conditions with AGX51 at P12 and euthanized them at P17 to measure the extent of neovascularization. Consistent with the Id1 and Id3 knockout data, intraocular injection of AGX51 significantly reduced retinal neovascularization (p < 0.01) (Figure 5F). These results are consistent with AGX51 targeting the Id proteins for degradation in regions of CNV, which, in turn, phenocopies our genetic loss of expression studies.
Because AGX51 has one chiral center, we sought to determine the relative activity of the two AGX51 enantiomers (called AGX51E1 and E2). Stereospecific synthesis of the two enantiomers was performed and X-ray crystallization studies identified AGX51E1 and AGX51E2 as the R and S forms of the molecule, respectively. The effects of i.p. injection of the racemic mix, AGX51E1, AGX51E2, and vehicle control were compared in the CNV assay. As shown in Figures 6A and 6B, only the racemic mix and AGX51E2 reduced CNV area significantly relative to the vehicle control (p < 0.05 and p = 0.0014, respectively). A dose titration of AGX51E2 in the intravitreal injection assay demonstrated significant efficacy at the 30 μg and 10 μg dose (p = 0.03) relative to fellow eye (FE) but not with the 3 μg or 1 μg dose (Figure 6C). Interestingly, AGX51E2 showed greater activity than AGX51E1 in the CD assay (Figure S7), further supporting the idea that it is the more active enantiomer.
Figure 6. Assessing the Effects of AGX51 Enantiomers and Combination Treatments with Aflibercept.

(A) Laser-induced CNV was induced in mice, and they were treated with DMSO, AGX51 racemate, or the enantiomers (AGX51E1 or AGX51E2); n = 10 mice per group. Mice were treated by i.p. injection with 50 μL of vehicle or 10 μg/mL compound bid. On day 14, the animals were euthanized, and area of CNV was measured as described (*p < 0.05, **p = 0.0014 by ANOVA and Bonferroni correction for multiple comparisons; error bars represent SEM).
(B) Representative Griffonia-simplicifolia-lectin (vascular cell marker)-stained choroidal flat mounts of eyes from mice treated in (A) (bar represents 100 μm).
(C) Eight mice per group were treated by intravitreal injection with 1, 3, 10, or 30 μg AGX51E2 on days 1 and 7 following laser-induced CNV. On day 14, the mice were euthanized, and the area of CNV was measured. Representative Griffonia-simplicifolia-lectin-stained choroidal flat mounts of eyes are shown. The quantification of data is plotted with *p < 0.05 by ANOVA; error bars represent SEM (bar represents 100 μm).
(D) Laser-induced CNV was induced in mice, and on days 1 and 7, the mice were treated by intravitreal injection with DMSO, AGX51 (10 μg), aflibercept (A) (40 μg), or AGX51+aflibercept; n = 10 mice per group. On day 14, the animals were euthanized, and the area of CNV was measured as described. FE refers to “fellow eye” and is defined as the untreated eye in an animal in which both eyes received the laser treatment (*p < 0.05, **p = 0.0014, and ***p < 0.0001 by ANOVA; error bars represent SEM).
See also Figure S7.
Current clinically approved treatment for AMD includes aflibercept, a VEGF trap, which inhibits the growth of neovessels. To determine the relative efficacy of AGX51E2 and aflibercept, we carried out head-to-head and combination treatment in the CNV assay. AGX51E2 significantly reduced CNV in this assay relative to the FE (p = 0.0014) but aflibercept, although showing inhibitory activity, failed to reach statistical significance under these conditions. In addition, the AGX51+aflibercept combination treatment worked better than aflibercept alone (p < 0.05) (Figure 6D). Overall, these results suggest that AGX51 targeting of Id proteins in pathologic neovascularization through systemic or intravitreal administration could be a valuable therapeutic approach.
Characterization of the AGX51 Derivative AGXA
We have recently started to carry out structure activity relationship (SAR) analyses on AGX51 by generating a structurally related small-molecule AGX51 derivative library. Thus far, we have identified AGXA (Figure 7A), which has greater activity than AGX51 in CD (Figure 7B) and NanoBRET assays (Figure S3C). In cell-based assays, AGXA showed about a 4-fold reduction in half maximal inhibitory concentration (IC50) values and reduced Id protein levels at lower concentrations than AGX51 (Figures 7C and 7D). Furthermore, AGXA worked better than AGX51 in the CNV assay at the 1 μg dose (Figure 7E).
Figure 7. Characterization of the AGX51 Derivative AGXA.

(A) The chemical structure of AGXA.
(B) Circular dichroism (CD) spectra of AGXA (0–110 μM in DMSO) and Id1.
(C) Western blot for ID1 on whole-cell lysates from HCT116 cells treated with 0–10 μM AGXA for 24 h. The IC50 is indicated. The IC50 of AGX51 was 22.28 μM.
(D) Western blot for Id1 on whole-cell lysates from 4T1 cells treated with 0–20 μM AGXA for 24 h. The IC50 is indicated. The IC50 of AGX51 was 26.66 μM.
(E) Laser-induced choroidal neovascularization (NV) was induced in mice, and they were treated by intravitreal injection with DMSO, 1 or 5 μg AGXA, or 1 or 5 μg AGX51. On day 14, the animals were euthanized, and the area of CNV was measured (**p < 0.01, ***p < 0.0001 by ANOVA; error bars represent SEM).
DISCUSSION
We report here that the genetic loss of Id proteins reduces neovascularization in two models of ocular neovascular disease and characterize the therapeutic potential of a small-molecule antagonist of the Id protein family, AGX51. This molecule was identified in an in silico screen for compounds that could interact with a hydrophobic pocket within the highly conserved loop region of the Id HLH dimerization motif. CD data demonstrated that AGX51 interacts with Id1 and Id3, and not E47, and alters the Id1 2° structure. The interaction with Id1 occurs in the 20 μM range, consistent with our EMSA data and coimmuno-precipitation (coIP) data. The concentrations of AGX51 required to see effects against Id proteins are similar to those required for Myc-Max inhibition (~20–50 μM) (Fletcher and Prochownik, 2015), the dimer consisting of two bHLH proteins with adjacent leucine zippers that interact to form a four-helix bundle. Importantly, the concentrations of AGX51 used in our in vitro and cell-based assays are in the micromolar range, which was achieved in the serum of mice after i.p. injection with no associated toxicity, thus suggesting the feasibility of systemic anti-Id therapies. The absence of an effect on bone marrow function, and the observation of no overt toxicities in general, are likely due to Id proteins primarily being required by adult stem cells when these cells are called into cycle in response to stress or injury. For example, it has been reported that Id proteins are required when the progenitors of long-term hematopoietic stem cells (LT-HSCs) are ablated and following serial transplantation of LT-HSCs (Jankovic et al., 2007; Perry et al., 2007). In addition, the loss of Id proteins in colonic stem and progenitor cells has little effect on gut homeostasis until the colonic epithelium is injured chemically (Zhang et al., 2014).
Our analyses show that AGX51 disrupts the endogenous Id1-E47 PPI in cells, consistent with the proposed model in which AGX51, by binding to a highly conserved and functional loop domain of the Id family, disrupts its ability to associate with E proteins. It is noteworthy that in vitro, cell lysate is required to observe the perturbation of the Id1/E47 interaction, suggesting that cellular factors (possibly ubiquitinylation of Id1 upon AGX51 binding) are required to facilitate destabilization of the PPI. Importantly, soon after this PPI is broken in cell culture, there is a steady decline in ID protein levels, which, at least for ID1, is due to an increase in ubiquitin-mediated proteolysis. This destabilization of Id proteins is consistent with genetic analyses in which co-expression of E proteins was shown to dramatically increase the stability of Id3 (Bounpheng et al., 1999; Deed et al., 1996). It is noteworthy that none of the other 13 proteins downregulated in response to AGX51 by SILAC analysis (R.B., P.W. unpublished) are known substrates for the Id1 deubiquitinase (USP1), making USP1 an unlikely target of the drug. As AGX51 appears to act as an Id protein antagonist and degrader in cell culture and tissues, the levels of Id proteins in tissues or circulating Id-expressing cells could potentially serve as biomarkers of AGX51 activity.
Although the loss of ID proteins in response to AGX51 treatment both in cells and in animals clearly indicates that they are drug targets, we hypothesize that ID proteins are the critical targets in AGX51-induced phenotypes. If true, one would expect the compound to recapitulate Id loss-of-function mutation effects, and this prediction has been borne out in multiple assays: AGX51 inhibits cell proliferation, inducing a G0/G1 arrest; inhibits ocular neovascularization in mouse models of AMD and ROP; and phenocopies the effects of Id1 and Id3 loss in a variety of cancer models, including ROS production and metastasis suppression (R.B., P.W., unpublished). In addition, the partial reduction of Id1 and Id3 with short hairpin RNAs (shRNAs) reduces the IC50 of AGX51 in cells, and cell killing is severely attenuated in quiescent cells in which Id proteins are undetectable (R.B., P.W., unpublished). We cannot yet rigorously rule out the possibility that other unintended targets also contribute to the phenotypes observed.
Results from CD, crosslinking, and NanoBRET, assays as well as crystal lattice perturbation presented here, support direct target engagement between ID1 and AGX51 both in vitro and in cells. Furthermore, preliminary SAR analyses on a library of structurally related AGX51 derivatives shows a correlation between target engagement and activity, namely, AGXA works at lower concentrations than AGX51 to effect secondary changes in Id1 in CD assays and to compete for tracer binding in the NanoBRET assay; correspondingly, AGXA degrades Id proteins in cells at lower concentrations than AGX51, has a lower IC50, and has stronger activity in the CNV assay. Conversely, higher concentrations of AGX8 (compound B in Figure 2) were required to perturb Id1 in the EMSA assay and degrade Id proteins in cells and showed a higher IC50 in cell viability assays (data not shown). Together, these data support the idea that AGX51 directly engages the target ID proteins.
Intravitreal and/or systemic administration of AGX51 suppressed ocular neovascularization in two models of neovascular ocular disease: AMD and ROP. Importantly, efficacy in the ROP model may also be predictive of that in diabetic retinopathy (Liu et al., 2017). Furthermore, we show that AGX51 performed as well as the currently available AMD treatment, aflibercept and combination therapy worked better than aflibercept alone. The effectiveness of AGX51 as an anti-neovascular agent is likely due to its effects on blocking cell proliferation and vascular network disruption. The ability of systemic delivery of AGX51 to inhibit neovascularization in the retina is consistent with intravitreal delivery and with the possibility that Id-dependent circulating endothelial progenitors (Ciarrocchi et al., 2007; Ding et al., 2010; Gao et al., 2008; Lyden et al., 2001) are contributing to the phenotype. Although the molecular signals promoting neovascularization are not necessarily identical in all organs, the involvement of Id proteins in ocular neovascularization and tumor angiogenesis suggests that AGX51 may have therapeutic potential in other diseases complicated by neovascularization.
In conclusion, we have identified a first-in-class Id protein antagonist and degrader, AGX51, that phenocopies Id genetic loss in pathologic states, suggesting that in addition to being a useful biologic tool for studying Id proteins, it can also be developed into a therapeutic agent that may provide clinical benefit in a variety of Id-related human pathologies.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Robert Benezra (benezrar@mskcc.org). Transfer of materials may require a material transfer agreement (MTA) to be signed.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In Vivo Animal Studies
Animal studies were carried out in accordance with institutional regulations (MO16M130 for the CNV neovascularization studies and M016M138 for the ROP studies) in a non-blinded fashion.
Pharmacokinetic Analyses
To determine the pharmacokinetic parameters of AGX51, eight-week old male BALB/c mice (Taconic farms) were dosed once by i.p. injection with 30 mg/kg, 50 mg/kg or 100 mg/kg AGX51 prepared in 70% DMSO (n = 3 mice per group). Another set of three mice was dosed with 100 mg/kg AGX51 prepared in 100% DMSO. Blood was collected at 30 minutes, 1 hour, 3 hours, 6 hours and 24 hours post AGX51 administration and plasma analyzed via LC-MS as per the protocol previously validated in the MSKCC Antitumor Assessment Core Facility. Following blood collection, the mice were sacrificed via CO2 asphyxiation and the eyes from the 30 mg/kg treatment group were collected and flash frozen for analysis. Data obtained from LC-MS was analyzed via WinNonLin software (version 8.1) for pharmacokinetic parameters.
Toxicity Analyses
To assess for toxicity female athymic nude mice (Envigo), age 6–8 weeks, were dosed i.p. with either a control vehicle (70% DMSO in water) or AGX51 at 60 mg/kg twice daily for 14 consecutive days. Mice were sacrificed 24 hours after the last test administration. Gross and complete necropsy, along with clinical pathology analysis was conducted on all mice. Clinical Chemistry parameters measured were: BUN, Creatine, ALP, ALT, AST, GGT, Bilirubin, Total Protein, Albumin, Globulin, Phosphorus, Glucose, Cholesterol, Phosphorus, Calcium, Sodium, Potassium, Chloride. Hematology parameters measured were: White blood cells (lymphocytes, monocytes, eosinophils, basophils, neutrophils), red blood cells, hemoglobin, hematocrit, MCV, MCH, MCHC, RDW, platelets. Organs/Tissues analyzed were: Lung, heart, thymus, kidneys, liver, spleen, gall bladder, pancreas, duodenum, jejunum, ileum, cecum, colon, bone marrow, femur, tibia, sternum, brain, eyes, ears, nasal and oral cavities, teeth, and mesenteric and tracheal lymph nodes.
Mouse Models of Ocular Neovascularization
CNV was induced as previously described (Tobe et al., 1998). Briefly, 4–6 week-old female Id1−/− or Id3−/− mice and littermate control Id1+/+ or Id3+/+ mice (all in C57BL/6 background) had laser-induced rupture of Bruch’s membrane at three locations in each eye and after 14 days mice were euthanized, eyes were removed, and choroidal flat mounts were stained with FITC-labeled Griffonia simplicifolia lectin (Vector Laboratories, Burlingame, CA) which selectively stains vascular cells (n = 12 mice per group). Flat mounts were examined by fluorescence microscopy and the area of each CNV was measured by image analysis with Image-Pro Plus software (Media Cybernetics, Silver Spring, MD) by an observer masked with respect to experimental groups. In other experiments, wild-type 4–6 week-old, female, C57BL/6 mice had rupture of Bruch’s membrane in each eye followed by intravitreal injection of 1–30 μg of AGX51 (racemic, E1 or E2) or vehicle in one eye immediately and after 7 days or mice were given twice daily i.p. injections of 500 μg of AGX51 or vehicle for 14 days (n = 7–10 mice per group). The area of CNV was measured 14 days after rupture of Bruch’s membrane. For aflibercept and AGXA treatment experiments, rupture of Bruch’s membrane was carried out and then mice were treated with 40 μg of aflibercept, 10 μg of AGX51E2, 1–5 μg of AGX51, 1–5 μg of AGXA, DMSO or combinations. The mice were 4–6 week-old female C57BL/6. After 14 days the mice were euthanized and CNV was measured as described above.
In the ROP experiment 26 C57BL/6, 36 Id−/− and 22 Id3−/− pups were place in 75% O2 at P7. On P12 the mice were returned to room air and at P17 they were euthanized and retinal neovascularization was measured as described above. In the AGX51 ROP experiment, C57BL/6 pups were place in 75% O2 at P7. On P12 the mice were returned to room air and injected in the eye with 10 μg AGX51 or DMSO in the FE (N = 15 mice/group). On P17 they were euthanized and retinal neovascularization was measured as described above.
Cell Lines and Bacterial Strains
Cell Lines
HCT116 (male), 4T1 (female) and 293T (female) cell lines were purchased from ATCC (Manassas, VA, USA) and grown in RPMI (HCT116) or DME (4T1 and 293T) media supplemented with 10% FBS (fetal bovine serum), 1% penicillin-streptomycin and 1% L-Glutamine. HUVECs were purchased from Corning (sex not specified) (Oneonta, NY, USA) and grown in EGM-2 media (Lonza, Walkersville, MD, USA). Cells were cultured at 37°C.
Bacterial Strains
Id1 and Id3 were purified from Rosetta 2 (DE3) Competent Cells (Sigma Millipore).
METHOD DETAILS
Id Protein Purification
pGEV-PSP-mId1 and mId3 expression constructs were transformed into Rosetta 2(DE3) Competent Cells (Sigma Millipore) for protein expression. To produce recombinant GST-tagged protein, a 50mL LB/Ampicillin (100 μg/m)+Chloramphenicol (25 μg/mL) culture was grown overnight at 37°C. Early the next day, the overnight culture was diluted 1:100 into 6L LB/Ampicillin+Chloramphenicol and cultured at 37°C for about four hours until OD600 = 0.7. Cultures were induced with 1mM IPTG and incubated at 16°C for 16–18 hours. Cells were harvested by centrifugation at 4000 rpm for 15 minutes at 4°C and the pellet was resuspended in lysis buffer (50mM Tris pH 8.0; 400mM NaCl; 0.5mM TCEP, Protease Inhibitor Cocktail) (25mL/L of culture). To lyse the cells, Triton X-100 and lysozyme were added to 0.1% and 10 μg/mL respectively, incubated for 30 minutes on ice and sonicated. Lysates were spun at 17,000 rpm for 30 minutes at 4°C and supernatants were filtered through a 0.45 μM filter before incubation with glutathione Sepharose for two hours at 4°C. The Sepharose with bound protein was run through a polypropylene column and washed twice with 50 mL wash buffer (50 mM Tris pH 8.0; 400 mM NaCl) before protein was eluted with 10 mL 50mM glutathione elution buffer, pH 8.0. Protein was subjected to buffer exchange with PD10 columns (Sigma) into 12 mL storage buffer (50 mM Tris.HCl pH 8.0, 400 mM NaCl, 10 mM EDTA, 1 mM DTT, 10% Glycerol).
GST-tag was cleaved with PreScission Protease (0.1 μL of 0.3 μg/mL PreScission Protease cleaves 10 μg of GST-mId1 when incubated at 4°C for four hours) and incubated with Glutathione Sepharose to separate out the cleaved GST-tag. The cleaved mId1 protein was further cleaned by incubation with GST antibodies (Abcam #ab9085–200 μL anti-GST rabbit polyclonal; ThermoFisher # MA4–004 anti-GST mouse monoclonal) before Coomassie gel analysis. Cleaved protein was concentrated to desired strength using Amicon 3000MCO Ultra-4 centrifugal columns (UFC800308).
Crystallization
Crystals of mouse Id1 (51–104) were grown by the hanging drop vapor diffusion method at 4°C. Aliquots (1.5 μL) of the protein at 2.8 mg/mL concentration in 20 mM Tris buffer (pH 8.0), 0.25 M NaCl and 5 mM DTT were mixed with 1.5 μL of reservoir buffer containing 0.1 M sodium citrate (pH 6.5), 0.2 M magnesium acetate, 10% PEG8000. Crystals were harvested, cryoprotected by stepwise transfer to a solution containing 0.1 M sodium citrate (pH 6.5), 0.2 M magnesium acetate, 11% PEG8000, 30% ethylene glycol and flash-frozen in liquid nitrogen. Crystals of mouse Id1 (58–104) were grown by the sitting drop vapor diffusion method at 4°C. Aliquots of protein at 2 mg/mL (2 μL) in 20 mM Tris buffer (pH 8.0), 0.25 M NaCl and 9% ethanol were mixed with 2 μL of reservoir buffer containing 0.1 M MES (pH 6.5), 0.2 M sodium acetate. Crystals were harvested, cryoprotected by transfer to a solution containing 0.1 M MES (pH 6.5), 0.2 M sodium acetate, 10% PEG8000 and 30% ethylene glycol and then flash-frozen in liquid nitrogen.
Crystals of mouse Id1 (51–104) - human E47 (348–399) complex were grown by the hanging drop vapor diffusion method at 22°C. Aliquots (1 μL) of the protein at 9 mg/mL concentration in 20 mM MES buffer (pH 6.5), 0.3 M NaCl and 5 mM DTT were mixed with 1 μL of reservoir buffer containing 0.1 M potassium phosphate (pH 6.0), 0.25 M NaCl, 22.5% PEG8000. Crystals were harvested, cryoprotected by transfer to a solution containing 0.1 M potassium phosphate (pH 6.0), 0.25 M NaCl, 23% PEG8000, 16% ethylene glycol and flash-frozen in liquid nitrogen.
Structure Determination
Diffraction data were collected from single crystals at beam line BNL-X9A for Id1 (51–104) and Id1-E47 to 1.8 and 1.9Å resolution, respectively. For Id1 (58–104) the data were collected at CHESS to 1.5Å resolution. Indexing and merging of the diffraction data were performed in HKL2000 (Otwinowski and Minor, 1997). The structure of Id1 (51–104) was solved by molecular replacement using PDB entry 1MDY as a search model. The search model was truncated to match the length of the construct used for crystallization. The structures of Id1 (58–104) and Id1-E47 complex were solved by molecular replacement using the refined structure of Id1 (51–104) as the search model. Molecular replacement, model building and refinement were accomplished in Phenix (Adams et al., 2010). Diffraction data collection and refinement statistics are summarized in Table S1.
In Silico Screening
Initial docking studies were performed on the Id1-E47 X-ray structure (Deposition ID: D_1000223931 PDB ID: (6MGN)). Compiled lists of commercially available compounds (libraries available from ChemBridge, ChemDiv, Maybridge, and Salor) were screened using a beta release of Autodock 4.0 (The Scripps Research Institute. Molecular Graphics Laboratory. La Jolla, California 92037) using standard settings. For the docking, a cleft adjacent to the loop region of Id1 present in the Id1-E47 heterodimer was targeted. Docking studies were performed on a Sun Microsystems (Menlo Park, CA 94025) workstation running Linux. The Monte Carlo simulation for the Id1-small-molecule complex was run for 1×106 steps and 100 conformations were collected and analyzed. The complex conformation with the best score and lowest total energy was selected for further analysis. 3000 compounds that provided promising docking scores, >6.0, were further computationally filtered by computed physical properties: ClogP < 5 (the 1-octanol-water partition coefficient), tPSA > 80 (topological polar surface area), MW < 600 and chemical and biochemical stability. The resulting computational hits, 364 compounds, were purchased from the vendor and screened for their ability to interfere with Id1-E47 homodimerization.
In Silico Modeling
All ligand preparation and docking calculations used the Schrödinger Suite version 2016–1 using default settings unless otherwise noted. Small-molecules were prepared for docking from sketched 2D structures using LigPrep (Schrödinger, LLC, 2016a). 3D structures were generated using the OPLS3 forcefield (Schrödinger, LLC, 2016a; Harder et al., 2016), and ionization states were determined using Epik (Schrödinger, LLC, 2016a, 2016b; Greenwood et al., 2010; Shelley et al., 2007) at pH 7.0+/−2.0. Id1 monomer, residues 58–104, was prepared from crystallographic coordinates using Protein Preparation Wizard (Schrödinger, LLC, 2016b; Harder et al., 2016; Sastry et al., 2013). Protein protonation states were assigned for pH 7.0 using PROPKA (2016a). The protein was minimized using the OPLS3 force field (Harder et al., 2016). A binding pocket within the Id1 monomer, independent of the in silico screening stage, was identified using SiteMap (Schrödinger, LLC, 2016c; Halgren, 2007, 2009) with default settings, and used to generate a receptor grid for use in docking with Glide (Schrödinger, LLC, 2016d; Friesner et al., 2004, 2006; Halgren et al., 2004) where hydroxyl hydrogens (Tyr66, Ser67, Thr75, and Ser83) were allowed to rotate. Docking was performed with Glide using extra precision (Wang et al., 2000), flexible ligand sampling, and ring conformation and nitrogen inversion sampling. Torsion sampling was biased for amides to penalize nonplanar conformations.
Compound Synthesis
AGX51 was synthesized through a contract research organization following a five step procedure which is delineated below (Scheme 1L). Briefly, Cinnamic acid derivative 2 was condensed with phenol 1 in trifluoroacetic acid to provide dihydrocoumarin 3. The lactone ring in 3 was opened by benzylamine and the resulting phenolic hydroxyl group was converted into the corresponding methyl ether. Reduction of the amide bond to amine 4 which was treated with propionyl chloride to afford the desired amide 5; AGX51.
Scheme 1.

Synthesis of racemic AGX51
AGX51 Spectral data: 1H NMR (DMSO-d6, 600 MHz) δ 7.30–7.18 (m, 4H), 7.15 (m, 1H), 7.08 (m, 2H), 6.92–6.88 (m, 2H), 6.84–6.74 (m, 2H), 6.68 (m, 1H), 5.93 (m, 2H), 4.54–4.40 (m, 2H), 4.18 (t, J = 7.9 Hz, 1H), 3.73, 3.72 (s, s, 3H), 3.16–2.98 (m, 2H), 2.54–2.05 (m, 4 H), 0.95 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 173.91, 173.83, 156.68, 147.66, 147.41, 145.91, 145.59, 138.34, 137.96, 137.47, 137.21, 132.79, 132.26, 128.80, 128.45, 128.24, 127.58, 127.44, 127.33, 127.19, 127.16, 126.34, 120.83, 120.81, 120.72, 120.68, 110.72, 110.66, 108.56, 108.48, 108.11, 107.95, 100.87, 100.70, 55.41, 55.37, 51.48, 48.05, 45.99, 45.36, 40.90, 40.50, 33.61, 32.54, 26.55, 26.02, 9.67, 9.48.
The BRET probe derived from AGX51 (AGX51 tracer) was prepared in 3 steps from advanced intermediate amine 4 by reaction with commercial N-Boc-aminoPEG2-NHS ester, removal of Boc protecting group, and reaction with Bodipy-558/568-NHS ester.
Circular Dichroism
Far UV Circular dichroism (CD) measurements were done on a Jasco J-1500 spectropolarimeter at room temperature using a 0.1 cm cell and a 0.1 mg/mL protein solution in the absence of presence of AGX51, AGX51E1, AGX51E2 or AGXA. Due to the presence of DMSO in the samples, the lowest wavelength to be scanned is limited.
NanoBRET™ Target Engagement Assay
A N-terminal NanoLuc® luciferase ID1 fusion protein was synthesized by Genewiz (South Plainfield, NJ, USA) into the pUC57 backbone and then cloned into the pcDNA3.1 plasmid with EcoRI-HF and XbaI (both enzymes from New England Biolabs, Ipswich, MA, USA). The assay was performed essentially as described in the NanoBRET™ TE Intracellular BET BRD Assay kit manual (Promega Corporation, Madison, WI, USA). Briefly cells were transfected and after 24 hours were plated onto flat bottom, non-binding surface, white, polystyrene 96-well plates (Corning Incorporated, Kennebunk, ME, USA). The AGX51 tracer was then added (0–4 μM), followed by digitonin (Sigma, St. Louis, MO, USA) to permeabilize the cells (50 μg/mL). The NanoBRET™ Nano-Glo® Substrate (Promega) was then added and readings taken using a GloMax Discover System instrument (Promega). For the competition assays, cells were treated with 2 μM of AGX51 tracer and 0–60 μM of AGXA or AGX51.
Covalent Binding of AGX51 Derivative to Id1
An analog of AGX51 (AGX51-XL2) was used which contains a benzophenone photoreactive moiety. 1 μg of purified Id1 (aa59–104) and 19.7ng of AGX51-XL2 (dissolved in DMSO) were combined, in the dark, and then exposed to UV light for 20 minutes, a negative control without UV exposure was also included. The samples were then run in the dark on a 15% denaturing gel, silver stained according to the manufacturers protocol (SilverQuest Staining Kit, Invitrogen, Grand Island, NY, USA) and bands were excised for mass spectrometry as described below. The above experiment was repeated with the addition of another sample that also had an 10-fold excess of AGX51, relative to AGX51-XL2, to assess the ability of AGX51 to compete with AGX51-XL2.
In-Gel Digestion for Mass Spectrometry
In-gel digestion was performed using the method by Shevchenko et al. (2006). Briefly, gel bands were excised, washed with 1:1 (acetonitrile: 100 mM ammonium bicarbonate) for 30 minutes, dehydrated with 100% acetonitrile for 10 minutes until gel slices shrunk and excess acetonitrile was removed and slices were dried in a speed-vac for 10 minutes without heat. Gel slices were reduced with 5 mM DTT for 30 minutes at 56°C in a thermostotated mixer with gentle mixing, removed, allowed to cool to room temperature then alkylated with 11 mM IAA for 30 minutes in the dark. Gel slices were washed with 100 mM ammonium bicarbonate and 100% acetonitrile for 10 minutes each. Excess acetonitrile was removed and the slices dried in a speed-vac for 10 minutes without heating. Gel slices were then rehydrated in a solution of 25 ng/μL trypsin in 50 mM ammonium bicarbonate on ice for 30 minutes. Digestions were performed overnight at 37°C in a thermostated heater with gentle mixing. Digested peptides were collected and further extracted from gel slices in extraction buffer (1:2 vol/vol) 5% formic acid/50% acetonitrile) at high speed mixing. Extractions were combined and dried down in a vacuum centrifuge. Peptides were desalted with C18 resin-packed stage-tips, lyophilized to dryness, then reconstituted in 3% acetonitrile/0.1% formic acid for LC-MS/MS analysis.
LC-MS/MS Analysis
LC-MS/MS was performed using a Waters NanoAcquity LC system (with a 100-μm inner diameter × 10 cm length C18 column (1.7 μm BEH130; Waters) configured with a 180 μm × 2 cm trap column coupled to a Thermo Q-Exactive Plus orbitrap mass spectrometer. Trapping was performed at 15 μL/min 0.1% formic acid (Buffer A) for 1 minute. The LC gradient was 0.5% to 50% B (100% acetonitrile; 0.1% formic acid) over 90 minutes at 300 nL/min. MS data were collected in data dependent acquisition (DDA) mode utilizing a top ten precursor ion selection for HCD fragmentation. Full MS scans were performed with the following parameters: Resolution: 70,000; AGC target: 1e6; Maximum IT: 50 ms; Scan Range: 400 to 1600 m/z. DDA parameters were as follows: Resolution: 17,500; AGC target 5e4; Maximum IT: 50 ms; Isolation window: 1.5 m/z; NCE: 27; Minimum AGC target: 2e3; Intensity Threshold: 4e4; Dynamic Exclusion: 15 s; Charge exclusion: unassigned, 1, 6–8, >8.
Crosslinked Peptide Identification Analysis
MS .raw files were processed using Byonic version 2.5 (Protein Metrics, San Carlos, USA) by searching against the mouse ID1 custom database. Search criteria include 10 ppm mass tolerance for MS spectra, 40 ppm mass tolerance for MS/MS spectra, a maximum of two allowed missed cleavages, fixed carbamidomethyl-cysteine modifications, variable methionine oxidation, deamidation on glutamine and asparagine, N-terminal protein acetylation, and the monoisotopic mass of the AGX51-XL2 cross linked product (419.1885 Da). Pep 2D significance threshold of 0.005 or lower were considered significant. Cross-linked peptides were further inspected by visual analysis.
Electrophoretic Mobility Shift Assays
To test the activity of compounds identified in the in silico screen, full-length E47 was purified from bacteria and mixed with a P32 labeled E-box sequence derived from the muscle creatine kinase (MCK) enhancer, BSA, DTT, poly dI-dC, salmon sperm, and HeLa nuclear extract, in the presence or absence of purified full-length Id1. Increasing concentrations of the various test compounds dissolved in DMSO or DMSO alone were added to the reaction mixes for 30 minutes and resolved on a 5% non-denaturing polyacrylamide gel and autoradiographed. Electrophoretic mobility shift assays (EMSAs) were carried out on whole cell lysates from AGX51-treated cells and the EMSA was performed as described previously (Tournay and Benezra, 1996).
Immunoblotting
For immunoblotting, cells were collected by trypsinization, washed with PBS and lysed in homogenization buffer (0.3 M sucrose, 10 mM Tris (pH 8.0), 400 mM sodium chloride, 3 mM magnesium chloride, 0.5% NP40/IGEPAL, 100 μg/mL Aprotinin+Protease inhibitor cocktail (Roche # 11 836 153 001). Proteins were separated by SDS-PAGE, transferred to a membrane (LI-COR), probed with primary antibodies overnight at 4°C, and probed with secondary antibodies (LI-COR) for 1–2 hours at room temperature. Proteins were visualized using the LI-COR Odyssey Infrared Imaging detection system. The following primary antibodies were used Id1, Id2, Id3, Id4 (195–14, 9-2-8, 17–3, 82–12, respectively, all from Biocheck), Cyclin D1 (2978, Cell Signaling), Actin (A2066, Sigma), Tubulin (T4026, Sigma). Western blot quantification was carried out using channel 700 and channel 800 intensity data from Odyssey application software version 3.0.30 (LI-COR), subtracting blank values and normalizing to Tubulin.
Immunoprecipitation
Cells were lysed in NP40 lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP40, 1.5 mM Na3VO4, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 10 mM β-glycerolphosphate and EDTA free protease inhibitor cocktail (Roche)) or RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP40, 0.5% Sodium Deoxycholate, 0.1% Sodium dodecyl sulfate, 1.5 mM Na3VO4, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 10 mM β-glycerolphosphate and EDTA free protease inhibitor cocktail (Roche)). Lysates were cleared by centrifuge at 15,000 rpm for 15 minutes at 4°C. For immunoprecipitation, cell lysates were incubated with primary antibody (FLAG M2 affinity gel, Sigma, F2426; ID1 (C-20), Santa Cruz, sc-488; E2A (N-649), Santa Cruz, sc-763) and protein G/A beads (Santa Cruz, sc-2003) at 4°C overnight. Beads were washed with lysis buffer four times and eluted in 2X SDS sample buffer. Protein samples were separated by SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane. Membranes were blocked in TBS containing 5% non-fat milk and 0.1% Tween20, and probed with primary antibodies. Antibodies and working concentrations are: ID1 1:500 (C-20, sc-488) and E2A 1:1000 (N-649, sc-763), obtained from Santa Cruz Biotechnology; HA 1:1000 (C29F4, #3724), obtained from Cell Signaling Technology; β-actin 1:8000 (A5441), Vinculin 1:8000 (V9131) and FLAG M2 1:500 (F1804) obtained from Sigma. Secondary antibodies horseradish-peroxidase-conjugated were purchased from Pierce and ECL solution (Amersham) was used for detection.
Ubiquitylation Assay
HCT116 cells were transfected with pcDNA3-ID1-Flag and pcDNA3-HA-Ubiquitin using lipofectamine 3000 (ThermoFisher). 36 hours after transfection, cells were treated with 60 μM AGX51 for two hours followed by 20 μM MG132 (EMD Millipore) for an additional six hours. After washing with ice-cold PBS twice, cells were lysed in 100 μL of TBS (50 mM Tris-HCl, pH 8.0, 150 mM NaCl) containing 2% SDS and boiled at 100°C for 10 minutes. Lysates were diluted with 900 μL of TBS containing 1% NP40 and EDTA free protease inhibitor cocktail (Roche) and were cleared by centrifuge at 15,000 rpm for 15 minutes at 4°C. Immunoprecipitation was performed using 1 mg of cellular lysates with FLAG M2 affinity gel (Sigma, F2426). Ubiquitinylated proteins were analyzed by immunoblot using indicated antibodies.
qRT-PCR
RNA was extracted using the RNeasy kit (QIAGEN, Valencia, CA, USA) and cDNA was generated from 1 μg of RNA using SuperScript IV First-Strand Synthesis System (Invitrogen, Grand Island, NY, USA). Quantitative PCR was performed using SYBR Green QuantiTect Primer Assay (QIAGEN) according to manufacturer’s instructions in a 7900HT Fast-Real Time PCR System Instrument (Applied Biosystems, Grand Island, NY, USA). Primer pairs for the individual genes were obtained from the bioinformatically validated QuantiTect Library and are as follows: ID1 (QT00230650), ID3 (QT01673336), and GAPDH (QT01192646). The fold changes in gene expression were calculated using the delta-delta CT method.
Cell Viability Assays
Cell lines were seeded in a 96 well plate (5000 cells per well). After overnight incubation, cells were treated with AGX51 and incubated for 24 hours then MTT reagent (5 mg/mL) was added and the cells were incubated for four hours. Following incubation, media was aspirated and 200 μL DMSO was added per well. Absorbance was then measured at 570 nm using a plate reader (Synergy 2, BioTek). Cell growth profiles were determined by seeding 38,000 cells in a 24-well plate, in triplicate for each time point, and counting the cells on days 1, 3 and 5 after seeding, using trypan blue exclusion of dead cells.
Cell Cycle Analysis
Cells were treated with AGX51 or DMSO, collected by trypsinization, washed with 1X PBS, resuspended in 500 μL 1X PBS and then diluted with 6 mL 70% ethanol and stored at −20°C until analysis. For cell cycle analysis cells were centrifuged 1000 rpm for 5 minutes, washed with 1X PBS and then resuspended in 0.5 mL PI/RNase staining buffer (550825, BD Biosciences), incubated for 15 minutes at room temperature and analyzed by flow cytometry (LSR II).
HUVEC Cell Branching Assays
For branching assays, 350 μL of Matrigel was loaded into each well of a 24-well plate on ice and incubate the plate for 30 minutes at 37°C to allow the Matrigel to solidify. 80,000 HUVECs in 0.5 mL of EGM-2 medium with the indicated AGX concentration was plated on the solidified Matrigel. At 18–20 hours of incubation when the tube formation has peaked, media from the well was carefully removed and fixed with 10% buffered formalin for 15 minutes. Each well was washed with DPBS. The morphology of capillary like structures was visualized using an inverted microscope and photographed with a digital camera at 10x magnification. To quantify the tube network, ImageJ with the Angiogenesis Analyzer plugin (public domain Java-based image-processing program) was installed, and the analysis for the number of nodes, junctions, meshes, and total branching length was performed as per the instruction. The statistical data analyses were performed using the Wilcoxon test.
HUVEC Scratch Assay
HUVECs were seeded on 24-well plate coated with 0.1% fibronectin. After 24 hours when cells were grown to confluency, cells were serum starved for 4 hours in Endothelial Basal Medium (EBM, Lonza) and scraped with a sterile P200 pipette tip to generate a cell free zone. Cells were washed with PBS and stimulated with EGM-2 medium with the indicated AGX concentration for 24 hours. The scratched area at 0 and at 24 hours was visualized using an inverted microscope and photographed with a digital camera at 20x magnification.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments can be found in the figure legends, Results, and STAR Methods details sections. Three replicates were generally used for each experimental condition for in vitro experiments and 5 mice per group were typically used in each mouse experiment. The sample sizes were determined based on an expected large effect size. With 3 replicated per condition, an effect size as small as 3 can be detected with 80% power at a two-sided significance level of 0.05 using a two-sample t test. With 5 mice per group, an effect size as small as 2 can be detected with 80% power at a two-sided significance level of 0.05 using a two-sample t test. Additional experiments may be performed when larger variation in data was observed, and data were pooled for analysis. In general, Welch’s t test was used to examine differences between two groups. ANOVA was used to examine differences across multiple experimental groups. Data may be transformed to ensure the underlying normality assumptions were met. Weighted linear regression analysis was used when heteroscedasticity was observed and data points in each group were typically weighted by the reciprocal of the standard deviation of data in each group. For data pooled from multiple experiments, the model included both experiments and experiments by treatment group interaction as covariates to account for potential differences in experiments. Significance of linear contrasts of interest was assessed based on estimates obtained from the weighted least-squares. Q-Q plot of the residuals was examined to ensure the underlying model assumptions were met. P value < 0.05 was considered statistically significant.
DATA AND CODE AVAILABILITY
Crystal structures were deposited in the Protein Database (http://www.rscb.org); PDB: 6U2U, 6MGM, and 6MGN (see Table S1). This study did not generate/analyze additional datasets/code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Id1 | Biocheck | Cat# 195–14 |
| Id2 | Biocheck | Cat# 9-2-8 |
| Id3 | Biocheck | Cat# 17–3 |
| Id4 | Biocheck | Cat# 82–12 |
| Cyclin D1 | Cell Signaling Technology | Cat# 2978 |
| Actin | Sigma | Cat# A2066 |
| Tubulin | Sigma | Cat# T4026 |
| FLAG | Sigma | Cat# F2426 |
| ID1 | Santa Cruz | Cat# sc-2003 |
| E2A | Santa Cruz | Cat# sc-763 |
| HA | Cell Signaling Technology | Cat# 3724 |
| Vinculin | Sigma | Cat# V9131 |
| Beta actin | Sigma | Cat# A5441 |
| FLAG | Sigma | Cat# F1804 |
| GST | Thermo Fisher | Cat# MA4–004 |
| GST | Abcam | Cat# ab9085 |
| Bacterial and Virus Strains | ||
| Bacteria: Rosetta 2 (DE3) Competent Cells | Sigma Millipore | Cat# 71400–4 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AGX51 (N-(3-(benzo[d][1,3]dioxol-5-yl)-3-(2-methoxyphenyl) propyl)-N-benzylpropionamide) | This paper | N/A |
| AGXA (6-chloro-2-methyl-N-(4-propoxyphenyl)quinolin-4-amine hydrochloride) | This paper | N/A |
| AGX8 (N’-(4-isopropylphenyl)benzo[b]thiophene-2-carbohydrazide) | This paper | N/A |
| AGX-XL2 (4-benzoyl-N-(3,3-diphenylpropyl)bezamide) | This paper | N/A |
| AGX51 tracer (3-(17-(benzo[d][1,3]dioxol-5-yl)-14-benzyl-17-(2-methoxyphenyl)-3,13-dioxo-7,10-dioxa-4,14-diazaheptadecyl)-5,5-difluoro-7-(1H-pyrrol-2-yl)-5H-5λ4-dipyrrolo[1,2-c:2’,1’-f][1,3,2]diazaborinin-4-ium) | This paper | N/A |
| Digitonin | Sigma | Cat# D141 |
| MG132 | EMD Millipore | Cat# 474790 |
| MTT | Thermo Fisher | Cat# M-6494 |
| Critical Commercial Assays | ||
| NanoBRET™ TE Intracellular BET BRD Assay | Promega | Cat# N2130 |
| SilverQuest Staining Kit | Invitrogen | Cat# LC6070 |
| Lipofectamine 3000 | Invitrogen | Cat# L3000001 |
| RNeasy kit | QIAGEN | Cat# 74104 |
| Superscript IV First-Strand Synthesis System | Invitrogen | 18091050 |
| Deposited Data | ||
| Id1 (51–104) structure | This paper | PDB ID: 6MGM |
| Id1 (58–104) structure | This paper | PDB ID: 6U2U |
| Id1-E47 structure | This paper | PDB ID: 6MGN |
| Experimental Models: Cell Lines | ||
| Human: HCT116 | ATCC | Cat# CCL-247 |
| Mouse: 4T1 | ATCC | Cat# CRL-2539 |
| Human: 293T | ATCC | Cat# CRL-1573 |
| Human: HUVEC | Corning | Cat# 354151 |
| Experimental Models: Organisms/Strains | ||
| Mouse: BALB/c mice Balb/cJ | Taconic | 000651 |
| Mouse: Athymic nude mice Hsd:Athymic Nude-Foxn1nu | Envigo | 069(nu)/070(nu/+) |
| Oligonucleotides | ||
| EMSA sense oligonucleotide: 5’-CCCCAACACCTGCCTGCCTGA-3’ | Eurofins | N/A |
| EMSA antisense oligonucleotide: 5’-GGGGTTGTGGACGGACGGACT-3’ | Eurofins | N/A |
| ID1 primers | QIAGEN | QT00230650 |
| ID3 primers | QIAGEN | QT01673336 |
| GAPDH primers | QIAGEN | QT01192646 |
| Recombinant DNA | ||
| Plasmid: NanoLuc® luciferase ID1 fusion protein | This paper | N/A |
| Plasmid: pcDNA3-ID1-Flag | This paper | N/A |
| Plasmid: pcDNA3-HA-Ubiquitin | This paper | N/A |
| Plasmid: pGEV-PSP-mld1 | This paper | N/A |
| Plasmid: pGEV-PSP-mld3 | This paper | N/A |
| Software and Algorithms | ||
| WinNonLin (version 8.1) | Certara | N/A |
| Image-Pro Plus software | Media Cybernetics | N/A |
| Phenix | Adams et al., 2010 | N/A |
| Schrodinger Suite version 2016–1 | Schrodinger | N/A |
| Byonic version 2.5 | Protein Metrics | N/A |
| Odyssey application software version 3.0.30 | LI-COR | N/A |
| Prism | GraphPad Software | N/A |
Highlights.
Genetic knockdown of Id proteins inhibits ocular neovascularization in mouse models
In silico screen and in vitro assays identified AGX51, an anti-Id small molecule
AGX51 treatment lead to pan-Id degradation, cell cycle arrest, and cell death
AGX51 phenocopies genetic Id loss, inhibiting pathologic ocular neovascularization
ACKNOWLEDGMENTS
The authors are grateful to members of the Benezra Lab, past and present, for helpful discussions throughout the course of this work. We are also grateful to Andrea Rizzi for additional supportive modeling work of AGX51 interactions and Drs. Gary Cook and Gary Novack for expert guidance on the ophthalmic studies. We thank Elisa de Stanchina and the MSKCC Antitumor Assessment Core Facility for assistance with pharmacokinetic and bioavailability assays, Nicholas Socci and the MSKCC Bioinformatics Core Facility for additional statistical analyses, and Jorge Gandara of the Weill Cornell Medical College Microbiome Core for assistance with the GloMax instrument. For expert guidance in implementing and executing the NanoBRET assay, we wish to thank Dan McKay and William Forrester at Novartis and James Vasta at Promega. R.B. and J.D.C. gratefully acknowledge support from the Sloan Kettering Institute and NIH grant P30 CA008748. R.B. is also supported by grants from the NCI (PO1 CA094060) and The Breast Cancer Research Foundation. A.J.D. is supported by funding from The Breast Cancer Research Foundation. S.K.A. was supported by the Louis V. Gerstner, Jr. Graduate School of Biomedical Sciences. Work was partially supported by 1R43CA150448-01 from NCI/NIH to W.A.G. and NIH grants to A.I. (R01CA101644 and R01CA131126). M.G.-C. was funded by fellowships from the Human Frontier Science Program (HFSP) and the Spanish Ministry of Education and Science (MEC). P.M.W. is a recipient of a Postdoctoral Training Award from the Fonds de Recherche du Quebec-Sante; (FRQS). M.G.E. was supported by fellowships issued by the Valencian Government of Spain (GVA) and the European Social Fund (ACIF/2018/004 and BEFPI/2018/029). Financial support was also provided by Angiogenex, Inc. to the R.B. Laboratory.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.073.
DECLARATION OF INTERESTS
R.B., W.A.G., J.C., and G.S. own shares of Angiogenex, Inc., which holds the patent for the use and development of AGX51. R.B. is the chief scientific officer of Angiogenex, Inc. and a board member, both positions unpaid. G.Y. is listed as an inventor on patents that were filed by MSKCC. O.O. is listed as an inventor and receives royalties from patents that were filed by MSKCC. O.O. is an unpaid member of the SAB of Angiogenex, Inc. and owns shares in Angiogenex, Inc. J.D.C. was on the Scientific Advisory Board for Schrödinger during part of this research and now serves on the Scientific Advisory Board of OpenEye Scientific.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadpour F, Ghirlando R, De Jong AT, Gloyd M, Shin JA, and Guarné A (2012). Crystal structure of the minimalist Max-E47 protein chimera. PLoS One 7, e32136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin R, Puklin JE, and Frank RN (1994). Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 35, 3178–3188. [PubMed] [Google Scholar]
- Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, Rodón L, Folch G, Carmona MA, Prieto-Sánchez RM, Barba I, Martínez-Sáez E, Prudkin L, et al. (2010). TGF-β Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 18, 655–668. [DOI] [PubMed] [Google Scholar]
- Benezra R (1994). An intermolecular disulfide bond stabilizes E2A homodimers and is required for DNA binding at physiological temperatures. Cell 79, 1057–1067. [DOI] [PubMed] [Google Scholar]
- Benezra R, Davis RL, Lockshon D, Turner DL, and Weintraub H (1990). The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell 61, 49–59. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, and Baker NE (2011). A network of broadly expressed HLH genes regulates tissue-specific cell fates. Cell 147, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bounpheng MA, Dimas JJ, Dodds SG, and Christy BA (1999). Degradation of Id proteins by the ubiquitin-proteasome pathway. FASEB J 13, 2257–2264. [PubMed] [Google Scholar]
- Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, Homayoun F, Ma Y, Patel N, Yell P, et al. (2016). Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 539, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W, Carvo I, Signoretti S, Bruick RK, Josey JA, et al. (2016). On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 539, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarrocchi A, Jankovic V, Shaked Y, Nolan DJ, Mittal V, Kerbel RS, Nimer SD, and Benezra R (2007). Id1 restrains p21 expression to control endothelial progenitor cell formation. PLoS One 2, e1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deed RW, Armitage S, and Norton JD (1996). Nuclear localization and regulation of Id protein through an E protein-mediated chaperone mechanism. J. Biol. Chem 271, 23603–23606. [DOI] [PubMed] [Google Scholar]
- Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, Mittal V, Kobayashi H, Shido K, Lyden D, et al. (2010). Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 468, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Omari K, Hoosdally SJ, Tuladhar K, Karia D, Hall-Ponselé E, Platonova O, Vyas P, Patient R, Porcher C, and Mancini EJ (2013). Structural basis for LMO2-driven recruitment of the SCL:E47bHLH heterodimer to hematopoietic-specific transcriptional targets. Cell Rep 4, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher S, and Prochownik EV (2015). Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 1849, 525–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, and Mainz DT (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, and Mittal V (2008). Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science 319, 195–198. [DOI] [PubMed] [Google Scholar]
- Gao H, Bu Y, Wu Q, Wang X, Chang N, Lei L, Chen S, Liu D, Zhu X, Hu K, and Xiong JW (2015). Mecp2 regulates neural cell differentiation by suppressing the Id1 to Her2 axis in zebrafish. J. Cell Sci 128, 2340–2350. [DOI] [PubMed] [Google Scholar]
- Greenwood JR, Calkins D, Sullivan AP, and Shelley JC (2010). Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des 24, 591–604. [DOI] [PubMed] [Google Scholar]
- Halgren T (2007). New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des 69, 146–148. [DOI] [PubMed] [Google Scholar]
- Halgren TA (2009). Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model 49, 377–389. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, and Banks JL (2004). Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- Hamdy FC, Fadlon EJ, Cottam D, Lawry J, Thurrell W, Silcocks PB, Anderson JB, Williams JL, and Rees RC (1994). Matrix metalloproteinase 9 expression in primary human prostatic adenocarcinoma and benign prostatic hyperplasia. Br. J. Cancer 69, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, et al. (2016). OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput 12, 281–296. [DOI] [PubMed] [Google Scholar]
- Heier JS, Brown DM, Chong V, Korobelnik JF, Kaiser PK, Nguyen QD, Kirchhof B, Ho A, Ogura Y, Yancopoulos GD, et al. ; VIEW 1 and VIEW 2 Study Groups (2012). Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 119, 2537–2548. [DOI] [PubMed] [Google Scholar]
- Heier JS, Bressler NM, Avery RL, Bakri SJ, Boyer DS, Brown DM, Dugel PU, Freund KB, Glassman AR, Kim JE, et al. ; American Society of Retina Specialists Anti-VEGF for Diabetic Macular Edema Comparative Effectiveness Panel (2016). Comparison of Aflibercept, Bevacizumab, and Ranibizumab for Treatment of Diabetic Macular Edema: Extrapolation of Data to Clinical Practice. JAMA Ophthalmol 134, 95–99. [DOI] [PubMed] [Google Scholar]
- Henke E, Perk J, Vider J, de Candia P, Chin Y, Solit DB, Ponomarev V, Cartegni L, Manova K, Rosen N, and Benezra R (2008). Peptide-conjugated antisense oligonucleotides for targeted inhibition of a transcriptional regulator in vivo. Nat. Biotechnol 26, 91–100. [DOI] [PubMed] [Google Scholar]
- Jankovic V, Ciarrocchi A, Boccuni P, DeBlasio T, Benezra R, and Nimer SD (2007). Id1 restrains myeloid commitment, maintaining the self-renewal capacity of hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 104, 1260–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S (2004). An overview of the basic helix-loop-helix proteins. Genome Biol 5, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee BL (2009). E and ID proteins branch out. Nat. Rev. Immunol 9, 175–184. [DOI] [PubMed] [Google Scholar]
- Kjørholt C, Akerfeldt MC, Biden TJ, and Laybutt DR (2005). Chronic hyperglycemia, independent of plasma lipid levels, is sufficient for the loss of beta-cell differentiation and secretory function in the db/db mouse model of diabetes. Diabetes 54, 2755–2763. [DOI] [PubMed] [Google Scholar]
- Klein R, Klein BE, and Linton KL (1992). Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology 99, 933–943. [DOI] [PubMed] [Google Scholar]
- Lambert V, Lecomte J, Hansen S, Blacher S, Gonzalez ML, Struman I, Sounni NE, Rozet E, de Tullio P, Foidart JM, et al. (2013). Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat. Protoc 8, 2197–2211. [DOI] [PubMed] [Google Scholar]
- Lasorella A, Benezra R, and Iavarone A (2014). The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 14, 77–91. [DOI] [PubMed] [Google Scholar]
- Ling F, Kang B, and Sun XH (2014). Id proteins: small molecules, mighty regulators. Curr. Top. Dev. Biol 110, 189–216. [DOI] [PubMed] [Google Scholar]
- Liu CH, Wang Z, Sun Y, and Chen J (2017). Animal models of ocular angiogenesis: from development to pathologies. FASEB J 31, 4665–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo A, Guanga GP, and Rose RB (2008). Crystal structure of E47-NeuroD1/beta2 bHLH domain-DNA complex: heterodimer selectivity and DNA recognition. Biochemistry 47, 218–229. [DOI] [PubMed] [Google Scholar]
- Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O’Reilly R, Bader BL, Hynes RO, Zhuang Y, Manova K, and Benezra R (1999). Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature 401, 670–677. [DOI] [PubMed] [Google Scholar]
- Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, et al. (2001). Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med 7, 1194–1201. [DOI] [PubMed] [Google Scholar]
- Mern DS, Hasskarl J, and Burwinkel B (2010a). Inhibition of Id proteins by a peptide aptamer induces cell-cycle arrest and apoptosis in ovarian cancer cells. Br. J. Cancer 103, 1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mern DS, Hoppe-Seyler K, Hoppe-Seyler F, Hasskarl J, and Burwinkel B (2010b). Targeting Id1 and Id3 by a specific peptide aptamer induces E-box promoter activity, cell cycle arrest, and apoptosis in breast cancer cells. Breast Cancer Res. Treat 124, 623–633. [DOI] [PubMed] [Google Scholar]
- Mistry H, Hsieh G, Buhrlage SJ, Huang M, Park E, Cuny GD, Galinsky I, Stone RM, Gray NS, D’Andrea AD, and Parmar K (2013). Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer Ther 12, 2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase R, Kawamura R, Singer E, Pakdel A, Sarma P, Judkins J, Elwakeel E, Dayal S, Martinez-Martinez E, Amere M, et al. (2014). Targeting multiple cannabinoid anti-tumour pathways with a resorcinol derivative leads to inhibition of advanced stages of breast cancer. Br. J. Pharmacol 171, 4464–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Peddada S, Yasui DH, and LaSalle JM (2006). Inhibitors of differentiation (ID1, ID2, ID3 and ID4) genes are neuronal targets of MeCP2 that are elevated in Rett syndrome. Hum. Mol. Genet 15, 2003–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry SS, Zhao Y, Nie L, Cochrane SW, Huang Z, and Sun XH (2007). Id1, but not Id3, directs long-term repopulating hematopoietic stem-cell maintenance. Blood 110, 2351–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesce S, and Benezra R (1993). The loop region of the helix-loop-helix protein Id1 is critical for its dominant negative activity. Mol. Cell. Biol 13, 7874–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzinova MB, Schoer RA, Gerald W, Egan JE, Pandolfi PP, Rafii S, Manova K, Mittal V, and Benezra R (2003). Effect of angiogenesis inhibition by Id loss and the contribution of bone-marrow-derived endothelial cells in spontaneous murine tumors. Cancer Cell 4, 277–289. [DOI] [PubMed] [Google Scholar]
- Saishin Y, Saishin Y, Takahashi K, Lima e Silva R, Hylton D, Rudge JS, Wiegand SJ, and Campochiaro PA (2003). VEGF-TRAP(R1R2) suppresses choroidal neovascularization and VEGF-induced breakdown of the blood-retinal barrier. J. Cell. Physiol 195, 241–248. [DOI] [PubMed] [Google Scholar]
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, and Sherman W (2013). Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des 27, 221–234. [DOI] [PubMed] [Google Scholar]
- Schrödinger LLC (2016a). Schrödinger Release 2016–1: Lig Prep, version 3.7 (Schrödinger: LLC) [Google Scholar]
- Schrödinger LLC (2016b). Schrödinger Release 2016–1: Epik, version 3.5 (Schrödinger: LLC) [Google Scholar]
- Schrödinger LLC (2016c). Schrödinger Release 2016–1: SiteMap, version 3.8 (Schrödinger: LLC) [Google Scholar]
- Schrödinger LLC (2016d). Small-Molecule Drug Discovery Suite 2016–1: Glide, version 7.0 (Schrödinger: LLC) [Google Scholar]
- Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, and Uchi-maya M (2007). Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des 21, 681–691. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, and Mann M (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc 1, 2856–2860. [DOI] [PubMed] [Google Scholar]
- Soroceanu L, Murase R, Limbad C, Singer E, Allison J, Adrados I, Kawamura R, Pakdel A, Fukuyo Y, Nguyen D, et al. (2013). Id-1 is a key transcriptional regulator of glioblastoma aggressiveness and a novel therapeutic target. Cancer Res 73, 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh HC, Ji M, Gooya J, Lee M, Klarmann KD, and Keller JR (2009). Cell-nonautonomous function of Id1 in the hematopoietic progenitor cell niche. Blood 114, 1186–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobe T, Ortega S, Luna JD, Ozaki H, Okamoto N, Derevjanik NL, Vinores SA, Basilico C, and Campochiaro PA (1998). Targeted disruption of the FGF2 gene does not prevent choroidal neovascularization in a murine model. Am. J. Pathol 153, 1641–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournay O, and Benezra R (1996). Transcription of the dominant-negative helix-loop-helix protein Id1 is regulated by a protein complex containing the immediate-early response gene Egr-1. Mol. Cell. Biol 16, 2418–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LH, and Baker NE (2015). E Proteins and ID Proteins: Helix-Loop-Helix Partners in Development and Disease. Dev. Cell 35, 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LJ, Kong XD, Zhang HY, Wang XP, and Zhang J (2000). Enhancement of the activity of l-aspartase from Escherichia coli W by directed evolution. Biochem. Biophys. Res. Commun 276, 346–349. [DOI] [PubMed] [Google Scholar]
- Yang S, Zhao J, and Sun X (2016). Resistance to anti-VEGF therapy in neovascular age-related macular degeneration: a comprehensive review. Drug Des. Devel. Ther 10, 1857–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MY, Clawson GA, Olivieri NF, Bell LL, Begley CG, and Miller BA (1997). Expression of SCL is normal in transfusion-dependent Diamond-Blackfan anemia but other bHLH proteins are deficient. Blood 90, 2068–2074. [PubMed] [Google Scholar]
- Zhang N, Yantiss RK, Nam HS, Chin Y, Zhou XK, Scherl EJ, Bosworth BP, Subbaramaiah K, Dannenberg AJ, and Benezra R (2014). ID1 is a functional marker for intestinal stem and progenitor cells required for normal response to injury. Stem Cell Reports 3, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystal structures were deposited in the Protein Database (http://www.rscb.org); PDB: 6U2U, 6MGM, and 6MGN (see Table S1). This study did not generate/analyze additional datasets/code.