

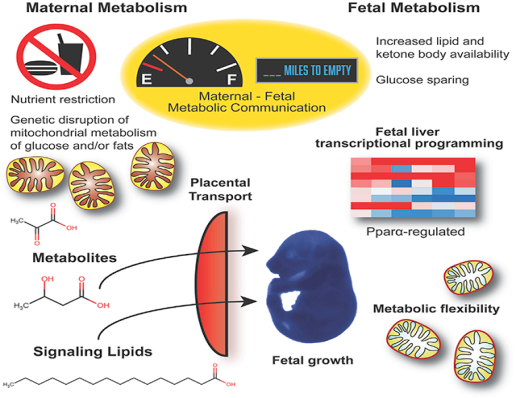
SUMMARY
The extreme metabolic demands of pregnancy require coordinated metabolic adaptations between mother and fetus to balance fetal growth and maternal health with nutrient availability. To determine maternal and fetal contributions to metabolic flexibility during gestation, pregnant mice with genetic impairments in mitochondrial carbohydrate and/or lipid metabolism were subjected to nutrient deprivation. The maternal fasting response initiates a fetal liver transcriptional program marked by upregulation of lipid- and peroxisome proliferator-activated receptor alpha (Pparα)-regulated genes. Impaired maternal lipid metabolism alters circulating lipid metabolite concentrations and enhances the fetal response to fasting, which is largely dependent on fetal Pparα. Maternal fasting also improves metabolic deficits in fetal carbohydrate metabolism by increasing the availability of alternative substrates. Impairment of both carbohydrate and lipid metabolism in pregnant dams further exacerbates the fetal liver transcriptional response to nutrient deprivation. Together, these data demonstrate a regulatory role for mitochondrial macronutrient metabolism in mediating maternal-fetal metabolic communication, particularly when nutrients are limited.
In Brief
Pregnancy is a time of incredible metabolic demand that necessitates metabolic communication between mother and fetus. Using multiple mouse models with impaired carbohydrate and/ or fatty acid metabolism, Bowman et al. show the maternal requirements during nutrient deprivation that drive metabolic and transcriptional programming in the fetus.
Graphical Abstract
INTRODUCTION
Pregnancy exerts an extreme metabolic demand that necessitates coordinated adaptations between mother and fetus to ensure that the energetic and biosynthetic requirements of the rapidly growing fetus are met without compromising maternal health and fecundity. Genetic and environmental perturbations that disrupt this intimate communication can lead to maladaptive metabolic responses and serious adverse consequences for both mother and fetus. While hormonal cues from both mother and conceptus can affect nutrient mobilization, the metabolic demands of the growing fetus can also directly modify maternal metabolism and behavior (Freinkel, 1980; Yamashita et al., 2000). The metabolic demands of pregnancy are incredibly high compared to non-pregnant states and nutrient withdrawal is especially challenging, particularly during late gestation when the conceptus is large enough to challenge maternal energy reserves. Fasting in late gestation during religious observation or illness results in earlier and more dramatic shifts to alternative oxidative substrates such as circulating ketone bodies, one hallmark of the accelerated starvation response characteristic of pregnancy (Boden, 1996; Freinkel, 1980).
Even though oxygen tension in utero is low relative to atmospheric levels, fetal oxidative metabolism is critical for fetal growth and development as evidenced by the host of physiological adaptations in place to ensure the adequate transport of glucose and oxygen to the conceptus (Blackburn, 2007). Whereas anaerobic metabolism dominates in early development, once placental exchange matures and fetal mitochondrial biogenesis accelerates (Ebert and Baker, 2013), the fetus is poised to utilize oxidative metabolism for energy production. The shift from anaerobic to aerobic metabolism continues to accelerate in late gestation to prepare the fetus for the oxidative environment of postnatal life.
Previously, we developed models of impaired mitochondrial carbohydrate and fatty acid metabolism that exhibit maladaptive responses to nutrient deprivation. The selective loss of fatty acid oxidation from the liver by the hepatocyte-specific deletion of carnitine palmitoyltransferase 2 results in mice (Cpt2L−/−) with elevated circulating lipids that cannot generate ketone bodies during a fast and that, remarkably, maintain fasting euglycemia via extrahepatic gluconeogenesis (Lee et al., 2016b, 2017). Additionally, we developed a mouse model of mitochondrial pyruvate carrier (MPC) deficiency by generating a hypomorphic knockin (KI) allele of Mpc1 that results in early perinatal lethality (Bowman et al., 2016). While the complete loss of Mpc1 is not compatible with embryonic development, a surprisingly small quantity of the MPC is sufficient for mammalian development due to an unexpected metabolic flexibility of the growing fetus. Among the alternative substrates that contributed to meeting metabolic demand during late gestation were amino acids and lipids, and defects in embryonic development could be rescued by feeding dams a ketogenic diet (Sussman et al., 2013; Vanderperre et al., 2016). Given that the ketogenic diet mimics fasting, we sought to determine the metabolic flexibility required during maternal nutrient deprivation to support fetal growth and development by utilizing models with genetically tractable metabolic perturbations in key regulatory nodes in mother and fetus.
Here, we have shown that defects in fetal pyruvate metabolism can be rescued by fasting-induced changes to the maternal metabolic milieu. The loss of hepatic fatty acid oxidation from the maternal but not fetal compartment affects fetal metabolic and transcriptional programming. Additionally, the lipid-mediated control of fetal transcriptional programming is phenocopied in an orthogonal model of deficient fatty acid catabolism, peroxisome proliferator-activated receptor alpha (Pparα) knockout (KO) dams, that also revealed a dependence on fetal Pparα signaling. Finally, the combined loss of mitochondrial pyruvate metabolism and hepatic fatty acid oxidation further exaggerated the fetal metabolic and transcriptional response to maternal nutrient stress. These data show the critical role of the maternal metabolic environment in dictating fetal metabolic and transcriptional programming.
RESULTS
Maternal Response to Nutrient Deprivation with Impaired Mitochondrial Pyruvate Transport
The rapidly growing fetus requires a continuous delivery of glucose and oxygen that places an incredible demand on maternal metabolism. The metabolism of glucose and oxygen converge at the transport of pyruvate into mitochondria via the MPC. We previously showed that late-gestation (e17.5) fetuses with a hypomorphic KI allele of Mpc1 exhibited impaired growth and lactic acidosis (Bowman et al., 2016). To understand the role of impaired pyruvate metabolism on the maternal and fetal response to a metabolic challenge during late gestation, we subjected both wild-type (WT) and Mpc1 KI heterozygous (Mpc1KI/+) pregnant dams to a 24-h period of food deprivation from 16.5 to 17.5 days post coitum (dpc). Relative to WT controls, Mpc1KI/+ dams exhibited a 50% reduction and Mpc1KI/KI fetuses exhibited undetectable Mpc1 and Mpc2 protein expression in liver in both the fed and fasted states (Figure 1A). The 24-h fasting paradigm induced an 8% loss in maternal body weight in both genotypes (Figure 1B) and was sufficient to stunt fetal growth of WT and Mpc1KI/+ littermates while Mpc1KI/KI fetal body weights were not further perturbed by maternal fasting (Figure 1C). Fasting induced a marked upregulation of circulating β-hydroxybutyrate in both WT and Mpc1KI/+ dams (Figure 1D), consistent with the accelerated fasting response observed during pregnancy (Freinkel, 1980; Metzger et al., 1982). Serum non-esterified free fatty acid (NEFA) levels were also upregulated by fasting, and Mpc1KI/+ dams exhibited a slight decrease in circulating NEFA concentrations and liver triglyceride content relative to WT dams (Figure 1D). Together, these data suggest that Mpc1KI/+ dams rely more on the utilization of fatty acids as an alternative energy source upon impaired mitochondrial pyruvate metabolism, as others have implicated in non-pregnant rodent models of MPC deficiency (Gray et al., 2015; Li et al., 2016; McCommis et al., 2015; Zou et al., 2018).
Figure 1. Maternal Fasting Ameliorates Metabolic Derangements in Mpc1-Deficient Fetuses.

(A) Immunoblot for Mpc1 and Mpc2 in maternal liver and fetal liver, 17.5 dpc, fed or 24 h fasted.
(B) Maternal body weights and changes in weight from 16.5 to 17.5 dpc.
(C) Late-gestation (e17.5) fetal body weights from fed or fasted Mpc1KI/+ dams.
(D) Maternal blood glucose, serum β-hydroxybutyrate, and serum non-esterified fatty acid (NEFA) concentrations (mean ± SEM, n = 5–6). Maternal liver triglyceride content normalized to protein content (mean ± SEM, n = 5–6).
(E) Steady-state metabolite abundances from e17.5 fetal liver, determined by 1H-NMR. Normalized to protein content and shown relative to WT fed (mean ± SEM, n = 6).
(F) Lactate concentrations from fetal brain and heart measured enzymatically (mean ± SEM, n = 6).
(G) Intracellular lactate concentrations from Mpc1 deletion (D/D) mouse embryonic fibroblasts (MEFs) under standard culture conditions ± 1 mM β-hydroxybutyrate (βHB) for 24 h, determined by 1H-NMR (mean ± SEM, n = 3).
(H) Triglyceride content of placenta and liver at e17.5, determined enzymatically (mean ± SEM, n = 5–8). Data from WT fetuses of WT dams are shaded green. Statistically significant differences (p < 0.05) for pairwise comparisons after ANOVA indicated by letters.
Maternal Fasting Rescues Metabolic Dysfunction in Mpc1-Deficient Fetuses
To better characterize the nutrient allocation between the maternal and fetal compartments, we compared steady-state maternal serum metabolites and maternal liver and fetal liver metabolites by 1H-NMR metabolomics. Consistent with the decrease in blood glucose and increase in serum β-hydroxybutyrate upon fasting, concentrations of these metabolites in maternal liver were similarly regulated (Table S1). As expected, Mpc1KI/KI fetal livers exhibited elevated lactate relative to WT littermates. Interestingly, maternal fasting rescued the lactic acidosis in Mpc1KI/KI fetal liver by reducing lactate concentrations to WT levels (Figure 1E). Mpc1KI/KI fetal brain lactate concentrations were also ameliorated by maternal fasting, while lactate levels in heart tissue were not regulated by maternal nutritional status (Figure 1F). Fetal liver aspartate was also high in Mpc1KI/KI fetuses and was suppressed by fasting similar to liver lactate concentrations (Figure 1E). Fasting induced a greater than 5-fold increase in β-hydroxybutyrate (βHB) levels in fetal liver, and Mpc1KI/KI fetal livers had higher concentrations than WT littermates (Figure 1E). To test whether βHB may have contributed to the fasting-induced suppression of lactic acidosis, WT and Mpc1 deletion (Mpc1D/D) mouse embryonic fibroblasts (MEFs) were cultured in the presence or absence of 1 mM βHB for 24 h, and steady-state intracellular lactate concentrations were measured. Consistent with its role as a fasting-induced alternative oxidative substrate, βHB treatment was sufficient to rescue lactate accumulation in cells lacking the MPC (Figure 1G). These data are consistent with the finding that a maternal ketogenic diet can rescue fetal lactic acidosis and developmental delay in another model of impaired MPC function (Vanderperre et al., 2016). Together, these data show that maternal fasting can ameliorate fetal defects in mitochondrial pyruvate metabolism.
Fasting induces adipose triglyceride mobilization and subsequent hepatic ketone generation, and both of these processes are increased during late gestation. To begin to understand the role of enhanced lipid mobilization during pregnancy (Elliott, 1975; Knopp et al., 1970), we measured several indicators of fetal lipid accrual during maternal fasting. First, we observed that placental triglyceride content was increased upon fasting independent of fetal genotype (Figure 1H), concurrent with increased maternal circulating NEFA during a fast (Figure 1D). In addition, fasting partially rescued lower liver triglyceride levels previously observed in Mpc1KI/KI fetuses (Figure 1H) (Bowman et al., 2016). These data demonstrate that maternal fasting can affect lipid content within the fetal compartment.
Maternal Lipid Metabolism Drives the Fetal Response to Fasting
To determine the contribution of maternal lipid metabolism to the fetal response to fasting, we used a genetic model of impaired fasting—liver-specific loss of mitochondrial β-oxidation of long-chain fatty acids via Cpt2 (Lee et al., 2016b, 2017). Cpt2 catalyzes an obligate step in the mitochondrial import of long-chain fatty acids and is encoded by a single gene, which makes it a genetically tractable target to block mitochondrial β-oxidation. Mice with liver-specific loss of Cpt2 (Cpt2L−/−) have massive hepatic lipid accumulation and impaired ketogenesis upon fasting with, surprisingly, no defects in systemic gluconeogenic capacity (Lee et al., 2016b). Female mice homozygous for the floxed Cpt2 allele (ff) were crossed to males with albumin-driven Cre recombinase expression (Cpt2L−/−), and Cpt2L−/− females were crossed to ff males, such that both WT (ff) and Cpt2L−/− fetuses could be studied from WT or mutant Cpt2L−/− dams (Figure 3B). WT and Cpt2L−/− dams were subjected to 24-h food deprivation from 16.5 to 17.5 dpc, and, remarkably, Cpt2L−/− dams were able to maintain blood glucose concentrations to the same level as fasted WT dams (Figure 2A). Consistent with a defect in liver fatty acid oxidation, however, Cpt2L−/− dams could not produce βHB during a fast, and circulating βHB levels in fasted Cpt2L−/− dams were even lower than fed WT controls (Figure 2A). Fasting also induced serum NEFA and triglyceride accumulation in Cpt2L−/− maternal serum (Figure 2A). Altogether, the impaired ketogenesis and elevated circulating lipid metabolite concentrations of Cpt2L−/− dams provide a useful model of impaired maternal fasting to better understand the fetal response to maternal nutrient deprivation.
Figure 3. The Maternal Response to Fasting Drives the Fetal Transcriptional Program.

(A) Volcano plot of RNA-seq data comparing WT fetal liver derived from fetuses gestating in either WT or Cpt2L−/− 24-h-fasted dams. Red points are significant with a threshold of z ≥ |3|.
(B) Experimental scheme.
(C) mRNA abundance of Cpt2 and select fastingregulated genes Acot1, Acot2, and Pdk4 in e17.5 liver from all genotypes (mean ± SEM, n = 6). Statistically significant differences (p < 0.05) for pairwise comparisons after ANOVA indicated by letters, ***p < 0.001.
See also Figure S1.
Figure 2. Maternal Lipid Metabolism Provides Substrates for Fetal Metabolism.

(A) Blood glucose, serum β-hydroxybutyrate, NEFA, and triglyceride levels from fed and 24-h-fasted liver-specific Cpt2 KO pregnant dams (Cpt2L−/−) and Cpt2f/f (WT) controls at 17.5 dpc (mean ± SEM, n = 6–8).
(B) Fetal body weights of control and Cpt2L−/− progeny from control and Cpt2L−/− pregnancies (mean ± SEM, sample size indicated).
(C) Fetal liver β-hydroxybutyrate concentrations determined by 1H-NMR (mean ± SEM, n = 6).
(D) Triglyceride content of placenta and liver at e17.5, determined enzymatically (mean ± SEM, n = 5–6).
(E) Maternal blood acylcarnitine abundances at 17.5 dpc determined from dried blood spots after extraction, butylation, and tandem mass spectrometry (mean ± SEM, n = 5–8).
(F) Fetal liver acylcarnitine abundances at e17.5 (mean ± SEM, n = 5). For each pair of colored bars, the left bar represents control fetuses and the right hatched bar represents Cpt2L−/− fetuses from the maternal genotype-nutritional combinations represented in (E).
Statistically significant differences (p < 0.05) for pairwise comparisons after ANOVA indicated by letters.
Fasting reduced late-gestation Cpt2L−/− fetal body weights to the same extent as had been observed in WT litters, with no effect of fetal or maternal genotype (Figure 2B). Importantly, loss of mitochondrial β-oxidation of long-chain fatty acids in fetal liver did not alter βHB concentrations as determined by 1H-NMR, but instead, fetal liver βHB levels reflected maternal circulating concentrations such that fetuses from fasted Cpt2L−/− dams had levels as low as fetuses from fed dams of either genotype (Figure 2C). Fetal liver and placental triglyceride content increased upon fasting and was similarly regulated independent of fetal genotype (Figure 2D). These data show that maternal fasting increases lipid metabolite delivery to the fetus but fetal liver has little capacity for ketogenesis.
To further determine the effects of altered maternal lipid metabolism on the fetal response, we characterized the abundance of a class of molecules that can serve as substrates for the enzymatic function of Cpt2: the long-chain acylcarnitines. Cpt2 catalyzes the transacylation of long-chain acylcarnitines to become long-chain acyl-CoAs in the mitochondrial matrix; therefore, loss of Cpt2 would be expected to increase long-chain acylcarnitine concentrations. We measured both maternal blood acylcarnitines and fetal liver acylcarnitines from WT and Cpt2L−/− dams in both the fed and 24-h fasted states (Figures 2E and 2F). Similar to Cpt2L−/− mice on a ketogenic diet, pregnant Cpt2L−/− mice exhibited elevated circulating long-chain acylcarnitine concentrations during fasting and suppressed free carnitine and short-chain acylcarnitines such as C2 (acetylcarnitine) (Lee et al., 2016b). Additional short-chain acylcarnitines (C4 and C5), which are derived from amino acid catabolism, were also downregulated in Cpt2L−/− circulation (Figure 2E). Long-chain acylcarnitine abundances were upregulated in fetal liver from fasted WT dams, consistent with increases in maternal circulating concentrations in response to fasting, and there were no effects of fetal genotype (Figure 2F). Interestingly, however, long-chain acylcarnitine concentrations in fetal livers from Cpt2L−/− dams were not increased to the extent that is seen in maternal blood. Increased maternal oxidation of long-chain acylcarnitines or impaired fetal fatty acid utilization in litters from Cpt2L−/− dams (independent of fetal genotype) could contribute to this pattern of acylcarnitine levels. These data show that fetal liver fatty acid oxidation plays a negligible role in maintaining fetal metabolic homeostasis during a fast, but maternal lipid metabolism determines fetal lipid metabolite concentrations, particularly during an acute nutrient stress.
Maternal Fasting Response Drives the Fetal Liver Transcriptional Program
The loss of hepatic Cpt2 in adult mice results in dyslipidemia coupled with dramatic increases in the expression of pro-catabolic fatty-acid-responsive genes in the liver and distant organs such as adipose and kidney (Lee et al., 2016b, 2017). To characterize the putative endocrine impact of impaired maternal lipid catabolism on the fetal liver transcriptional response, we performed an RNA sequencing (RNA-seq) analysis of WT fetal liver derived from fetuses exposed in utero to the metabolic milieu of either WT or Cpt2L−/− 24-h fasted dams (Figure S1). In this way, the effects of in utero exposure to impaired maternal lipid metabolism on the fetal liver transcriptome were determined independent of fetal genotype. We found metabolic genes such as enoyl-coenzyme A, hydratase/3-hydroxyacyl coenzyme A dehydrogenase (Ehhadh), and pyruvate dehydrogenase kinase 4 (Pdk4) were induced, and angiopoietin-like 8 (Angptl8) and acyl-coenzyme A amino acid N-acyltransferase 1 (Acnat1) were suppressed in fetal liver from Cpt2L−/− dams by 2.5-fold or greater, demonstrating a robust effect of maternal metabolism on fetal gene expression (Figure 3A). In order to validate and expand the genes identified, we compared a subset of these genes under all combinations of genetic and environmental exposure: WT and Cpt2L−/− genotypes from both maternal and fetal compartments under both fed and fasted conditions (Figure 3B). The mRNA abundances of several fasting-regulated genes (acyl-CoA thioesterase 1 (Acot1), Acot2, and Pdk4) were increased in fetal liver in response to maternal nutrient deprivation (Figure 3C). While fetuses from fasted WT dams responded similarly to Cpt2L−/− fetuses from WT fasted dams, both WT and Cpt2L−/− fetuses from fasted Cpt2L−/− dams exhibited significantly higher induction of these fastingregulated genes than litters from WT dams (Figure 3C). Here, we have shown that the fetal liver transcriptional response to maternal fasting was highly dependent on maternal but not fetal genotype, i.e., the fetal environment was the main determinant for the transcriptional response independent of the role of fetal fatty acid catabolism. This suggests that a metabolic or endocrine signal altered in fasted Cpt2L−/− dams promotes an exacerbated fasting transcriptional program in fetuses exposed to this intrauterine environment.
The Fetal Response to Fasting Is Pparα-Dependent and Regulated by Maternal Metabolism
To gain a better understanding of the maternal metabolic environment and potential metabolite mediators of the endocrine response that led to changes in fetal hepatic transcription, we performed global untargeted metabolomics on serum from 24-h-fasted control and Cpt2L−/− female non-pregnant and pregnant mice (Figures 4 and S2). Additionally, we analyzed serum from 24-h-fasted pregnant Pparα KO mice, Mpc1KI/+ mice, and Mpc1KI/+; Cpt2L−/− (double knockout [DKO]) dams to identify conserved metabolic alterations that could account for the fetal response to maternal fasting in these models (Figure 4A). A principle component analysis showed that the serum metabolomes of pregnant and non-pregnant mice are readily distinguishable based on pregnancy alone (Figure 4B). In addition, Cpt2L−/− and Pparα KO dams, despite both having defects in fatty acid oxidation, exhibit unique serum metabolomes (Figure 4B). Consistent with the requirement of hepatic fatty acid β-oxidation for ketone body production, all Cpt2L−/− mice exhibited a dramatic reduction in β-hydroxybutyrate, and Pparα KO dams exhibited a significant reduction as well (Figure 4C). Pregnant mice had higher corticosterone levels than non-pregnant mice, as expected, but there was no effect of maternal genotype (Figure 4D). Other pregnancy-specific changes were seen, such as increased circulating lactate and depleted glycine (Figure 4D). As we have demonstrated with targeted metabolomics in Cpt2L−/− mice (Figure 2E), many short-chain acylcarnitine species were suppressed in Cpt2L−/− but not Pparα KO dams (Figure 4E). One of the most striking effects seen in both Cpt2L−/− and Pparα KO dams was the overall increase in circulating lipid metabolites (Figure S2). For example, most long-chain free fatty acids, including docosahexaenoic acid (DHA) and palmitate, were increased in both models (Figures 4E and 4F). Broad classes of fatty acids and their metabolites were increased in Cpt2L−/− and Pparα KO dams. This includes known signaling metabolites such as endocannabinoids and putative Pparα ligands (Figures 4E and 4F). These data suggest that increasing signaling lipids in response to maternal nutrient stress or genetic perturbations can signal from dam to fetus to alter fetal liver transcriptional programming.
Figure 4. Untargeted Metabolomics Reveals Pregnancy- and Genotype-Regulated Serum Metabolites.

(A) Unsupervised clustering of all serum metabolites.
(B) Principle component analysis of the serum metabolome data.
(C) Relative concentrations of serum β-hydroxybutyrate in pregnant (17.5 dpc) and non-pregnant mice of the genotypes indicated (mean ± SEM, n = 6).
(D) Pregnancy-specific changes in metabolites.
(E) Lipid metabolite changes as a consequence of genotype.
(F) Broad classes of lipids derived from the fatty acid palmitate that are regulated by genotype (mean ± SEM, n = 6).
Statistically significant differences (p < 0.05) for pairwise comparisons after ANOVA compiled in Table S2.
See also Figure S2.
The metabolomics analysis demonstrated an induction of circulating Pparα ligands, and many of the genes observed to be transcriptionally regulated in fetal liver in response to impaired mitochondrial pyruvate metabolism (Bowman et al., 2016) or in response to maternal fasting are canonical Pparα target genes. Therefore, we next turned our attention to mice with a deletion of the lipid-dependent transcriptional regulator and so-called master regulator of fatty acid catabolism, Pparα. Upon subjecting dams to 24-h food deprivation, e17.5 fetuses with heterozygous or homozygous Pparα deletion from Pparα−/− dams exhibited a decrease in body weight comparable to fasted WT litters (Figure 5A). Moreover, Pparα+/− and Pparα+/− fetuses from Pparα+/− dams were not different in size than the same fetal genotypes from Pparα+/− dams. Therefore, the loss of Pparα in the maternal or fetal compartment did not affect fetal growth, similar to what was observed for Cpt2L−/− mice (Figure 2B).
Figure 5. Pparα-Dependent Fetal Response to Maternal Fasting Is Potentiated by Impaired Maternal Lipid Metabolism.

(A) e17.5 body weights from WT dams, Pparα−/− dams carrying heterozygous or homozygous Pparα deletion litters (shaded in yellow), or all three fetal genotypes (WT, +/−, and −/−) from Pparα+/− intercrosses (shaded in orange). Circles are from fed litters and squares are from fasted litters.
(B) Volcano plot of RNA-seq data comparing Pparα+/− fetal liver derived from fetuses gestating in either Pparα+/ or Pparα−/− 24-h-fasted dams.
(C) Volcano plot of RNA-seq data comparing Pparα−/− fetal liver from Pparα−/− 24-h-fasted dams to Pparα+/− fetuses from Pparα+/− dams. Red points are significant with a threshold of z ≥ |3|.
(D) mRNA abundance of fasting-regulated genes Acot1, Acot2, Pdk4, Ehhadh, Apoa4, and Epn3 in fetal liver, same genotypes as described in (A). Open bars are for fed samples; filled bars are for fasted samples (mean ± SEM, n = 6).
Statistically significant differences (p < 0.05) for pairwise comparisons after ANOVA compiled in Table S2.
To examine the dependence of the fetal transcriptional response to maternal Pparα, we performed RNA-seq on Pparα+/− fetal livers that were derived from fasted Pparα+/− or Pparα−/− dams. Similar to Cpt2L−/− dams, the loss of maternal Pparα and induction of lipid signaling molecules in maternal circulation was sufficient to induce Pparα-dependent genes in Pparα+/− fetal livers (Figure 5B). Additionally, we performed RNA-seq analysis on Pparα+/− fetal livers from Pparα+/− dams to understand the dependence of these changes on fetal Pparα. We identified many genes that were induced upon fasting in Pparα+/− fetuses gestating in Pparα−/− dams that were dependent upon fetal Pparα signaling—that is, genes with lower expression in Pparα−/− fetuses (Figures 5C and S1). To expand and validate these changes, we examined fasting-regulated gene induction in fed and fasted WT, Pparα+/−, and Pparα−/− fetal livers from WT, Pparα+/−, and Pparα−/− dams (Figure 5D). The genes shown in Figure 5D demonstrated Pparα-dependent fasting induction in that Pparα−/− livers failed to induce expression of these genes (Acot1, Acot2, Pdk4, Ehhadh, Apoa4, and Epn3) upon fasting. While the fetal liver transcriptional response to fasting requires fetal Pparα, the effect of the intrauterine environment is also critical for eliciting these responses, as the fetal response to fasting in Pparα−/− dams was most robust (Figure 5D). These data again suggest that maternally derived lipid signals from models of impaired fatty acid catabolism can potentiate the fetal liver response to maternal nutrient deprivation and that the fetal response is at least partially Pparα dependent. This is consistent with the Cpt2L−/− model in which fetal liver from fasted Cpt2L−/− dams had higher liver expression of fastingregulated transcripts than fetal liver from fasted WT control dams (Figure 3).
Combined Deficiencies of Carbohydrate and Fatty Acid Metabolism further Potentiate the Fetal Response to Maternal Fasting
To understand the effects of maternal lipid metabolism on fetuses with impaired glucose metabolism, we crossed Mpc1KI/+ and Cpt2L−/− mice to generate dams with defective hepatic fatty acid oxidation and heterozygosity for Mpc1 deficiency (Cpt2L−/−; Mpc1KI/+). These dams carried fetuses that were either WT or Mpc1 deficient (Mpc1KI/KI) from mating to Cpt2f/f; Mpc1KI/+ males. Mpc1 heterozygosity did not further potentiate defects in Cpt2L−/− maternal serum chemistry (Figures 6A and 6B). Following a 24-h fast Cpt2L−/−; Mpc1KI/+ 17.5 dpc dams maintained normal fasting blood glucose, exhibited elevated NEFA and triglycerides, and showed an absence of βHB, as expected (Figure 6A). Additionally, 24-h-fasted Cpt2L−/−; Mpc1KI/+ 17.5 dpc dams exhibited decreased blood carnitine and short-chain acylcarnitines with elevated long-chain acylcarnitines similar to Cpt2L−/− dams (Figure 6B). These data show that late-gestation Cpt2L−/−; Mpc1KI/+ dams exhibit a similar metabolic dysfunction to Cpt2L−/− dams, including a loss in βHB and increased circulating lipids, consistent with global untargeted metabolomics (Figure 4).
Figure 6. Combined Deficiencies of Carbohydrate and Fatty Acid Oxidation Further Potentiate the Fetal Response to Maternal Fasting.

(A) Maternal blood glucose, blood lactate, and serum β-hydroxybutyrate, NEFAs, and triglyceride (TG) concentrations after 24-h fast from 16.5 to 17.5 dpc (mean ± SEM, n = 5–8).
(B) Maternal 24-h-fasted blood acylcarnitine abundances shown relative to control dam (mean ± SEM, n = 5).
(C) Fetal brain lactate concentrations measured enzymatically (mean ± SEM, n = 5).
(D) Steady-state metabolite abundances from e17.5 fetal liver, determined by 1H-NMR. Normalized to protein content and shown relative to WT fetus from MpcKI/+ dam (mean ± SEM, n = 5).
(E) Fetal liver transcriptional response determined by qRT-PCR (mean ± SEM, n = 5–6).
Statistically significant differences (p < 0.05) for pairwise comparisons after ANOVA indicated by letters.
Next, we measured the lactate concentration in WT and Mpc1KI/KI e17.5 fetal brains from Mpc1KI/+ dams with (Cpt2f/f; Mpc1KI/+) and without (Cpt2L−/−; Mpc1KI/+) hepatic fatty acid oxidation following a 24-h fast. As expected, the loss of maternal hepatic fatty acid oxidation eliminated βHB accumulation in both WT and Mpc1KI/KI fetal liver (Figure 6D). The loss of ketone body production in Cpt2L−/−; Mpc1KI/+ dams modestly affected lactate accumulation in Mpc1KI/KI e17.5 fetal brains (Figure 6C). By profiling fetal liver metabolites by 1H-NMR, the loss of maternal and fetal Mpc1, in addition to the loss of maternal βHB, resulted in significant suppressions in glucose and tricarboxylic acid (TCA) cycle intermediates (Figure 6D). The duel impairment of maternal and fetal carbohydrate and lipid mitochondrial metabolism created significant metabolic limitations for the developing fetus.
Finally, we assayed fetal transcriptional programming in the liver of fetuses exposed to these disparate maternal environments. As we have shown before, Cpt2L−/− dams potentiate the fetal response to fasting. Consistent with the increased metabolic dysfunction in the fetal liver, the loss of one copy of Mpc1 in Cpt2L−/− dams greatly exacerbated the fetal response to fasting as Acot1, Acot2, Cpt1b, and Pdk4 were further increased in fetal livers of Cpt2L−/−; Mpc1KI/+ dams (Figure 6E). In fact, fetal liver Pck1 expression from fasted Cpt2L−/−; Mpc1KI/+ dams was induced ~40-fold over fed WT controls. As we showed previously, fetal loss of Mpc1 further potentiated this transcriptional response independently by ~2-fold. These data show that the compound loss of Mpc1 and Cpt2 greatly enhanced the fetal transcriptional response to maternal fasting above what was observed when either was deleted alone. Altogether, the metabolic and transcriptional changes observed in these genetic models provide evidence for robust metabolic communication between mother and fetus, particularly in the context of nutrient restriction.
DISCUSSION
The fetus is thought to utilize nutrients much like the adult mammalian brain; that is, it preferentially utilizes glucose but can switch to ketone bodies following prolonged fasting (Girard et al., 1977). Mutations in the MPC result in pyruvate and lactate acidosis in mouse models and human inborn errors that are not compatible with sustained independent life (Bricker et al., 2012; Herzig et al., 2012). In utero, these effects can be somewhat mitigated by maternal metabolite clearance, but MPC mutations result in developmental defects (Bowman et al., 2016; Vanderperre et al., 2016). Incredibly, simply feeding pregnant mice a ketogenic diet from 8.5 dpc can rescue many of the developmental defects seen in MPC mutant fetuses (Sussman et al., 2013; Vanderperre et al., 2016). As the ketogenic diet is a fasting mimetic, we sought to understand the effects of maternal fasting on fetal metabolism and have shown that fasting is sufficient to reverse many of the metabolic alterations in MPC mutant fetuses. This is remarkable given the putative requirements of the fetus for glucose and suggests an incredible and previously unrecognized metabolic flexibility in the rapidly growing fetus.
Due to the incredible demand for glucose during gestation, fasting during pregnancy results in an accelerated starvation response that is characterized by an earlier and more exaggerated shift to ketone body utilization (Boden, 1996; Freinkel, 1980). We have demonstrated that ketone bodies derived from maternal liver fatty acid oxidation, in combination with increased serum concentrations of triglyceride, phospholipids, and NEFAs, may help mediate this metabolic adaptation. Levels of these metabolites increase over the course of gestation, and circulating triglyceride levels, for example, increase more than 250% by the end of pregnancy, which is almost twice the post-prandial concentration in a non-pregnant individual (Darmady and Postle, 1982; Haggarty, 2010). The increase in concentrations of these lipids and lipid-derived metabolites is so dramatic that overnight fasting (14–18 h) in normal late-term pregnancy leads to serum metabolite concentrations that rival the effects of 2–3 days of starvation in non-pregnant individuals (Boden, 1996; Metzger et al., 1982). Fasting during pregnancy results in an earlier than normal shift from glucose to fat and ketone utilization by maternal tissues to spare glucose and amino acids for fetal uptake (Metzger et al., 1982), and we have shown that mitochondrial metabolism is an important regulator of these changes in substrate utilization.
Deleting Cpt2 and therefore fatty acid β-oxidation from the liver renders mice incapable of generating sufficient ketone bodies and increases serum lipid levels (Lee et al., 2016b, 2017). We took advantage of this model to understand the contribution of maternal lipid metabolism to the fetal fasting response, including the requirement for ketone body delivery to the fetus. We have previously shown that adult Cpt2L−/− mice exhibit an incredible induction of pro-catabolic genes in their livers following a fast (Lee et al., 2016b). Here, we have shown that fetal liver β-hydroxybutyrate levels closely reflect maternal circulating concentrations, and the loss of fatty acid β-oxidation does not affect transcriptional programming in a fetal-autonomous manner. However, the loss of Cpt2 from maternal liver is critically important for fetal ketone body delivery and fetal transcriptional programming in response to fasting.
Following a fast, adult Cpt2L−/− mice induce a pro-catabolic endocrine program in non-hepatic tissues to maintain systemic energy homeostasis (Lee et al., 2016b). Similarly, defects in maternal hepatic fatty acid oxidation or a loss of maternal Pparα generates an endocrine signal that affects fetal liver transcriptional programming. Classic maternal-fetal endocrine loops have been identified, such as glucocorticoid signaling, that are critical for fetal development, maturation, and parturition during late gestation (Liggins, 1968; Liggins et al., 1967; Rando et al., 2016). We have shown that maternal nutrient deprivation or defects in maternal lipid catabolism can signal to the fetus presumably through the dramatic increase in circulating maternal fatty acid Pparα ligands. Untargeted and targeted metabolomics revealed broad classes of signaling lipids that are induced, including Pparα ligands and endocannabinoids such as acyle-thanolamines and acyltaurines. In contrast to proteinaceous hormones, many of the hydrophobic metabolites that we identified would be predicted to cross the hemochorial placenta, although detailed pharmacokinetics have not been performed. While we have shown that the loss of fetal Pparα abrogates much of the transcriptional response, there is clearly Pparα-independent regulation as well. This may be mediated by other classes of metabolites, for example, the endocannabinoids (Correa et al., 2016). The loss of these critical metabolic nodes in central carbon metabolism affects many pathways, as we have shown in the metabolomics analysis, and some of the transcriptional effects may be quite indirect by affecting placental metabolism or transport.
The acute fasting paradigm employed here should be contrasted with chronic nutritional deprivation caused by poor nutrition, limited access to food, or placental insufficiency. Acute fasting is not an absence of nutrients but a dramatic shift in the type of nutrients available, many being lipids or lipid-derived metabolites. The types of genes affected by fasting and potentiated by a lack of maternal lipid catabolism suggest that the fetus is accelerating its ability to utilize oxidative metabolism. The fetus increases the expression of genes involved in oxidative metabolism throughout pregnancy as oxygen becomes more available and to prepare the fetus for the consumption of postpartum milk fat. By our model, we propose that fasting in late gestation may hasten this progression in preparation for parturition.
Together, our genetic mouse models of impaired mitochondrial pyruvate and fatty acid metabolism demonstrate the importance of mitochondrial oxidative metabolism in regulating both the maternal and fetal responses to nutrient deprivation. Despite the nutritionally sensitive nature of this stage in mammalian development, there exists remarkable metabolic flexibility and crosstalk among these major pathways of central carbon metabolism. Maternal-fetal metabolic communication helps ensure adequate nutrient exchange and is particularly important during times of nutrient limitation. Miscommunication may result in metabolic complications during pregnancy including gestational diabetes, and promoting healthy maternal-fetal metabolic communication may prove an important therapeutic approach for metabolic disorders of pregnancy.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
All unique/stable reagents generated in this study are available from the Lead Contact without restriction. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Wolfgang (mwolfga1@jhmi.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Generation of Genetic Mouse Models
Mpc1 knock-in mice were generated by targeting loxP sequences to introns flanking exons 3 to 5 of the mouse Mpc1 gene and targeting a transcriptional reporter construct to the 3′ untranslated region of Mpc1 by homologous recombination in C57BL/6J embryonic stem cells by standard methods, as previously described (Bowman et al., 2016). A conditional knock-out (KO) allele of mouse Cpt2, carnitine palmitoyltransferase 2, was generated by the same approach (Gonzalez-Hurtado et al., 2017, 2018; Kim et al., 2017; Lee et al., 2015, 2016a, 2016b; Nomura et al., 2016). To generate mice with a liver-specific loss of mitochondrial fatty acid β-oxidation, Cpt2 floxed mice were crossed to Albumin-Cre (Alb-Cre) transgenic mice (Postic et al., 1999) to generate animals (Cpt2L−/−) that cannot oxidize long-chain fatty acids in hepatocytes (Lee et al., 2016b, 2017). PPARα−/− mice (stock number 008154) were obtained on a C57BL/6J background from The Jackson Laboratory and were crossed to wild-type (WT) mice for studies of heterozygotes (Lee et al., 1995).
All mutant mice and littermate controls were housed in a facility with ventilated racks on a 14h light/10h dark cycle with ad libitum access to a standard rodent chow (2018SX Teklad Global, 18% protein). For timed matings, the presence of a copulatory plug in the morning was designated as 0.5 days post coitum (dpc), and pregnant females were housed separately. Female mice pregnant with their first litter were 12–18 weeks old at the end of late gestation studies. Non-pregnant controls were age matched. For 24h fasting experiments, food was removed from pregnant dams at 16.5 dpc between 13:00–15:00, and tissues were collected 24h later at 17.5 dpc.
Ethical Statement
All procedures were performed in accordance with the NIH’s Guide for the Care and Use of Laboratory Animals and under the approval of the Johns Hopkins Medical School Animal Care and Use Committee.
Cell Culture
Mouse embryonic fibroblasts (MEFs) were obtained from timed pregnant dams by standard methods. Primary MEFs were maintained in DMEM (25mM glucose) supplemented with 10% fetal bovine serum and 1% pen/strep antibiotic (Invitrogen) at 37°C in a humidity-controlled incubator at 10% CO2. For 1H-NMR of cell extracts, cells were switched to serum-free DMEM containing 2.5mM glucose, 2.5mM pyruvate, and 2mM glutamine in the presence or absence of 1mM D,L-β-hydroxybutyrate (Sigma-Aldrich, H6501) for 24h before collection of cell extracts for steady-state metabolite measurements as described above.
METHOD DETAILS
Serum and Tissue Metabolite Measurement
Serum metabolites were determined by enzymatic, colorimetric assays, according to the manufacturer’s instructions: β-hydroxybutyrate (Stanbio B-HB LiquiColor Assay, EKF Diagnostics, Boerne, TX), NEFA (NEFA-HR(2), Wako Diagnostics, Richmond, VA), triglyceride (TR0100, Sigma-Aldrich, St. Louis, MO). Placental and liver triglyceride content was also determined by enzymatic measurement after chloroform: methanol (2:1 (v/v)) extraction, drying, and resuspension in 3:1:1 (v/v/v) t-butanol: methanol: Triton X-100. Triglyceride levels were normalized to protein content as determined by bicinchoninic acid (BCA) assay (Thermo Fisher Scientific). Blood glucose and lactate were measured with a Nova Max Plus glucometer and Lactate Plus meter (Nova Biomedical, Waltham, MA). Tissue lactate concentrations were determined enzymatically after homogenizing frozen tissues in lactate assay buffer on ice and were normalized to protein content (MAK065, Sigma-Aldrich, St. Louis, MO).
For 1H-NMR of tissue and serum metabolites, extraction was performed as previously reported (Lee et al., 2017). For 1H-NMR of cell extracts, mouse embryonic fibroblasts (MEFs) were washed with ice-cold PBS and scraped into ice-cold methanol and snap frozen in liquid nitrogen before two additional freeze-thaw rounds with methanol extraction (Bowman et al., 2017). Cell extracts were subjected to a final spin at 15,000 g for 1 min at 4°C, then the supernatant was dried under vacuum and the extract resuspended in 20 mM phosphate buffer, pH 7.4 ± 0.1, with 0.1 mM TMSP as an internal reference and 0.1 mM sodium azide. 1H spectra were recorded on a Bruker Avance III 500MHz (Bruker Instruments, Germany) NMR spectrometer, operating at 499.9MHz and equipped with room temperature quadruple nuclei probe. Typical 1H spectra were acquired using presaturation solvent suppression pulse sequence (noesyprld). Acquisition parameters were set as follows; spectral width of 8012.820 with 64K data points, 512 scans, with a relaxation delay of 7 s for a total collection time of 1.14h. Samples were automatically tuned and matched, and shimmed to TMSP signal. Spectra were exported into Bruker format and were processed with Chenomx NMR Suit 8.2 Professional (Chenomx Inc, Edmonton, Alberta, Canada). TMSP signal (0.0 ppm) was used as a reference peak, spectra were manually phase corrected and spline function was applied for the baseline correction. Metabolites were profiled and quantified using built-in Chenomx 500MHz library. Metabolite concentrations were normalized to protein content. Untargeted metabolomics was done as previously described (Bowman et al., 2016).
Immunoblotting
Tissue lysates for SDS-PAGE were prepared by homogenization of tissue in RIPA buffer (50mM Tris-HCl, pH 7.4, 150mM NaCl, 1mM EDTA, 1% Triton X-100, 0.25% deoxycholate) with protease inhibitor cocktail (Roche) and PhosSTOP phosphatase inhibitor (Roche), followed by pelleting of insoluble debris at 13,000 g for 15 min at 4°C. Protein concentrations of lysates were determined by BCA assay (Thermo Scientific), and 30μg of lysate was separated by Tris-Glycine SDS-PAGE (15% polyacrylamide), unless otherwise noted. Proteins were transferred to PVDF membranes (Immobilon), blocked in 5% nonfat-milk TBST, and incubated with primary antibodies overnight. Primary antibodies used include: We generated and validated affinity-purified rabbit polyclonal antibodies against mouse Mpc1 and Mpc2 (Bowman et al., 2016); heat shock chaperone 70 (Hsc70) mouse monoclonal at 1:1000 (Santa Cruz sc-7298); Cy3-conjugated anti-mouse (Invitrogen) or Cy5-conjugated anti-rabbit (Invitrogen) secondary antibodies at 1:1500, or horseradish peroxidase (HRP)-conjugated anti-rabbit (GE Healthcare NA934V) secondary antibodies at 1:2000 were incubated with washed membranes, and proteins were visualized with Amersham Prime enhanced chemiluminescent substrate (GE Healthcare) or epifluorescence on an Alpha Innotech MultiImage III instrument. Protein abundance was quantified using Alpha Innotech FluorChem Q software (Santa Clara, CA) and was normalized to Hsc70 expression.
Quantitative Real-Time PCR
Total RNA was extracted using Trizol reagent, per manufacturer’s recommendations (Life Technologies) and was further purified using the RNeasy Mini kit (QIAGEN). RNA was quantified by NanoDrop, and cDNA was synthesized using random primers and MultiScribe High-Capacity cDNA reverse transcription kit (Applied Biosystems). RT-PCR was performed using 10 ng of template cDNA in a 20μL reaction using Bio-Rad SsoAdvanced SYBR Green master mix with primers specific to the genes of interest (Table S3). All PCR reactions were carried out in a Bio-Rad CFX Connect Thermocycler and were concluded with a melt-curve determination step. Expression data from mouse tissues was normalized to the average Ct values for 4 reference genes: 18S, Rpl22, β-actin, and Gapdh, and data are expressed as 2−dCt and shown relative to wild-type or fed controls, as appropriate.
RNA Sequencing and Analysis
Total RNA was extracted from frozen liver tissue from wild-type, Pparα+/−, or Pparα−/− e17.5 mice (n = 4, 2 male, 2 female) from fasted WT, Cpt2L−/−, Pparα+/−, or Pparα−/− dams using Trizol reagent and purified using the RNeasy Mini kit (QIAGEN) as described above. RNA quality was assessed, and Illumina TruSeq stranded mRNA library was prepared for sequencing single 75bp reads using an Illumina NextSeq 500 sequencing platform. CLC Genomics Server v10.0.1 was used to assemble and align reads to the mouse reference genome GCRm38.p6, and determine FPKM. Log2 transformed FPKM values were quantile normalized with Partek Genomics Suite v7.0. Differential expression between samples was calculated using one-way ANOVA. Significance threshold was set as a binned z-score of 3 for transcripts that were found in minimum 2 animals per comparison genotype, where z = (Xgene—Xpopulation /σpooled). Use of z-score threshold accounts for total variance (σ2) in Volcano plots of NCBI protein coding genes were generated using GraphPad Prism.
Acylcarnitine Analysis of Blood and Liver
Acylcarnitine abundances were determined from dried blood spot samples (Chace et al., 1997; Sandlers et al., 2012) or from frozen liver, as previously described (Lee et al., 2016b, 2017). Tissues acylcarnitine concentrations were normalized to frozen tissue weight. Briefly, samples were methanol extracted and butylated in the presence of acid, further extracted, and analyzed on an API 3200 (AB SCIEX) operated in positive ion mode, with a precursor ion scan for m/z 85, which is a characteristic product ion of butyl ester acylcarnitine species.
Pathway Analysis
Pathway analysis was carried out using quantile normalized RNA-seq values. Indicated ANOVA values were taken from the one-way ANOVA described in RNA sequencing and analysis. Genotypic transcript levels were calculated by normalizing an individual FPKM count to the average count for a given gene. The PCA plot was generated using the web-based data analysis tool MetaboAnalyst. The normalized expression heatmap was generated from the genes with the 200 lowest grand p values from the one-way ANOVA. The Pparα target heatmap was generated using the log2 fold change over control values as determined by one-way ANOVA. Target genes were obtained from PPARgene (Fang et al., 2016). Heatmaps were visualized using GraphPad Prism 8. KEGG pathway analysis was carried out using DAVID on differentially expressed transcripts relative to control genotypes as determined by the significance threshold of z ≥ |3| in a one-way ANOVA (Huang da et al., 2009). Metabolomics pathway analysis was performed via MetaboAnalyst. (Chong et al., 2018).
QUANTIFICATION AND STATISTICAL ANALYSIS
One-way and two-way ANOVA were used to evaluate statistical significance for quantitation of body weights and steady-state metabolite concentrations. For global serum metabolomics, unsupervised hierarchical clustering and principle component analysis (PCA) were conducted using the web-based metabolomics data analysis tool MetaboAnalyst (Chong et al., 2019).
DATA AND CODE AVAILABILITY
Accession Numbers
The Gene Expression Omnibus (GEO) accession number for the RNA-seq data reported in this paper is GSE129368.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mpc1, rabbit polyclonal | Bowman et al., 2016 | N/A |
| Mpc2, rabbit polyclonal | Bowman et al., 2016 | N/A |
| Hsc70, heat shock chaperone 70, mouse monoclonal | Santa Cruz | Cat#sc-7298; RRID: AB_627761 |
| Deposited Data | ||
| RNaseq data | This paper | GEO GSE129368 |
| Experimental Models: Cell Lines | ||
| Mpc1 deletion mouse embryonic fibroblasts (MEFs) | Bowman et al., 2016 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Mpc1 knock-in (KI) hypomorphic allele in C57BL/6J mice | Bowman et al., 2016 | N/A |
| Mouse: Cpt2 floxed allele in C57BL/6J mice with Albumin-Cre transgene | Lee et al., 2016b, 2017 | N/A |
| Mouse: Pparα knock-out (KO) in C57BL/6J mice | Lee et al., 1995; The Jackson Laboratory | JAX: 008154 |
| Oligonucleotides | ||
| See Table S3 | N/A | N/A |
Highlights.
Maternal fasting rescues deficits in fetal pyruvate metabolism
Maternal lipid metabolism drives the fetal response to fasting
Deficits in maternal lipid catabolism potentiate the fetal response to fasting
The fetal response to fasting is Pparα dependent
ACKNOWLEDGMENTS
This work was supported in part by NIH grants R01NS072241 and R01DK116746 to M.J.W. and R01NS110808 and R01NS111230 to S.S. C.E.B. was supported in part by a fellowship from the American Heart Association (15PRE25090309).
Footnotes
DECLARATION OF INTERESTS
The authors have no competing financial interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.09.053.
REFERENCES
- Blackburn ST (2007). Maternal, Fetal, and Neonatal Physiology: A Clinical Perspective, Third Edition (Saunders Elsevier; ). [Google Scholar]
- Boden G (1996). Fuel metabolism in pregnancy and in gestational diabetes mellitus. Obstet. Gynecol. Clin. North Am 23, 1–10. [DOI] [PubMed] [Google Scholar]
- Bowman CE, Zhao L, Hartung T, and Wolfgang MJ (2016). Requirement for the Mitochondrial Pyruvate Carrier in Mammalian Development Revealed by a Hypomorphic Allelic Series. Mol. Cell. Biol 36, 2089–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman CE, Rodriguez S, Selen Alpergin ES, Acoba MG, Zhao L, Hartung T, Claypool SM, Watkins PA, and Wolfgang MJ (2017). The Mammalian Malonyl-CoA Synthetase ACSF3 Is Required for Mitochondrial Protein Malonylation and Metabolic Efficiency. Cell Chem. Biol 24, 673–684.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, et al. (2012). A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chace DH, Hillman SL, Van Hove JL, and Naylor EW (1997). Rapid diagnosis of MCAD deficiency: quantitative analysis of octanoylcarnitine and other acylcarnitines in newborn blood spots by tandem mass spectrometry. Clin. Chem 43, 2106–2113. [PubMed] [Google Scholar]
- Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, and Xia J (2018). MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46 (W1), W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J, Yamamoto M, and Xia J (2019). MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolites 9, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa F, Wolfson ML, Valchi P, Aisemberg J, and Franchi AM (2016). Endocannabinoid system and pregnancy. Reproduction 152, R191–R200. [DOI] [PubMed] [Google Scholar]
- Darmady JM, and Postle AD (1982). Lipid metabolism in pregnancy. Br. J. Obstet. Gynaecol 89, 211–215. [DOI] [PubMed] [Google Scholar]
- Ebert SN, and Baker CN (2013). Development of aerobic metabolism in utero: Requirement for mitochondrial function during embryonic and fetal periods. OA Biotechnol. 2, 16. [Google Scholar]
- Elliott JA (1975). The effect of pregnancy on the control of lipolysis in fat cells isolated from human adipose tissue. Eur. J. Clin. Invest 5, 159–163. [DOI] [PubMed] [Google Scholar]
- Fang L, Zhang M, Li Y, Liu Y, Cui Q, and Wang N (2016). PPARgene: A Database of Experimentally Verified and Computationally Predicted PPAR Target Genes. PPAR Res. 2016, 6042162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freinkel N (1980). Banting Lecture 1980. Of pregnancy and progeny. Diabetes 29, 1023–1035. [DOI] [PubMed] [Google Scholar]
- Girard JR, Ferré P, Gilbert M, Kervran A, Assan R, and Marliss EB (1977). Fetal metabolic response to maternal fasting in the rat. Am. J. Physiol 232, E456–E463. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hurtado E, Lee J, Choi J, Selen Alpergin ES, Collins SL, Horton MR, and Wolfgang MJ (2017). The loss of macrophage fatty acid oxidation does not potentiate systemic metabolic dysfunction. Am. J. Physiol Endocrinol Metab 312, E381–E393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hurtado E, Lee J, Choi J, and Wolfgang MJ (2018). Fatty acid oxidation is required for active and quiescent brown adipose tissue maintenance and thermogenic programing. Mol. Metab 7, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray LR, Sultana MR, Rauckhorst AJ, Oonthonpan L, Tompkins SC, Sharma A, Fu X, Miao R, Pewa AD, Brown KS, et al. (2015). Hepatic Mitochondrial Pyruvate Carrier 1 Is Required for Efficient Regulation of Gluconeogenesis and Whole-Body Glucose Homeostasis. Cell Metab. 22, 669–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty P (2010). Fatty acid supply to the human fetus. Annu. Rev. Nutr 30, 237–255. [DOI] [PubMed] [Google Scholar]
- Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, and Martinou JC (2012). Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Kim SP, Li Z, Zoch ML, Frey JL, Bowman CE, Kushwaha P, Ryan KA, Goh BC, Scafidi S, Pickett JE, et al. (2017). Fatty acid oxidation by the osteoblast is required for normal bone acquisition in a sex- and diet-dependent manner. JCI Insight 2, e92704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopp RH, Herrera E, and Freinkel N (1970). Carbohydrate metabolism in pregnancy. 8. Metabolism of adipose tissue isolated from fed and fasted pregnant rats during late gestation. J. Clin. Invest 49, 1438–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ellis JM, and Wolfgang MJ (2015). Adipose fatty acid oxidation is required for thermogenesis and potentiates oxidative stress-induced inflammation. Cell Rep. 10, 266–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Choi J, Aja S, Scafidi S, and Wolfgang MJ (2016a). Loss of Adipose Fatty Acid Oxidation Does Not Potentiate Obesity at Thermoneutrality. Cell Rep. 14, 1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Choi J, Scafidi S, and Wolfgang MJ (2016b). Hepatic Fatty Acid Oxidation Restrains Systemic Catabolism during Starvation. Cell Rep. 16, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Choi J, Selen Alpergin ES, Zhao L, Hartung T, Scafidi S, Riddle RC, and Wolfgang MJ (2017). Loss of Hepatic Mitochondrial Long-Chain Fatty Acid Oxidation Confers Resistance to Diet-Induced Obesity and Glucose Intolerance. Cell Rep. 20, 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, and Gonzalez FJ (1995). Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell. Biol 15, 3012–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Li Y, Han G, Li X, Ji Y, Fan Z, Zhong Y, Cao J, Zhao J, Mariusz G, et al. (2016). Establishment of mitochondrial pyruvate carrier 1 (MPC1) gene knockout mice with preliminary gene function analyses. Oncotarget 7, 79981–79994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggins GC (1968). Premature parturition after infusion of corticotrophin or cortisol into foetal lambs. J. Endocrinol 42, 323–329. [DOI] [PubMed] [Google Scholar]
- Liggins GC, Kennedy PC, and Holm LW (1967). Failure of initiation of parturition after electrocoagulation of the pituitary of the fetal lamb. Am. J. Obstet. Gynecol 98, 1080–1086. [DOI] [PubMed] [Google Scholar]
- McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, Kletzien RF, Burgess SC, and Finck BN (2015). Loss of Mitochondrial Pyruvate Carrier 2 in the Liver Leads to Defects in Gluconeogenesis and Compensation via Pyruvate-Alanine Cycling. Cell Metab. 22, 682–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger BE, Ravnikar V, Vileisis RA, and Freinkel N (1982). “Accelerated starvation” and the skipped breakfast in late normal pregnancy. Lancet 1, 588–592. [DOI] [PubMed] [Google Scholar]
- Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, and Finkel T (2016). Fatty acid oxidation in macrophage polarization. Nat. Immunol 17, 216–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, and Magnuson MA (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem 274, 305–315. [DOI] [PubMed] [Google Scholar]
- Rando G, Tan CK, Khaled N, Montagner A, Leuenberger N, Bertrand-Michel J, Paramalingam E, Guillou H, and Wahli W (2016). Glucocorticoid receptor-PPARα axis in fetal mouse liver prepares neonates for milk lipid catabolism. eLife 5, e11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandlers Y, Moser AB, Hubbard WC, Kratz LE, Jones RO, and Raymond GV (2012). Combined extraction of acyl carnitines and 26:0 lysophosphatidylcholine from dried blood spots: prospective newborn screening for X-linked adrenoleukodystrophy. Mol. Genet. Metab 105, 416–420. [DOI] [PubMed] [Google Scholar]
- Sussman D, Ellegood J, and Henkelman M (2013). A gestational ketogenic diet alters maternal metabolic status as well as offspring physiological growth and brain structure in the neonatal mouse. BMC Pregnancy Childbirth 13, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderperre B, Herzig S, Krznar P, Hörl M, Ammar Z, Montessuit S, Pierredon S, Zamboni N, and Martinou JC (2016). Embryonic Lethality of Mitochondrial Pyruvate Carrier 1 Deficient Mouse Can Be Rescued by a Ketogenic Diet. PLoS Genet. 12, e1006056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita H, Shao J, and Friedman JE (2000). Physiologic and molecular alterations in carbohydrate metabolism during pregnancy and gestational diabetes mellitus. Clin. Obstet. Gynecol 43, 87–98. [DOI] [PubMed] [Google Scholar]
- Zou S, Lang T, Zhang B, Huang K, Gong L, Luo H, Xu W, and He X (2018). Fatty acid oxidation alleviates the energy deficiency caused by the loss of MPC1 in MPC1+/− mice. Biochem. Biophys. Res. Commun 495, 1008–1013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession Numbers
The Gene Expression Omnibus (GEO) accession number for the RNA-seq data reported in this paper is GSE129368.